")
Back to Journals » Drug Design, Development and Therapy » Volume 10
Selective use of sorafenib in the treatment of thyroid cancer
Authors Pitoia F, Jerkovich F
Received 17 October 2015
Accepted for publication 22 February 2016
Published 11 March 2016 Volume 2016:10 Pages 1119—1131
DOI https://doi.org/10.2147/DDDT.S82972
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Wei Duan
Fabián Pitoia, Fernando Jerkovich
Division of Endocrinology, Hospital de Clinicas – University of Buenos Aires, Buenos Aires, Argentina
Abstract: Sorafenib is a multiple kinase inhibitor (MKI) approved for the treatment of primary advanced renal cell carcinoma and advanced primary liver cancer. It was recently approved by several health agencies around the world as the first available MKI treatment for radioactive iodine-refractory advanced and progressive differentiated thyroid cancer. Sorafenib targets C-RAF, B-RAF, VEGF receptor-1, -2, -3, PDGF receptor-β, RET, c-kit, and Flt-3. As a multifunctional inhibitor, sorafenib has the potential of inhibiting tumor growth, progression, metastasis, and angiogenesis and downregulating mechanisms that protect tumors from apoptosis and has shown to increase the progression-free survival in several Phase II trials. This led to the Phase III trial (DECISION) which showed that there was an improvement in progression-free survival of 5 months for patients on sorafenib when compared to those on placebo. Adverse events with this drug are common but usually manageable. The development of resistance after 1 or 2 years is almost a rule in most patients who showed partial response or stabilization of the disease while on sorafenib, which makes it necessary to think of a plan for subsequent therapies. These may include the use of another MKI, such as lenvatinib, the second approved MKI for advanced differentiated thyroid cancer, or include patients in clinical trials or the off-label use of other MKIs. Given sorafenib’s earlier approval, most centers now have access to its prescription. The goal of this review was to improve the care of these patients by describing key aspects that all prescribers will need to master in order to optimize outcomes.
Keywords: multiple kinase inhibitor, differentiated thyroid cancer, progression-free survival, radioiodine
Introduction
Differentiated thyroid cancer (DTC) includes the papillary, follicular, and poorly differentiated histological types. Its incidence has raised rapidly worldwide, especially in women in the last three decades.1 Long-term survival of patients with DTC is usually excellent. A good prognosis, considering mortality and recurrence, generally applies to patients younger than 60 years old with no local gross extension and no distant metastases.2 Most of these patients may experience 10-year survival rates as high as 85%.2,3 Generally, patients with a diagnosis of a DTC undergo surgical treatment of the primary tumor.4 The extension of the surgery (hemithyroidectomy, total thyroidectomy, or total thyroidectomy associated to lymph node dissection) may vary according to the presurgical risk of recurrence. The same situation occurs with radioactive iodine (RAI) administration after total thyroidectomy. Currently, there is a selective approach for the use of remnant ablation: it is not routinely indicated in low-risk patients, may be considered in intermediate-risk patients, and is an absolute indication in those with high risk of recurrence.4
Local or distant metastases can occur in nearly 10% of patients with DTC; for these cases, there are multiple therapeutic options that may include the use of several RAI doses administered consecutively, metastasectomy, and/or the use of external beam radiotherapy, among other therapeutic modalities.4 Despite these treatments, between one-third to two-thirds of patients with metastatic DTC will become RAI-refractory.5,6 RAI-refractoriness represents <5% of patients with clinical thyroid cancer. This subgroup of patients has a poor overall prognosis, with 10-year survival rates of only 10% and a median survival from the discovery of metastases of only 3–5 years.5–7 The American Thyroid Association, among other societies in the world, currently provides recommendations to include patients with advanced, progressive, and RAI-refractory DTC as candidates for being treated with multiple kinase inhibitors (MKIs).4,8,9 Until now, US Food and Drug Administration and European Medicines Agency (among other health agencies around the world) approved only two drugs, sorafenib and lenvatinib, for this indication,4 but the practical details of managing these patients often vary between physicians and countries.10 Given its earlier approval, most centers will have access to sorafenib, and now, 2 years later, we aim to improve the care of these patients by describing key aspects that all prescribers will need to master in order to optimize patient outcomes.
RAI-refractory status and defining which patients are candidates for systemic treatment
First, it is important to reach a common definition of 131I-refractory disease, both for clinical trials and in community practice. To some degree, a consensus has been evolving over the past 5 years.11 At the least, 131I-refractory disease should be defined as the presence of a lesion that does not uptake 131I detected at imaging or clinical evidence that 131I is no longer beneficial as in the case of progression despite visible uptake or, for example, progression despite a cumulative activity >600 mCi.4,5,12,13 In all cases, tumor progression has been defined according to Response Evaluation Criteria in Solid Tumors (RECIST)14 occurring within ~12–14 months, and so active surveillance imaging should be performed every 3–12 months according to the knowledge of the activity of a patient’s disease.10,13 On occasion, RAI resistance might be difficult to define when mixed response is present, but progressing RAI-refractory lesions that are not amenable for local treatment (due to location or multiple locations) should always be considered for systemic therapy.
Molecular biology of DTC: pathways involved in tumor aggressiveness and progression
In the last three decades, there has been an increase in our understanding of the impact of somatic gene alterations in the outcome of patients with DTC. Most of these genetic rearrangements and mutations have an impact on tumor initiation but not on tumor progression.15–18 RET/PTC rearrangements were one of the first genetic alterations described in DTC.14,19 The RET/PTC 1 and RET/PTC 3 are the most common rearrangements found, being RET/PTC 1 characteristic of young patients and associated with a high frequency of lymph node metastasis.15,19 On the other side, RET/PTC 3 rearrangement is more prevalent in childhood, and it is associated with a past history of radiation exposure.15,19 RAS oncogenes codify for three proteins (H-, K-, and N-RAS). Point mutations in codons 12 or 61 are the most commonly found in DTC.20,21 Constitutive activation of this protein increases thyroid cell proliferation and decreases the expression of thyroglobulin, thyroperoxidase, and NIS protein.20,21 RAS mutations are found with similar frequency in thyroid adenomas, DTC, and anaplastic tumors.20,22 Nearly 15%–20% of papillary thyroid tumors may present RAS oncogene mutations, mainly those with follicular variant, which are also encapsulated with a low frequency of lymph node metastasis.23 Recently, a high prevalence of this mutation was shown in patients with DTC and RAI-avid distant metastasis.24 Despite a seemingly preserved ability to concentrate iodine, RAI seems to be ineffective in achieving cure in most patients with RAI-avid metastatic DTC and RAS-mutant disease.24 However, the presence of RAS mutation might be a predictor of MKI redifferentiation therapy with selumetinib in patients with RAI-refractory DTC.25 The association between RAS mutations and a more aggressive behavior of DTC has also been described.26
B-RAF mutations are found in 30%–70% of patients with papillary thyroid cancer (PTC). There is a hot spot T1796A that generates a substitution from valine to glutamate at residue 600 (V600E).27,28 Most of the studies published thus far show that tumors harboring a B-RAF mutation have higher rates of extrathyroidal extension, higher frequency of lymph node metastasis, higher frequency of structural recurrences, and lower RAI uptake.29–34 It was also described that B-RAF mutation may present with a heterogeneous distribution in the same tumor.35–38 The outcome in the long-term follow-up of B-RAF-positive patients may be more related to the percentage of allelic mutations in a given tumor than the only presence of B-RAF positivity.35–38
The phosphoinositide 3-kinase (PI3K) pathway regulates growth, motility, and survival of cells. Activating mutations of PI3K are almost exclusive of follicular thyroid cancer (FTC) and anaplastic thyroid cancer (ATC). However, the amplification of PI3K pathway may be observed in 13% of follicular adenomas, 16% of PTC, 30% of FTC, and 50% of ATC.39,40
Recently, telomerase reverse transcriptase gene (TERT), which plays an important role in the immortality of cells keeping the telomere length at the end of the chromosomes, was found to be overexpressed in multiple tumors, including DTC. Also, somatic point mutations were found that were shown to increase the telomerase activity. TERT mutations are found in 11% of FTC and 16%–40% of PTC (and frequently associated to B-RAF mutations).41–43 The presence of TERT overexpression/mutations has been related to more aggressive tumors when associated with B-RAF mutations. These patients might be considered as presenting a high risk of recurrence of the disease.4 Finally, vascular endothelial growth factor (VEGF) is overexpressed in both tumor cells and the blood vessels within tumors; its main receptor VEGFR-2 is generally upregulated in DTC, and it is implicated in neoplastic growth, progression, and aggressiveness.44 This is likely the primary target of many of the MKIs in use to treat RAI-refractory DTC today.
Sorafenib: pharmacology and pharmacokinetics
Sorafenib is a MKI approved for the treatment of primary kidney cancer (advanced renal cell carcinoma), advanced primary liver cancer (hepatocellular carcinoma), and advanced and progressive DTC.45–47 Sorafenib targets C-RAF, B-RAF, VEGF receptor (VEGFR)-1, -2, -3, PDGF receptor (PDGFR)-β, RET, c-kit, and Flt-3.48–51 As a multifunctional inhibitor, sorafenib has the potential of inhibiting tumor growth, progression, metastasis, and angiogenesis, as well as downregulating mechanisms that protect tumors from apoptosis.48–50
Pharmacology and pharmacokinetics
Sorafenib is absorbed at a moderate rate after the first dose, and maximum concentration observed (Cmax) occurred at 2.5–12.5 hours after administration.50–52 The mean relative bioavailability of sorafenib tablets is 38%–49%, relative to an oral solution.50,51 The absolute bioavailability of sorafenib has yet to be determined.51 The bioavailability of sorafenib is reduced by ≈30% when the drug is administered with a high-fat meal (50% fat) rather than in the fasted state, but not after a moderate-fat meal (30% fat).50,51 Therefore, sorafenib should be taken without food or with a low- or moderate-fat meal.
Subsequently, after oral administration, plasma concentrations of sorafenib decrease slowly. There is no observable dose dependency in the plasma concentration–time profiles after the first dose of 100–800 mg. Substantial accumulation in plasma following multiple twice daily (bid) administrations is observed. Intake of food before dosing had no relevant impact on the pharmacokinetics of sorafenib except for slightly prolonging time of maximum concentration. Mean half-life ranged from 24 hours to 38 hours. Similar to the values observed after single dosing, area under the curve and Cmax values are highly variable following multiple doses of sorafenib bid. Multiple dosing of sorafenib for 7 days resulted in a 2.5-fold to sevenfold accumulation, compared to single-dose administration. Steady-state plasma sorafenib concentrations are achieved within 7 days, with a peak-to-trough ratio of mean concentrations <2.52,53
Sorafenib metabolism
Metabolism of sorafenib occurs mainly in the liver via cytochrome P450 (CYP) 3A4-mediated oxidation and uridine diphosphate glucuronosyltransferase 1A9-mediated glucuronidation.50,51 Sorafenib comprises ≈70%–85% of the circulating analytes at steady state. Of the eight identified sorafenib metabolites, five are present in plasma. The pyridine-N-oxide (M-2) is the major circulating metabolite in plasma, accounting for ≈9%–16% of circulating analytes at steady state. In vitro, the pyridine-N-oxide has demonstrated similar potency to that of sorafenib.50,51 Considering an oral administration of 100 mg of a solution formulation of sorafenib, 96% of the dose was recovered within 14 days, with 77% of it excreted in feces and 19% in urine as glucuronidated metabolites. Also, 51% of the dose accounted for unchanged sorafenib; it was excreted in the feces but not in urine, suggesting that biliary excretion of unchanged drug might contribute to the elimination of sorafenib.50,53
Age, sex, and race did not influence the pharmacokinetics of sorafenib. Subjects with normal renal function and those with mild, moderate, or severe renal impairment do not show any relationship between sorafenib exposure and renal function.53
Sorafenib exposure was shown to be higher in patients with DTC than in patients with renal cancer and hepatocellular carcinoma; however, the ranges overlap significantly.54 The elevated sorafenib exposure in patients with thyroid cancer did not appear to be due to CYP3A4 inhibition because plasma concentration of the sorafenib metabolite M-2 was higher in patients with thyroid cancer than in patients with renal hepatocellular carcinoma (CYP3A4 inhibition would lead to decreased M-2 levels). Elevated M-2 would also appear to preclude the levothyroxine metabolite T3 (a known CYP3A4 inhibitor) as the cause of the elevated sorafenib exposure (through effect on CYP3A4) in patients with DTC in the DECISION study, most of whom were receiving levothyroxine. The reason for increased sorafenib exposure in the DECISION study has not yet been elucidated. There was no clear, clinically relevant correlation between sorafenib exposure and either progression-free survival (PFS) or adverse effect (AE) incidence/severity in patients with DTC.54 However, lower doses of sorafenib have been commonly associated with decreased efficacy of the agent in the clinic (M Brose, personal communication, 2016).
Efficacy studies of sorafenib in patients with thyroid cancer
The first trial to suggest the efficacy of sorafenib in patients with iodine-refractory metastatic thyroid cancer was published in an abstract version in the ASCO Annual Meeting 2006.55 Later, Gupta-Abramson et al56 published a Phase II trial including 30 patients, 18 with PTC, nine with FTC, one with medullary thyroid cancer (MTC), and two with poorly differentiated and ATC. All patients received sorafenib at a dose of 400 mg orally twice a day. The median duration of treatment was 6.7 months. The authors reported partial response (PR) by RECIST criteria in seven patients (23.3%) and stable disease (SD) in 16 patients (53.3%) with a clinical benefit rate (PR + SD) of 77%. Only two patients with poorly differentiated thyroid cancer and ATC showed progressive disease (PD). Considering patients with DTC only, a median PFS of 21 months was observed with no difference between PTC and FTC. The median PFS in the entry cohort was 18 months.56
Another Phase II study57 including 41 patients with PTC (33 chemotherapy naïve) showed a PR in 15%, with a clinical benefit rate of 56%. Median PFS was 15 months. In this trial, there was no correlation between the serum Tg response and the radiological response. Ten fine-needle aspiration samples obtained before and 8 weeks after initiation of sorafenib were analyzed for levels of immunoactive VEGFR, VEGF expression, and ERK and AKT phosphorylation by immunohistochemistry. An inhibitory action on RAS-RAF kinase, as well as in the angiogenic signaling pathway, was firmly demonstrated. The impact on B-RAF mutation in predicting the MKI response could not be analyzed due to the small number of B-RAF mutation-negative patients. Regarding the use of fluorodeoxyglucose-positron emission tomography/computed tomography (18F-FDG-PET/CT) scan in the follow-up of patients taking sorafenib, it is important to highlight that this study found no clear correlation between the percentage of changes in standardized uptake value maximum (SUVmax) and tumor responses according to RECIST criteria.
In 2009, Brose et al58 showed a PFS of 21 months in 55 patients with thyroid cancer (47% PTC, 36% FTC/Hürthle cell variant, 8% MTC, 9% poorly differentiated/ATC) receiving sorafenib 400 mg twice a day. Considering 16 patients with PTC and FTC in whom genotyping of B-RAF was possible, the PFS was longer in those with B-RAF V600E mutation compared to B-RAF wild type (21 months vs 13 months, P=0.028).
The aim of the trial of Hoftijzer et al59 was to assess the reinduction of RAI uptake in 31 patients with RAI-resistant DTC by using sorafenib 800 mg bid. A 59% of clinical benefit rate was obtained, 25% achieving PR. The PFS was 14.5 months. However, a reinduction of RAI uptake was not demonstrated. In this study, patients with bone metastasis had a worse response to sorafenib treatment (P=0.004) and a shorter PFS (0.046). A specific tissue response to sorafenib therapy was assessed by Cabanillas et al,60 including 13 patients on sorafenib 800 mg bid. The objective response rates were similar to the previous reports. When analyzing the response by organ site, the reduction of target lesions was significantly greater in lungs (median change, 22%; range 38%–21%) than in lymph nodes (median change, 0%; range 18%–33%). Bone metastases were again refractory to MKI therapy. Nevertheless, the two patients who had irradiated bony metastases before initiation of targeted therapy had SD and the other two patients with bone metastasis without radiation therapy had a PR (despite having concomitant good response in their lung metastases). This observation would suggest that external beam radiation prior to MKI treatment could avoid progression of the bony target lesions. Also, the two patients with pleural effusion had a PD while on sorafenib treatment.
In the 2011 ASCO annual meeting, the first Phase II clinical trial reporting OS in patients treated with sorafenib was presented.61 This study including 55 patients (85% DTC, 9% ATC, and 6% MTC) showed a PFS of 23 months (24 months in the poorly differentiated/DTC group) and an OS of 35 months.61
Another important Phase II trial on sorafenib treatment was the study of Marotta et al,62 which included 17 progressive RAI-refractory DTC. The best response was observed in lymph nodes and lungs as previously observed60 but with a better response in the lymph nodes. The use of 18FDG-PET/CT assessment at baseline and in the early initial response was correlated with the final radiological response, contrary to a previous trial.57 Therefore, in the opinion of the authors, 18F-FDG-PET/CT could be helpful for the timely identification of nonresponding patients, in view of the fact that the early reduction in average SUVmax in patients during sorafenib treatments was greater in responding subjects.57
A long-term outcome study on sorafenib was presented by Schneider et al.63 The median follow-up period was 25 months (range 3.5–39 months) and median sorafenib treatment period was 9.2 months (range 0.1–39 months). In the first 6 months of treatment, eight patients (31%) had a PR. At the end of follow-up, four patients (15%) had PR, three (12%) showed SD, and 15 (58%) evidenced PD. The presence of a B-RAF V600E mutation was not related to disease progression. The PFS was 18 months (95% confidence interval [CI] 7–29 months), and the median overall survival (OS) was 34.5 months. The OS and PFS were shorter in patients with bone metastasis (23 months and 12 months, respectively). Similarly to other studies,56,59,60 thyroglobulin measurement during sorafenib therapy reflected the radiological response.
There are a few trials analyzing sorafenib on MTC. In 2010, Lam et al64 published a trial including 21 patients with MTC (16 sporadic and five hereditary). Only two patients achieved a PR, 18 had a SD (nine of whom had a durable SD ≥15 months), and one patient had a clinical PD but was not evaluable for RECIST response criteria. The PFS was 17.9 months for the sporadic group and was not reported in the hereditary group. Although calcitonin levels decreased in the majority of patients, neither the timing nor the degree of the calcitonin levels had correlation with the degree or duration of the objective response. Similar rate responses were observed in the study of Ahmed et al,65 which included 15 patients with MTC. In this Phase II trial, two patients reached a PR and 13 had a SD, with an OS rate at 1 year of 100% and a PFS of 93%. Different from the previous study, a correlation between the tumor markers (calcitonin and carcinoembryonic antigen levels) and the radiological response was found. Also, a reduction by >50% of the mean calcitonin level was shown in the first month of sorafenib treatment followed by a further stabilization, showing the impact of this MKI on the calcitonin gene expression. The Spanish trial of Capdevila et al66 observed a PR rate of 47% and SD rate of 40% in 15 patients with MTC treated with sorafenib 400 mg bid with a PFS of 10.5 months. These data differ from previous studies in which patients with MTC treated with sorafenib are supposed to have PR rates <15%, and the majority of the cohort is expected to show a SD. In any case, although a Phase III trial is needed, the available data suggest that sorafenib is effective in MTC and could be an option in patients in whom the approved MKIs vandetanib and cabozantinib are not available.
In July 2014, Brose et al47 published the first randomized, double-blind, placebo-controlled study (DECISION) analyzing the efficacy of a standard dose of sorafenib 800 mg in patients with RAI-refractory locally advanced or metastatic DTC. The population included 417 patients (207 in the sorafenib arm and 210 in the placebo group) with a median time of follow-up of 16.2 months (range 0.03–33.2). The PFS was 5 months longer in the group of patients on sorafenib treatment (10.8 months vs 5.8 months, hazard ratio [HR] 0.587, 95% CI 0.454–0.758; P<0.0001). The improvement in the PFS was proved independently of age, sex, geographical region, histologic subtype, site of metastasis, and tumor size. The median OS was not reached, and the OS did not differ significantly between the two arms of the study (HR 0.80, 95% CI 0.54–1.19; P=0.14). However, it is appropriate to consider that 71.4% of patients in the placebo arm crossed over to receive open-label sorafenib at progression. The clinical benefit rate was 54% (against 33.8% in the placebo group, P<0.0001), with a PR rate of 12.2% (against 0.5% in the placebo group) and a SD >6 months of 41.8% (against 33.2% in the placebo group). Similar to previously reported trials, no patients showed a complete response (disappearance of all target lesions). Another point to consider in the DECISION study is the assessment of the predictive value of the biomarkers in the management of DTC. In patients harboring both B-RAF and RAS mutations, sorafenib significantly improved the PFS in comparison with wild-type patients. However, neither B-RAF nor RAS mutation was predictive of this improvement itself due to the similar HR between the sorafenib and the placebo group for each mutational subgroup. The authors of the DECISION study did not recommend to use the biomarker analysis to select patients who are candidates for sorafenib therapy. Eventually, the researchers analyzed the thyroglobulin concentrations. In patients receiving placebo, thyroglobulin levels increased, while in all patients treated with sorafenib, thyroglobulin concentrations had an initial decrease followed by an increase, stability, or decrease according to the objective response (PD, SD, or PR, respectively). Although other trials59,62 also showed that the thyroglobulin value paralleled the radiological response to sorafenib treatment, Brose et al47 concluded that the use of this tumor marker to monitor the MKI treatment is not well established yet.
In all the previously mentioned studies, sorafenib was administered at a starting dose of 400 mg twice a day. Considering the AEs of the MKI, it is important to highlight attempts to use lower doses. Dadu et al67 performed a study that aimed to compare 51 patients receiving sorafenib at a starting dose of 800 mg with 24 patients with a starting dose <800 mg/d. The initial dose in the latter group was 400 mg/d in almost all patients, except for one who started sorafenib at 200 mg/d. The efficacy was assessed by median time to progression (time from the start of treatment to discontinuation because of progression), which is similar to the PFS. The time to progression was not statistically different between the two groups: 11 months in group 1 and 8 months in group 2 (P=0.354). Chen et al68 enrolled nine patients treated with sorafenib 200 mg twice a day. Of them, 33% showed a PR and 44% had a SD. The mean PFS was 10.5 months. Regarding that the mean administered dose in the sorafenib arm in the DECISION study was 651 mg/d revealing the frequent need of dose reductions, further studies clarifying this point would be of great value.
In 2014, Shen et al69 published the first meta-analysis on sorafenib treatment of patients with RAI-resistant DTC. The publication of this research was done before the DECISION study. The seven trials included those previously cited in this review56,57,59,62,63,65,66 and involved 211 patients. This group of patients showed a PR rate of 22% (range 15%–33%), a SD rate of 52% (range 41%–82%), a PFS of 12.4 months (range 4.5–19.6 months), and an OS between 10 months and 37.5 months. Any complete response was reported. A more recent meta-analysis70 was published analyzing MKI in patients with all histologic subtypes of thyroid cancer. This work included the majority of clinical trials that participate in the meta-analysis of Shen et al69 and incorporated the DECISION Phase III clinical trial. Among patients with DTC treated with sorafenib, the authors reported a PR rate of 17% and a clinical benefit rate of 53%.70
More recently, additional published studies have shown similar response rates as previously reported.71,72 In Argentina, Pitoia73 published his clinical experience in eight patients with DTC who received off-label sorafenib. One patient exhibited a maintained PR for 16 months, five patients had a SD for 8±3 months, and two patients showed SD for 8 months and 12 months, respectively, and then have a PD at final follow-up.73
Currently, there is an ongoing observational study to understand the use of sorafenib in the clinical setting after approval.74 The results of the clinical trials mentioned earlier are summarized in Table 1.
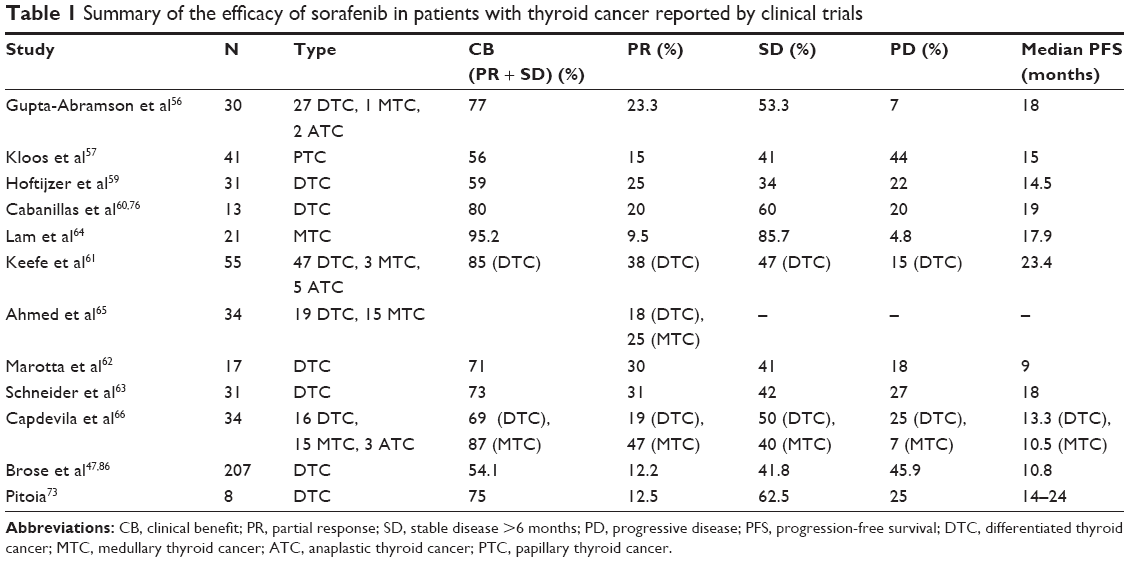 | Table 1 Summary of the efficacy of sorafenib in patients with thyroid cancer reported by clinical trials |
In November 2013, sorafenib was approved for the treatment of refractory advanced DTC by the US Food and Drug Administration75 and by the European Medicines Agency in May 2014.
A few trials evaluated the combination of sorafenib with another targeted agent. Cabanillas et al76 and Hong et al77 treated patients with advanced thyroid cancer with a combination of sorafenib and tipifarnib, a farnesyltransferase inhibitor that inactivates RAS, showing similar tumor responses as with sorafenib alone.76,77 A combined therapy with sorafenib and an mammalian target of rapamycin (mTOR) inhibitor was suggested by Sherman et al78 using temsirolimus. More recently, the same authors evaluated 38 patients with thyroid cancer (ten MTC and 28 DTC) and showed that the combination of sorafenib and everolimus, another mTOR inhibitor, achieves better clinical responses than for sorafenib alone: PR 55% and SD 37%.79 Brose et al80 enrolled 35 patients with evidence of progression by RECIST criteria on sorafenib treatment and demonstrated an additional median PFS of 13.7 months, a SD ≥6 months of 54%, and a PR of 3% adding everolimus to sorafenib.80
Safety and tolerability
In the meta-analysis of Shen et al,69 among 211 patients, the most commonly reported AEs were, in order of incidence, hand–foot syndrome (HFS) 80%, diarrhea 68%, fatigue 67%, rash 66%, weight loss 52%, and hypertension 31%. Similar results were obtained by the DECISION trial47: HFS 76.3%, diarrhea 68.6%, alopecia 67.1%, rash 50.2%, weight loss 46.9%, and hypertension 40.6%. The HFS was also the most frequent Common Terminology Criteria for Adverse Events81 grade 3 or 4 AEs (20.3%), followed by hypertension (9.7%), hypocalcemia (9.2%), weight loss (5.8%), diarrhea (5.8%), and fatigue (5.8%). Secondary malignancies were reported in 4.3%. Twelve deaths occurred in patients receiving sorafenib, only one of them was attributable to sorafenib (myocardial infarction).47 The incidence of fatal events in the meta-analysis performed by Schutz et al82 was 1.4% in the sorafenib group of patients. The most common fatal events were hemorrhage, followed by myocardial infarction, abnormal liver function or failure, sepsis, ischemic stroke, pulmonary embolism, dehydration, and sudden death.82 A by-cycle treatment analysis of the adverse events of the DECISION study performed by Worden et al83 showed that most of them had a highest incidence during the first two cycles of treatment (one cycle =28 days), increased their prevalence over the first two to six cycles, then stabilizing or declining with the exception of weight loss that tended to rise in prevalence in later cycles. A summarized list of the adverse events, rates of dose reductions, interruptions, and withdrawals of sorafenib reported in different clinical trials is presented in Table 2. The Common Terminology Criteria for Adverse Events for some of the most frequent AEs are detailed in Table 3.
 | Table 2 Adverse effects of sorafenib, fatal events, dose reductions, interruptions, and withdrawals reported in each trial |
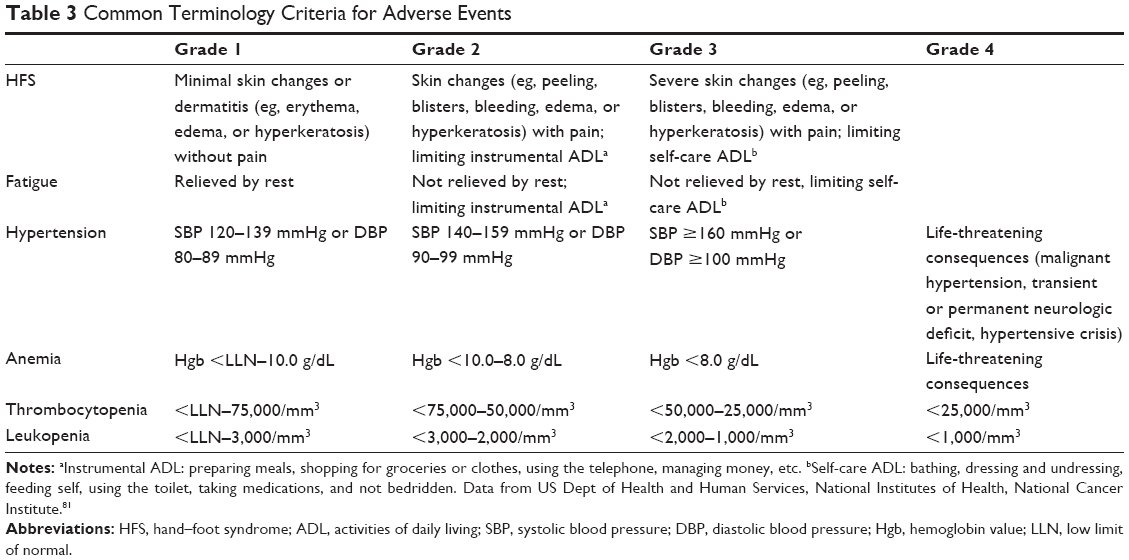 | Table 3 Common Terminology Criteria for Adverse Events |
HFS and other skin reactions
HFS was the most common reason for dose reduction (33.8%), interruption (26.6%), and withdrawal (5.3%).47 This skin reaction is characterized by hyperkeratotic plaques with erythematous borders that are most pronounced on weight-bearing areas but can also affect the dorsum of the hands, lateral slides of the soles, and the shin. The intensity varies from mild erythema to severe hyperesthesia. The median time of onset was 18.4 days since the initiation of sorafenib,84 and the severity tends to decrease after the sixth month.85 The physiopathology is attributed to inhibition of growth and repair pathways in the mentioned areas mediated by the VEGF and platelet-derived growth factor receptors (PDGFR). Management includes keeping hands and feet well hydrated, probably the use of urea-based creams, removing calluses, wearing comfortable shoes, avoiding hot water, and using pain medication as needed. With grades 1 and 2 HFS, dose reduction is not necessary. Patients having grade 2 HFS that does not improve with supportive measures, or after the prescription of ibuprofen at doses of 600 mg three times a day, or grade 3 HFS require dose reductions and short interruptions (ie, for 1 week) as seen in Figure 1. Of note, most of the investigators on DECISION were giving sorafenib for the first time. It is generally accepted that with experience, dose holidays, and earlier intervention, the incidence of grades 3 and 4 is significantly lower (~5%).86
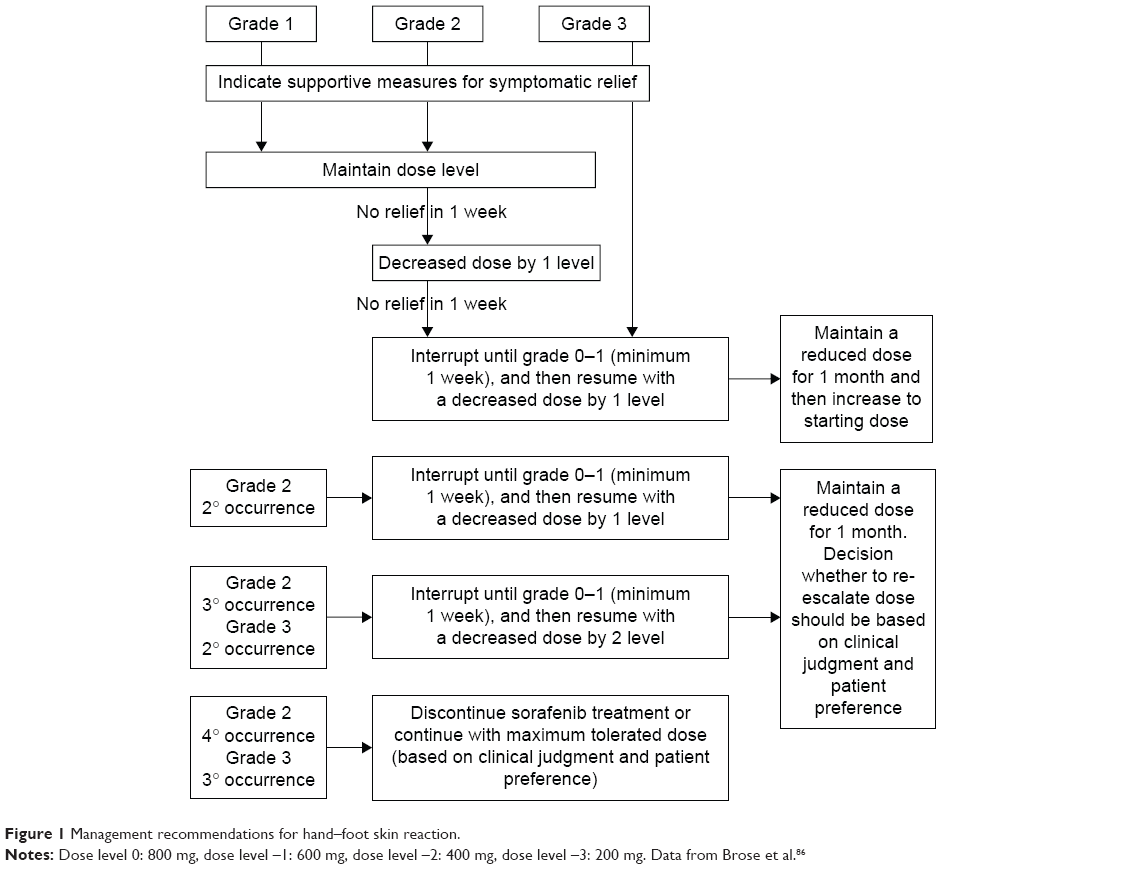 | Figure 1 Management recommendations for hand–foot skin reaction. |
A macular or papular rash over the extremities and the trunk is frequently observed during the first month of treatment and follows a clinical behavior similar to HFS. Most cases are mild and solve without specific therapy, but topical corticosteroids and antihistamines may be considered.86
Diarrhea and gastrointestinal complaints
Diarrhea is the second more common disorder related with sorafenib therapy.47 It occurs mostly as a grade 1 (increase of less than four stools per day over baseline) or grade 2 (increase of four to six stools per day over baseline) adverse event, with an incidence of grade 3 or 4 diarrhea (increase of more than seven stools per day over baseline with life-threatening consequences) of 5%–15%.69 It often begins at the third or fourth month of treatment, has a slow onset, may be worst later, and is particularly intermittent (two or three episodes per week) in patients with DTC.86 The proposed toxicity mechanisms are an effect on intestinal receptors such as c-kit expressed on interstitial cells of Cajal that regulate peristaltic movements, inhibition of VEGFR leading to a microcirculation damage, and a high concentration of the drug with direct irritation of the bowel mucosa. Grade 1 or 2 diarrhea is, in the majority of patients, successfully managed with dietary adjustments, usually accompanied by loperamide that can be used intermittently or prophylactically in schedules individualized for the patient. In case of grade 3 or 4 gastrointestinal disorders, a dose reduction or interruption until diarrhea returns to grade 1 is needed. Other gastrointestinal issues such as mucositis and dyspepsia are common in patients receiving sorafenib and are usually controlled by standard treatment.86
Fatigue
Fatigue is a common symptom in patients taking sorafenib and has a high incidence during the first months of treatment, remaining stable or in cases improving during the treatment.83 To rule out depression, decrease in physical activity and nutritional issues is recommended. This symptom tended to improve after the first few months of treatment on its own. However, ongoing fatigue can be improved with weight-bearing exercise, and additional attention to nutritional status and control of diarrhea is essential.86
Hypertension and other vascular events
The incidence of hypertension in clinical trials varies from 30% to 43%.47,56,59,63,64 It tends to appear early during the sorafenib treatment and be stable over time. In the DECISION study,47 hypertension occurred as a grade 3 AE in 9.7% of patients and in any patients as a grade 4 adverse event. Hypertension is the known result of the VEGF pathway inhibition and has been seen across MKIs that inhibit VEGFR. This results in reduction in nitric oxide and prostaglandins levels, a possible increase in endothelin-1 production, microvascular rarefaction, local thrombosis, and other endothelial insults leading to vasoconstriction, and an increase in vascular resistance.87 The VEGF blockade also limits lymphatic growth in response to salt intake causing sodium retention and increase in extracellular fluid causing a rightward shift in the chronic pressure–natriuresis relationship.87 The management of this condition requires blood pressure monitoring weekly or biweekly during the first 6 weeks of treatment to instruct the patients in changes in lifestyle and to assess other cardiovascular risks.86 The choice of the antihypertensive drug should be individualized.87 No clinical trial comparing different antihypertensive agents in patients with DTC under MKI therapy has been published. Angiotensin-converting enzyme inhibitors, calcium channel blockers,87 and beta-blockers86 have been suggested as initial therapy.
The VEGF pathway inhibition and consequently the vascular damage have been associated with the risk of developing congestive heart failure,88 proteinuria,89 hemorrhage, and cardiac infarction.90 The meta-analysis of Qi et al,90 including 10,553 patients with cancer treated with VEGFR-MKIs, showed an incidence of all-grade and high-grade congestive heart failure of 3.2% and 1.4%, respectively. On the other hand, the incidence of proteinuria was assessed in another meta-analysis89 reporting an overall incidence of 11.6% and 0.9% considering the high-grade cases. However, proteinuria was not a common adverse event as reported in the DECISION study and, therefore, may be less frequent with sorafenib.
Thyroid dysfunction
An adjustment in thyroid hormone in postthyroidectomy patients with thyroid carcinoma was required in many patients treated with sorafenib. Phase II clinical trials previously mentioned with an incidence range between 12% and 76% of the respective cohorts.56,59,62,63,65 In the DECISION trial, 33.3% of patients in the sorafenib arm showed an increase in serum thyroid-stimulating hormone.47 The hypothesis for this phenomenon includes: 1) a MCT8 inhibition, the most prominent thyroid hormone transport protein, leading to an impairment of the levothyroixine absorption in the intestine91 and/or reducing the pituitary and hypothalamic thyroid hormone feedback,91 2) an enhancement in peripheral T4 and T3 metabolism due to increase in deiodinase 3 activity,92 and 3) a reduction in thyroid-stimulating hormone clearance.93 A destruction of the thyroid gland due to capillary dysfunction related with VEGFR inhibition has been described,94 but it only applies for patients treated with sorafenib for other neoplasias such as hepatocarcinoma or renal cell carcinoma who conserved the thyroid. Transient hyperthyroidism has also been reported in this group of patients.94
Hematologic toxicities
Since VEGFR, FLT-3, and c-kit are expressed in hematopoietic precursors, bone marrow toxicity is expected when using MKIs. Schutz et al,95 in a meta-analysis for patients with nonthyroid cancer treated with sorafenib, reported an all-grade anemia, neutropenia, thrombocytopenia, and lymphopenia incidence of 43.9%, 18.0%, 25.3%, and 34.1%, respectively. The incidences of high-grade anemia, neutropenia, thrombocytopenia, and lymphopenia were 2.0%, 5.1%, 4.0%, and 13.1%, respectively.95 Alterations in blood cell count were described in Phase II and III clinical trials: anemia (range 25%–38%),59,63,64 thrombocytopenia (range 10%–29%),59,63,64 lymphocytopenia (14%), and neutropenia (33%).64 In all cases, these adverse events were grade 1 or 2 with the exception of one case of febrile neutropenia65 and one case of grade ≥3 thrombocytopenia reported in the study of Lam et al.64 Pitoia et al96 have recently published one clinical case of grade 3 thrombocytopenia, which was solved by dose reduction of sorafenib. Other alterations in serum chemistry such as hypocalcemia (range 18.8%–57%),47,59,63,64 which is more frequent and severe in patients with thyroid cancer probably due to the presence of hypoparathyroidism, and elevated transaminases levels have been reported.47,59,64
Interpretation of studies and future directions
Phase II trials of several MKIs showed antitumor activity and delayed cancer progression in patients with DTC refractory to RAI. These data led to study sorafenib in a multicenter randomized double-blind placebo-controlled Phase III trial (DECISION).47 In addition to being the first Phase III trial completed and reported in patients with DTC, the trial demonstrated improved PFS and overall response rate. The toxicity was increased compared to placebo, health-related quality of life was slightly reduced, and OS benefit was not proven.47
Although the types of AEs observed with sorafenib were similar to those seen in other cancers, a higher proportion of patients with DTC had more severe grades of toxicity. The unequivocal antitumor effects, the safety of the drug, and lack of treatment options for patients with DTC refractory to RAI support the clinical use of sorafenib.
Future directions will likely include approaches with combination therapy for patients with RAI-refractory thyroid cancer at earlier time points or for those patients who have become resistant to sorafenib. While international guidelines currently suggest to stop RAI administration in patients with metastatic disease and a cumulative activity of 600 mCi 131I,4 new published evidence suggests that pretreatment with redifferentiating agents such as selumetinib or drabrafenib might reinduce RAI uptake in patients, and PR to treatment could be observed in two-thirds of those respondent subjects.25,97 Although these are very preliminary data, when new Phase III trials with these drugs are available, we could be thinking of different steps to manage patients with RAI-resistant advanced DTC, taking as first step the indication of redifferentiating agents, and when no RAI uptake is reinduced or no response to treatment according to RECIST 1.1 criteria is observed, move to a second step of treatment where sorafenib or lenvatinib, followed by sequential MKIs or combinations of drugs, depending on the personal experience and availability in each region.
Conclusion
Not long ago, there were no effective approved treatments for patients with RAI-refractory progressive DTC. Sorafenib is the first MKI approved for the treatment of these patients, and it has shown its efficacy against DTC in numerous clinical trials, resulting mainly in disease stabilization. Over the past few years, we have made great strides in the optimal use of sorafenib as well as other MKIs for the treatment of our patients. Together, these new agents will surely result in a more individualized approach in patients with RAI-resistant DTC, which will improve their prognosis and may in time be shown to increase the OS.
Acknowledgment
The authors would like to thank Marcia Brose for the critical reading of this manuscript.
Disclosure
Fabián Pitoia is a consultant for Bayer. Fernando Jerkovich reports no conflicts of interest in this work.
References
Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61(2):69–90. | ||
Hundahl SA, Fleming ID, Fremgen AM, Menck HR. A National Cancer Data Base report on 53,856 cases of thyroid carcinoma treated in the U.S., 1985–1995. Cancer. 1998;83(12):2638–2648. | ||
Eustatia-Rutten CF, Corssmit EP, Biermasz NR, Pereira AM, Romijn JA, Smit JW. Survival and death causes in differentiated thyroid carcinoma. J Clin Endocrinol Metab. 2006;91(1):313–319. | ||
Haugen BR, Alexander EK, Bible KC, et al. 2015 American Thyroid Association Management Guidelines for adult patients with thyroid nodules and differentiated thyroid cancer. Thyroid. 2016;26:1–133. | ||
Durante C, Haddy N, Baudin E, et al. Long-term outcome of 444 patients with distant metastases from papillary and follicular thyroid carcinoma: benefits and limits of radioiodine therapy. J Clin Endocrinol Metab. 2006;91(8):2892–2899. | ||
Hodak SP, Carty SE. Radioiodine-resistant differentiated thyroid cancer: hope for the future. Oncology. 2009;23(9):775–776. | ||
Robbins RJ, Wan Q, Grewal RK, et al. Real-time prognosis for metastatic thyroid carcinoma based on 2-[18F]fluoro-2-deoxyd-glucose-positron emission tomography scanning. J Clin Endocrinol Metab. 2006;91(2):498–505. | ||
American Thyroid Association (ATA) Guidelines Taskforce on Thyroid Nodules and Differentiated Thyroid Cancer, Cooper DS, Doherty GM, et al. Revised American Thyroid Association Management Guidelines for patients with thyroid nodules and differentiated thyroid cancer. Thyroid. 2009;19(11):1167–1214. | ||
Pitoia F, Ward L, Wohllk N, et al. Recommendations of the Latin American Thyroid Society on diagnosis and management of differentiated thyroid cancer. Arq Bras Endocrinol Metabol. 2009;53(7):884–887. | ||
Brose MS, Smit J, Capdevila J, et al. Approach to the patient with advanced differentiated thyroid cancer. Expert Rev Anticancer Ther. 2012;12(9):1137–1147. | ||
Schlumberger M, Brose M, Elisei R, et al. Definition and management of radioactive iodine-refractory differentiated thyroid cancer. Lancet Diabetes Endocrinol. 2014;13(2):70215–70218. | ||
Pacini F, Ito Y, Luster M, Pitoia F, Robinson B, Wirth L. Radioactive iodine-refractory differentiated thyroid cancer: unmet needs and future directions. Expert Rev Endocrinol Metab. 2012;7(5):541–554. | ||
Eisenhauer EA, Therasse P, Bogaerts J, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer. 2009;45(2):228–247. | ||
Kim A, Balis FM, Widemann BC. Sorafenib and sunitinib. Oncologist. 2009;14(8):800–805. | ||
Marotta V, Guerra A, Sapio MR, Vitale M. RET/PTC rearrangement in benign and malignant thyroid diseases: a clinical standpoint. Eur J Endocrinol. 2011;165(4):499–507. | ||
Fagin JA. Challenging dogma in thyroid cancer molecular genetics – role of RET/PTC and BRAF in tumor initiation. J Clin Endocrinol Metab. 2004;89(9):4264–4266. | ||
Santoro M, Melillo RM, Fusco A. RET/PTC activation in papillary thyroid carcinoma: European Journal of Endocrinology Prize Lecture. Eur J Endocrinol. 2006;155(5):645–653. | ||
Xing M. BRAF mutation and thyroid cancer recurrence. J Clin Oncol. 2015;33(22):2482–2483. | ||
Minoletti F, Butti MG, Coronelli S, et al. The two genes generating RET/PTC3 are localized in chromosomal band 10q11.2. Genes Chromosomes Cancer. 1994;11(1):51–57. | ||
Suarez HG, du Villard JA, Severino M, et al. Presence of mutations in all three ras genes in human thyroid tumors. Oncogene. 1990;5(4):565–570. | ||
Garcia-Rostan G, Zhao H, Camp RL, et al. Ras mutations are associated with aggressive tumor phenotypes and poor prognosis in thyroid cancer. J Clin Oncol. 2003;21(17):3226–3235. | ||
Ezzat S, Zheng L, Kolenda J, Safarian A, Freeman JL, Asa SL. Prevalence of activating ras mutations in morphologically characterized thyroid nodules. Thyroid. 1996;6(5):409–416. | ||
Vasko VV, Gaudart J, Allasia C, et al. Thyroid follicular adenomas may display features of follicular carcinoma and follicular variant of papillary carcinoma. Eur J Endocrinol. 2004;151(6):779–786. | ||
Sabra MM, Dominguez JM, Grewal RK. Clinical outcomes and molecular profile of differentiated thyroid cancers with radioiodine-avid distant metastases. J Clin Endocrinol Metab. 2013;98(5):E829–E836. | ||
Ho AL, Grewal RK, Leboeuf R. Selumetinib-enhanced radioiodine uptake in advanced thyroid cancer. N Engl J Med. 2013;368(7):623–632. | ||
Howell GM, Hodak SP, Yip L. RAS mutations in thyroid cancer. Oncologist. 2013;18(8):926–932. | ||
Kimura ET, Nikiforova MN, Zhu Z, Knauf JA, Nikiforov YE, Fagin JA. High prevalence of BRAF mutations in thyroid cancer: genetic evidence for constitutive activation of the RET/PTC-RAS-BRAF signaling pathway in papillary thyroid carcinoma. Cancer Res. 2003;63(7):1454–1457. | ||
Xing M. BRAF mutation in thyroid cancer. Endocr Relat Cancer. 2005;12(2):245–262. | ||
Xing M, Westra WH, Tufano RP, et al. BRAF mutation predicts a poorer clinical prognosis for papillary thyroid cancer. J Clin Endocrinol Metab. 2005;90(12):6373–6379. | ||
Xing M. BRAF mutation in papillary thyroid cancer: pathogenic role, molecular bases, and clinical implications. Endocr Rev. 2007;28(7):742–762. | ||
O’Neill CJ, Bullock M, Chou A, et al. BRAF(V600E) mutation is associated with an increased risk of nodal recurrence requiring reoperative surgery in patients with papillary thyroid cancer. Surgery. 2010;148(6):1139–1145. | ||
Wang W, Zhao W, Wang H, et al. Poorer prognosis and higher prevalence of BRAF (V600E) mutation in synchronous bilateral papillary thyroid carcinoma. Ann Surg Oncol. 2012;19(1):31–36. | ||
Grabellus F, Worm K, Schmid KW, Sheu SY. The BRAF V600E mutation in papillary thyroid carcinoma is associated with glucose transporter 1 overexpression. Thyroid. 2012;22(4):377–382. | ||
Joo JY, Park JY, Yoon YH, et al. Prediction of occult central lymph node metastasis in papillary thyroid carcinoma by preoperative BRAF analysis using fine-needle aspiration biopsy: a prospective study. J Clin Endocrinol Metab. 2012;97(11):3996–4003. | ||
Xing M. BRAFV600E mutation and papillary thyroid cancer: chicken or egg? J Clin Endocrinol Metab. 2012;97(7):2295–2298. | ||
Guerra A, Sapio MR, Marotta V, et al. The primary occurrence of BRAF(V600E) is a rare clonal event in papillary thyroid carcinoma. J Clin Endocrinol Metab. 2012;97(2):517–524. | ||
Guerra A, Fugazzola L, Marotta V, et al. A high percentage of BRAFV600E alleles in papillary thyroid carcinoma predicts a poorer outcome. J Clin Endocrinol Metab. 2012;97(7):2333–2340. | ||
de Biase D, Cesari V, Visani M, et al. High-sensitivity BRAF mutation analysis: BRAF V600E is acquired early during tumor development but is heterogeneously distributed in a subset of papillary thyroid carcinomas. J Clin Endocrinol Metab. 2014;99(8):E1530–E1538. | ||
Saji M, Ringel MD. The PI3K-Akt-mTOR pathway in initiation and progression of thyroid tumors. Mol Cell Endocrinol. 2010;321(1):20–28. | ||
Ricarte-Filho JC, Ryder M, Chitale DA, et al. Mutational profile of advanced primary and metastatic radioactive iodine-refractory thyroid cancers reveals distinct pathogenetic roles for BRAF, PIK3CA, and AKT1. Cancer Res. 2009;69(11):4885–4893. | ||
Landa I, Ganly I, Chan TA, et al. Frequent somatic TERT promoter mutations in thyroid cancer: higher prevalence in advanced forms of the disease. J Clin Endocrinol Metab. 2013;98(9):E1562–E1566. | ||
Liu X, Qu S, Liu R, et al. TERT promoter mutations and their association with BRAF V600E mutation and aggressive clinicopathological characteristics of thyroid cancer. J Clin Endocrinol Metab. 2014;99(6):E1130–E1136. | ||
Malaguarnera R, Chen KY, Kim TY, et al. Switch in signaling control of mTORC1 activity after oncoprotein expression in thyroid cancer cell lines. J Clin Endocrinol Metab. 2014;99(10):E1976–E1987. | ||
Bunone G, Vigneri P, Mariani L, et al. Expression of angiogenesis stimulators and inhibitors in human thyroid tumors and correlation with clinical pathological features. Am J Pathol. 1999;155:1967–1976. | ||
Escudier B, Eisen T, Stadler WM, et al. Sorafenib for treatment of renal cell carcinoma: final efficacy and safety results of the phase III treatment approaches in renal cancer global evaluation trial. J Clin Oncol. 2009;27(20):3312–3318. | ||
Llovet JM, Ricci S, Mazzaferro V, et al; SHARP Investigators Study Group. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359(4):378–390. | ||
Brose MS, Nutting CM, Jarzab B, et al; DECISION investigators. Sorafenib in radioactive iodine-refractory, locally advanced or metastatic differentiated thyroid cancer: a randomised, double-blind, phase 3 trial. Lancet. 2014;384(9940):319–328. | ||
Adnane L, Trail PA, Taylor I, Wilhelm SM. Sorafenib (BAY 43-9006, Nexavar), a dual-action inhibitor that targets RAF/MEK/ERK pathway in tumor cells and tyrosine kinases VEGFR/PDGFR in tumor vasculature. Methods Enzymol. 2006;407:597–612. | ||
Wilhelm SM, Carter C, Tang L, et al. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 2004;64(19):7099–7109. | ||
Bayer HealthCare Pharmaceuticals Inc [webpage on the Internet]. Prescribing Information for Sorafenib (Nexavar). USA: Bayer HealthCare Pharmaceuticals Inc; 2011. Available from: http://www.accessdata.fda.gov/drugsatfda_docs/label/2013/021923s016lbl.pdf. Accessed September 14, 2015. | ||
European Medicines Agency [homepage on the internet]. Nexavar (Sorafenib): Summary of Product Characteristics. United Kingdom: European Medicines Agency; 2014. Available from: http://www.ema.europa.eu. Accessed September 14, 2015. | ||
Strumberg D, Richly H, Hilger RA, et al. Phase I clinical and pharmacokinetic study of the novel Raf kinase and vascular endothelial growth factor receptor inhibitor BAY 43-9006 in patients with advanced refractory solid tumors. J Clin Oncol. 2005;23(5):965–972. | ||
Fallahi P, Ferrari SM, Santini F, et al. Sorafenib and thyroid cancer. BioDrugs. 2013;27(6):615–628. | ||
Bastholt L, Brose MS, Jarzab B, et al. Population PK modeling and exposure-response analyses of sorafenib in patients with radioactive iodine-refractory differentiated thyroid cancer (RAI-rDTC) in the phase III DECISION trial. J Clin Oncol. 2014;32(suppl 5):6061. | ||
Kloos RMR, Knopp M, Heverhagen J, et al. Significant clinical and biologic activity of RAF/VEGF-R kinase inhibitor BAY 43-9006 in patients with metastatic papillary thyroid carcinoma (PTC): updated results of a phase II study. J Clin Oncol. 2006;24(suppl 288):5534. | ||
Gupta-Abramson V, Troxel AB, Nellore A, et al. Phase II trial of sorafenib in advanced thyroid cancer. J Clin Oncol. 2008;26(29):4714–4719. | ||
Kloos RT, Ringel MD, Knopp MV, et al. Phase II trial of sorafenib in metastatic thyroid cancer. J Clin Oncol. 2009;27(10):1675–1684. | ||
Brose MS, Troxel AB, Redlinger M, et al. Effect of BRAFV600E on response to sorafenib in advanced thyroid cancer patients. J Clin Oncol. 2009;27(suppl 15):6002. | ||
Hoftijzer H, Heemstra KA, Morreau H, et al. Beneficial effects of sorafenib on tumor progression, but not on radioiodine uptake, in patients with differentiated thyroid carcinoma. Eur J Endocrinol. 2009;161(6):923–931. | ||
Cabanillas ME, Waguespack SG, Bronstein Y, et al. Treatment with tyrosine kinase inhibitors for patients with differentiated thyroid cancer: the M.D. Anderson experience. J Clin Endocrinol Metab. 2010;95(6):2588–2595. | ||
Keefe SM, Troxel AB, Rhee S, et al. Phase II trial of sorafenib in patients with advanced thyroid cancer. J Clin Oncol. 2011;29(suppl 15):5562. | ||
Marotta V, Ramando V, Camera L, et al. Sorafenib in advanced iodine-refractory differentiated thyroid cancer: efficacy, safety and exploratory analysis of role of serum thyroglobulin and FDG-PET. Clin Endocrinol (Oxf). 2012;78(5):760–767. | ||
Schneider TC, Abdulrahman RM, Corssmit EP, Morreau H, Smit JW, Kapiteijn E. Long-term analysis of the efficacy and tolerability of sorafenib in advanced radio-iodine refractory differentiated thyroid carcinoma: final results of a phase II trial. Eur J Endocrinol. 2012;167(5):643–650. | ||
Lam ET, Ringel MD, Kloos RT, et al. Phase II clinical trial of sorafenib in metastatic medullary thyroid cancer. J Clin Oncol. 2010;28(14):2323–2330. | ||
Ahmed M, Barbachano Y, Riddell A, et al. Analysis of the efficacy and toxicity of sorafenib in thyroid cancer: a phase II study in a UK based population. Eur J Endocrinol. 2011;165(2):315–322. | ||
Capdevila J, Iglesias L, Halperin I, et al. Sorafenib in metastatic thyroid cancer. Endocr Relat Cancer. 2012;19(2):209–216. | ||
Dadu R, Waguespack SG, Sherman SI, et al. Efficacy and tolerability of different starting doses of sorafenib in patients with differentiated thyroid cancer. Oncologist. 2014;19(5):477–482. | ||
Chen L, Shen Y, Luo Q, Yu Y, Lu H, Zhu R. Response to sorafenib at a low dose in patients with radioiodine-refractory pulmonary metastases from papillary thyroid carcinoma. Thyroid. 2011;21(2):119–124. | ||
Shen CT, Qiu ZL, Luo QY. Sorafenib in the treatment of radioiodine-refractory differentiated thyroid cancer: a meta-analysis. Endocr Relat Cancer. 2014;21(2):253–261. | ||
Klein Hesselink EN, Steenvoorden D, Kapiteijn E, et al. Therapy of endocrine disease: response and toxicity of small-molecule tyrosine kinase inhibitors in patients with thyroid carcinoma: a systematic review and meta-analysis. Eur J Endocrinol. 2015;172(5):R215–R225. | ||
Massicotte MH, Brassard M, Claude-Desroches M, et al. Tyrosine kinase inhibitor treatments in patients with metastatic thyroid carcinomas: a retrospective study of the TUTHYREF network. Eur J Endocrinol. 2014;170(4):575–582. | ||
Gallo M, Michelon F, Castiglione A, et al. Sorafenib treatment of radioiodine-refractory advanced thyroid cancer in daily clinical practice: a cohort study from a single center. Endocrine. 2015;49(3):726–734. | ||
Pitoia F. Response to sorafenib treatment in advanced metastatic thyroid cancer. Arq Bras Endocrinol Metabol. 2014;58(1):37–41. | ||
Clinicaltrials.gov [webpage on the Internet]. Nexavar® versus Placebo in Locally Advanced/Metastatic RAI-Refractory Differentiated Thyroid Cancer. USA: National Institutes of Health; 2015. Available from: https://clinicaltrials.gov/ct2/show/NCT00984282?term=NCT00984282&rank=1. Accessed October 14, 2015. | ||
Tuttle RM, Haddad RI, Ball DW, et al. Thyroid carcinoma, version 2.2014. J Natl Compr Canc Netw. 2014;12(12):1671–1680. | ||
Cabanillas ME, Kurzrock R, Sherman SI, et al. Phase I trial of combination sorafenib and tipifarnib: the experience of advanced differentiated thyroid cancer (DTC) and medullary thyroid cancer (MTC). J Clin Oncol. 2010;28(suppl 15):5586. | ||
Hong DS, Cabanillas ME, Wheler J, et al. Inhibition of the Ras/Raf/MEK/ERK and RET kinase pathways with the combination of the multikinase inhibitor sorafenib and the farnesyltransferase inhibitor tipifarnib in medullary and differentiated thyroid malignancies. J Clin Endocrinol Metab. 2011;96(4):997–1005. | ||
Sherman EJ, Ho AL, Fury MG, et al. A phase II study of temsirolimus/sorafenib in patients with radioactive iodine (RAI)-refractory thyroid carcinoma. J Clin Oncol. 2012;30(suppl 15):5514. | ||
Sherman EJ, Ho AL, Fury MG, et al. Phase II study of everolimus and sorafenib for the treatment of metastatic thyroid cancer. J Clin Oncol. 2013;31(suppl):6024. | ||
Brose MS, Troxel AB, Yarchoan M, et al. A phase II study of everolimus (E) and sorafenib (S) in patients (PTS) with metastatic differentiated thyroid cancer who have progressed on sorafenib alone. J Clin Oncol. 2015;33(suppl):6072. | ||
US Dept of Health and Human Services, National Institutes of Health, National Cancer Institute [webpage on the Internet]. Common Terminology Criteria for Adverse Events v4.0 (CTCAE). US Dept of Health and Human Services, National Institutes of Health, National Cancer Institute; 2006. Available from: http://ctep.cancer.gov/protocolDevelopment/electronic_applications/ctc.htm. Accessed September 25, 2015. | ||
Schutz FA, Je Y, Richards CJ, Choueiri TK. Meta-analysis of randomized controlled trials for the incidence and risk of treatment-related mortality in patients with cancer treated with vascular endothelial growth factor tyrosine kinase inhibitors. J Clin Oncol. 2012;30(8):871–877. | ||
Worden F, Fassnacht M, Shi Y, et al. Safety and tolerability of sorafenib in patients with radioiodine-refractory thyroid cancer. Endocr Relat Cancer. 2015;22(6):877–887. | ||
Lee WJ, Lee JL, Chang SE, et al. Cutaneous adverse effects in patients treated with the multitargeted kinase inhibitors sorafenib and sunitinib. Br J Dermatol. 2009;161(5):1045–1051. | ||
Flaherty KT, Brose MS. Sorafenib-related hand-foot skin reaction improves, not worsens, with continued treatment. Clin Cancer Res. 2009;15(24):7749. | ||
Brose MS, Frenette CT, Keefe SM, Stein SM. Management of sorafenib-related adverse events: a clinician’s perspective. Semin Oncol. 2014;41(suppl 2):S1–S16. | ||
Robinson ES, Khankin EV, Karumanchi SA, Humphreys BD. Hypertension induced by vascular endothelial growth factor signaling pathway inhibition: mechanisms and potential use as a biomarker. Semin Nephrol. 2010;30(6):591–601. | ||
Kamba T, McDonald DM. Mechanisms of adverse effects of anti-VEGF therapy for cancer. Br J Cancer. 2007;96(12):1788–1795. | ||
Zhang ZF, Wang T, Liu LH, Guo HQ. Risks of proteinuria associated with vascular endothelial growth factor receptor tyrosine kinase inhibitors in cancer patients: a systematic review and meta-analysis. PLoS One. 2014;9(3):e90135. | ||
Qi W, Shen Z, Tang L, Yao Y. Congestive heart failure risk in cancer patients treated with VEGFR-MKIs: a systematic review and meta-analysis of 36 clinical trials. Br J Clin Pharmacol. 2014;78(4):748–762. | ||
Braun D, Kim TD, le Coutre P, Köhrle J, Hershman JM, Schweizer U. Tyrosine kinase inhibitors noncompetitively inhibit MCT8-mediated iodothyronine transport. J Clin Endocrinol Metab. 2012;97(1):E100–E105. | ||
Abdulrahman RM, Verloop H, Hoftijzer H, et al. Sorafenib-induced hypothyroidism is associated with increased type 3 deiodination. J Clin Endocrinol Metab. 2010;95(8):3758–3762. | ||
Verloop H, Smit JW, Dekkers OM. Sorafenib therapy decreases the clearance of thyrotropin. Eur J Endocrinol. 2013;168(2):163–167. | ||
Illouz F, Laboureau-Soares S, Dubois S, Rohmer V, Rodien P. Tyrosine kinase inhibitors and modifications of thyroid function tests: a review. Eur J Endocrinol. 2009;160(3):331–336. | ||
Schutz FA, Je Y, Choueiri TK. Hematologic toxicities in cancer patients treated with the multi-tyrosine kinase sorafenib: a meta-analysis of clinical trials. Crit Rev Oncol Hematol. 2011;80(2):291–300. | ||
Pitoia F, Abelleira E, Jerkovich F, Urciuoli C, Cross G. Partial response to sorafenib treatment associated with transient grade 3 thrombocytopenia in a patient with locally advanced thyroid cancer. Arch Endocrinol Metab. 2015;59(4):347–350. | ||
Rothenberg SM, McFadden DG, Palmer EL, Daniels GH, Wirth LJ. Redifferentiation of iodine-refractory BRAF V600E-mutant metastatic papillary thyroid cancer with dabrafenib. Clin Cancer Res. 2015;21(5):1028–1035. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.