")
Back to Journals » Drug Design, Development and Therapy » Volume 14
Safety, Tolerability, and Pharmacokinetics of Adrenomedullin in Healthy Males: A Randomized, Double-Blind, Phase 1 Clinical Trial
Authors Kita T , Kaji Y, Kitamura K
Received 29 July 2019
Accepted for publication 13 December 2019
Published 6 January 2020 Volume 2020:14 Pages 1—11
DOI https://doi.org/10.2147/DDDT.S225220
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Qiongyu Guo
Toshihiro Kita,1 Yoshikazu Kaji,2 Kazuo Kitamura1
1Division of Circulatory and Body Fluid Regulation, Department of Internal Medicine, Faculty of Medicine, University of Miyazaki, Miyazaki, Japan; 2Souseikai Hakata Clinic, Fukuoka, Japan
Correspondence: Toshihiro Kita
Division of Circulatory and Body Fluid Regulation, Department of Internal Medicine, Faculty of Medicine, University of Miyazaki, 5200 Kihara, Miyazaki 889-1692, Japan
Tel +81-985-85-0872
Fax +81-985-85-6596
Email [email protected]
Background: Adrenomedullin (AM), an endogenous vasodilative peptide, has immunomodulative effects and acts as an accelerator of mucosal regeneration in the digestive tract. AM has shown beneficial effects in rodent models of inflammatory bowel disease and patients with ulcerative colitis. The present study aimed to evaluate the pharmacodynamic properties and safety of AM in healthy male adults in a phase 1 clinical trial.
Methods: This phase 1, randomized, double-blind, single-center study was conducted on healthy males aged 20–65 years. Subjects received either a placebo, 3 ng/kg/min AM, 9 ng/kg/min AM, or 15 ng/kg/min AM via continuous 12-h intravenous infusion. Other subjects received either placebo or 15 ng/kg/min AM for 8 h per day for 7 days. Adverse events (AEs), vital signs, physical examinations, laboratory tests, electrocardiograms (ECG), and pharmacokinetics were assessed.
Findings: All 24 subjects in the single-dose test completed the study. Of the 12 subjects in multiple dosing test, one from the AM group withdrew owing to a headache. No serious AEs were reported. Hemodynamic parameters were well maintained in all subjects. Slight ECG abnormalities were observed in the single-dose test. The plasma concentration of AM progressively increased in a dose-dependent manner and reached Cmax at the end of administration. Plasma AM rapidly returned to baseline concentrations after termination, with a T1/2 of under 60 min.
Interpretation: This is the first phase 1 trial in healthy men evaluating the safety of AM. Our results demonstrate the safety and tolerability of AM for subsequent Phase 2 trials.
Keywords: adrenomedullin, Phase 1 clinical trial, clinical pharmacology, inflammatory bowel disease
Introduction
Inflammatory bowel disease (IBD), including ulcerative colitis (UC) and Crohn’s disease (CD), is a group of intractable diseases that cause chronic inflammation in the gastrointestinal tract. IBD is relatively common in Western countries, but the number of patients diagnosed with IBD has drastically increased in Asian countries in the last three decades.1 In recent decades, many new medications and technologies have been introduced for the treatment of IBD, such as immunosuppressants, biologicals including anti-tumor necrosis factor agents and anti-integrin antibodies, and leukocyte apheresis. These new therapies have contributed to increased remission rates and a reduced risk of complications in patients with IBD.2 However, there are still many patients who cannot be effectively treated by existing therapies.
Adrenomedullin (AM) is a potent hypotensive peptide that is found ubiquitously in tissues and organs, especially in cardiovascular tissues, kidneys, lungs, and endocrine glands.3 AM has multiple functions in a wide range of tissues and acts mainly as a vasodilative and anti-arteriosclerotic factor.3 In addition, AM inhibits inflammation and supports tissue homeostasis through the suppression of pro-inflammatory cytokine synthesis and the induction of wound healing.4 The expression of AM and its receptor has been detected in the digestive tract, where it acts as a stimulator of mucosal regeneration. Previous studies have confirmed the beneficial effects of AM in experimental rodent models of IBD.5–10 Furthermore, AM previously displayed considerable effects in seven patients with UC in an exploratory clinical study.11 AM has also shown high efficacy in the treatment of intractable Crohn’s disease with a loss of response to infliximab.12 These data strongly suggest that AM is a novel potent therapeutic agent for IBD.13 Additionally, AM is an endogenous peptide; thus, it is expected to be safe for patients without the need for excessive immune-suppression and antibody production.
Preclinical studies, including toxicity studies, have been performed by Hubit Genomix Co. (Osaka, Japan) for the development and research of AM to treat myocardial infarctions (unpublished data). In this study, we performed an investigator-initiated trial of AM to treat refractory UC due to the lack of support from industrial companies. Using a health labour sciences research grant, we produced an active AM compound and placebo that were compliant with the specific criteria of good manufacturing practice (GMP). We then prepared a protocol for a phase 1 study and obtained funding from the Japan agency for medical research and development (AMED), and performed an investigator-initiated phase 1 trial using healthy male volunteers in compliance with good clinical practice (GCP). An important factor for developing the phase 1 protocol is that AM showed anti-inflammatory and wound healing effects in IBD patients within sub-depressor dose.11,12 Therefore, the development of an AM formulation as a therapeutic agent for IBD, and confirmation of the upper safe and tolerable limits of AM, particularly the limit for an acceptable vasodilating influence, are crucial. This paper reports the results of the phase 1 trial, which included single-dose and multiple-dose administration tests using AM in healthy males through a placebo-controlled double-blinded study. The objectives of the study were to evaluate the safety, tolerability, and pharmacokinetic (PK) effects of intravenous AM administration.
Materials and Methods
Drug Formulation
AM is a peptide containing 52 amino acid residues with a ring structure formed by one intramolecular disulphide bond and an amidated structure formed by a C-terminal tyrosine.14 We have produced original AM formulation for this investigator-initiated clinical trial. Bulk AM powder was chemically synthesized by the Peptide Institute (Osaka, Japan) according to GMP. Bulk AM powder was dissolved in water containing D-mannitol and formulated as a freeze-dried material by established pharmaceutical company (Fuji Yakuhin, Toyama, Japan) according to GMP. A vial of AM for injection contained 500 μg AM and 50 mg D-mannitol. A vial of placebo contained 50 mg D-mannitol only. The vials were stored at 2–8 °C.
Study Design
This phase 1, randomized, double-blind, single-center, dose-escalation study was conducted at the Souseikai Hakata Clinic (Fukuoka, Japan). The investigator-initiated trial was performed on healthy male Japanese volunteers, and consisted of single-administration and 7-day multiple-administration studies. After approval by the Pharmacological and Medical Device Agency (PMDA), ethical approval for this study was obtained from Institutional Review Board of University of Miyazaki and the Souseikai Hakata Clinic. This clinical trial was conducted in compliance with ethical principles based on the Declaration of Helsinki, GCP of the Japanese Ministerial Ordinance, and other related regulation requirements. The trial were registered in the JAPIC clinical trials information; Japic CTI-194891 (190729917784) and Japic CTI-194890 (190729217785).
Subjects
Subjects eligible for inclusion in the study were healthy males aged 20–65 years. The inclusion criteria included 1) no abnormal findings in medical history and physical examination, 2) normal laboratory tests (hematology evaluations, blood chemistry and urinalysis), vital signs (systolic and diastolic blood pressure, pulse rate (PR) and temperature) and ECG record, 3) not having participated in another clinical trial during the 3 months before starting the current trial and 4) not having donated blood during the 3 months prior to starting the current trial. Key exclusion criteria included a history of malignancy and/or myocardial infarction, systolic blood pressure (SBP) under 100 mmHg, PR under 45 bpm, and body weight over 90 kg. Also, subjects with drug allergy and positive serology for hepatitis B, C, or for HIV, and those who took regular medication and any other medication in a 7-day period prior to the trial, with history or clinical evidence of chronic diseases were excluded. Potential subjects were identified from a volunteer panel at the Souseikai Hakata Clinic and through advertising. All subjects provided written informed consent for all study-related procedures.
Randomization and Masking
Suitable subjects were enrolled for the study by the principal investigator or designee based on the above inclusion and exclusion criteria. Subjects were randomized at a ratio of 1:3 to receive either placebo or increasing doses of AM (3, 9, and 15 ng/kg/min) in a single administration study. The randomization list was prepared by an independent contract research organization (CRO), Adjust (Sapporo, Japan); within each group, a block size of eight was used to attain a 1:3 ratio of randomization to placebo or AM, respectively. The same CRO prepared the medication (vials containing AM or placebo were indistinguishable) according the list. The remaining subjects were randomized at a ratio of 1:1 to receive either placebo or AM (15 ng/kg/min) in a 7-day multiple administration study. The same CRO prepared a randomization list using a block size of twelve to attain a 1:1 ratio of randomization to placebo or AM, respectively, and then prepared the medication. The randomization lists were kept under rigid control and opened after the completion of each study procedure and data confirmation.
Procedures
Single-Dose Administration Study
Screening tests, including physical examination, investigation of medical history, measurement of vital signs, 12-lead electrocardiogram (ECG), and blood and urine examinations, were conducted within 1 month prior to administration. Eligible subjects were randomized to receive placebo or escalating doses of AM (3, 9, and 15 ng/kg/min) intravenously. Eight subjects from each group were successively hospitalized from the day before administration until the day after administration. Continuous intravenous infusion was initiated in the morning and performed over 12 h (±30 min). Subjects received safety examinations and were discharged the following morning. Dose escalation was performed after approval by the principal investigator (Dr. Kaji) based on the safety results on the day after administration in all eight subjects in each group.
Multiple-Dose Administration Study
The same screening tests described above were conducted within 1 month prior to administration. Eligible subjects were randomized to the placebo or AM (15 ng/kg/min) groups. All subjects in both groups were hospitalized from the day before administration until the day after the final administration. Continuous intravenous infusion was initiated in the morning and performed over 8 h (±30 min) every day for 7 days. Subjects received safety examinations and were discharged on the morning following the final administration.
Assessments and Analyses
Safety Assessments
Adverse events (AEs) and serious adverse events (SEAs) were monitored throughout the study. Vital signs, clinical laboratory tests including changes in humoral factors (adrenocorticotropic hormone, cortisol, aldosterone, atrial natriuretic peptide, and brain natriuretic peptide), physical examinations, and 12-lead ECG were performed at specified times after administration. Additionally, monitor ECG was performed from the beginning of testing until 24 h after drug administration. Based on the collected AE information, proper terms were selected from the Medical Dictionary for Regulatory Activities/Japanese version (MedDRA/JTM).
PK Assessments
In the single-dose study, blood samples were drawn pre-dose; as well as 5, 10, 20, and 30 min; and 1, 2, 4, 6, 8, 10, and 12 h after administration. After the termination of infusion, additional blood samples were drawn after 2, 5, 10, 20, and 30 min, and after 1, 2, and 12 h. In the multiple-dose study, blood samples were drawn pre-dose; as well as 5, 10, 20, and 30 min; and 1, 2, 4, 6, and 8 h after dosing, and then at 2, 5, 10, 20, 30 min, as well as 1 and 2 h after completing infusion on the first and seventh days of administration. On all other days, blood samples were drawn pre-dose and 8 h after dosing; a final blood sample was taken on the morning of the final day of hospitalization. Blood samples were collected in tubes containing 1·5 mg/mL ethylenediaminetetraacetic acid (EDTA-2Na) and 500 U/mL aprotinin and centrifuged at 4 °C within 2 h after sampling. Supernatant plasma was transferred to polypropylene tubes containing 20% w/v CHAPS PBS solution (3-[(3-cholamidopropyl)-dimethylammonio]-1-propanesulfonate in phosphate buffered salt solution) and stored at −25 °C until assayed. The concentrations of AM were measured using an automated enzyme immune assay analyser (AIA-1800, Toso, Tokyo, Japan) at the Bozo Research Center (Tsukuba, Japan). We only measured the mature form of AM because exogenous AM is composed of the mature form only.15 The calculated PK parameters included the maximum measured plasma concentration (Cmax); the time to maximum measured plasma concentration (Tmax); the cumulative area under the plasma concentration-time curve (AUC) from time 0 to time t, where time t is the time of the last quantifiable plasma concentration; and terminal plasma elimination half-life (T1/2). The Cmax and Tmax values were obtained directly from the data and the AUC and T1/2 were calculated using Phoenix WinNonlin software 6.1 (Pharsight, CA, USA).
Data Collection and Statistical Analysis
All data, except for the PK parameters, were collected and managed by Intellim (Tokyo, Japan), an independent CRO. Additional statistical analyses of BP, PR, and PK parameters were carried using SPSS version 22 (IBM Japan, Tokyo, Japan). After confirming the normal distribution of all variables, significant differences were evaluated using a paired t-test or analysis of variance (ANOVA) followed by Fisher’s multiple comparison test. PK data were collected and analyzed by Bozo Research Center, as stated above. Data were expressed as the mean ± standard deviation (SD). A value of P < 0.05 was considered statistically significant.
Results
Subjects
The single-dose administration study was conducted between November 12 and December 23, 2015. A total of 52 volunteers were eligible for testing. Of the 52 subjects, 24 underwent the experimental treatment, and all subjects completed the study. The multiple-dose administration study was conducted between June 15 and July 18, 2016. A total of 19 volunteers were eligible for testing. Of the 19 subjects, 12 received the experimental treatment and 11 completed the study. One subject in the AM group dropped out after 2 days of drug administration due to headaches. All 12 subjects were included in the safety population, and 11 subjects were included in the PK study and hemodynamic assessment.
The demographics and basal characteristics of the 24 subjects in the single-dose administration study and the 12 subjects in the multiple-dose administration study are illustrated in Table 1. All subjects were Japanese men in good general health based on medical history, physical examination, vital signs, and laboratory data.
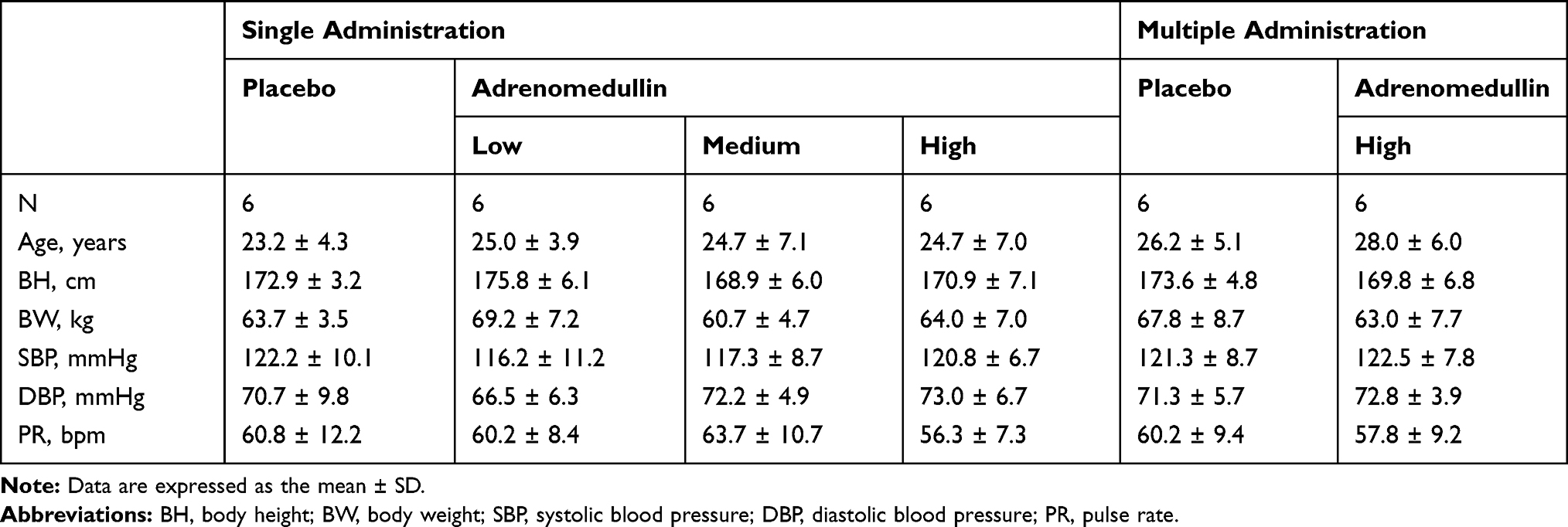 |
Table 1 Baseline Characteristics |
Adverse Effects and Compliance
A total of six (25%) subjects experienced at least one AE during the single-dose administration study (Table 2). Headaches, a common AE from AM, occurred in one subject who received a medium dose of AM, one subject who received a high dose of AM, and one subject who received the placebo. Wenckebach type atrioventricular block, which is not a known AE from AM, occurred in two subjects who received a medium dose of AM and one subject who received a high dose of AM. All Wenckebach type atrioventricular blocks occurred in the middle of the night after the termination of AM administration and were transitory (within 1 min), as determined through an investigation of monitor ECG. There were no discontinuations or withdrawals from the study due to AEs, and no deaths or SAEs were reported (Table 2).
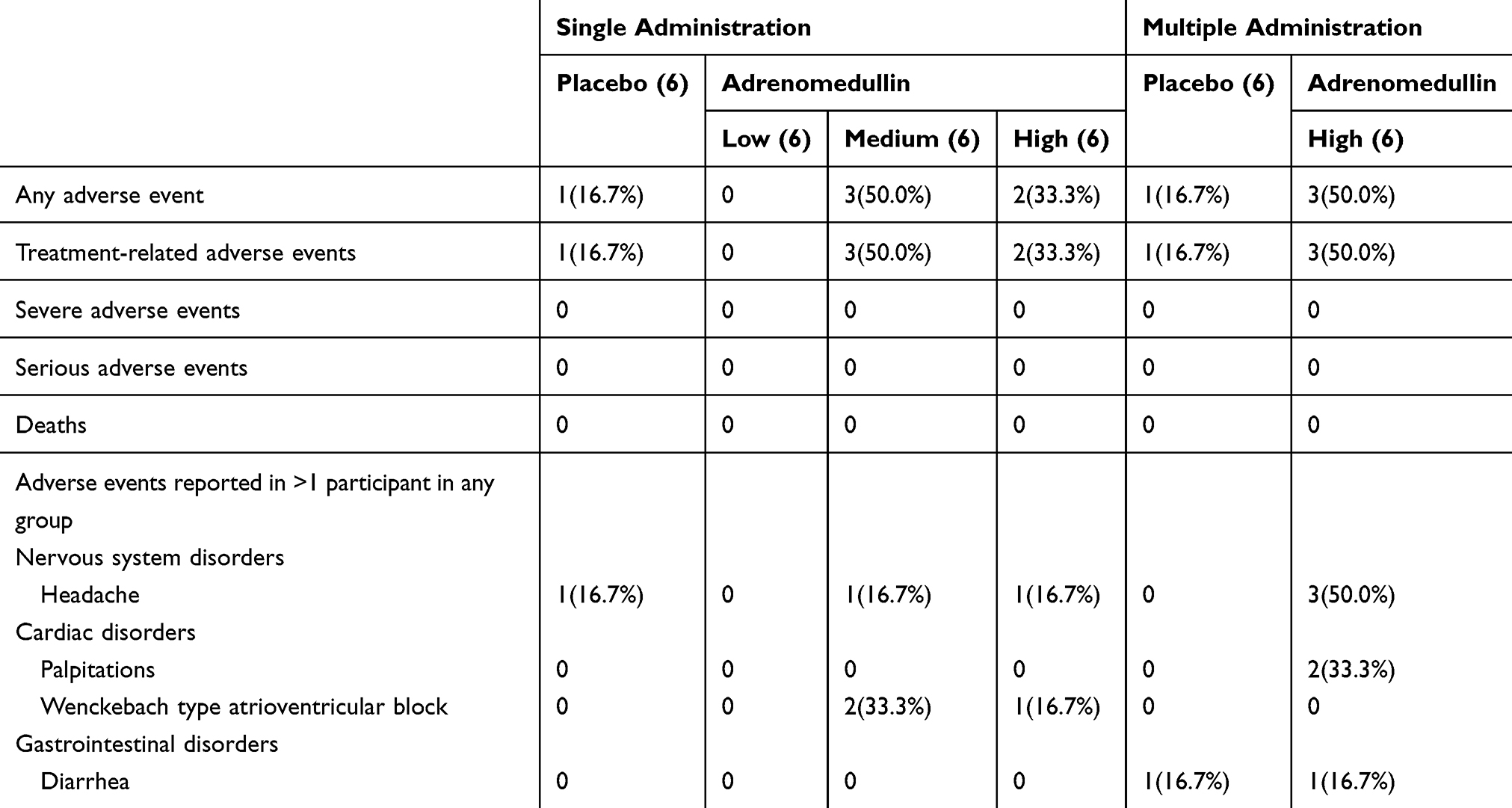 |
Table 2 Summary of Reported Adverse Events During Treatment |
A total of four (33%) subjects experienced at least one AE during the multiple-dose administration study (Table 2). Headaches repeatedly occurred in three of six subjects who received a high dose of AM, and two of them concurrently experienced palpitations, another common AE from AM. Diarrhea, an uncommon AE from AM, occurred in one subject who received AM and one subject who received placebo. ECG abnormalities, including monitor ECG, were not reported in these groups. There was one withdrawal from the study due to headache, and no deaths or SAEs were reported (Table 2).
Hemodynamic Effects of AM
AM is one of the strongest vasodilators among the known bioactive substances; therefore, we closely monitored BP and PR. In the single-dose administration study, continuous decreases in SBP were observed in the medium-dose group (9 ng/kg/min), but slight increases, followed by slight decreases, in SBP were observed in the high-dose group (15 ng/kg/min). Slight decreases in SBP were also observed in the placebo group (Table 3). Slight but meaningful decreases in diastolic BP (DBP) and increases in PR were observed in the medium- and high-dose groups (Table 3). However, the same tendency was observed in the placebo group. The average BP and PR values across the 7 days in the multiple-dose administration study are summarized in Table 4. SBP was maintained at a stable level throughout the multiple-dose administration study in all groups. Small but significant decreases in DBP and increases in PR were observed in the multiple-dose AM group. A similar tendency was also observed for DBP and PR in the placebo group, but significant differences were observed between the AM and placebo groups.
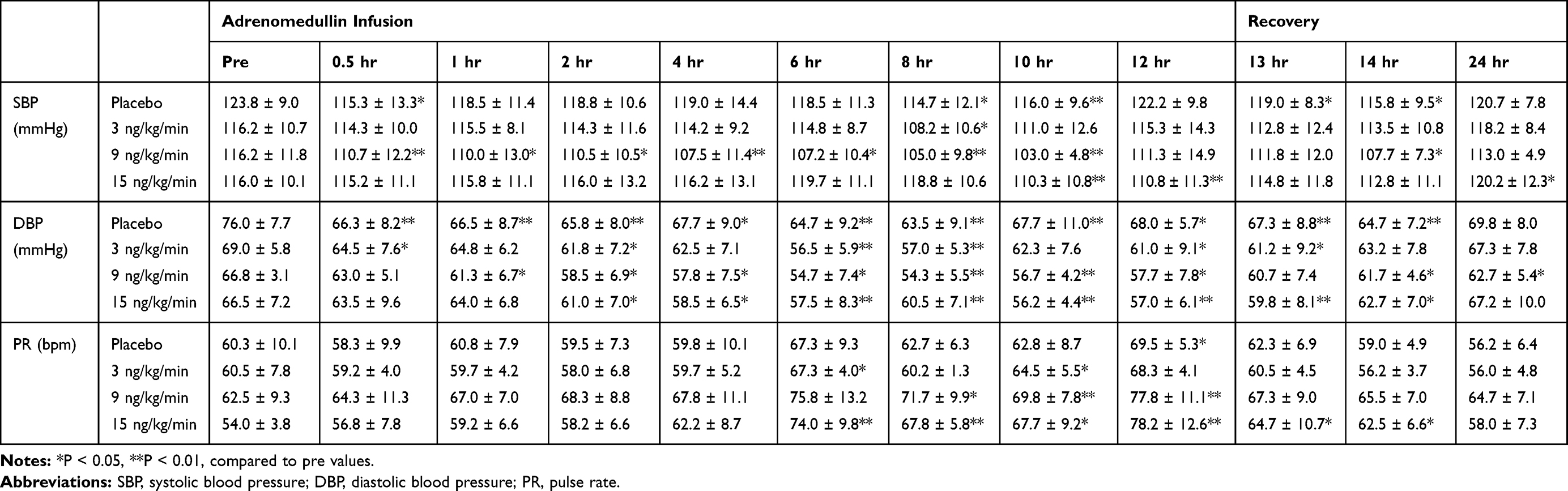 |
Table 3 Hemodynamic Parameters of Participants Treated with the Single Dose Administration Test |
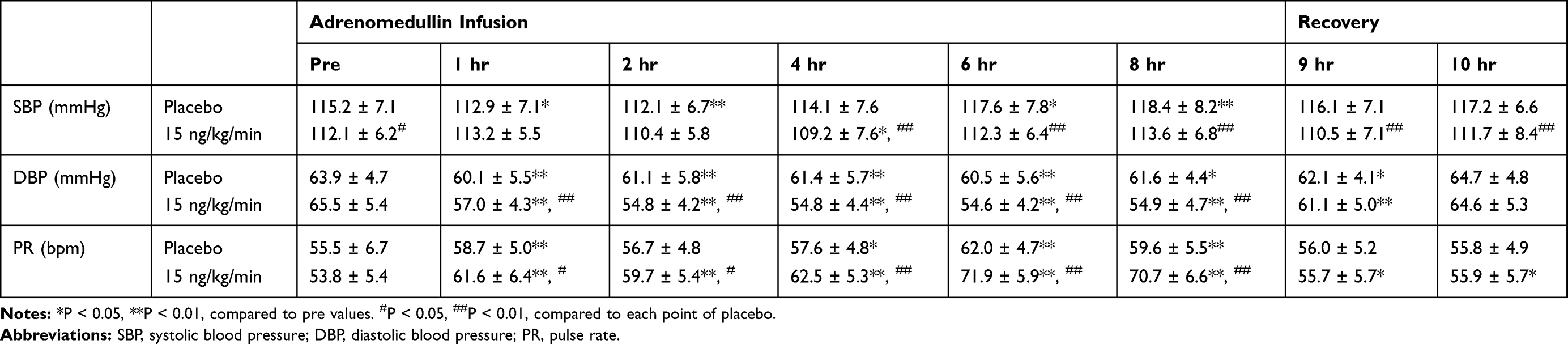 |
Table 4 Hemodynamic Parameters of Participants Treated with the Multiple Doses Administration Test |
Clinical Laboratory Tests
Although slight variations were found in the average and median values of most test parameters throughout the study, these variations were generally small and not clinically significant. In addition, there were no meaningful differences in the average and median values between treatment groups. There were no changes in the laboratory test values associated with different doses of AM. Therefore, it was concluded that there were no changes due to the direct effect of AM.
Pharmacokinetics
AM is an endogenous bioactive peptide; therefore, basal values were detectable before AM administration. The control plasma concentration of AM was 7·2 ± 1·4 pg/mL (n = 23; one subject was below the lower limit of quantification) in the single-dose administration study, and 7·0 ± 0·9 pg/mL (n = 11; one withdrawal subject was excluded) in the multiple-dose administration study. The plasma concentration of AM increased in a time-dependent manner during the continuous infusion of AM, and reached Cmax at the end of AM administration (Figures 1 and 2). Cmax was increased in a dose-dependent manner, and increased by sevenfold compared to the basal concentration of AM in the high-dose AM group (Table 5 and Figure 1). AM concentration was reduced to baseline levels soon after the termination of AM infusion, with a T1/2 of less than 60 min (Table 5, Figures 1 and 2). In the multiple-dose administration study, the AUC0-10 was significantly increased after repeated administration of AM (Table 5 and Figure 2).
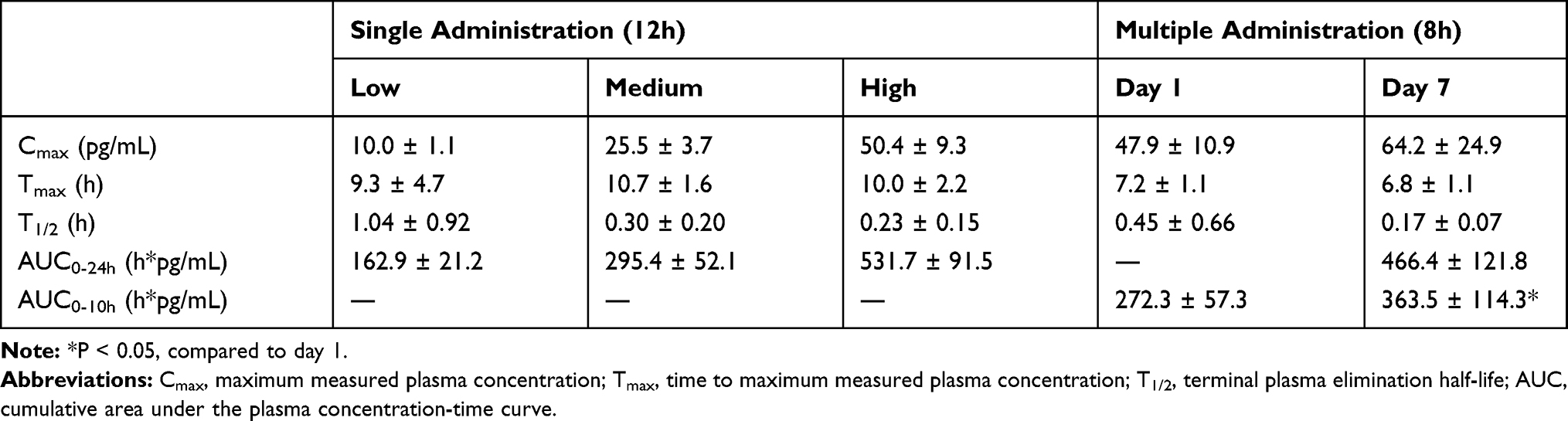 |
Table 5 Adrenomedullin Pharmacokinetic Parameters |
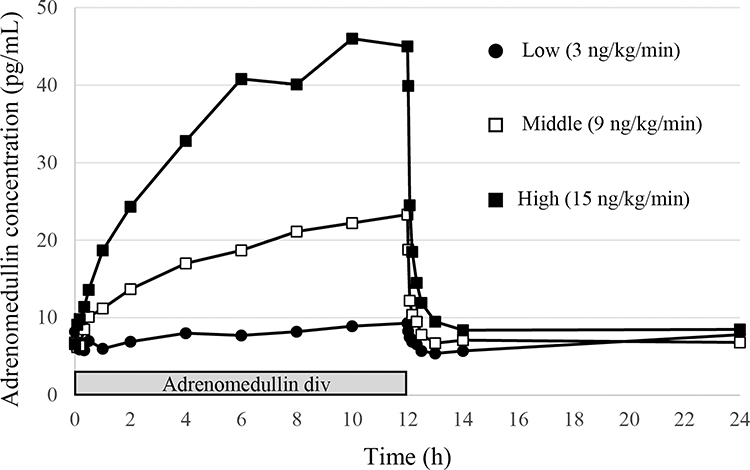 |
Figure 1 Mean plasma concentration-time profiles of adrenomedullin following infusion with three different doses. |
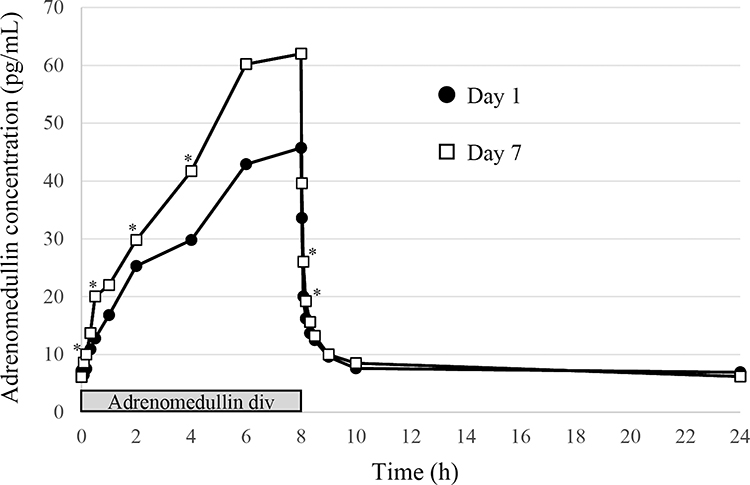 |
Figure 2 Mean plasma concentration-time profiles of adrenomedullin at day 1 and day 7 of repeated infusion. *P < 0.05 compared to each time point at day 1. |
Discussion
The purpose of this phase 1 clinical trial was to evaluate the clinical safety, tolerability, and PK parameters of AM in healthy male volunteers for the successful transition to phase 2 trials. In spite of several mild AEs that were tolerable without any medical treatment, continuous AM administration was possible and hemodynamic parameters were maintained at stable levels throughout AM administration. Based on the results of this phase 1 trial, the continuous intravenous administration of AM at concentrations of up to 15 ng/kg/min is safe and tolerable, indicating that AM is safe for phase 2 trials.
In the single-dose administration study, all 24 subjects completed the study and well tolerated the 12-h administration of AM. The vasodilative effect of AM caused a headache in each subject given a medium and high dose of AM. However, these headaches were mild and tolerable without any required medical treatment and recovered spontaneously. Headaches were also reported in the placebo group. Headaches were an expected effect based on the known pharmacological features of AM and the results of previous reports.16–18 We were concerned that severe headaches might have hindered the continuous administration of AM, but this study demonstrated that the headaches remained tolerable at up to 15 ng/kg/min AM. However, Wenckebach type atrioventricular blocks were an unexpected effect. The administration of AM was terminated at around 21h00, and atrioventricular blocks occurred late at night (04h23 and 06h03 in the medium-dose group, and 02h57 in the high-dose group). The duration of the atrioventricular block was very short (within 1 min) and caused no symptoms; therefore, the blocks were only found by observing the monitor ECG. There has been no previous evidence of atrioventricular blocks due to AM. Additionally, the plasma concentration of AM should have returned to control levels before the atrioventricular blocks occurred, because the T1/2 of AM after termination of administration was 23–30 min. Therefore, it is unclear whether this was directly caused by AM. In healthy Japanese subjects, Holter ECGs previously detected Wenckebach type atrioventricular blocks in 13 out of 161 (8%) patients, and these blocks mostly occurred late at night.19 Therefore, it is possible that these Wenckebach type atrioventricular blocks occurred by chance during the study period. We concluded that the atrioventricular blocks were an acceptable phenomenon that does not hinder the progression of the trials. However, we set up strict measures to care for atrioventricular blocks in the multiple-dose administration study.
In the multiple-dose administration study, one subject in the AM group withdrew from the study owing to headaches. Although the patient had undergone infusion of AM for two full days of the study with no need for treatment, he then requested to withdraw from the study because of headaches. Including the withdrawn subject, three out of six subjects in the AM group showed repeated headaches or palpitations. AM infusion was performed between 09h00 and 17h00; the AEs occurred between 12h30 and 16h00, and many disappeared between 17h30 and 19h00. These effects were consistent with the increased plasma concentration of AM, and may have been caused by the vasodilatation activity of AM. However, the reported AEs were all mild, and it was possible to continue to administer AM without treatment. Based on this, the safety of AM was confirmed at up to the maximum dose (15 ng/kg/min). Diarrhea was observed in two subjects, one of whom was treated with the placebo. This was a continuous symptom irrespective of the dosage time, and was therefore considered to be unrelated to AM. Notably, the Wenckebach type atrioventricular blocks observed in the single-dose study were not observed in the multiple-dose study.
We further examined the hemodynamic effects of AM in the patients. In a previous study, AM induced a clear decrease in BP at a dose of 30 ng/kg/min after 90 min of intravenous infusion in normotensive and hypertensive patients.20 Furthermore, the prolonged infusion of AM at a dose of 15 ng/kg/min for 27 h decreased the BP of patients in a previous report, especially late at night.21 Based on these results, we chose the upper limit of AM administration as 15 ng/kg/min for 12 h, allowing the infusion to be terminated before midnight. Indeed, the safety of hemodynamic parameters was confirmed in a previous exploratory clinical study, where AM was infused at a dose of 9 ng/kg/min for 8 h in patients with UC.11 In the single-dose administration study, significant increases in PR were observed in the medium- and high-dose AM groups, but this did not appear to be dose-dependent (Table 2). SBP and DBP were decreased during the latter half of the infusion process, but similar decreases were observed in the placebo group. As expected, changes in SBP were not dose-dependent in patients treated with low, medium, and high doses of AM. These data indicated that the hemodynamic effects of AM were negligible at up to 15 ng/kg/min AM infusion. A significant decrease in DBP and increase in PR were observed in average values of the 7-day administration process in the multiple-dose administration study (Table 4). However, SBP was well maintained within AM infusion (Table 4). These data further indicated that the hemodynamic effects of AM were negligible after the repeated administration of AM.
The plasma concentration of AM increased in proportion to the treatment dose and decreased promptly after the end of infusion (Figure 1), similar to the results of previous reports. In the multiple-dose study, plasma AM concentrations were generally higher on day 7 compared to those on day 1. Figure 3 shows the plasma concentration of AM at each day just before the end of AM infusion; it was found to be similar to the measured Cmax. A significant increase in the plasma concentration of AM was observed only on day 7. The exact reason for this increase was not clear; potentially, a decrease in AM clearance may have been attenuated by the repeated administration of AM. The AM receptor controls its clearance in pulmonary circulation.22,23 Previously, clearance was shown to be decreased by pre-treatment with high concentrations of AM agonist, or by sepsis, where the plasma concentration of AM was markedly increased.24,25 These data indicated that the saturation of the pulmonary AM receptor induced a deterioration in AM clearance. Therefore, the repeated administration of AM may saturate the AM receptor in lungs. However, adverse events such as headaches did not increase or aggravate after the repeated administration of AM. Furthermore, plasma AM concentrations decreased rapidly after the end of the 7-day AM administration, and accumulation was not observed. We have also confirmed the safety of the repeated infusion of AM (8 h per day for 4 weeks) in rat and dog models (unpublished data).
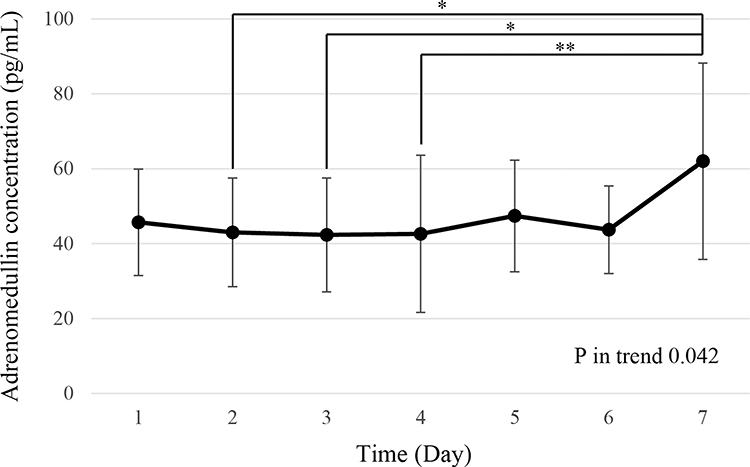 |
Figure 3 Mean plasma concentration of adrenomedullin at end of each day’s infusion in the multiple-dose administration study. *P < 0.05, **P < 0.01. |
In conclusion, we have reported the first evidence, in full compliance with GCP, that AM is a safe and clinically tolerable therapeutic agent when administered intravenously. Based on these results, presently, we are conducting phase 2 clinical trials in Japan with AM for intractable UC and CD.
Data Sharing Statement
The authors do not intend to share substantial data of this study, but they are ready to share the file of substantial data and all other study-related documents (in Japanese), at any specific time for any period, if the editorial board requires.
Acknowledgement
We would like to thank Editage for English language editing.
Disclosure
Kazuo Kitamura and Toshihiro Kita report a patent US 10335455 B2 issued. Kazuo Kitamura reports a patent PCT/JP2012/051010 licensed to Himura AM Pharma Corp.; holds stocks in Himuka AM Pharma Corp and is an advisor of Himura AM Pharma Corp. Toshihiro Kita holds stocks in Himuka AM Pharma Corp. The authors report no other conflicts of interest in this work.
References
1. Prideaux L, Kamm MA, De Cruz PP, Chan FK, Ng SC. Inflammatory bowel disease in Asia: a systematic review. J Gastroenterol Hepatol. 2012;27:1266–1280. doi:10.1111/j.1440-1746.2012.07150.x
2. Masaki T, Kishiki T, Kojima K, Asou N, Beniya A, Matsuoka H. Recent trends (2016–2017) in the treatment of inflammatory bowel disease. Ann Gastroenterol Surg. 2018;2:282–288. doi:10.1002/ags3.2018.2.issue-4
3. Eto T, Kato J, Kitamura K. Regulation of production and secretion of adrenomedullin in the cardiovascular system. Regul Pept. 2002;112:61–69. doi:10.1016/S0167-0115(03)00023-5
4. Cheung BM, Tang F. Adrenomedullin: exciting new horizons. Recent Pat Endocr Metab Immune Drug Discov. 2012;6:4–17. doi:10.2174/187221412799015263
5. Ashizuka S, Ishikawa N, Kato J, et al. Effect of adrenomedullin administration on acetic acid-induced colitis in rats. Peptides. 2005;26:2610–2615. doi:10.1016/j.peptides.2005.05.007
6. Gonzales-Rey E, Fernandez-Martin A, Chorny A, Delgado M. Therapeutic effect of urocortin and adrenomedullin in a murine model of Crohn’s disease. Gut. 2006;55:824–832. doi:10.1136/gut.2005.084525
7. Talero E, Sánchez-Fidalgo S, de la Lastra CA, Illanes M, Calvo JR, Motilva V. Acute and chronic responses associated with adrenomedullin administration in experimental colitis. Peptides. 2008;29:2001–2012. doi:10.1016/j.peptides.2008.07.013
8. Ashizuka S, Inagaki-Ohara K, Kuwasako K, Kato J, Inatsu H, Kitamura K. Adrenomedullin treatment reduces intestinal inflammation and maintains epithelial barrier function in mice administered dextran sulphate sodium. Microbiol Immunol. 2009;53:573–581. doi:10.1111/mim.2009.53.issue-10
9. Talero E, Alvarez de Sotomayor M, Sánchez-Fidalgo S, Motilva V. Vascular contribution of adrenomedullin to microcirculatory improvement in experimental colitis. Eur J Pharmacol. 2011;670:601–607. doi:10.1016/j.ejphar.2011.09.032
10. Hayashi Y, Narumi K, Tsuji S, et al. Impact of adrenomedullin on dextran sulfate sodium-induced inflammatory colitis in mice: insights from in vitro and in vivo experimental studies. Int J Colorectal Dis. 2011;26:1453–1462. doi:10.1007/s00384-011-1254-0
11. Ashizuka S, Inatsu H, Kita T, Kitamura K. Adrenomedullin therapy in patients with refractory ulcerative colitis: a case series. Dig Dis Sci. 2016;61:872–880. doi:10.1007/s10620-015-3917-0
12. Ashizuka S, Kuroishi N, Nakashima K, Inatsu H, Kita T, Kitamura K.Adrenomedullin: a novel therapy for intractable Crohn’s disease with a loss of response to infliximab. Intern Med.2019;58:1573–1576. doi:10.2169/internalmedicine.1791-18
13. Ashizuka S, Inatsu H, Inagaki-Ohara K, Kita T, Kitamura K. Adrenomedullin as a potential therapeutic agent for inflammatory bowel disease. Curr Protein Pept Sci. 2013;14:246–255. doi:10.2174/13892037113149990044
14. Kitamura K, Kangawa K, Kawamoto M, et al. Adrenomedullin: a novel hypotensive peptide isolated from human pheochromocytoma. Biochem Biophys Res Commun. 1993;192:553–560. doi:10.1006/bbrc.1993.1451
15. Ohta H, Tsuji T, Asai S, et al. One-step direct assay for mature-type adrenomedullin with monoclonal antibodies. Clin Chem. 1999;45:244–251.
16. Nagaya N, Nishikimi T, Uematsu M, et al. Haemodynamic and hormonal effects of adrenomedullin in patients with pulmonary hypertension. Heart. 2000;84:653–658. doi:10.1136/heart.84.6.653
17. Troughton RW, Lewis LK, Yandle TG, Richards AM, Nicholls MG. Hemodynamic, hormone, and urinary effects of adrenomedullin infusion in essential hypertension. Hypertension. 2000;36:588–593. doi:10.1161/01.HYP.36.4.588
18. McGregor DO, Troughton RW, Frampton C, et al. Hypotensive and natriuretic actions of adrenomedullin in subjects with chronic renal impairment. Hypertension. 2001;37:1279–1284. doi:10.1161/01.HYP.37.5.1279
19. Yokoi A, Hishida H, Ohashi S, Mizuno Y, Kajihara K, Shimada Y.Wenckebach A-V block documented by 24 hour ambulatory electrocardiographic monitoring in healthy subjects. Shindenzu.1983;3:71–77. doi:10.5105/jse.3.71
20. Kita T, Suzuki Y, Kitamura K. Hemodynamic and hormonal effects of exogenous adrenomedullin administration in humans and relationship to insulin resistance. Hypertens Res. 2010;33:314–319. doi:10.1038/hr.2009.236
21. Kita T, Tokashiki M, Kitamura K. Aldosterone antisecretagogue and antihypertensive actions of adrenomedullin in patients with primary aldosteronism. Hypertens Res. 2010;33:374–379. doi:10.1038/hr.2010.8
22. Dupuis J, Caron A, Ruël N. Biodistribution, plasma kinetics and quantification of single-pass pulmonary clearance of adrenomedullin. Clin Sci. 2005;109:97–102. doi:10.1042/CS20040357
23. Dschietzig T, Azad HA, Asswad L, et al. The adrenomedullin receptor acts as clearance receptor in pulmonary circulation. Biochem Biophys Res Commun. 2002;294:315–318. doi:10.1016/S0006-291X(02)00474-6
24. Ornan DA, Chaudry IH, Wang P. Saturation of adrenomedullin receptors plays an important role in reducing pulmonary clearance of adrenomedullin during the late stage of sepsis. Biochim Biophys Acta. 2002;1586:299–306. doi:10.1016/S0925-4439(01)00108-9
25. Ornan DA, Chaudry IH, Wang P. Pulmonary clearance of adrenomedullin is reduced during the late stage of sepsis. Biochim Biophys Acta. 1999;1427:315–321. doi:10.1016/S0304-4165(99)00032-X
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.