")
Back to Journals » Drug Design, Development and Therapy » Volume 12
Rucaparib: a novel PARP inhibitor for BRCA advanced ovarian cancer
Authors Colombo I, Lheureux S, Oza AM
Received 30 November 2017
Accepted for publication 17 January 2018
Published 21 March 2018 Volume 2018:12 Pages 605—617
DOI https://doi.org/10.2147/DDDT.S130809
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Qiongyu Guo
Ilaria Colombo, Stephanie Lheureux, Amit Manulal Oza
Division of Medical Oncology and Hematology, Princess Margaret Cancer Centre, University Health Network, Toronto, ON, Canada
Abstract: Rucaparib is a potent small-molecule inhibitor of poly (ADP-ribose) polymerase (PARP) proteins (PARP-1, PARP-2 and PARP-3) that play an important role in repairing DNA damage and maintaining genomic stability. Tumors with mutations in BRCA1/2 or other homologous recombination deficiency (HRD) genes are particularly sensitive to PARP inhibitors because of “synthetic lethality”, whereby a therapeutic agent can take advantage of an intrinsic weakness in DNA repair. Rucaparib has been investigated in several preclinical and clinical studies showing promising activity in BRCA-mutant and BRCA–wild-type epithelial ovarian cancers (EOCs). Dose-escalation Phase I studies have established the recommended Phase II dose to be 600 mg twice a day for oral rucaparib. Phase II and III studies have defined its role as treatment for BRCA-mutant recurrent high-grade EOC and as maintenance treatment for platinum-sensitive relapsed EOC following response to platinum-based chemotherapy. Genomic loss of heterozygosity has also been investigated as a potential signature of HRD and as a potential predictive biomarker of response. Treatment-induced adverse events (AEs) have been observed in almost all patients treated with rucaparib, but mainly lower grade; with the most common being nausea, vomiting, asthenia/fatigue, anemia and transient transaminitis. The majority of AEs occurred early in treatment, were transient and have been easily managed with supportive treatment, dose interruption or discontinuation. This review will analyze the results of clinical trials investigating efficacy and safety of rucaparib in patients with ovarian cancer.
Keywords: rucaparib, ovarian cancer, BRCA mutations, homologous recombination deficiency, maintenance treatment, PARP inhibitor
Introduction
Ovarian cancer is the most lethal gynecological cancer and the fifth leading cause of cancer death among women in the USA.1 No effective screening tests are available and more than 70% of patients are diagnosed at advanced stage.2 Epithelial ovarian cancer (EOC) accounts for >90% of all subtypes of ovarian cancer and the most common histological subtype is high-grade serous, representing ~70% of all epithelial ovarian malignancies.3 Similar to the majority of cancers, EOC is characterized by the presence of acquired or inherited mutations in different DNA repair pathways.4 DNA double strand breaks (DSBs) can be repaired by nonhomologous end-joining (NHEJ) pathway without the need to copy an intact DNA template that is prone to errors,5 or by homologous recombination repair system that is an error-free pathway requiring an homologous DNA template to function.6 DNA single strands breaks (SSBs) are corrected by base excision repair, nucleotide excision repair or mismatch repair systems, using the other DNA strand as guide.7 The repair of DNA damage is necessary to maintain genomic stability, promote cell survival and replication which is regulated by different enzymes. BRCA1, BRCA2 and other homologous recombination proteins are recruited to repair DNA DSBs.8,9 Tumors with BRCA1/2 mutations or other defects in homologous recombination repair system genes (eg, EMSY, RAD51, ATM, ATR, Fanconi Anemia, BARD1, BRIP1, PALB2) rely on alternative mechanisms to repair DNA damage, like the “error prone” NHEJ recombination. Poly (ADP-ribose) polymerase (PARP) enzymes, particularly PARP-1 and -2, play a critical role in the repair of DNA SSBs through the base excision repair and other SSB pathways.10 Inhibition of PARP leads to accumulation of SSBs causing collapse of replication forks and accumulation of DSBs that are commonly repaired by homologous recombination enzymes.11 Tumors with BRCA1/2 mutations or other defects in the homologous recombination repair system are particularly sensitive to PARP inhibitors due to accumulation of SSBs leading to DSBs that cannot be repaired due to homologous recombination deficiency (HRD), and ultimately result in cell death.12 This has also been reported as “synthetic lethality” to describe the phenomenon of cell death due to mutation or lack of function of two or more genes, whereas the defect in only one gene does not alter cell survival.12 PARP inhibitors also elicit their activity through other different mechanisms such as interfering with NHEJ DNA repair pathway, which is upregulated when homologous recombination pathways are deficient13 or causing trapping of PARP-1 and -2 at the level of the DNA break, resulting in obstruction of the replication fork that requires an intact homologous recombination pathway to repair the damage.14
Fifty percent of all high-grade serous ovarian carcinomas (HGSOCs) present HRD with 22% harboring germinal or somatic mutation in BRCA1 or BRCA2 (due to genomic or epigenetic events, eg, promoter methylation or activation of inhibitors).15,16 Germline or somatic mutations in homologous recombination genes are usually associated with increased response to platinum-based chemotherapy, longer disease-free interval and better prognosis;16 however, some HGSOCs show similar clinical behavior without identifiable mutations in BRCA1/2 or other homologous recombination genes.17–19
Several PARP inhibitors (olaparib, niraparib and rucaparib) have been investigated and are now available for the treatment or maintenance therapy of patients with HGSOC.20–26 PARP inhibitors have also showed promising results in other solid tumors harboring BRCA1/2 mutations, such as HER-2 negative breast cancer and metastatic pancreatic and castration-resistant prostate cancers.20,27,28
The present review will focus on the role of the PARP inhibitor rucaparib in ovarian malignancies. In December 2016, the US Food and Drug Administration (FDA) granted accelerated approval of rucaparib for the treatment of patients with HGSOC carrying deleterious germline (gBRCA) or somatic BRCA (sBRCA) mutation previously treated with two or more lines of chemotherapy.
Clinical trials of rucaparib in ovarian cancer
Rucaparib (CO-338, formerly known as AG-014669 and PF-01367338) is a potent small-molecule inhibitor of PARP-1, PARP-2 and PARP-3 that has shown preclinical and clinical activity in ovarian carcinoma as well as other types of solid tumors; it has been extensively investigated in solid tumors harboring BRCA1/2 mutations or HRD. Two formulations of rucaparib have been initially developed: the phosphate salt (intravenous [iv] formulation) and the camphorsulfonic acid salt (oral formulation in the form of tablets), known as rucaparib cammsylate. Different studies24,33,34,36,42,46 have been designed to explore the activity and safety of rucaparib in ovarian cancer, some of which are still active and enrolling patients (Figure 1, Table 1).
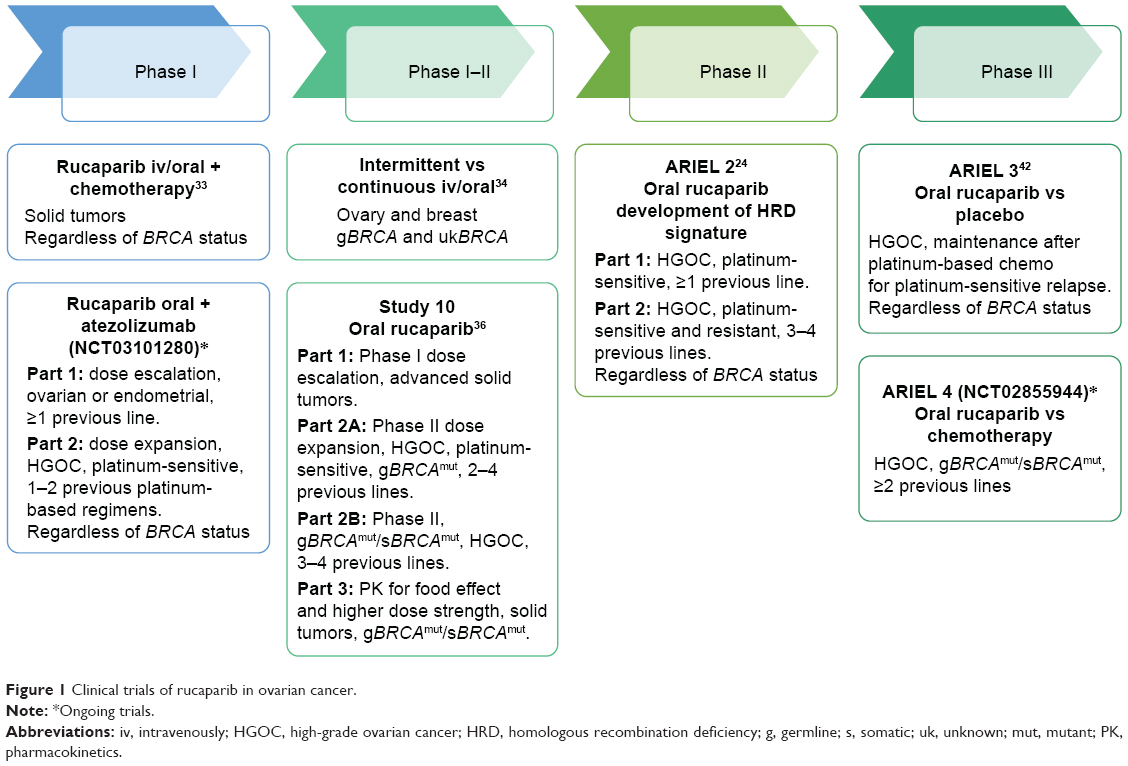 | Figure 1 Clinical trials of rucaparib in ovarian cancer. |
 | Table 1 Summary of rucaparib efficacy in ovarian cancer |
Preclinical studies
Rucaparib has shown activity in BRCA-mutant ovarian and breast cancer cell lines, xenografts and also in cell lines with other homologous recombination genes knocked-down (eg, RAD51, FANCM, PALB2, ATR and BARD1), confirming the hypothesis of synthetic lethality.29 As observed with other PARP inhibitors, another mechanism of action of rucaparib is related to PARP-1 and -2 trapping, which has been confirmed in preclinical models.14 In an animal model, dose-related bone marrow and gastrointestinal (diarrhea, vomiting, appetite reduction and weight loss) toxicities have been observed and were self-limiting.30 No cardiac or neurological toxicities occurred.30
Preliminary antiproliferative activity of rucaparib has been demonstrated in breast and pancreatic cancer cell lines carrying mutated or epigenetically silenced BRCA1/2 or other HRD genes. Rucaparib was also able to reduce the growth of xenograft tumors with mutated or silenced BRCA1/2.29 Subsequently, the activity of rucaparib was investigated in 39 ovarian cancer cell lines characterized by BRCA1/2 mutation, BRCA1/2 promoter methylation or other DNA repair gene mutations, to define predictors of response.31 This study demonstrated in vitro activity of rucaparib in cancer cells carrying HRD beyond BRCA1/2 mutations, supporting the role of further evaluation in sporadic ovarian cancer.31 In addition, rucaparib showed in vitro synergistic activity with chemotherapy agents, such as topotecan, carboplatin, doxorubicin, paclitaxel and gemcitabine.31 Chemotherapy agents, like platinum, induce DNA damages that cannot be repaired when PARP is inactivated, resulting in increasing cytotoxicity.32
Phase 1
Oral and IV rucaparib in combination with chemoteharpy
To investigate the synergistic role between DNA damaging agents and PARP inhibitors, a Phase I study (NCT01009190) has been designed to investigate incremental doses of iv/oral rucaparib in combination with different chemotherapy regimens (carboplatin, paclitaxel + carboplatin, pemetrexed + cisplatin, epirubicin + cyclophosphamide) in patients with solid tumors irrespective of BRCA status.33 A total of 85 patients have been enrolled: 25.9% (22/85) with diagnosis of breast cancer, 17.6% (15/85) ovarian/primary peritoneal cancer, 9.4% (8/85) lung cancer, 4.7% (4/85) pancreatic or rectal cancer, 36.5% (31/85) other primary cancers and 1.2% (1/85) unknown primary cancer. The recommended Phase II dose (RP2D) for the combination of carboplatin and oral rucaparib was area under the curve (AUC)5 every 3 weeks and 240 mg once a day, respectively.33 Combining chemotherapy with a PARP inhibitor resulted in expected increased toxicity, especially hematological. Across all cohorts, grade (G) ≥3 adverse events (AEs) have been observed in 75% patients (64/85), and the more frequent were neutropenia (27.1%), thrombocytopenia (18.8%), fatigue (12.9%), anemia (11.8%) and nausea (7.1%).33 Seventy-seven patients were evaluable for response: one patient (1.2%) had complete response (CR), nine (10.6%) partial response (PR) and 43 (50.6%) stable disease (SD). Pharmacokinetic analysis showed proportional kinetics, half-life of ~12 hours and good bioavailability (36%) of rucaparib, not influenced by administration of iv chemotherapy.33
Phase I/II
Intermittent versus continuous schedule
A multicenter, two-stage Phase I/II study (NCT00664781) was designed to investigate intermittent versus continuous schedule of single agent iv rucaparib administered for 5 days every 21 days in patients with locally advanced/metastatic breast or ovarian cancer and gBRCA mutations.34 Given the lack of data on rucaparib for the treatment of gBRCA breast and ovarian carcinoma, a dose-escalation phase was incorporated in the trial. Fourteen patients have been enrolled in the dose-escalation phase (stage 1) receiving 4–18 mg/m2 of iv rucaparib, and 18 mg/m2 was established as RP2D. In stage 2, patients received iv rucaparib at RP2D as established in stage 1 and were stratified in four groups according to the type of BRCA mutations (BRCA 1 or 2) and the tumor type (breast or ovary). After preliminary analysis of the first 28 patients, the continuous schedule was found to be associated with improved activity. After oral rucaparib became available, the study was amended, and the new patients enrolled received escalating doses of oral rucaparib (from 92 mg once a day [od] to 600 mg twice a day [bid]) at increased duration (7, 14 or 21 days) according to six different dose levels. A subsequent amendment allowed the enrollment of patients with unknown gBRCA status. In total, 78 patients were enrolled (47 receiving iv rucaparib and 31 oral) with the majority harboring gBRCA1/2 mutation (n=74/78). Twenty-seven patients had breast cancer and 51 ovarian cancer. A total of 73 patients were evaluable for response. The intermittent iv dosing of rucaparib showed minimal activity with objective response rate (ORR) of only 2% and SD ≥12 weeks of 41%. ORR for all six dose levels of oral rucaparib was observed in 12% of patients and SD ≥12 weeks in 63% with increased benefit observed in patients receiving a continuous dose of rucaparib, with diagnosis of ovarian cancer and with the longest platinum-free interval (PFI, time between the last dose of platinum-based chemotherapy and documented disease progression).34 Overall, the ORR observed was lower than expected, likely due to reduced activity of rucaparib if administered intermittently, the low dose used in the dose-escalation part (600 mg bid dose was administered only in one patient) and the inclusion of patients with platinum resistant (PFI <6 months) ovarian cancer, who are less likely to respond to PARP inhibitors.35 The maximum administered dose of rucaparib was 600 mg bid for 21 days continuously; this dose was assessed only on one patient and RP2D was not defined.34
Study 10
Study 10 (NCT01482715) is a three-part Phase I/II study of oral rucaparib designed to define RP2D of rucaparib and preliminary efficacy. Part 1 investigated escalating doses of continuous oral rucaparib (40 to 500 mg od and 240 to 840 mg bid) in patients with advanced solid tumors.36 A total of 56 patients were enrolled (64.3% gBRCA), 48.2% with breast cancer and 35.7% with ovarian cancer. The treatment was safe and tolerable with the most common (≥20% of patients) AEs being fatigue, nausea, vomiting, diarrhea, anemia, thrombocytopenia, neutropenia, decreased appetite, elevation of aspartate transaminase (AST) and/or alanine transaminase (ALT). The maximum tolerated dose was not reached and 600 mg bid was established as RP2D based on the safety and pharmacokinetic assessment. This study also confirmed that rucaparib can be taken with or without food. The RP2D was then assessed in an expanded cohort (Part 2A) of 42 patients with diagnosis of platinum-sensitive (PFI ≥6 months) high-grade (serous and endometrioid) ovarian cancer (HGOC) carrying gBRCA mutation who progressed after two to four previous lines of treatment regimens. Objective responses were observed in 59.5% of patients with a median duration of response of 7.8 months. G3 or 4 AEs occurred in 76.2% of patients (32/42) with the most common being fatigue, nausea, anemia and ALT/AST elevation. Four (9.5%) patients discontinued treatment due to AEs and 29 (69%) required a dose reduction.36 Study 10 Part 1 and 2A defined 600 mg bid as the RP2D of rucaparib and showed promising activity in gBRCA-mutated platinum-sensitive HGOC with manageable toxicity profile. Part 2B of this trial enrolled patients with platinum-sensitive or -resistant/refractory (PFI <6 months) HGOC with gBRCA or sBRCA mutation who had received two to four previous lines of treatment. Part 3 was designed to investigate pharmacokinetics and safety of higher dose tablets (300 mg) of rucaparib in patients with relapsed solid tumor and germline or somatic BRCA1/2 mutation.36 For Part 2B and 3, recruitment is completed and results are pending.
Phase II
ARIEL 2
ARIEL 2 (NCT01891344) is a multicenter, two-part Phase II open-label study assessing rucaparib in relapsed/progressive high-grade serous or endometrioid ovarian carcinoma. Part 1 enrolled patients with platinum-sensitive (PFI ≥6 months) HGOC having progressed after at least one previous platinum-based chemotherapy. Part 2 enrolled patients who had received at least three but no more than four previous chemotherapy regimens, including patients with platinum-sensitive (PFI ≥6 months), platinum-resistant (PFI <6 months) and platinum-refractory disease (PFI <2 months). However, a PFI ≥6 months following the first line of platinum treatment was required for all patients. In addition, all patients had measurable disease according to RECIST 1.172 criteria and underwent baseline tumor biopsy. Rucaparib was administered continuously at 600 mg bid in a 28-day cycle. The aim of Part 1 was to develop a tumor-based molecular signature of HRD capable of predicting rucaparib activity in patients with relapsed high-grade serous or endometrioid ovarian cancer beyond germline or somatic BRCA mutations. Indeed, as a consequence of HRD, genomic scars accumulate and may be measured as extension of genomic loss of heterozygosity (LOH).37 An LOH score measuring HRD was assessed in previous studies and showed an ability to predict HRD regardless of the underlying causal mechanism.37–39 In the ARIEL 2 study, the percent of LOH has been measured on baseline and archival tissue with the Foundation Medicine Next-Generation Sequencing (NGS) assay40 using a cutoff of 14%, based on data from the analysis of The Cancer Genome Analysis.15 Between October 2013 and December 2014, 206 patients were enrolled in ARIEL 2 Part 1 study and 204 patients received rucaparib.24 One-hundred and ninety-two patients were assigned to one of three HRD categories assessed on the most recent collected tumor sample (baseline tumor biopsy if available or most recent archival tissue when biopsy was not performed): 40 deleterious germline or somatic BRCA-mutant (BRCAmut) patients; 82 BRCA–wild-type and LOH high (BRCAwt/LOHhigh); and 70 BRCA–wild-type and LOH low (BRCAwt/LOHlow). Median progression-free survival (PFS) was significantly longer in BRCAmut subgroup (12.8 months; HR 0.27, 95% CI 0.16–0.44, p<0.0001) and in BRCAwt/LOHhigh (5.7 months; HR 0.62, 95% CI 0.42–0.90, p=0.011) compared to BRCAwt/LOHlow subgroup (5.2 months). Response rate by RECIST 1.1 criteria was higher in the BRCAmut (80%, 95% CI 64%–91%, p<0.0001) and in BRCAwt/LOHhigh (29%, 95% CI 20%–40%, p=0.0033) than BRCAwt/LOHlow subgroup (10%, 95% CI 4%–20%). Genomic LOH was shown to be a better predictor of response to rucaparib in patients with BRCA wild-type tumors with a sensitivity of 78%, compared to mutation in other HRD genes (sensitivity 11%, p<0.0001) or methylation of BRCA1 or RAD51C (sensitivity 48%, p<0.021). However, by combining mutations in HRD genes and methylation of BRCA1 or RAD51C, no statistically different sensitivity was observed (sensitivity 59%, p=0.13).24 All 204 patients were evaluable for toxicity and the most common G3 AEs were anemia (22% patients) and increase in ALT/AST (12%). Thirty-nine percent of the patients required dose reduction, mainly due to anemia and nausea. Discontinuation occurred in 9% of the patients as a consequence of AEs, with the most common cause being fatigue. LOH has been assessed on archival tissue and baseline biopsy with high concordance (Fisher’s exact test, r=0.86, p<0.0001). Seventeen of 50 patients (34%) had a change in LOH from low in archival tissue to high in baseline biopsy and five achieved a partial response. No change of LOH from high to low was identified.24 ARIEL 2 Part 1 trial confirmed the feasibility of a tumor-based molecular signature of HRD to select patients most likely to benefit from PARP inhibitor treatment, using an algorithm that combines a measure of genomic LOH and BRCA mutations assessed with tumor-based next-generation sequencing assay.24 A retrospective analysis of these results has established that the cutoff ≥16% to define LOH high is more appropriate to discriminate improvement in PFS and ORR in BRCA–wild-type patients.41 Other trials have further investigated the potential predictive role of genomic LOH score: ARIEL 2 Part 2 study included patients with platinum-resistant ovarian cancer and the prospective randomized Phase III trial (ARIEL 3, NCT01968213)42 was designed to confirm the cutoff and validate the assay in the maintenance setting.
In addition, a preliminary analysis on 18 patients enrolled in ARIEL 2 Part 1 study showed that variations in TP53 mutant allele fraction detected on circulating tumor DNA (ctDNA) from plasma samples collected before, during treatment and at the end of treatment (total 65 samples) correlated with response to rucaparib.43 Wild-type p53 acts as checkpoint molecule and helps in maintaining genomic stability.44 Mutation in TP53 or lack of its expression is the most frequent genomic aberration in all tumor types and in HGOC represents an early and common event, likely seen in precursor lesions.45 For 18 patients, ctDNA isolated during screening, at day 1 of each cycle and at the end of treatment with rucaparib, and TP53 mutations were concordant between tumor tissue and ctDNA. Responses according to RECIST 1.1 and Gynecologic Cancer InterGroup (GCIG) combined RECIST and cancer antigen 125 (CA-125) criteria were evaluable in 14 patients. Nine patients (9/14; 64%) had >50% reduction in TP53 mutant allele fraction, among whom seven showed a partial response according to RECIST 1.1 criteria (7/9, 78%). No objective response was observed among patients with <50% reduction in TP53 mutant allele fraction. Further analysis of all patients enrolled on this trial is ongoing to confirm the role of ctDNA as a predictive biomarker.43
Joint analysis of Study 10 and ARIEL 2
An integrated efficacy analysis of Study 10 (Part 2A) and ARIEL 2 (Parts 1 and 2) has been performed to further define the efficacy and safety of rucaparib 600 mg bid as treatment in patients with HGOC and deleterious germline (Study 10) or somatic BRCA1/2 mutation (Study 10 and ARIEL 2) previously treated with at least two lines of chemotherapy.46 For the integrated safety analysis, patients enrolled in Parts 1, 2A and 3 of Study 10 and in Parts 1 and 2 of ARIEL 2 and who had received at least one dose of rucaparib 600 mg were included regardless of the BRCA1/2 status.46 The integrated efficacy analysis included a total of 106 patients: 42 from Study 10 (Part 2A) and 64 from ARIEL 2 (Parts 1 and 2). Deleterious BRCA1/2 mutations were present in all patients: 83% germline and 17% somatic; among them, 63.2% were in BRCA1 and 36.8% in BRCA2. Sixty-one percent of the patients were treated with ≥3 previous lines of chemotherapy and 74.5% had a PFI ≥6 months from the last platinum-based chemotherapy. Investigator-assessed ORR per RECIST 1.1 criteria was 53.5% (95% CI 43.8–63.5) and 70.8% (95% CI 61.1–79.2) per GCIG combined RECIST and CA-125 response criteria. Median duration of response was 9.2 months (range 1.7–19.8 months, 95% CI 6.6–11.6 months). Higher ORR was observed in less pretreated patients (2 lines vs ≥3: 68.3% vs 44.6%), in subjects with PFI >12 months (73.9%) compared to PFI 6–12 months (62.5%) or <6 months (18.5%) and if sensitive (65.8%) to the most recent platinum-based chemotherapy compared to resistant (25%). Investigator-assessed PFS was 10 months (95% CI 7.3–12.5) with 47% of patients still receiving rucaparib at the time of data cutoff.46 The integrated safety analysis included 377 patients (62 from Study 10 [Parts 1, 2A and 3] and 315 from ARIEL 2 [Parts 1 and 2]) and confirmed the manageable toxicity profile of rucaparib. All patients had at least one treatment-related AE and 60.7% of patients had >G3 AEs. The most common AEs (all grades) were nausea, asthenia/fatigue, vomiting and anemia. Increase of AST/ALT and creatinine was commonly observed, usually in the first weeks of treatment followed by stabilization, and in the majority of cases was reversible and asymptomatic.46 The most common >G3 AEs were anemia, AST/ALT increase and asthenia. One patient developed myelodysplastic syndrome (MDS) and another one acute myeloid leukemia (AML). Both patients had BRCA–wild-type tumors and received 12 cycles of platinum-based chemotherapy prior to the trial. Rucaparib was interrupted in 58.6% of patients and dose reduced in 45.9% due to treatment-related AEs. In 9.8% of patients, AEs lead to rucaparib discontinuation.46
Phase III
ARIEL 3
ARIEL 3 (NCT01968213) is a multicenter, randomized, double-blind, placebo-controlled Phase III study assessing efficacy and safety of rucaparib as maintenance treatment following response to platinum-based chemotherapy for platinum-sensitive relapse of high-grade serous or endometrioid epithelial ovarian, primary peritoneal or fallopian tube cancer.42 Patients must have had achieved a radiological CR or PR as per RECIST1.1 criteria or CA-125 response by GCIG criteria (when disease was not measurable per RECIST1.1) following the last platinum-based chemotherapy, with all the patients required to have CA-125 below the upper limit of normal.47 Patients experiencing partial response after platinum-based chemotherapy and with measurable residual disease were suitable for enrollment. Patients were randomized 2:1 to receive rucaparib 600 mg bid or placebo in a 28-day cycle, stratified by HRD status on archival tissue, interval between penultimate platinum-based chemotherapy, radiological previous PFI (6–12 months vs >12 months) and response to last platinum-based chemotherapy (CR vs PR or CA-125 GCIG response). Randomization had to occur within 8 weeks from the last dose of platinum-based chemotherapy. Primary objective was to assess PFS in three different nested predefined cohorts: 1) BRCA-mutated patients (germline or somatic); 2) HRD (BRCA-mutant or BRCA–wild-type and high LOH) assessed using the Foundation Medicine T5 NGS assay and 16% cutoff as defined following the results of ARIEL 2 Part 1 study; and 3) intention-to-treat (ITT) population. Secondary objectives were overall survival, safety, patient-reported outcomes and response rate in patients with measurable disease at study entry. A total of 564 patients were enrolled (375 in rucaparib arm and 189 in placebo arm). Rucaparib significantly improved investigator-assessed median PFS in all three groups: 1) deleterious BRCA mutation: 16.6 months in the rucaparib group (n=130) versus 5.4 months in the placebo (n=66) (HR 0.23, 95% CI 0.16–0.34, p<0.0001); 2) HRD: 13.6 months in the rucaparib group (n=236) versus 5.4 months in placebo (n=118) (HR 0.32, 95% CI 0.2–0.42, p>0.0001); 3) ITT: 10.8 months in the rucaparib arm versus 5.4 months in the placebo (HR 0.36, 95% CI 0.30–0.45, p<0.0001). A preplanned subgroup analysis confirmed the statistically significant improvement of PFS with rucaparib versus placebo in all the subgroups: BRCA mutation types (BRCA 1 vs 2, germline vs somatic), BRCA wild-type and LOH high versus low (LOH high: PFS 9.7 months with rucaparib vs 5.4 months with placebo [HR 0.44], LOH low: 6.7 months vs 5.4 months [HR 0.58], respectively), measurable disease at baseline (present vs absent and bulky vs non-bulky), number of previous lines of chemotherapy (2 vs ≥3), previous use of bevacizumab (yes vs no), PFI to penultimate platinum-based chemotherapy (6–12 months vs >12 months), response to last platinum-based chemotherapy (CR vs PR or GCIG CA-125 response). Response rate according to RECIST1.1 criteria was assessed in patients with measurable disease at the time of enrollment (n=207 in the ITT population); 18% of the patients in the rucaparib arm (26/141) achieved a confirmed objective response (7% complete, 10/141) and 8% (5/66) in the placebo group (2% complete, 1/66).42 Overall survival (OS) data are not yet mature. A safety analysis was conducted in 372 patients receiving rucaparib and 189 receiving placebo. Three patients in the rucaparib arm withdrew consent before the first dose of study medication. Treatment-emergent AEs were observed in 100% of patients in the rucaparib group and 96% in the placebo group. The majority of side effects were easily managed and the most common were consistent with the ones reported in previous studies,24,34,36 including nausea, fatigue, dysgeusia, anemia, constipation and vomiting.42 G ≥3 treatment-emergent AEs occurred in 54% of the patients in the rucaparib arm and 15% in the placebo arm with the most common being anemia and transaminitis. Serious AEs occurred in 21% of patients receiving rucaparib and in 11% of patients receiving placebo. Treatment was discontinued due to AEs in 13% of patients receiving rucaparib and 2% receiving placebo. Four deaths occurred due to AEs, two of them were treatment related and due to AML and MDS. The ARIEL 3 study confirmed the role of PARP inhibitors as maintenance treatment following response to platinum-based chemotherapy in patients with platinum-sensitive relapse of ovarian carcinoma.42 Rucaparib has shown to be active not only in patients with BRCA deleterious mutation but also in patients with non-BRCA-related HRD; however, the overall population enrolled in the present trial, including BRCA–wild-type and LOH-low patients, achieved a benefit from rucaparib. Whilst the Foundation Medicine T5 NGS LOH score could be used to select patients who would benefit the most from rucaparib, it cannot be incorporated in routine practice as a predictive biomarker of response given improvement in PFS has also been observed in patients with BRCA–wild-type and LOH-low tumors.42 Results on patient-reported health outcomes are not available yet and will be the subject of a separate publication.
Ongoing trials in ovarian cancer
Numerous trials are ongoing to further investigate rucaparib activity and safety and to confirm the predictive role of the HRD assay and LOH algorithm developed in the ARIEL 2 trial.
ARIEL 2 Part 2
ARIEL 2 Part 2 (NCT01891344) has been designed to explore the role of rucaparib regardless of PFI and to further assess the role of LOH genomic score to predict rucaparib activity. Patients with high-grade serous or endometrial carcinoma who have progressed after three or four previous lines of chemotherapy have been enrolled. The study was designed to enroll 300 patients including at least 80 with gBRCA or sBRCA mutations. The primary objective is to define ORR by molecular subgroup (BRCAmut, BRCAwt/LOHhigh, BRCAwt/LOHlow). Secondary objectives are PFS, OS and safety. Exploratory objectives include the assessment of efficacy end points according to the three molecular subgroups, the optimization of the LOH algorithm and the assessment of potential changes in HRD over time. Accrual has been completed and data analyses are awaited.
ARIEL 4
Despite increasing evidence of PARP inhibitors’ activity in patients with BRCA mutations, no direct comparisons with standard chemotherapy agents are available. ARIEL 4 (NCT02855944) is a multicenter, randomized Phase III study designed to compare efficacy and safety of rucaparib versus standard chemotherapy in patients with BRCA-mutant HGSOC or G2-3 endometrioid ovarian, primary peritoneal or fallopian tube cancer, and is currently enrolling patients. Patients with a deleterious BRCA1/2 mutation (germline or somatic) who had received at least two previous lines of chemotherapy can be enrolled and randomized in a 2:1 ratio to receive rucaparib 600 mg bid or weekly paclitaxel (if PFI after last dose of platinum-based chemotherapy was between 1 and 12 months) or platinum-based chemotherapy (if PFI ≥12 months). Patients with platinum-refractory disease are excluded from the study. Other histological subtypes can be considered if the patient harbors BRCA1/2 mutations. The primary objective is difference in investigator-assessed PFS between rucaparib and chemotherapy. Main secondary objectives are OS, ORR, patient-reported outcome and safety. Exploratory objectives will include assessment of molecular changes over time, cell-free tumor DNA as a marker of response and pharmacokinetic analysis.
Rucaparib + atezolizumab
In the last decade, significant advances have been obtained in the field of immunotherapy and anti-program death 1 (PD1)/program death ligand 1 (PD-L1) agents are under investigation in EOC as single agents or in combination. Preclinical data in cancer cell lines and xenografts demonstrated increased PD1-mediated immunosuppression following treatment with PARP inhibitors suggesting the rationale to combine anti-PD1/PDL1 agents with PARP inhibitors.48 A Phase Ib study (NCT03101280) is investigating the combination of rucaparib with the anti-PDL1 atezolizumab in patients with advanced gynecological cancer. In Part 1 (dose escalation) study, patients with advanced ovarian or endometrial cancer who had received at least one previous line of chemotherapy will receive an escalating dose of oral rucaparib starting from 400 mg bid up to 600 mg bid in a 21-day cycle and atezolizumab 1,200 mg iv every 3 weeks. In Part 2 (dose expansion) study, rucaparib will be administered at the RP2D as defined in the dose-escalation part in combination with atezolizumab (same dose and schedule as Part 1) in patients with platinum-sensitive relapsed HGSOC or G2–3 endometrioid ovarian carcinoma after at least one but no more than two platinum-based chemotherapy regimens. This study is currently open to accrual and recruiting patients.
Other studies
Other Phase I studies are ongoing to assess drug-to-drug interactions of rucaparib with caffeine, warfarin, omeprazole, midazolam, digoxin and vitamin K (NCT02740712) and to explore mass balance, absorption, metabolism and elimination of a single oral dose of 600 mg [14C] rucaparib in patients with advanced solid tumors (NCT02986100).
Mechanisms of resistance
Despite promising results obtained with the introduction of PARP inhibitors for the treatment of women with ovarian carcinoma, the majority of patients will ultimately develop disease progression, and different mechanisms of resistance have been proposed (Table 2). Reversion of BRCA1/2 mutations has been described as one of the possible mechanisms of resistance to chemotherapy and to PARP inhibitors.49–52 Given the increased evidence of the role of HRD in defining PARP inhibitor activity, investigations are ongoing to assess the role of somatic reversion mutations in HRD genes as a mechanism of resistance. Kondrashova et al have reported results from 12 patients with platinum-sensitive HGSOC treated with rucaparib as part of the ARIEL 2 Part 1 trial and with paired biopsies (pre-treatment and post-progression) available for analysis.53 From the six patients who had a germline or somatic mutation (four in BRCA1, one in RAD51C and one in RAD51D), five developed a secondary mutation restoring gene activity on the biopsy at disease progression, including RAD51C and RAD51D. Following secondary mutations, homologous recombination activity was restored resulting in rucaparib resistance, supporting the relevant role of non-BRCA1/2 HRD in PARP inhibitors’ sensitivity and resistance.53 In the absence of BRCA1/2 reversion, resistance to PARP inhibitors can be caused by hyperactivation of the NHEJ pathway. The TP53 binding 1 protein (53BP1) is responsible for the balance between NHEJ and homologous recombination in repairing DNA DSBs and is regulated by BRCA1. When HRD is present, 53BP1 promotes activity of the NHEJ pathway. When 53BP1 is lost, homologous recombination activity is restored and such resistance to PARP inhibitors is possible.54,55 Cell cultures with NHEJ deficiency showed resistance to rucaparib, whilst cultures with HRD and competent NHEJ were sensitive to rucaparib.56 Another reported mechanism of resistance is correlated with the loss of MLL3/4 complex protein whose role is to stall the replication forks favoring protection from DNA damage, thus causing resistance to PARP inhibitors or other DNA damaging agents.54,57,58 Further investigation is required to assess PARP inhibitors in combination with other agents that are able to overcome mechanisms of PARP resistance (Table 2).
 | Table 2 Potential mechanisms of resistance to PARP inhibitors |
Toxicity
Rucaparib has been shown to be manageable and relatively well tolerated with AEs commonly seen with other PARP inhibitors. At the dose of 600 mg bid, the most common AEs observed across the different trials were fatigue, nausea/vomiting, myelosuppression and ALT/AST elevation (Table 3). These side effects have been easily managed with supportive care and/or rucaparib dose reduction or delays (Table 4).
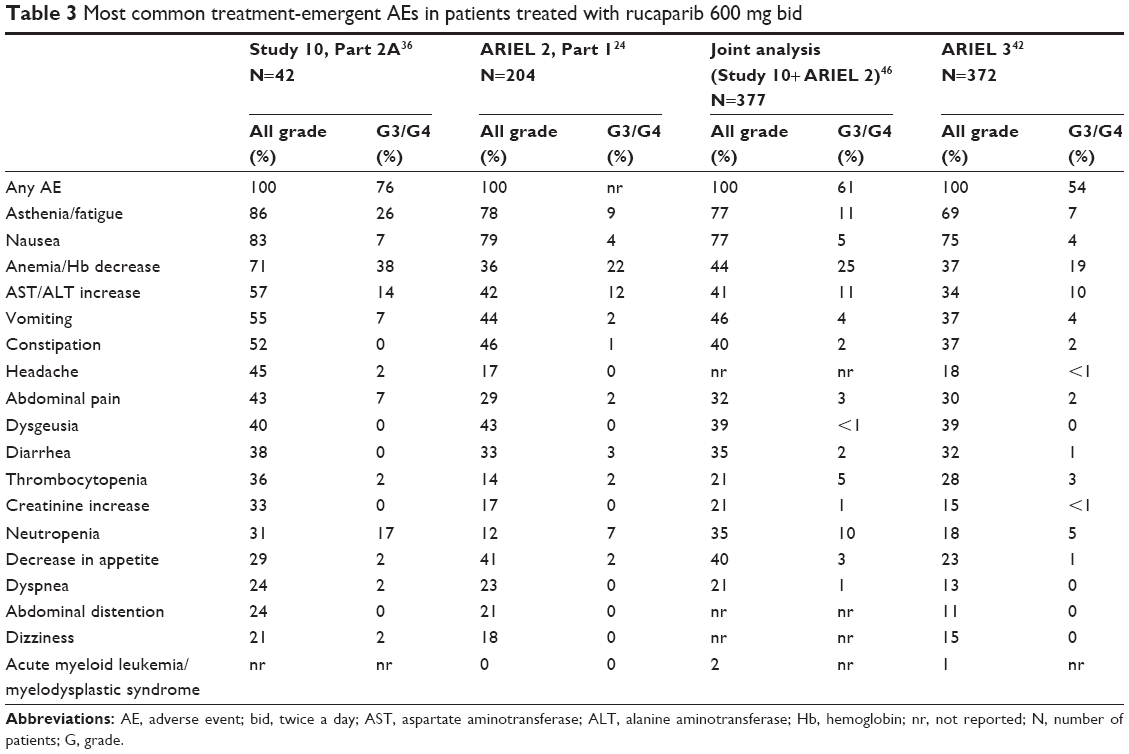 | Table 3 Most common treatment-emergent AEs in patients treated with rucaparib 600 mg bid |
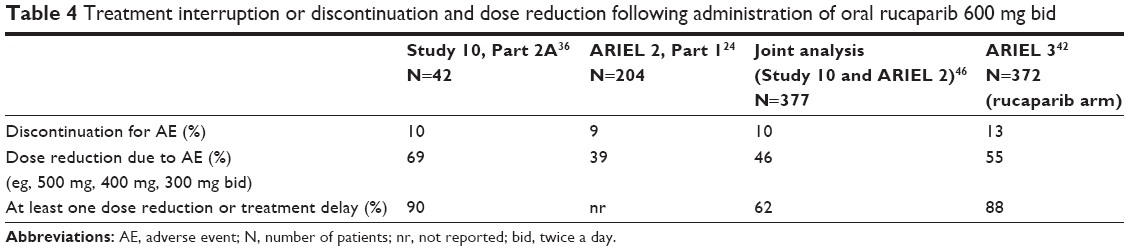 | Table 4 Treatment interruption or discontinuation and dose reduction following administration of oral rucaparib 600 mg bid |
Increase in serum creatinine level has been observed in patients treated with rucaparib, as with other PARP inhibitors. This AE may be due to reduction in creatinine secretion at the level of the proximal tubule as a consequence of inhibition of the tubule transporters MATE1, MATE2-K and OCT-2.71 No G3 or G4 creatinine increase has been reported with rucaparib. Increase of transaminase has also been observed, and the cause of this phenomena has not been well established yet. However, this AE is generally transient and managed with dose reduction or treatment delay and for asymptomatic <G3 transaminitis treatment, rucaparib can be continued.
MDS and AML have been rarely observed in patients treated with rucaparib in Phase II (Study 10 and ARIEL 2) and 3 (ARIEL 3) studies.24,36,42,46 In the ARIEL 3 trial, three patients had MDS or AML in the rucaparib arm and none in the placebo arm. Two patients had germline BRCA-mutant carcinoma and one BRCA–wild-type and LOH-low tumor. One patient died due to progressive MDS and one due to AML.42 MDS and AML have been reported with other PARP inhibitors. In the SOLO2 (NCT01874353) trial assessing maintenance olaparib versus placebo in patients with platinum-sensitive recurrence of HGOC and BRCA1/2 mutation, two patients developed AML, one chronic myelomonocytic leukemia and one MDS (total 4/195, 2%) in the olaparib arm and three MDS and one AML (total 4/99, 4%) in the placebo arm.22 In the NOVA (NCT01847274) trial investigating maintenance niraparib versus placebo in platinum-sensitive relapse of HGSOC regardless of BRCA status, five cases of MDS were observed in the niraparib group (5/367, 1.4%) and one MDS and one AML (2/179, 1.1%) in the placebo group.26 No definitive data are available on the possible causes of MDS/AML occurrence in patients treated with PARP inhibitors. However, previous exposure to chemotherapy or radiotherapy and the presence of gBRCA mutations might concur in increasing the risk of other malignancies.
Conclusion
Rucaparib is a potent inhibitor of PARP-1, PARP-2 and PARP-3 and has been investigated in different Phase II and 3 studies as treatment for patients with progressive EOC after at least two previous lines of chemotherapy and as maintenance treatment following response to platinum-based chemotherapy for platinum-sensitive relapse.24,42,46 The FDA has approved oral rucaparib 600 mg bid as treatment for patients with deleterious BRCA1/2-mutated (germline or somatic) EOC who had received ≥2 previous lines of chemotherapy. Along with rucaparib, the FDA also approved the first NGS-based companion diagnostic (FoundationFocus CDxBRCA test) to assess BRCA1/2 mutation in the tumor tissue to select the patients who could benefit the most from rucaparib.
Subsequently, the ARIEL 3 study has established the role of rucaparib as maintenance treatment following response to platinum-based chemotherapy for platinum-sensitive relapse of HGOC. PFS was significantly better in patients treated with rucaparib in the different cohorts: deleterious BRCA1/2 mutation, HRD and ITT population.42 The Foundation Medicine NGS assay was confirmed to be a useful tool to select patients who will benefit the most from rucaparib; however, it cannot be used as a predictive biomarker as benefit from rucaparib has been observed in patients with BRCA–wild-type and LOH-low tumors.42
Whilst different PARP inhibitors have been shown to be active, a direct comparative analysis of agents is not feasible. This is primarily due to the fact that trials assessing olaparib, niraparib, veliparib and rucaparib differ from one another in selection of patients (only BRCA1/2-mutated or also BRCA1/2–wild-type), HRD definition, study design, inclusion or exclusion of patients with residual bulky disease or abnormal CA-125 level for maintenance studies. Moreover, each PARP inhibitor has its characteristic toxicity profile.
Further research is warranted to define biomarkers that are able to predict PARP inhibitor activity, to overcome mechanisms of PARP inhibitors resistance and investigate the appropriate combination of PARP inhibitors with other agents like immune checkpoint inhibitors, cell cycle inhibitors or other DNA damaging agents such as chemotherapy and radiotherapy.
Disclosure
The authors report no conflicts of interest in this work.
References
Cancer Stat Facts: Ovarian Cancer. National Cancer Institute. Surveillance, Epidemiology, and End Results Program. Available from https://seer.cancer.gov/statfacts/html/ovary.html. Accessed September 10, 2017. | ||
Fleiming GF, Seidman J, Lengyel E. Epithelial ovarian cancer. In: Principles and Practice of Gynecologic Oncology. 6th ed. Philadelphia: Lippincott Williams and Wilkins; 2013:757–847. | ||
Seidman JD, Horkayne-Szakaly I, Haiba M, Boice CR, Kurman RJ, Ronnett BM. The histologic type and stage distribution of ovarian carcinomas of surface epithelial origin. Int J Gynecol Pathol. 2004;23(1):41–44. | ||
Hoeijmakers JH. DNA damage, aging, and cancer. N Engl J Med. 2009;361(15):1475–1485. | ||
Davis AJ, Chen DJ. DNA double strand break repair via non-homologous end-joining. Transl Cancer Res. 2013;2(3):130–143. | ||
Szostak JW, Orr-Weaver TL, Rothstein RJ, Stahl FW. The double-strand-break repair model for recombination. Cell. 1983;33(1):25–35. | ||
Cooper GM. The Cell: A Molecular Approach. 2nd ed. Sunderland, MA: Sinauer Associates; 2000. Available from: https://www.ncbi.nlm.nih.gov/books/NBK9900/. Accessed September 10, 2017. | ||
Moynahan ME, Chiu JW, Koller BH, Jasin M. Brca1 controls homology-directed DNA repair. Mol Cell. 1999;4(4):511–518. | ||
Moynahan ME, Pierce AJ, Jasin M. BRCA2 is required for homology-directed repair of chromosomal breaks. Mol Cell. 2001;7(2):263–272. | ||
Schreiber V, Dantzer F, Ame JC, de Murcia G. Poly(ADP-ribose): novel functions for an old molecule. Nat Rev Mol Cell Biol. 2006;7(7):517–528. | ||
Helleday T, Petermann E, Lundin C, Hodgson B, Sharma RA. DNA repair pathways as targets for cancer therapy. Nat Rev Cancer. 2008;8(3):193–204. | ||
Farmer H, McCabe N, Lord CJ, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434(7035):917–921. | ||
Helleday T. The underlying mechanism for the PARP and BRCA synthetic lethality: clearing up the misunderstandings. Mol Oncol. 2011;5(4):387–393. | ||
Murai J, Huang SY, Das BB, et al. Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer Res. 2012;72(21):5588–5599. | ||
Cancer Genome Atlas Research N. Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474(7353):609–615. | ||
Pennington KP, Walsh T, Harrell MI, et al. Germline and somatic mutations in homologous recombination genes predict platinum response and survival in ovarian, fallopian tube, and peritoneal carcinomas. Clin Cancer Res. 2014;20(3):764–775. | ||
Tan DS, Rothermundt C, Thomas K, et al. “BRCAness” syndrome in ovarian cancer: a case-control study describing the clinical features and outcome of patients with epithelial ovarian cancer associated with BRCA1 and BRCA2 mutations. J Clin Oncol. 2008;26(34):5530–5536. | ||
Turner N, Tutt A, Ashworth A. Hallmarks of “BRCAness” in sporadic cancers. Nat Rev Cancer. 2004;4(10):814–819. | ||
Konstantinopoulos PA, Spentzos D, Karlan BY, et al. Gene expression profile of BRCAness that correlates with responsiveness to chemotherapy and with outcome in patients with epithelial ovarian cancer. J Clin Oncol. 2010;28(22):3555–3561. | ||
Kaufman B, Shapira-Frommer R, Schmutzler RK, et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J Clin Oncol. 2015;33(3):244–250. | ||
Kim G, Ison G, McKee AE, et al. FDA approval summary: olaparib monotherapy in patients with deleterious germline BRCA-mutated advanced ovarian cancer treated with three or more lines of chemotherapy. Clin Cancer Res. 2015;21(19):4257–4261. | ||
Pujade-Lauraine E, Ledermann JA, Selle F, et al. Olaparib tablets as maintenance therapy in patients with platinum-sensitive, relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT-Ov21): a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2017;18(9):1274–1284. | ||
Ledermann J, Harter P, Gourley C, et al. Olaparib maintenance therapy in platinum-sensitive relapsed ovarian cancer. N Engl J Med. 2012;366(15):1382–1392. | ||
Swisher EM, Lin KK, Oza AM, et al. Rucaparib in relapsed, platinum-sensitive high-grade ovarian carcinoma (ARIEL2 Part 1): an international, multicentre, open-label, phase 2 trial. Lancet Oncol. 2017;18(1):75–87. | ||
Balasubramaniam S, Beaver JA, Horton S, et al. FDA approval summary: rucaparib for the treatment of patients with deleterious BRCA mutation-associated advanced ovarian cancer. Clin Cancer Res. 2017;23(3):7165–7170. | ||
Mirza MR, Monk BJ, Herrstedt J, et al. Niraparib maintenance therapy in platinum-sensitive, recurrent ovarian cancer. N Engl J Med. 2016;375(22):2154–2164. | ||
Robson M, Im SA, Senkus E, et al. Olaparib for metastatic breast cancer in patients with a germline BRCA mutation. N Engl J Med. 2017;377(6):523–533. | ||
Mateo J, Carreira S, Sandhu S, et al. DNA-repair defects and olaparib in metastatic prostate cancer. N Engl J Med. 2015;373(18):1697–1708. | ||
Drew Y, Mulligan EA, Vong WT, et al. Therapeutic potential of poly(ADP-ribose) polymerase inhibitor AG014699 in human cancers with mutated or methylated BRCA1 or BRCA2. J Natl Cancer Inst. 2011;103(4):334–346. | ||
Rucaparib tablet (camsylate form) for oral administration. Investigator’s Brochure v9. January 2017. Clovis Oncology, Inc.. | ||
Ihnen M, zu Eulenburg C, Kolarova T, et al. Therapeutic potential of the poly(ADP-ribose) polymerase inhibitor rucaparib for the treatment of sporadic human ovarian cancer. Mol Cancer Ther. 2013;12(6):1002–1015. | ||
Calabrese CR, Almassy R, Barton S, et al. Anticancer chemosensitization and radiosensitization by the novel poly(ADP-ribose) polymerase-1 inhibitor AG14361. J Natl Cancer Inst. 2004;96(1):56–67. | ||
Wilson RH, Evans TJ, Middleton MR, et al. A phase I study of intravenous and oral rucaparib in combination with chemotherapy in patients with advanced solid tumours. Br J Cancer. 2017;116(7):884–892. | ||
Drew Y, Ledermann J, Hall G, et al. Phase 2 multicentre trial investigating intermittent and continuous dosing schedules of the poly(ADP-ribose) polymerase inhibitor rucaparib in germline BRCA mutation carriers with advanced ovarian and breast cancer. Br J Cancer. 2016;114(12):e21. | ||
Fong PC, Yap TA, Boss DS, et al. Poly(ADP)-ribose polymerase inhibition: frequent durable responses in BRCA carrier ovarian cancer correlating with platinum-free interval. J Clin Oncol. 2010;28(15):2512–2519. | ||
Kristeleit R, Shapiro GI, Burris HA, et al. A phase I–II study of the oral PARP inhibitor rucaparib in patients with germline BRCA1/2-mutated ovarian carcinoma or other solid tumors. Clin Cancer Res. 2017;23(15):4095–4106. | ||
Abkevich V, Timms KM, Hennessy BT, et al. Patterns of genomic loss of heterozygosity predict homologous recombination repair defects in epithelial ovarian cancer. Br J Cancer. 2012;107(10):1776–1782. | ||
Telli ML, Timms KM, Reid J, et al. Homologous Recombination Deficiency (HRD) Score predicts response to platinum-containing neoadjuvant chemotherapy in patients with triple-negative breast cancer. Clin Cancer Res. 2016;22(15):3764–3773. | ||
Timms KM, Abkevich V, Hughes E, et al. Association of BRCA1/2 defects with genomic scores predictive of DNA damage repair deficiency among breast cancer subtypes. Breast Cancer Res. 2014;16(6):475. | ||
Frampton GM, Fichtenholtz A, Otto GA, et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol. 2013;31(11):1023–1031. | ||
Coleman RL, Swisher EM, Oza AM, et al. Refinement of prespecified cutoff for genomic loss of heterozygosity (LOH) in ARIEL2 part 1: a phase II study of rucaparib in patients (pts) with high grade ovarian carcinoma (HGOC). J Clin Oncol. 2016;34:abstract 5540. | ||
Coleman RL, Oza AM, Lorusso D, et al. Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum therapy (ARIEL3): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2017;390(10106):1949–1961. | ||
Piskorz A, Lin KK, Morris JA, et al. Feasibility of monitoring response to the PARP inhibitor rucaparib with targeted deep sequencing of circulating tumor DNA (ctDNA) in women with high grade serous carcinoma on the ARIEL2 trial. In: ASCO Annual Meeting. Chicago, June 2017. | ||
Soussi T, Wiman KG. Shaping genetic alterations in human cancer: the p53 mutation paradigm. Cancer Cell. 2007;12(4):303–312. | ||
Cole AJ, Zhu Y, Dwight T, et al. Comprehensive analyses of somatic TP53 mutation in tumors with variable mutant allele frequency. Sci Data. 2017;4:170120. | ||
Oza AM, Tinker AV, Oaknin A, et al. Antitumor activity and safety of the PARP inhibitor rucaparib in patients with high-grade ovarian carcinoma and a germline or somatic BRCA1 or BRCA2 mutation: integrated analysis of data from Study 10 and ARIEL2. Gynecol Oncol. 2017;147(2):267–275. | ||
Rustin GJ, Vergote I, Eisenhauer E, et al. Definitions for response and progression in ovarian cancer clinical trials incorporating RECIST 1.1 and CA 125 agreed by the Gynecological Cancer Intergroup (GCIG). Int J Gynecol Cancer. 2011;21(2):419–423. | ||
Jiao S, Xia W, Yamaguchi H, et al. PARP inhibitor upregulates PD-L1 expression and enhances cancer-associated immunosuppression. Clin Cancer Res. 2017;23(14):3711–3720. | ||
Norquist B, Wurz KA, Pennil CC, et al. Secondary somatic mutations restoring BRCA1/2 predict chemotherapy resistance in hereditary ovarian carcinomas. J Clin Oncol. 2011;29(22):3008–3015. | ||
Barber LJ, Sandhu S, Chen L, et al. Secondary mutations in BRCA2 associated with clinical resistance to a PARP inhibitor. J Pathol. 2013;229(3):422–429. | ||
Christie EL, Fereday S, Doig K, Pattnaik S, Dawson SJ, Bowtell DDL. Reversion of BRCA1/2 germline mutations detected in circulating tumor DNA from patients with high-grade serous ovarian cancer. J Clin Oncol. 2017;35(12):1274–1280. | ||
Lheureux S, Bruce JP, Burnier JV, et al. Somatic BRCA1/2 recovery as a resistance mechanism after exceptional response to poly (ADP-ribose) polymerase inhibition. J Clin Oncol. 2017;35(11):1240–1249. | ||
Kondrashova O, Nguyen M, Shield-Artin K, et al. Secondary somatic mutations restoring RAD51C and RAD51D associated with acquired resistance to the PARP inhibitor rucaparib in high-grade ovarian carcinoma. Cancer Discov. 2017;7(9):984–998. | ||
Jaspers JE, Kersbergen A, Boon U, et al. Loss of 53BP1 causes PARP inhibitor resistance in Brca1-mutated mouse mammary tumors. Cancer Discov. 2013;3(1):68–81. | ||
Bouwman P, Aly A, Escandell JM, et al. 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nat Struct Mol Biol. 2010;17(6):688–695. | ||
McCormick A, Donoghue P, Dixon M, et al. Ovarian cancers harbor defects in nonhomologous end joining resulting in resistance to rucaparib. Clin Cancer Res. 2017;23(8):2050–2060. | ||
Chaudhuri AR, Callen E, Ding X, et al. Erratum: replication fork stability confers chemoresistance in BRCA-deficient cells. Nature. 2016;539(7629):456. | ||
Patel AG, Sarkaria JN, Kaufmann SH. Nonhomologous end joining drives poly(ADP-ribose) polymerase (PARP) inhibitor lethality in homologous recombination-deficient cells. Proc Natl Acad Sci U S A. 2011;108(8):3406–3411. | ||
Johnson N, Johnson SF, Yao W, et al. Stabilization of mutant BRCA1 protein confers PARP inhibitor and platinum resistance. Proc Natl Acad Sci U S A. 2013;110(42):17041–17046. | ||
Rondinelli B, Gogola E, Yucel H, et al. EZH2 promotes degradation of stalled replication forks by recruiting MUS81 through histone H3 trimethylation. Nat Cell Biol. 2017;19(11):1371–1378. | ||
Samol J, Ranson M, Scott E, et al. Safety and tolerability of the poly(ADP-ribose) polymerase (PARP) inhibitor, olaparib (AZD2281) in combination with topotecan for the treatment of patients with advanced solid tumors: a phase I study. Invest New Drugs. 2012;30(4):1493–1500. | ||
Garcia TB, Snedeker JC, Baturin D, et al. A Small-molecule inhibitor of WEE1, AZD1775, synergizes with olaparib by impairing homologous recombination and enhancing dna damage and apoptosis in acute leukemia. Mol Cancer Ther. 2017;16(10):2058–2068. | ||
Karnak D, Engelke CG, Parsels LA, et al. Combined inhibition of Wee1 and PARP1/2 for radiosensitization in pancreatic cancer. Clin Cancer Res. 2014;20(19):5085–5096. | ||
Yazinski SA, Comaills V, Buisson R, et al. ATR inhibition disrupts rewired homologous recombination and fork protection pathways in PARP inhibitor-resistant BRCA-deficient cancer cells. Genes Dev. 2017;31(3):318–332. | ||
Murai J, Feng Y, Yu GK, et al. Resistance to PARP inhibitors by SLFN11 inactivation can be overcome by ATR inhibition. Oncotarget. 2016;7(47):76534–76550. | ||
Rottenberg S, Jaspers JE, Kersbergen A, et al. High sensitivity of BRCA1-deficient mammary tumors to the PARP inhibitor AZD2281 alone and in combination with platinum drugs. Proc Natl Acad Sci U S A. 2008;105(44):17079–17084. | ||
Oplustil O’Connor L, Rulten SL, Cranston AN, et al. The PARP inhibitor AZD2461 provides insights into the role of PARP3 inhibition for both synthetic lethality and tolerability with chemotherapy in preclinical models. Cancer Res. 2016;76(20):6084–6094. | ||
Cardnell RJ, Feng Y, Mukherjee S, et al. Activation of the PI3K/mTOR pathway following PARP inhibition in small cell lung cancer. PLoS One. 2016;11(4):e0152584. | ||
Rehman FL, Lord CJ, Ashworth A. The promise of combining inhibition of PI3K and PARP as cancer therapy. Cancer Discov. 2012;2(11):982–984. | ||
Du Y, Yamaguchi H, Wei Y, et al. Blocking c-Met-mediated PARP1 phosphorylation enhances anti-tumor effects of PARP inhibitors. Nat Med. 2016;22(2):194–201. | ||
Kikuchi R, Lao Y, Bow DA, et al. Prediction of clinical drug-drug interactions of veliparib (ABT-888) with human renal transporters (OAT1, OAT3, OCT2, MATE1, and MATE2K). J Pharm Sci. 2013;102(12):4426–4432. | ||
Eisenhauer EA, Therasse P, Bogaerts J, et al. New response evaluations criteria in solid tumours: Revised RECIST guideline (Version 1.1). Eur J Cancer. 2009;(45):228–247. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.