")
Back to Archived Journals » Reports in Organic Chemistry » Volume 6
Recent applications of Cinchona alkaloid-based catalysts in asymmetric addition reactions
Received 19 April 2016
Accepted for publication 2 June 2016
Published 8 September 2016 Volume 2016:6 Pages 47—75
DOI https://doi.org/10.2147/ROC.S73908
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Sean Kerwin
Girija S Singh, Elizabeth MO Yeboah
Chemistry Department, University of Botswana, Gaborone, Botswana
Abstract: This review documents recently developed asymmetric addition reactions catalyzed by natural Cinchona alkaloids and their derivatives. These reactions include direct nucleophilic additions across the carbon–oxygen double bond and the carbon–nitrogen double bond, 1,4-additions, and cycloadditions. Natural and modified Cinchona alkaloids with different functionalities, such as amino, alkoxy, hydroxyl, amido, urea, and thiourea, especially on position C9, have been employed as catalysts in these reactions. Mechanistic considerations are discussed in many cases. In some cases, the application of adducts in further synthesis is also described.
Keywords: Cinchona, asymmetric organocatalysis, nucleophilic additions, Michael additions, cycloadditions
Introduction
Stereocontrol in organic reactions is the most important aspect of synthetic organic chemistry. Among different techniques used for stereoinduction, asymmetric catalysis is becoming an increasingly popular strategy in asymmetric synthetic endeavors, due to design and development of several natural product-derived chiral molecular frameworks as chiral oragnocatalysts.1 The alkaloids of Cinchona species, which were once known for the popular antimalarial drug quinine, have emerged as the most powerful class of compounds in the realm of asymmetric organocatalysis during the last 2 decades.2,3 Apart from natural Cinchona alkaloids, many derivatives, such as those containing hydroxyl groups, amines, ureas, and thiourea functionalities, especially at the C9 position, either alone or in the presence of an additional catalyst that might be a simple achiral compound or metal salt, have been employed in diverse types of enantioselective syntheses by asymmetric catalysis. This can be attributed to the abundance of Cinchona alkaloids in nature, their commercial availability at reasonable prices, stability and easy handling in laboratory, and their convenient modification by simple reactions.
The Cinchona skeleton consists of two rigid rings: an aliphatic quinuclidine and an aromatic quinoline ring joined together by two carbon–carbon single bonds. There are five stereocenters in the molecule. The Cinchona alkaloids occur in pairs, which differ in configurations at C8, C9, and N1 positions. The eight major Cinchona alkaloids (Figure 1) are diastereomers, usually referred to as pseudoenantiomers because they offer enantiomeric products when used as catalysts. The structural features of Cinchona alkaloids and their derivatives responsible for their catalytic activity in terms of yields and diastereoselectivity and enantioselectivity of products have been discussed in our previous review article, and thus will not be repeated here.4 Several studies have shown that Cinchona compounds work as bifunctional catalysts. The quinuclidine ring, bearing a tertiary nitrogen atom, is 103 times more basic in comparison to quinoline nitrogen. The tertiary nitrogen is thus responsible for the basic character of the catalyst. The H-bonding groups, such as the hydroxyl group, urea, and thiourea at the C9 position, activate the electrophile by hydrogen bonding. The relative orientation of the two rings quinoline and quinuclidine creates a “chiral pocket” around the reactive site, forcing a particular approach of the substrates, resulting in enantioselective product formation.4
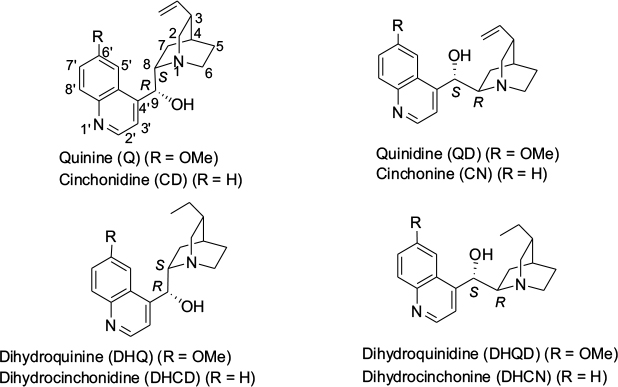 | Figure 1 Structures of eight major Cinchona alkaloids. |
In recent years, Stegbauer et al reviewed bifunctional organo/metal cooperative catalysis with Cinchona alkaloid scaffolds, while Duan and Li updated the applications of primary amines derived from Cinchona alkaloids in asymmetric organocatalysis.5,6 Other Cinchona-alkaloid derivatives employed in inducing chirality in organic reactions include mainly C9 primary amines, C9 ureas and thioureas, C9/C6′ hydroxyl, C9 amides, and C9 squaramides. The objective of this review paper is to discuss the development of selected asymmetric addition reactions catalyzed by Cinchona alkaloids and their derivatives over the last 5 years (2011–2015). These reactions include 1) nucleophilic 1,2-addition to the C=O bond, 2) nucleophilic 1,2 addition to the C=N bond, 3) nucleophilic conjugate/Michael addition, and 4) cycloaddition reactions.
Nucleophilic 1,2-addition to the C=O bond
Nucleophilic 1,2-additions to the C=O bond are the most extensively investigated reactions of carbonyl compounds, and among them, the aldol reaction constitutes the most common and powerful method for carbon–carbon bond formation involving two carbonyl compounds.7,8 The aldol adducts with a new stereocenter serve as power-building blocks in organic synthesis. For example, Franchino et al reported recently the application of asymmetric aldol reaction between p-nitrobenzaldehyde and benzhydryl-2-isocyanoacetate in the synthesis of (–)-chloramphenicol.9 Interestingly, a Cinchona alkaloid-derived amino-phosphine and silver oxide were used as catalysts in this enantioselective aldol reaction. In another report, Zheng and Deng reported the application of aldol adducts 4, obtained from asymmetric direct aldol reaction of alkyl azlactones 1 and aliphatic aldehydes 2, in the synthesis of β-hydroxy-α-amino acids 5-bearing alkyl substituents.10 Such compounds are structural motifs in many natural products. In this study as well, the aldol reaction was catalyzed by a Cinchona-based catalyst. Using the 6′-OH Cinchona alkaloid 3 as the most efficient catalyst under optimized conditions, the authors showed the versatility of the reaction by utilizing various azalactone and aliphatic aldehydes, including one with a C12 linear chain (Figure 2).
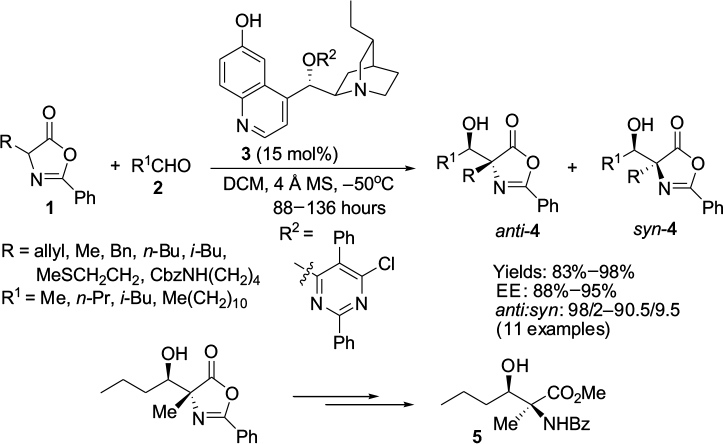 | Figure 2 Asymmetric aldol reaction of azlactones with aliphatic aldehydes. Abbreviations: DCM, dichloromethane; MS, molecular sieves; EE, enantiomeric excess. |
Our previous review described a number of Cinchona alkaloid-based phase-transfer catalysts and Cinchona alkaloid-proline derivatives as efficient organocatalysts for enantioselective aldol reactions.4 Subsequently, several Cinchona alkaloids and their derivatives have been developed as optimized catalysts for asymmetric aldol reactions of various substrates, which are described in following sections. The literature is arranged according to the C9-substitution pattern of the catalysts to showcase the main classes of Cinchona alkaloid-derived catalysts used in asymmetric addition reactions.
Application of C9 primary amine derivatives
A highly enantioselective aldol reaction of benzaldehydes with cyclohexanone in aqueous medium was reported by Wang et al, employing a heterogeneous, porous, and recyclable zirconium phosphonate-supported 9-amino-9-deoxy-epi-cinchonidine catalyst.11 The steric confinement effect of the inorganic backbone of zirconium phosphonate played an important role in determining the configuration and enantiomeric excess of adducts. Wan et al investigated the recycling aspect of some Cinchona alkaloid-based primary amine catalysts in an aldol reaction of cyclohexanone with benzaldehydes.12 They observed that the catalysts were highly effective over three cycles in terms of yields and stereoselectivity of the products on proper control of solubility in an aqueous–organic biphasic system through regulation of the pH of the aqueous phase and effecting the protonation and deprotonation of primary amino, tertiary amino, and pyridyl groups.
The first asymmetric cross-aldol reaction of enolizable aldehydes 2 and α-ketophosphonates 6 was reported by Perera et al.13 Application of quinine-derived primary amine 7 as an organocatalyst and 4-methoxybenzoic acid as an additive led to the highly enantioselective synthesis of tertiary β-formyl-α-hydroxyphosphonates 8 with anticancer activity (Figure 3). The reaction worked very well with acetaldehyde, which is a difficult substrate for aldol reaction. The authors established the configuration of the product as “R” on the basis of single crystal X-ray crystallography. This configuration led them to propose the mechanism in which the enamine attacks from the Si face of the ketophosphonates. The carbonyl groups in the latter are hydrogen-bonded to the protonated quinuclidine nitrogen.
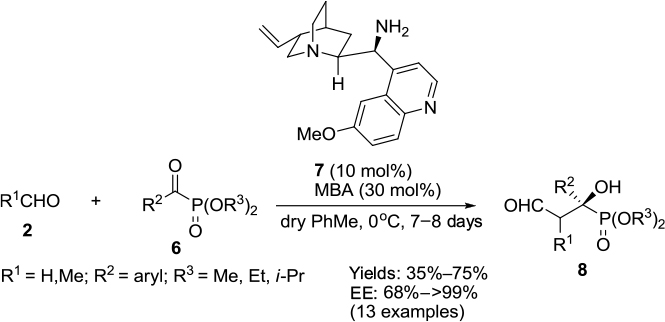 | Figure 3 Asymmetric cross-aldol reaction of enolizable aldehydes and α-ketophosphonates. Abbreviations: MBA, 4-methoxybenzoic acid; EE, enantiomeric excess. |
Isatins have emerged as one of the most powerful classes of compounds, due to the diverse biological properties of their derivatives and applications in synthesis of spirooxindoles during the last 15 years.14 The reactions of the C3 ketone group of isatins, including aldol reaction, have been explored for the synthesis of 3-substituted 3-hydroxy-2-oxindoles and spirooxindoles. Guo and Zhao reported the application of quinine-derived primary amine catalyst 10 in asymmetric aldol reaction of isatins 9 with acetaldehyde 2a.15 The corresponding adducts were obtained in high yields and with good enantioselectivity. However, they were not very stable and were immediately reduced to diols 11 (Figure 4). The use of other enolizable aldehydes and ketones in this reaction usually led to low enantioselectivity. According to the proposed mechanism, the primary amino group of the catalyst reacts with aldehyde to form an enamine intermediate. The cocatalyst benzoic acid may catalyze the enamine formation and also protonate the quinuclidine nitrogen atom, which in turn activates the C3 ketone group of isatin by H-bonding and also directs the approach of isatin. Attack of the enamine to the Re face of isatin led to the formation of major S-enantiomer.
 | Figure 4 Quinine-derived primary amine-catalyzed aldol reaction of isatins. Abbreviations: THF, tetrahydrofuran; EE, enantiomeric excess; equiv, equivalents. |
Kumar and Chimni reported an asymmetric aldol reaction of isatins 9 with pyruvic aldehyde dimethyl acetal 12 using pseudoenantiomeric catalysts 10 and 10a in the presence of trichloroacetic acid as an additive, resulting in the formation of enantiomeric 3-substituted 3-hydroxy-2-oxindoles 13 or 13a (Figure 5).16 The reaction of several isatins with either free NH or N-alkyl groups and electron-withdrawing groups at C5 or C7 or at both C5 and C7 takes place at room temperature in a dioxane:water solvent system to give products in excellent yields (87%–96%) and with high enantioselectivity (89%–97% enantiomeric excess [EE]). The catalyst 9-amino-9-dexoxy-epi-cinchonine induced the formation of S-enantiomer, whereas the 9-amino-9-dexoxy-epi-cinchonidine afforded the R-isomer.
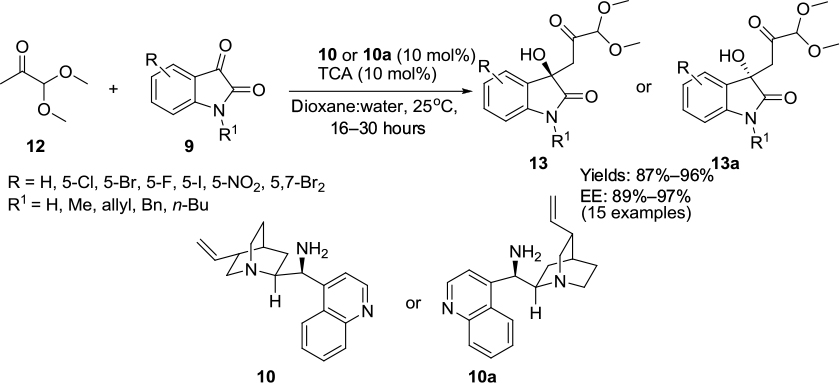 | Figure 5 Asymmetric aldol reaction of isatins with pyruvic aldehyde dimethyl acetal. Abbreviations: TCA, trichloroethane; EE, enantiomeric excess. |
Asymmetric aldol reaction of β,γ-unsaturated α-ketoesters has been the subject of many recent communications. In 2010, the asymmetric aldol reaction of acetone to β,γ-unsaturated α-ketoesters was investigated using 5 mol% of 9-amino 9-(deoxy)-epi-Cinchona alkaloid 10 as a catalyst and 4-nitrobenzoic acid as an additive, leading to the enantioselective synthesis of a chiral tertiary alcohol moiety with excellent yields and enantioselectivity.17 Another application of this catalyst in the presence of trifluoroacetic acid as an additive was reported in the asymmetric aldol reaction of β,γ-unsaturated α-ketoesters 15 and O-protected hydroxyacetones 14, leading to the formation of chiral glycerol derivatives 16 (Figure 6).18
 | Figure 6 Asymmetric aldol reaction of β,γ-unsaturated α-ketoesters and O-protected hydroxyacetones catalyzed by 9-amino-9-(deoxy)-epi-Cinchona. Abbreviations: THF, tetrahydrofuran; RT, room temperature; EE, enantiomeric excess. |
Application of C9 alkoxy/hydroxyl derivatives
Lu et al employed a unique Cinchona derivative 18 bearing a thiourea group at the C6′ position and benzyloxy group at the C9 position in an asymmetric aldol reaction of isocyanoesters 17 with β,γ-unsaturated α-ketoesters 15.19 After acidic hydrolysis, β-hydroxy-α-amino esters 20 were obtained in excellent yields (up to 95%) and high enantioselectivity (up to 92%) (Figure 7). According to the proposed mechanism, the thiourea moiety forms two H-bonds with the carbonyl group of ketoester, while the tertiary nitrogen atom of the catalyst activates the isocyanoacetate. An attack of enolized isocyanoester then occurs on the carbonyl group of the ketoester followed by intramolecular cyclization. The subsequent acidic hydrolysis of the oxazoline product 19 gives the β-hydroxy-α-amino acid derivatives.
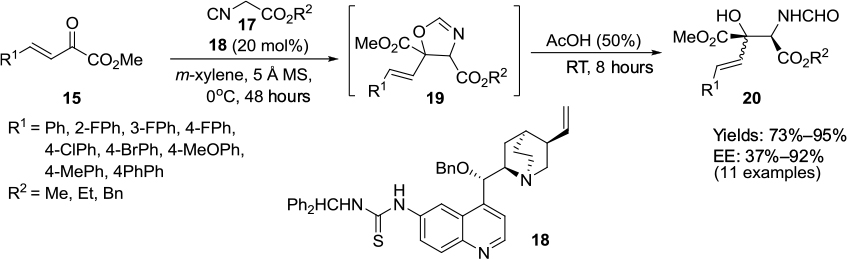 | Figure 7 Asymmetric aldol reaction of isocyanoesters with β,γ-unsaturated α-ketoesters. Abbreviations: MS, molecular sieves; RT, room temperature; EE, enantiomeric excess. |
A Cinchona-alkaloid derivative 23 bearing a hydroxyl group at C9 and an isopropoxy group at C6′ has been employed in a direct asymmetric aldol reaction of the sugar aldehyde 21 with a pyruvate ester 22.20 The aldol adduct anti-24, on treatment with acidic ion-exchange resin (Amberlyst 15) in methanol, afforded the cyclic form of the KDG ester 25 (Figure 8). The authors proposed initial deprotonation of the pyruvate ester by catalyst that also helped in the creation of an asymmetric environment at the reaction center through a network of hydrogen bonds. The pyruvate enolate attacks the aldehyde from the Si face, because the Re face of the latter is covered by the quinoline ring.
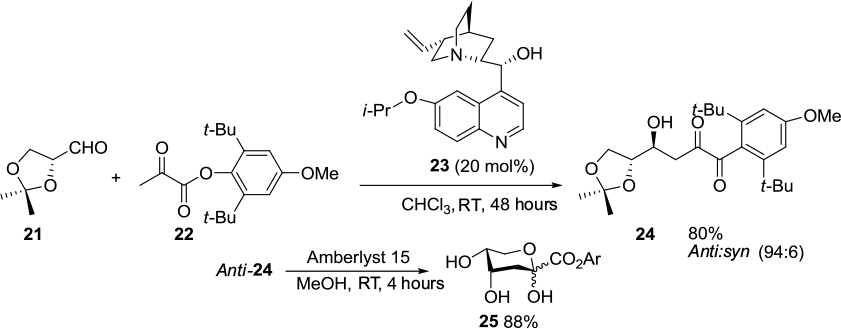 | Figure 8 Asymmetric aldol reaction of a pyruvate ester with a sugar aldehyde catalyzed by Cinchona-derived catalyst with C6′ isopropoxy group and C9 hydroxyl group. Abbreviation: RT, room temperature. |
Wang et al screened commercially available quinine 26 as the most effective catalyst in terms of offering enantioselectivity for the phospho-aldol reaction of isatins with diphenyl phosphite.21 A variety of N-alkylated isatin derivatives 9 undergo asymmetric phospho-aldol reaction with diphenyl phosphite, forming 3-hydroxyisatin-3-phosphonates 27 in good-to-excellent yields and with moderate to good enantioselectivity (Figure 9). The reaction was also sensitive to solvents: nonpolar solvents gave better enantioselectivity in comparison to polar solvents. A study of electronic effects on the isatin ring showed that electron-donating substituents offered better enantioselectivity than electron-withdrawing substituents. These observations led the authors to propose a transition state TS (Figure 9) involving a ternary complex between the phosphite, the isatin, and the catalyst, in which the isatin’s C3 ketone and phosphite were activated by H-bonding with the hydroxyl group of the catalyst.
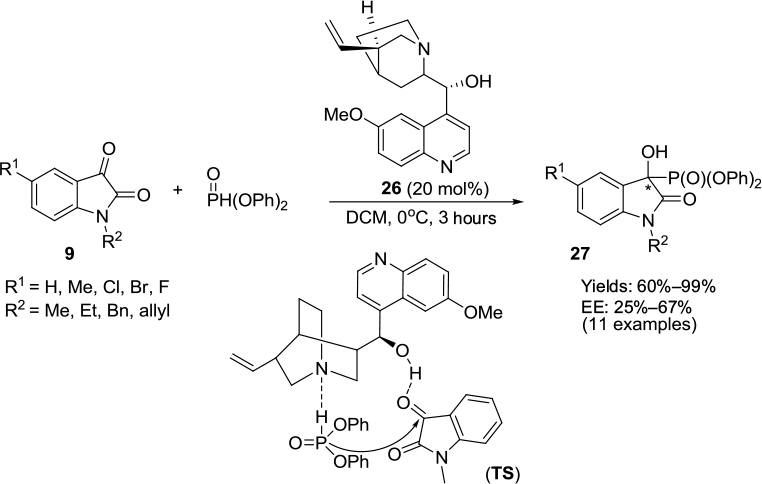 | Figure 9 Asymmetric phospho-aldol reaction of isatins catalyzed by Cinchona-based catalyst bearing C6′ methoxy and C9 hydroxyl groups. Abbreviations: DCM, dichloromethane; EE, enantiomeric excess; TS, transition state. |
Application of C9 urea/thiourea derivatives
Guo et al reported enolate-mediated asymmetric aldol reactions of isatins and inactivated ketones using quinidine-derived thioureas forming 3-substituted 3-hydroxy-2-oxindoles.22 Liu et al also employed Cinchona alkaloid-derived thioureas (Figure 10) in asymmetric aldol reactions of isatins with α,β-unsaturated ketones affording aldol adducts at moderate-to-good yields (18%–98%) and with moderate-to-high enantioselectivity (30%–97% EE).23 The authors proposed that the enones were deprotonated by the tertiary amine of the quinuclidine and isatin activated and oriented by hydrogen bonding of its carbonyl group with a thiourea moiety in the catalyst.
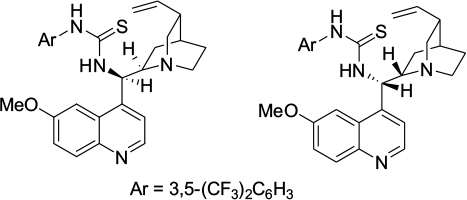 | Figure 10 C9 thiourea derivatives of Cinchona alkaloid used in aldol reaction of isatins. |
Allu et al reported the application of a C9 urea derivative 29 as the most efficient catalyst in the aldol reaction of isatins 9 with various inactivated and activated carbonyl compounds 28 furnishing 3-substituted 3-hydroxy-2-oxindoles 30 with good yields and enantioselectivity (Figure 11).24 The application of C9 amine derivative 10 in this reaction took much longer (10 hours) and offered relatively lower yields (up to 58%) and enantioselectivity (up to 90%). The mechanism proposed is similar to one proposed by Liu et al.23 The enolate is generated by abstraction of proton from ketones/aldehydes by quinuclidine nitrogen, and isatin is activated by H-bonding with urea moiety.
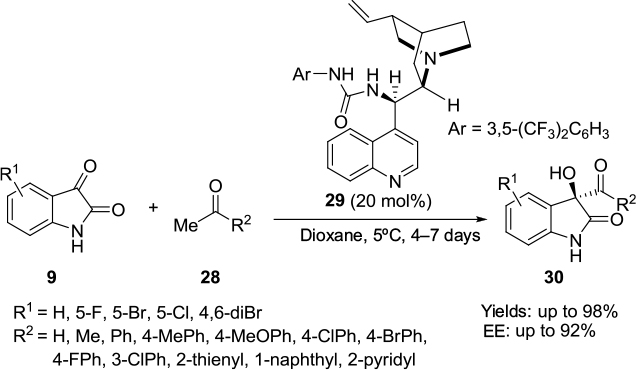 | Figure 11 Asymmetric aldol reaction of isatins catalyzed by Cinchona-derived urea. Abbreviation: EE, enantiomeric excess. |
Okumus et al reported an asymmetric aldol reaction of α-azido ketones 31 with ethyl pyruvate 32 mediated by a Cinchona-based bifunctional urea catalyst 33 (Figure 12).25 The reaction led to the first asymmetric synthesis of ethyl 4-aryl-3-azido-2-hydroxy-2-methyl-4-oxobutanoates 34 with high enantioselectivity (up to 91%) and diastereoselectivity (diastereomeric ratio [DR] 95:5 syn:anti), but in moderate yields (Table 1).
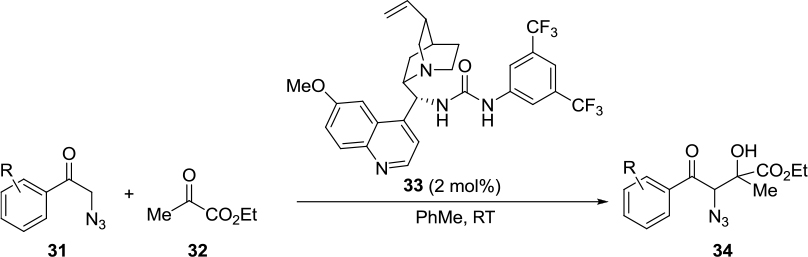 | Figure 12 Asymmetric synthesis of ethyl 4-aryl-3-azido-2-hydroxy-2-methyl-4-oxobutanoates. Abbreviation: RT, room temperature. |
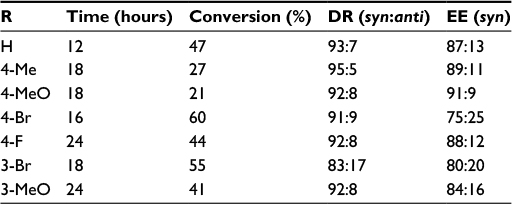 | Table 1 Substrate scope in the aldol reaction of ethyl pyruvate with α-azido ketones Abbreviations: DR, diastereomeric ratio; EE, enantiomeric excess. |
A review of the literature on asymmetric 1,2-addition on the C=O bond reveals that enantioselective aldol reaction of a number of inactivated/activated aldehydes or ketones and other carbonyl compounds has been realized using Cinchona alkaloid-based catalysts with NH2, OH, and alkoxy groups at the C9 position. A few applications of urea and thiourea derivatives are also reported. Among substrates, isatins and β,γ-unsaturated α-ketoesters have drawn considerable interest apart from common aldehydes and ketones. There are two principal mechanisms for aldol reactions. The primary amine catalysts interfere in the reaction through an enamine formation with aldehydes or ketones. The use of an acid cocatalyst can accelerate the formation of enamine, as well as protonate the quinuclidine nitrogen atom. The urea/thiourea catalysts interfere through an enol/enolate mechanism. The quinuclidine nitrogen abstracts the proton, and the thiourea moiety activates the carbonyl group through H-bonding. The network of H-bonding also creates asymmetry at the reaction center and determines the stereochemical outcome of the reaction. In the reaction of isatin with phosphite using quinine, the C9 hydroxyl group is proposed to activate the carbonyl group of isatin, whereas the tertiary amine activates the phosphite through H-bonding.
Nucleophilic 1,2-addition to the C=N bond
Azomethine compounds serve as important starting materials in organic synthesis, especially through 1,2-addition reactions. The important addition reactions of imines, in which stereocontrol is governed by chiral catalysts, include the Mannich reaction, Strecker reaction, β-lactam formation, allylation, and hydrosilylation. The Mannich reaction, involving reactions of imines with enolizable substrates, provides an easy methodology for the synthesis of several other important building blocks, such as amino alcohols, amino carbonyls, and their derivatives. Also, these compounds are important in pharmaceutical and agricultural fields. The application of natural Cinchona alkaloids, such as quinine, quinidine, cinchonine, and cinchonidine, Cinchona alkaloid-derived thioureas, and a 9-silyloxy derivative of quinine in asymmetric Mannich reactions has been explored previously.4 The following sections describe applications of Cinchona alkaloid-derived catalysts in Mannich, Strecker, and hydrosilylation reactions.
Application of C9 urea/thiourea derivatives
Han et al have recently reported a cinchonine-derived urea 29a as an efficient catalyst for the asymmetric Mannich reactions of 5H-oxazol-4-ones 35 with various alkyl and aryl sulfonamides 36, forming β-alkyl/aryl-substituted α-hydroxy-β-amino acids 37 at good yields and excellent diastereo- and enantioselectivity (Figure 13).26 The different substituents on the aromatic ring of imines did not affect the reaction rate or the stereoselectivity of the reaction. The substituents at the nitrogen atom of imines affected the diastereoselectivity: the N-triisopropylbenzenesulfonylimines offered the best diastereoselectivity. The reaction was able to be scaled up to the gram scale without compromising enantioselectivity. The absolute configuration was determined by single-crystal X-ray crystallographic studies of a product. The authors demonstrated the synthetic utility of this reaction by accessing an important α-methyl-α-hydroxy-β-amino acid derivative, an α-methylated C13 side chain of taxol, and taxotere, from the Mannich adduct.
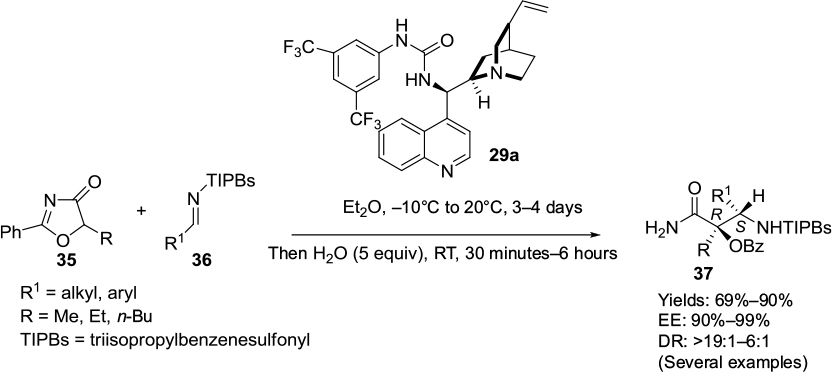 | Figure 13 Asymmetric Mannich reactions of 5H-oxazol-4-ones with alkyl and aryl sulfonamides catalyzed by Cinchona-based thiourea. Abbreviations: RT, room temperature; EE, enantiomeric excess; DR, diastereomeric ratio; equiv, equivalents. |
Fan et al reported the application of a Cinchona-based thiourea 40 in diastereo- and enantioselective nitro-Mannich reactions of N-phosphoryl imines 38 with α-substituted nitroacetates 39.27 This reaction resulted in the formation of the corresponding β-nitro ethylphosphoramidate 41 with adjacent quaternary and tertiary chiral centers in high yields (up to 86%), high diastereoselectivity (up to 99:1 anti-selectivity), and enantioselectivity (up to 99% EE) (Figure 14). Phosphoramidates are reported to have anticancer and antiviral activities. The position of substituents on the phenyl ring had no effect on the stereoselectivity of the reaction. Moderate yields were obtained with N-phosphoryl imines of furan-2-carbaldehyde and trans-cinnamaldehyde. All the imines took 17 hours for the reaction, except the imines of 3- and 2-chlorobenazaldehydes and propenal, which took 36–42 hours.
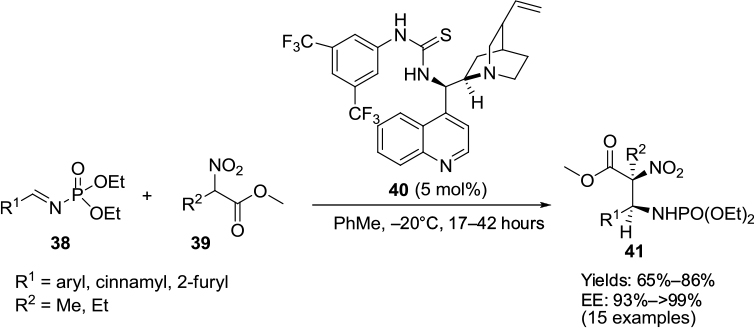 | Figure 14 Nitro-Mannich reactions of N-phosphoryl imines with α-substituted nitroacetates. Abbreviation: EE, enantiomeric excess. |
The plausible mechanism suggested by the authors involves a double-hydrogen-bonding interaction of thiourea N–H with P=O of imines. Another double-hydrogen-bonding interaction has been suggested between protonated tertiary amine nitrogen (HN+) of the catalyst, nitro, and carbonyl groups on the nitroacetates. Subsequently, the Cinchona-alkaloid catalyst scaffold restricts the Si face attack of electrophilic imines through the Si face of the enolates, affording the Mannich adducts with S-configuration on both chiral centers.
Recent years have seen considerable attention given to asymmetric synthesis of 3-substituted 3-amino-2-oxindoles,28 which have proved a valuable class of compounds for the synthesis of spirooxindoles.17 We wish to mention here some reports focusing on the synthesis of 3-substituted 3-amino-2-oxindoles by the addition of different nucleophiles to isatin imines using Cinchona alkaloid-based catalysts.
Cinchona alkaloid-derived thiourea 44 served as the most efficient catalyst in the asymmetric addition of α-aryl isocyanoacetates 43 to isatin imines 42.29 Most reactions were complete in 24 hours in chloroform, affording enantioenriched 3-substituted 3-amino-2-oxindoles 45 in good-to-excellent yields and moderate-to-good enantioselectivity (Figure 15). A wide variety of α-aryl isocyanoacetates and isatin imines with diverse electronic and steric environments were tolerated in this reaction. The electronic properties of substituents on isatin nitrogen also affected the reaction. Isatins with methyl and phenyl groups at the N-1 position gave better results in comparison to isatins with allyl, benzyl, or p-methoxybenzyl groups at this position. Isatin with free NH failed to react under standard reaction conditions. The authors demonstrated the application of products in the synthesis of spirooxindoles imidazolines.
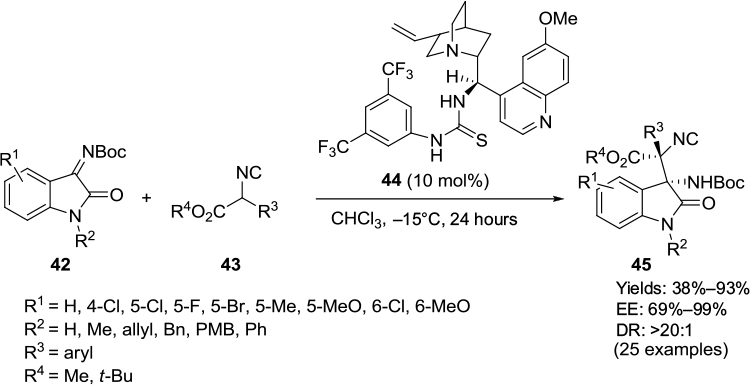 | Figure 15 Asymmetric addition of α-aryl isocyanoacetates to isatin imines catalyzed by Cinchona-derived thiourea. Abbreviations: PMB, p-methoxybenzyl; EE, enantiomeric excess; DR, diastereomeric ratio. |
Recently, Liu and Zhou and Wang et al independently reported an improved asymmetric Strecker reaction to synthesize chiral 3-cyano-3-amino-2-oxindoles 47 using a Cinchona alkaloid-derived thiourea catalyst.30,31 Simple hydrolysis and esterification of these products offer easy access to 2-oxindoles with a free amino group and an ester group at the C3 position. Liu and Zhou employed cinchonine-derived thiourea 40a in cyanation of N-Boc isatin imines to obtain products in high yields (81%–95%) and excellent enantioselectivity (90%–>99%) (Figure 16). Wang et al employed quinine-derived thiourea 44a as an organocatalyst catalyst in the reactions of N-Boc isatin imines 42 with trimethylsilyl cyanide 46, which afforded products in good-to-excellent yields (78%–98%) and with very good enantioselectivity (up to 94%) (Figure 16). The proposed transition state involves activation of imine by hydrogen bonding with the thiourea moiety and simultaneous generation of nucleophiles by the tertiary amine. The catalyst also provides proper orientations to both substrates to enhance enantioselectivity. Yan et al reported the application of quinine-derived thiourea 44a as a catalyst in the enantioselective Mannich-type addition of 1,3-diketones and malonates to N-Boc isatin imines, yielding products in 83%–99% yields and 75%–98% EE.32
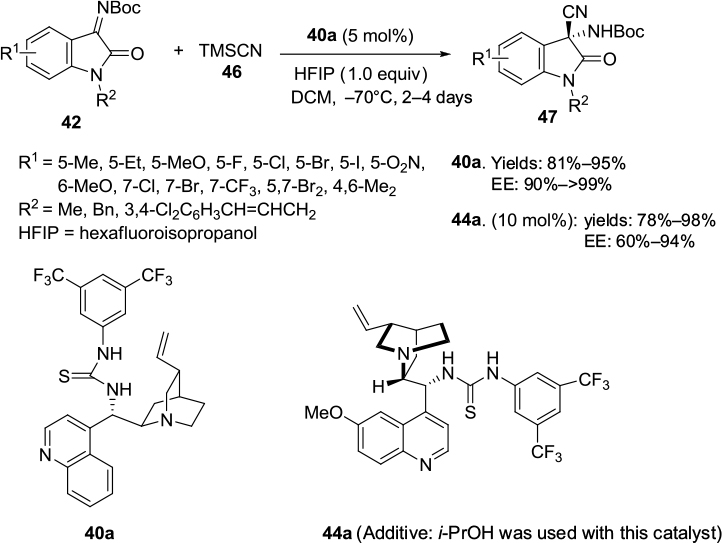 | Figure 16 Asymmetric Strecker reaction catalyzed by Cinchona-derived thioureas. Abbreviations: TMSCN, trimethylsilyl cyanide; DCM, dichloromethane; EE, enantiomeric excess; equiv, equivalents. |
Application of C9 benzyloxy derivatives
Guo et al reported the asymmetric synthesis of 3-substituted 3-amino-2-oxindoles 50 by direct vinylogous Mannich reactions of γ-butenolide 48 with isatin imines 42.33 Using a Cinchona alkaloid-derived catalyst 49 bearing a benzyloxy group at C9 and a hydroxyl group at C6′, several 3-aminooxindoles with adjacent tertiary and quaternary chiral centers with butenolide moiety at the C3 position were obtained in excellent yields (up to 97%) and high enantioselectivity (83%–96%) (Figure 17). Isatin imines with the bromo group at the C4 position failed to react, presumably due to steric factors. Butenolides are activated by the tertiary amine group on the catalyst, whereas the imines are activated by hydrogen bonding with the C6′ OH group. Steric factors led to attack on the Re face of the imines.
 | Figure 17 Asymmetric synthesis of 3-substituted 3-amino-2-oxindoles by Mannich reaction catalyzed by Cinchona-derived catalyst with C6′ OH and C9 alkoxy groups. Abbreviations: DCM, dichloromethane; EE, enantiomeric excess. |
Another synthesis of optically active 3-amino-2-oxindoles 52 with a nitroalkyl group at the C3 position was developed by Kumar et al by aza-Henry reaction of N-Boc-substituted isatin imines 42 with nitroalkanes 51 catalyzed by modified Cinchona-alkaloid catalyst 49a (Figure 18).34 Looking at the experimental observations and absolute configuration, the authors proposed a transition state for the reaction, involving a ternary complex between the catalyst and the substrates. The quinuclidine tertiary amine can abstract an α-proton from the nitroalkane, thus activating it for nucleophilic attack. The C6′ hydroxyl group of the catalyst may activate the imine by hydrogen bonding. The nucleophilic attack of nitroalkanes occurs from the Re face of imines, affording products with R-configuration.
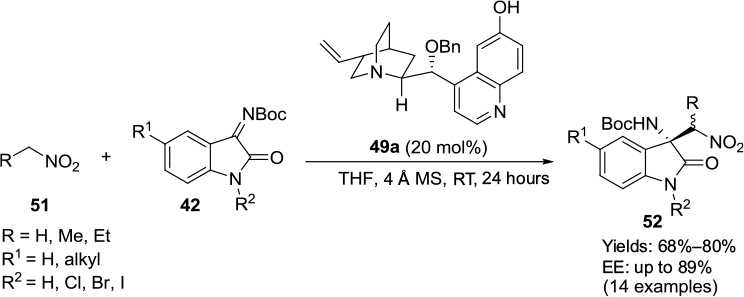 | Figure 18 Aza-Henry reaction of N-Boc-substituted isatin imines with nitroalkanes catalyzed by Cinchona-derived catalyst with C6′ OH and C9 alkoxy groups. Abbreviations: THF, tetrahydrofuran; MS, molecular sieves; RT, room temperature; EE, enantiomeric excess. |
Application of C9 sulfonamide and picolinamide derivatives
Hara et al reported an enantioselective decarboxylative Mannich-type addition of malonic acid half-thioesters 53 to the C=N bond of isatin 3-N-Boc-substituted imines 42 using the N-heteroarylsulfonamide derivative 54 of a Cinchona alkaloid as a catalyst (Figure 19).35 The application of this sulfonamide catalyst afforded 3-aminooxindoles 55 58%–92% yields and 75%–83% EE. X-ray crystallography of the catalyst showed that the hydrogen atom on the sulfonamide nitrogen formed hydrogen bonds with the quinoline and quinuclidine nitrogen atoms. The thioester gets activated by hydrogen bonding of its thiocarbonyl oxygen with sulfonamide hydrogen of the catalyst. The deprotonation and decarboxylation of the thioester is assisted by the quinuclidine nitrogen to give thioester enolate, which adds on to the Re face of isatin imines activated by protonated quinuclidine nitrogen to give a product with S-configuration.
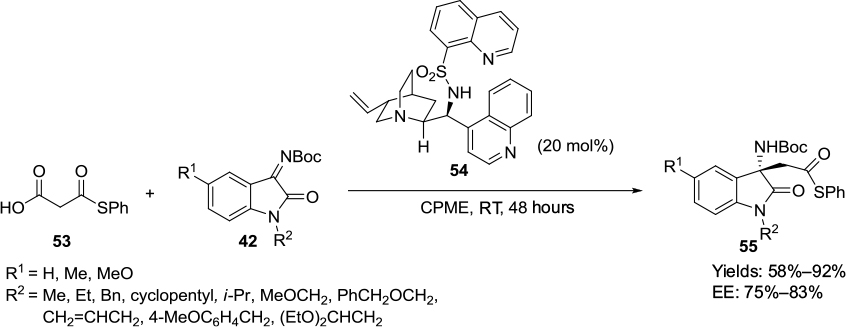 | Figure 19 Cinchona-derived sulfonamide-catalyzed decarboxylative Mannich-type addition. Abbreviations: CPME, cyclopentyl methyl ether; RT, room temperature; EE, enantiomeric excess. |
A cationic picolinamide catalyst 57, derived from epi-cinchonidine alkaloid by amide coupling and methylation, has been employed for the first time in the asymmetric reduction of imines by hydrosilylation.36 A wide range of aryl ketimines 56 was reduced to sec-amines 58 (Figure 20) with good stereochemical efficiency using HSiCl3. Relatively higher enantioselectivity was observed in imines with a 4-methoxyphenyl group on the nitrogen atom.
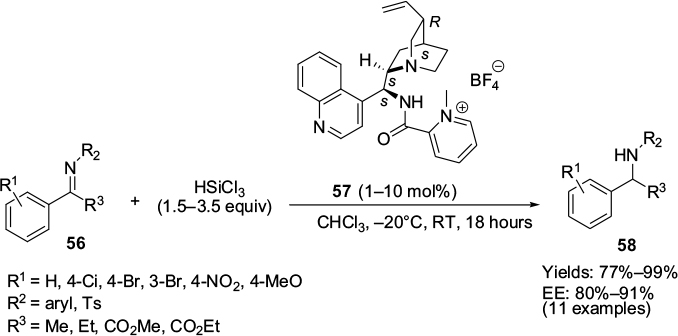 | Figure 20 Application of epi-cinchonidine alkaloid-derived cationic picolinamide catalyst in hydrosilylation of imines. Abbreviations: RT, room temperature; EE, enantiomeric excess. |
Cinchona alkaloid-derived catalysts are thus employed in the Mannich, Strecker, and hydrosilylation reactions of imines. Most of the catalysts employed have either a thiourea or a urea moiety at C9 position. One end of urea/thiourea bears a phenyl group with the CF3 group at C3 and C5 positions. There are also examples of successful applications of catalysts with a C9-benzyloxy and C6′-hydroxyl groups or amide or sulfonamide group at C9 position. Isatin imines, oxazolinones, and imines, with a strong electron-withdrawing group on nitrogen, serve as popular substrates. The thiourea/urea/hydroxyl/sulfonamide group at C9 or C6′ positions activates the imines, and the quinuclidine nitrogen abstracts the proton. As usual, the chiral scaffold of the catalysts compels attack on a particular side of imines to give enantioenriched products.
Michael-addition reactions
The asymmetric Michael/conjugate-addition reactions of nucleophiles to electron-deficient alkenes have become one of the most exploited reactions for the formation of C–C bonds in an enantioselective manner.37,38 As a result of the wide range of Michael donors and acceptors that can be generated by varying the electron-withdrawing groups, Michael reactions can be tailored to cover an expansive spectrum of reactions for the production of a wide range of highly functionalized biologically active compounds, each requiring different types of catalysts. This section of the review focuses on a wide range of optimized Cinchona alkaloid-derived catalysts utilized for diverse Michael reactions. The literature is arranged on the basis of frequently used classes of catalysts or substrates. Among a particular class of substrates, the literature is arranged according to the C9 functionality of the catalyst.
Application of C9 thioureas
The quinine-derived thiourea catalysts have been reported to be the most efficient catalysts in different Michael additions (Table 2). The organocatalytic enantioselective addition of malonates to 3-nitro-2H-chromenes afforded highly functionalized and synthetically versatile adducts with two adjacent stereogenic carbon atoms at high levels of enantioselectivity in the presence of catalyst 44a.39 An asymmetric synthesis of 2-amino-5-oxo-5,6,7,8-tetrahydro-4H-chromene-3-carboxylates was reported via a tandem Michael-cyclization reaction of 1,3-cyclohexanediones and alkylidene cyanoacetates using catalyst 44b.40 The products were obtained in high yields (up to 92%) and good enantioselectivity (up to 82%). The conjugate addition of thiols to α-substituted N-acryloyloxazolidin-2-ones followed by asymmetric protonation was reported to yield addition products.41 In addition, both of the enantiomers of the products were accessible with the same level of enantioselectivity using catalyst 44a and its pseudoenantiomer – 44b. Li et al developed the first catalytic asymmetric conjugate addition of 1,3-dicarbonyl compounds to nitroenynes.42 The 1,4-addition products were obtained in moderate-to-good yields (up to 93%) with good enantioselectivity (up to 99% EE) using Cinchona alkaloid-derived thiourea 44c. The protocol worked well with both aryl- and alkyl-substituted nitroenyne substrates, thus providing a conceptually different entry to the precursors of pharmaceutically important chiral β-alkynyl acid derivatives and synthetically useful chiral nitroalkynes.
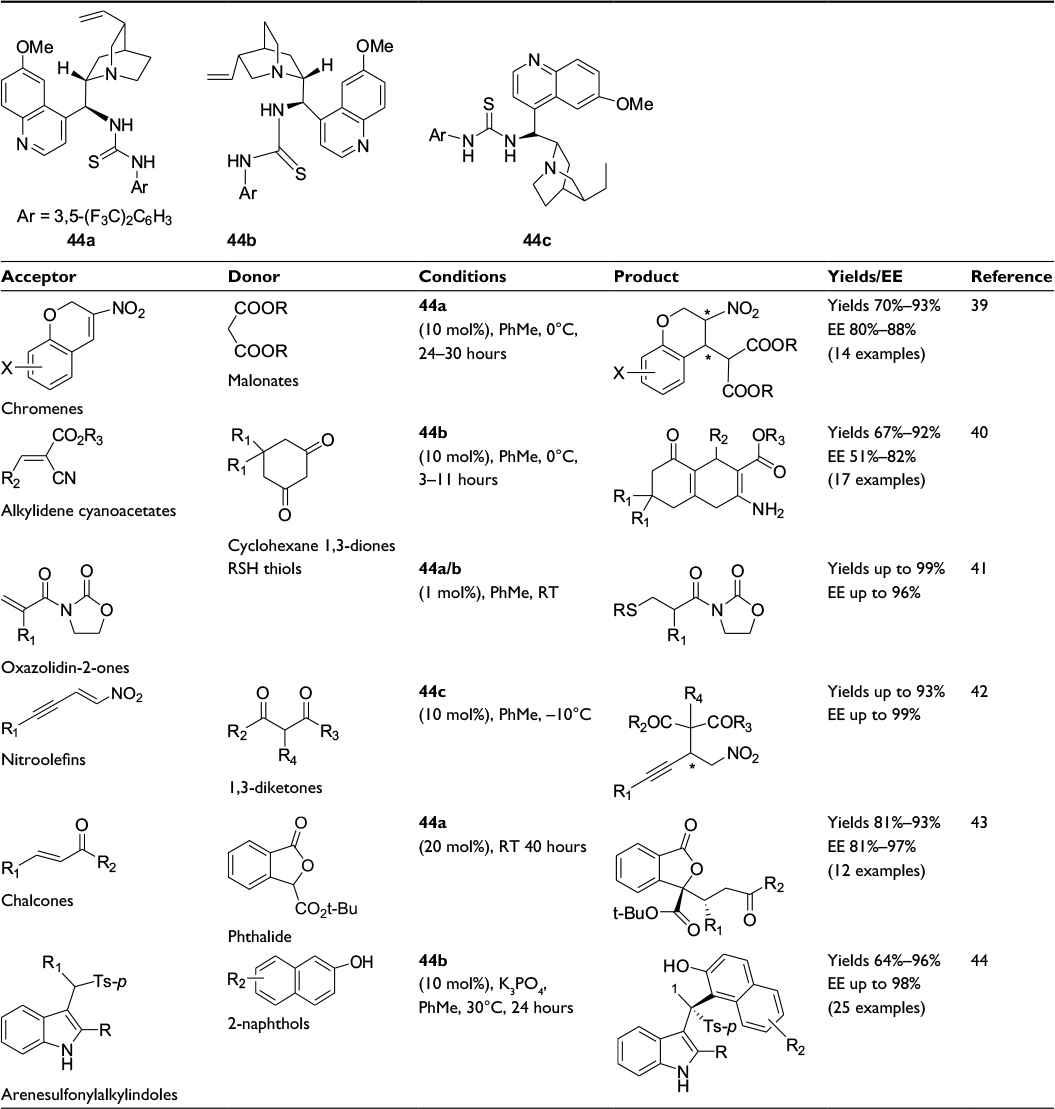 | Table 2 Scope of application of Cinchona-based thiourea catalysts 44a–44c in asymmetric Michael additions Abbreviations: EE, enantiomeric excess; RT, room temperature. |
The first asymmetric Michael addition of 3-substituted phthalide derivatives to chalcones was developed by Luo et al.43 The reaction, using quinine-derived thiourea 44a, afforded 3,3-disubstituted phthalide derivatives, bearing vicinal quaternary and tertiary stereogenic centers.
Yu et al found the diastereomer 44b of catalyst 44a to be the most effective catalyst for the Michael addition of 2-naphthols to alkylidene-indolinine intermediates, generated in situ from arenesulfonylalkylindoles, yielding the corresponding adducts as a series of optically active C3 alkyl-substituted indole derivatives containing phenolic hydroxyl groups.44
Zhao et al developed the cinchonidine-derived thiourea 40b-catalyzed asymmetric Michael addition of 3-substituted-N-Boc-oxindoles 59 to a vinyl bisphosphonate 60, affording the corresponding adducts 61 (Figure 21) bearing a quaternary carbon stereocenter and germinal bisphosphonate ester fragment at the C3 position of the oxindoles.45 In the plausible transition-state model, substrate 59 would be activated by tertiary amine thiourea in bifunctional mode, and the enolized oxindoles would attack the vinyl bisphosphonates from the Si face to give the corresponding adducts with R-configuration. Later on, the catalyst 40b was used by Cai et al in the Michael-addition reaction of oxazolones to vinylogous imine intermediates generated in situ from arylsulfonyl indoles.46
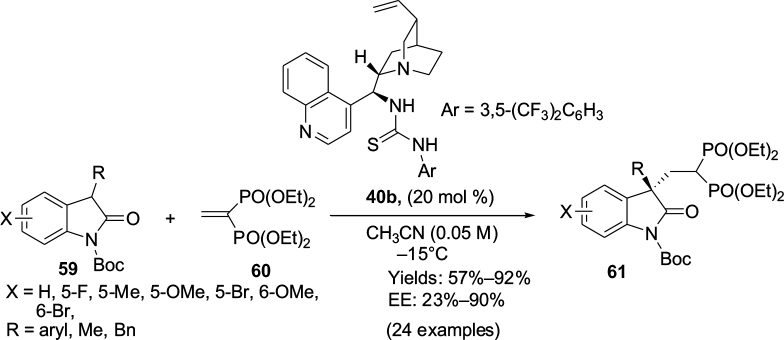 | Figure 21 Cinchonidine-derived thiourea-catalyzed asymmetric Michael addition of 3-aryl-N-Boc oxindoles to vinyl bisphosphonate. Abbreviation: EE, enantiomeric excess. |
Application of Cinchona-derived catalysts in Michael additions to nitroolefins
Asymmetric Michael additions of various Michael donors to nitroolefins have been reported in the presence of different classes of Cinchona-based catalysts. Enantioselective Michael additions of Meldrum’s acid,47 cyclohexanones,48 and aldehydes49 to nitroolefins catalyzed by different Cinchona-based thioureas were reported during 2010. An example of Michael addition to nitroolefins catalyzed by Cinchona-based thiourea has been described in the preceding section. In the Michael addition of acetylacetone 62 to nitroolefins 63, a thiourea catalyst 65 has been employed (Figure 22).50 Liu et al also reported the Michael-addition reaction of these substrates using a chiral 1,1′-bi-2-naphthol (BINOL)–quinine–squaramide catalyst 66, affording adducts 64 in good yields and enantioselectivity (Figure 22).51
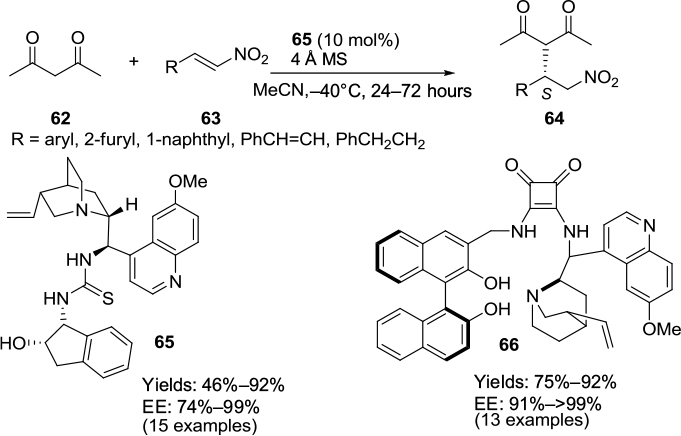 | Figure 22 Applications of Cinchona-based thiourea and BINOL–quinine–squaramide catalysts in Michael additions to nitroolefins. Abbreviations: BINOL, 1,1′-bi-2-naphthol; MS, molecular sieves; EE, enantiomeric excess. |
A new class of hydrogen-bond donor catalysts 68 based on the 1,1-diamino-2-nitroethylene scaffold has been introduced for the activation of trans-β-nitrostyrenes 63 toward reactions with a range of carbon-based nucleophiles 67, such as 2-hydroxynaphthalene-1,4-dione, 1,3-oxazolin-5-one, N-Boc-2-oxindole, benzophenone imine, and cyclopetanone-2-carboxylate, affording the corresponding adducts 69 in excellent yields (Figure 23). Importantly, this new set of organocatalysts is easily prepared from commercially available starting materials in mild reaction conditions.52 The reactions of pantane-2,4-dione and indole with trans-β-nitrostyrene showed moderate enantioselectivity (44% and 36% EE, respectively).
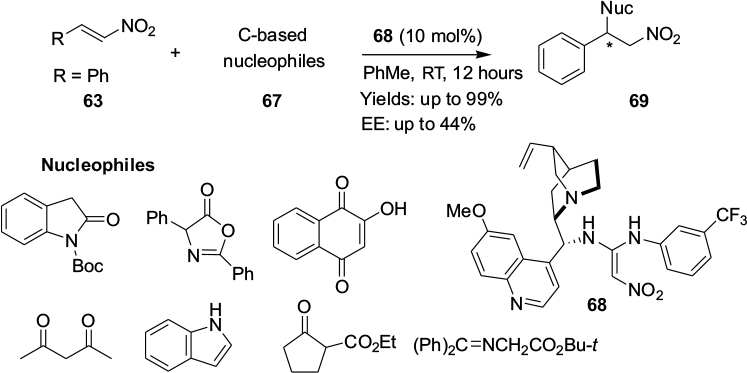 | Figure 23 Michael addition of trans-β-nitrostyrenes with carbon nucleophiles. Abbreviations: RT, room temperature; EE, enantiomeric excess. |
In another study, the enantioselective Michael addition of homoserine lactone-derived cyclic imino esters 70 to nitroolefins 63 was reported in the presence of commercially available bis-Cinchona alkaloid catalyst diydroquinidine anthraquinone ([DHQD]2AQN) 71.53 This reaction occurs under mild conditions, with a wide range of substrates affording the corresponding products 72 excellent diastereoselectivity (>95:5 DR) and good enantioselectivity (up to 87% EE) (Figure 24).
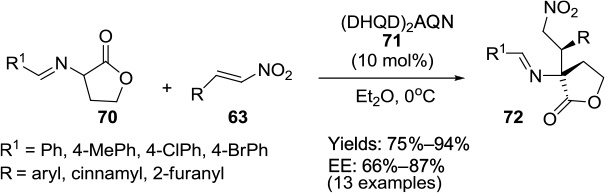 | Figure 24 Application of bis-Cinchona alkaloid catalyst (DHQD)2AQN in Michael additions to nitroolefins. Abbreviation: DHQD, dihydroquinidine; AQN, anthraquinone; EE, enantiomeric excess. |
Ashokkumar and Siva reported an asymmetric Michael addition of diethyl malonate 73 to (E)-2-nitorstyrene 63 using new pentaerythritol tetrabromide-based chiral quaternary ammonium salts 74 as catalysts.54 The reactions occurred at room temperature in the presence of very low concentrations of catalysts to afford products 75 in very good yields and with high enantioselectivity (Figure 25).
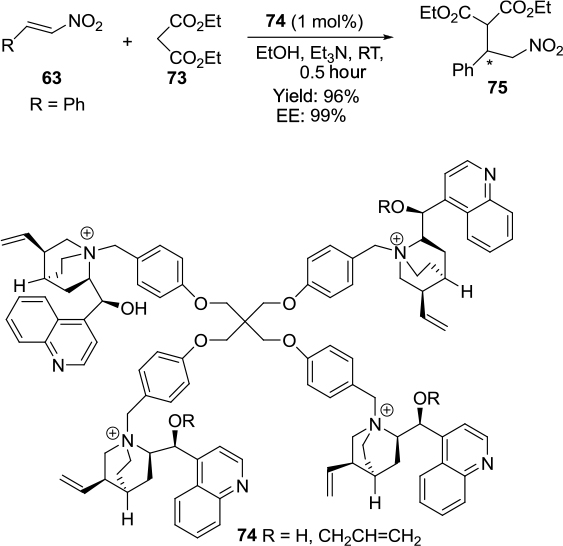 | Figure 25 Application of pentaerythritol tetrabromide-based chiral quaternary ammonium salt in Michael addition of diethyl malonate to (E)-2-nitrostyrene. Abbreviations: RT, room temperature; EE, enantiomeric excess. |
Application of Cinchona-derived catalysts in Michael additions of α,β-unsaturated carbonyl compounds
Thioureas, ureas, amines, squaramides, and 9-OH and 9-OBn derivatives of Cinchona alkaloids have been investigated and employed as optimized catalysts in enantioselective Michael additions involving α,β-unsaturated carbonyl compounds. Campbell et al developed an efficient synthetic approach to provide an asymmetric access to previously inaccessible chiral 3,5-dialkyl-2-pyrazolines.55 The synthesis was achieved through an asymmetric conjugate addition of hydrazide nucleophile 76 to aliphatic α,β-enones 77, followed by a deprotection cyclization of the 1,4-adduct 78 to access the desired 2-pyrazolines 79 (Figure 26). The reaction was best catalyzed by 9-epi-aminoquinine 7 and its pseudoenantiomer, 9-epi-aminoquinidine, thus yielding both enantiomers of the chiral pyrazolines.
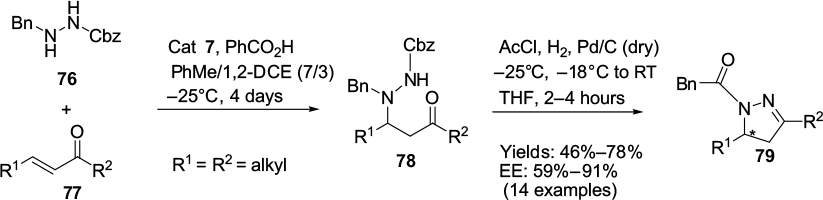 | Figure 26 Application of 9-epi-aminoquinine and its pseudoenantiomer 9-epi-aminoquinidine in synthesis of chiral 3,5-dialkyl-2-pyrazolines. Abbreviations: cat, catalyst; DCE, dichloroethane; RT, room temperature; THF, tetrahydrofuran; EE, enantiomeric excess. |
Liu et al reported the Cinchona-alkaloid primary amine 7 to be an efficient catalyst for the asymmetric conjugate addition reaction of nitroalkanes 80 to enones 77, forming the corresponding Michael adducts 81 in moderate-to-good yields and with high enantioselectivity (91%–99% EE) (Figure 27) for both acyclic and cyclic enones.56
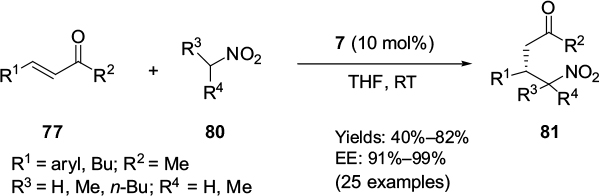 | Figure 27 Asymmetric conjugate addition of nitroalkanes to enones catalyzed by 9-epi-aminoquinine. Abbreviations: THF, tetrahydrofuran; RT, room temperature; EE, enantiomeric excess. |
Molleti et al designed a conjugate addition of cyclic 1,3-dicarbonyl compounds 83 to a range of β-substituted 2-enoylpyridines 82 (Figure 28).57 The reaction was efficiently catalyzed by cinchonidine/cinchonine-derived urea catalyst 29, producing the desired Michael adducts 84 in excellent yields and enantioselectivity. Additionally, by use of the pseudoenantiomeric pair of the catalyst 29, both enantiomers of the products were obtained at the same level of enantioselectivity.
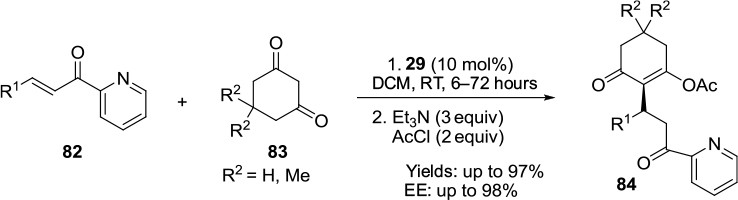 | Figure 28 Conjugate addition of 1,3-dicarbonyl compounds to β-substituted 2-enoylpyridines. Abbreviations: DCM, dichloromethane; RT, room temperature; EE, enantiomeric excess; equiv, equivalents. |
Michael-addition reactions of α-fluoro-β-ketoesters 85 and maleimides 86 forming the corresponding adducts 88 have been reported to be catalyzed efficiently by a Cinchona-derived catalyst 87 attached with a perfluoroalkyl tag (Figure 29). The catalyst was recovered by fluorous solid-phase extraction and reused for a second cycle.58
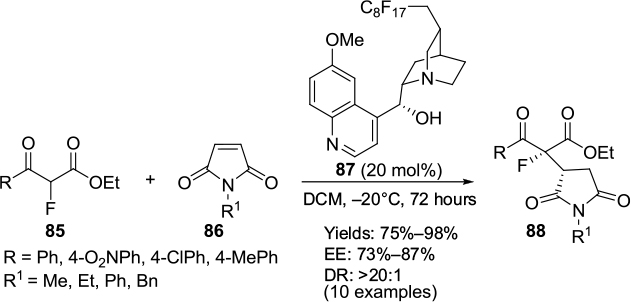 | Figure 29 Application of a Cinchona-derived catalyst attached with a perfluoroalkyl tag in Michael addition. Abbreviations: DCM, dichloromethane; DR, diastereomeric ratio; EE, enantiomeric excess. |
A direct organocatalytic enantioselective vinylogous Michael addition of γ-substituted butenolides 90 and α,β-unsaturated esters 89 has been reported to be catalyzed by quinine-derived catalyst 91.59 This reaction results in the formation of almost diastereo- and enantiomerically pure γ,γ-disubstituted butenolide products 92 with adjacent tertiary and quaternary stereocenters (Figure 30). The authors suggested that the phenolic hydroxyl group of the catalyst activated the Michael acceptor by H-bonding and the quinuclidine base deprotonated the acidic proton of butenolides.
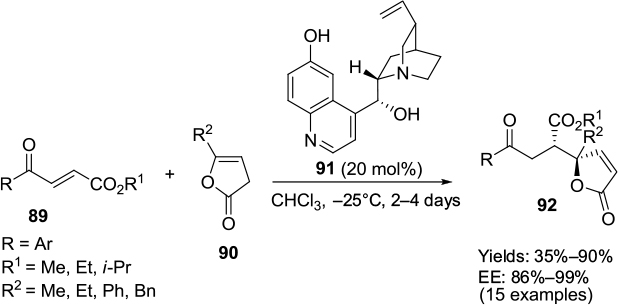 | Figure 30 Michael addition of γ-substituted butenolide to α,β-unsaturated esters catalyzed by quinine-derived catalyst. Abbreviation: EE, enantiomeric excess. |
Wang et al recently investigated the catalytic efficiency of 9-thiourea- and 9-squaramide-modified Cinchona alkaloids, quinidines, cinchonine, cinchonidine, (DHQ)2DQN, dihydroquinine, and quinine in the reactions of pyrazolones with p-benzoquinone.60 They observed quinine at an extremely low concentration (2 mol%) to be the most efficient catalyst. The reaction afforded products by asymmetric tandem Michael addition/oxidation at the C4 and C2 positions of pyrazolones and benzoquinone, respectively. The reaction of p-benzoquinone with 1-phenylpyrazol-5-ones bearing alkyl and aryl substituents on C3 and C4 in dichloromethane at room temperature (24 hours) afforded products with 27%–72% yields and enantioselectivity from 0% to 99%. The 1-phenylpyrazol-5-one, bearing a 2-thienyl group at C3 and a benzyl group at C4, gave the lowest yield of 27%. The maximum enantioselectivity (99% EE) was observed in the reaction of 4-allyl-1,3-diphenylpyrazol-5-one, while no enantiomeric excess was observed in the reaction of 4-benzyl-3-methyl-1-phenylpyrazol-5-one.
Asymmetric Michael addition of another biologically important diazaheterocycle, triketopiperazines to enones, in the presence of Cinchona alkaloid-derived catalysts was investigated by Cabanillas et al.61 A highly enantioselective formation of adducts was achieved using 10 mol% of O-benzyl-substituted 6′-hydroxycinchonine as a catalyst (Figure 31). The reactions were carried out in dichloromethane at –20°C for 3–6 hours. According to the proposed mechanism, the quinuclidine nitrogen activates the donor by enolate formation. The 6′-OH group and protonated quinuclidine nitrogen of the catalyst were H-bonded to the C=O of the enone moiety and enolate oxygen, respectively, in the transition state.
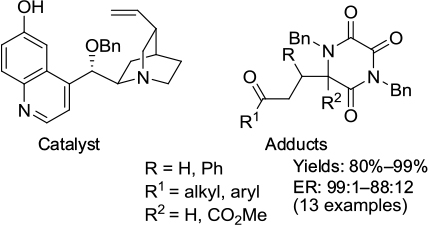 | Figure 31 Adducts from enantioselective Michael addition of triketopiperazines to enones and the catalyst used. Abbreviation: ER, enantiomeric ratio. |
Zhao et al reported asymmetric Michael addition of α-aryl isocyanoacetates 47 to β-trifluoromethylated enones 93 catalyzed by the Cinchona alkaloid squaramide 94.62 The corresponding adducts 95, with an adjacent chiral tertiary carbon center bearing a trifluoromethyl group and a quaternary carbon center, were obtained in moderate-to-good yields and with excellent stereoselectivity (Figure 32). The authors have successfully employed adducts 95 in the synthesis of biologically attractive chiral β-trifluoromethylated pyrroline carboxylates 96. Isocyanoacetates with no substituents or with an electron-withdrawing group usually gave higher yields in comparison to those with an electron-donating group. The yields and enantioselectivity also depended on the electronic and steric factors on the enones. β-Trifluoromethylated enones with an electron-withdrawing or weak electron-donating group on the para- or meta-position of the aromatic ring led to higher yields in comparison to those with a strong electron-donating group. The sterically hindered enones offered low yields and enantioselectivity, as expected.
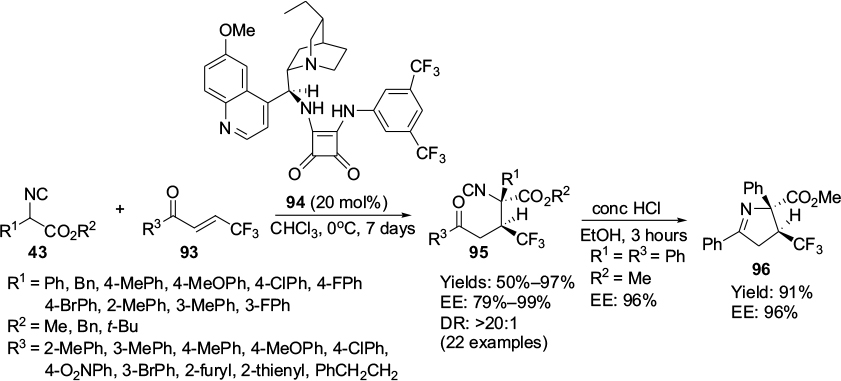 | Figure 32 Application of Cinchona alkaloid squaramide in Michael addition of α-aryl isocyanoacetates to β-trifluoromethylated enones. Abbreviations: DR, diastereomeric ratio; EE, enantiomeric excess; conc, concentrated. |
Application of Cinchona-derived catalysts in oxy- and sulfa-Michael additions
The Cinchona-based urea catalyst 33 has been employed in an intramolecular oxy-Michael addition of phenol derivatives 97 bearing easily available (E)-α,β-unsaturated ketones.63 The reaction led to the formation of asymmetric synthesis of 2-substituted chromans 98 in high yields (Figure 33). A substrate with the 4-bromophenyl group yielded the quantitative yield (99%) of the product with 83% EE. A substrate with R = Me offered moderate yield of 64% and low enantioselectivity (36% EE).
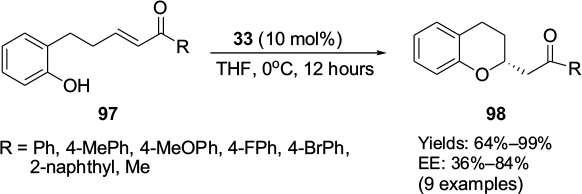 | Figure 33 Application of Cinchona alkaloid-based urea catalyst in oxy-Michael addition. Abbreviations: THF, tetrahydrofuran; EE, enantiomeric excess. |
The catalytic asymmetric sulfa-Michael addition of thiols to electron-deficient olefins represents a straightforward and versatile approach toward obtaining valuable optically active sulfur compounds of biological interest. Dai et al reported the enantioselective sulfa-Michael addition of a variety of thiols to trans-chalcones.64 They utilized Cinchona alkaloid-derived squaramide catalysts, first developed by Malerich et al,65 under mild conditions, getting the corresponding adducts in moderate-to-excellent yields and high enantioselectivity (up to 99%). Rana et al developed a sulfa-Michael addition of thioacids 99 to α,β-unsaturated ketones 100 to synthesize the chiral sulfa-containing frameworks, such as 101 (Figure 34), which are common in biologically active natural products and pharmaceutical agents.66 The pseudoenantiomeric quinine/quinidine-derived urea catalysts 33a/b catalyzed the reaction most efficiently, affording both enantiomers of the desired products equally in high yields and with same level of enantioselectivity.
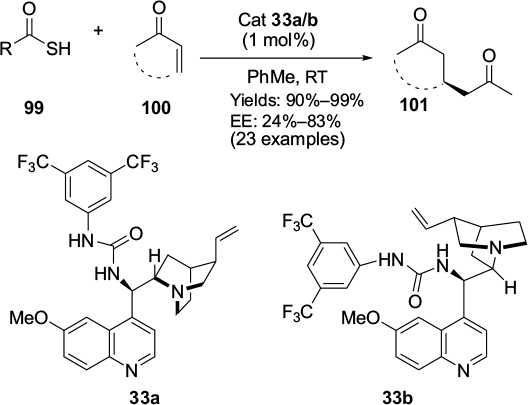 | Figure 34 Application of Cinchona alkaloid-based urea catalyst in sulfa-Michael addition. Abbreviations: cat, catalyst; RT, room temperature; EE, enantiomeric excess. |
Recently, Breman et al reported another example of sulfa-Michael addition.67 This group studied a range of Cinchona-alkaloid derived catalysts bearing a hydrogen bonding group at the C6′ position in the reaction of diverse thiols 103 with α,β-unsaturated N-acylated oxazolidin-2-ones 102 and related α,β-unsaturated α-amino acids. The catalysts with a sulfonamide group were observed to be superior, as they gave products 105 very good diastereoselectivity and high enantioselectivity (Figure 35). The C6′ sulfonamide catalyst 104 gave the main enantiomer with (S)-configuration in the reaction of α,β-unsaturated N-acylated oxazolidin-2-ones, while the C6′ thiourea gave the main enantiomer with (R)-configuration. The sulfa-Michael addition products were subsequently converted to β-functionalized cysteines 106 in high yields.
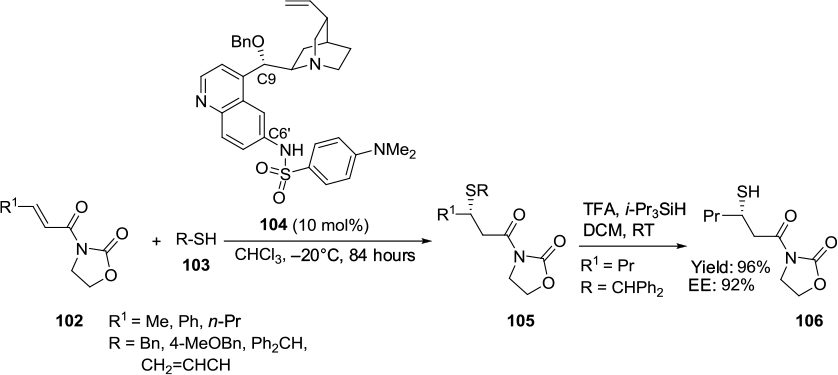 | Figure 35 Application of a Cinchona alkaloid-based catalysts bearing a C9 benzyloxy and C6′ sulfonamide group in sulfa-Michael addition. Abbreviations: TFA, trifluoroacetic acid; DCM, dichloromethane; RT, room temperature; EE, enantiomeric excess. |
It is evident from the discussions in preceding sections that asymmetric Michael additions are by far the most investigated reactions. Cinchona-derived thioureas have emerged as powerful catalysts for enantioselective Michael additions. Nitroolefins serve as the most common type of Michael receptor. This literature review reveals the development of some novel catalysts with sulfonamide and squaramide groups at C9, pentaerythritol tetrabromide-based chiral quaternary ammonium salts, and BINOL-quinine squaramide to achieve enantioselective Michael additions of different kinds of substrates. These catalysts have been successfully employed not only in C–C bond-forming reactions but also in C–O and C–S bond-forming reactions.
Cinchona alkaloid-derived primary amines in the presence of acids activate the α,β-unsaturated carbonyl compounds via iminium ion catalysis while interacting with nucleophiles, such as hydrazides, by H-bonding in a stereo-defining step. In the absence of an acid cocatalyst, enamine mechanism has been postulated. The thiourea and urea catalysts activate the electrophilic component by H-bonding through two coplanar protons. In these catalysts, the quinuclidine nitrogen activates the nucleophile. The C6′/C9 hydroxyl group also activates the electrophilic component in Michel-addition reactions.
Cycloaddition reactions
Cycloaddition reactions constitute the most versatile methodology in architecture of complex cyclic – both carbocyclic and heterocyclic – motifs. The most common and useful among them are the [2+2]-, [3+2]-, and [4+2]-cycloaddition reactions. Controlling the stereochemistry of cycloaddition reactions is a challenging endeavor.68 As in many other reactions, asymmetric catalysis is becoming an increasingly popular technology for controlling the stereochemistry of cycloaddition reactions. The application of Cinchona alkaloids and their derivatives in the induction of chirality in cycloaddition reactions was not very common till the end of the last decade. There were few papers reported when we wrote our last review article on the application of Cinchona alkaloids in organocatalysis.4 In recent years, however, several applications of modified Cinchona alkaloids in [2+2]-, [3+2]-, and [4+2]-cycloadditions have been reported. In particular, two substrates – allenic esters and 3-substituted 2-oxindoles – have drawn the considerable interest of the researchers.
Application of Cinchona-derived catalysts in [2+2]-cycloaddition reactions
An enantioselective formal [2+2]-cycloaddition of imines and allenoates in the presence of a Cinchona-alkaloid amide derivative as a chiral catalyst was reported by Zhu et al.69 This reaction, leading to the formation of a biologically important class of compounds, known as azetidines, constitutes the first example of an organocatalytic enantioselective [2+2]-cycloaddition of allenoates with N-sulfonyl imines. The reaction of aryl N-sulfonyl imines 107 with allenoates 108 in the presence of catalyst 109 bearing the N-Boc glycinamide group on C6′ and benzyloxy group on C9 afforded 2-alkylideneazetidines 110 in good yields with high enantioselectivity (Figure 36). It was observed that the electronic properties of the substituents on imine aryl groups did not influence enantioselectivity but did affect the yield, which in the case of an imine with 4-methoxyphenyl group was greatly diminished. The authors explained the formation of azetidines through the generation of a zwitterion 111 from the reaction of the catalyst with allenoate. The addition of the zwitterion to the imine may lead to the formation of intermediate 112, which may undergo 4-exo-trig cyclization to produce the cyclic intermediate 113. A hydride elimination from the latter intermediate may furnish the azetidine with concurrent regeneration of the catalyst. A possible transition-state model showed activation of imine by both CONH and Boc-NH through two hydrogen bonds, which in turn could be stabilized by a π–π interaction between the arenesulfonyl group and quinoline.
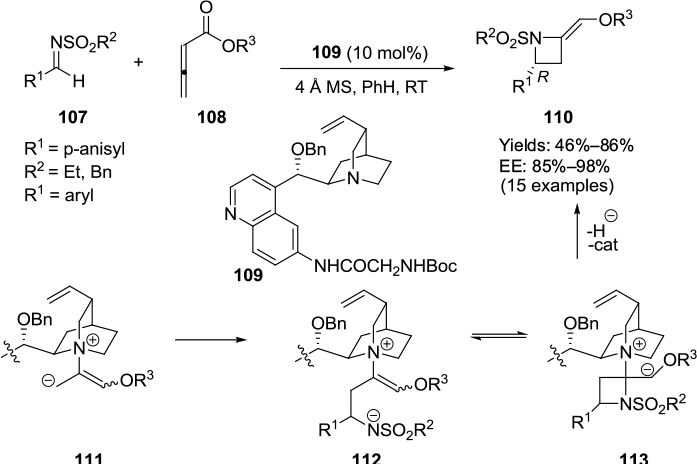 | Figure 36 Application of Cinchona alkaloid-based catalyst with N-Boc glycinamide on C6′ and benzyloxy groups on C9 in [2+2]-cycloaddition. Abbreviations: MS, molecular sieves; RT, room temperature; EE, enantiomeric excess. |
Application of Cinchona-derived catalysts in [4+2]-cycloaddition reactions
Pei and Shi developed a quinidine-derived catalyst 115 for the nucleophile-promoted asymmetric [4+2]-cycloaddition of allenic esters 108 with N-substituted imines of salicylaldehyde 114, forming the corresponding adducts 116 at up to 85% yield and 87% EE (Figure 37).70 The reaction was sensitive to solvents as well, and diethyl ether was the solvent of choice over a number of other ethers, haloforms, toluene, dioxane, ethyl acetate, and alcohols. Moreover, the addition of additives, such as diisopropylethylamine, p-toluenesulfonic acid, and benzoic acid, did not enhance enantioselectivity. Usually, the presence of an electron-donating group on the phenyl ring of the imines and an isopropyl group on ester gave relatively higher yield and enantioselectivity. The authors proposed a transition-state model in which the substrate was bound anti to the quinoline ring of the catalyst to minimize the steric interaction. The allenic ester may attack the imine from the Si face, affording adducts with predominantly (R)-configuration.
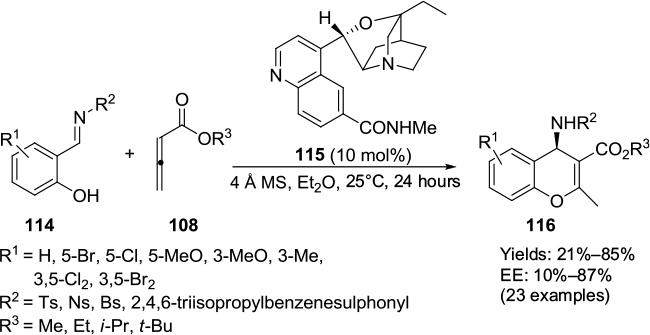 | Figure 37 Application of quinidine-derived catalyst for the nucleophile-promoted asymmetric [4+2]-cycloaddition. Abbreviations: MS, molecular sieves; EE, enantiomeric excess. |
Another example of a Cinchona-mediated enantioselective [4+2]-cycloaddition reported by Pei et al involved the reaction of an allenic ester with β,γ-unsaturated α-ketophosphonates, leading to the synthesis of highly functionalized phosphonate-substituted pyrans and dihydropyrans.71 In this case, a quinidine-derived catalyst identical to 115 but with a CONHPhOMe-4 group at the C6′ position instead of CONHMe was screened as the most efficient catalyst in acetonitrile solvent.
Ying et al reported an asymmetric [4+2]-cycloaddition reaction of β,γ-unsaturated α-ketoesters 15 with oxazolinones 1, affording a series of highly functionalized δ-lactones 118 with adjacent quaternary-β-tertiary stereocenters (Figure 38).72 After screening a number of quinine- and quinidine-derived catalysts, this group found the catalyst 117 to be the most efficient catalyst in terms of yields and enantioselectivity of the products. The configurations at quaternary and tertiary stereocenters were determined as S and R, respectively, by X-ray crystallography analysis. Diethyl ether was screened as the best solvent for the reaction. Esters with a broad range of aryl groups on the alkene moiety were well tolerated in the reaction. However, the ester bearing phenyl groups with strong electron-donating groups on alkenes appeared to give better enantioselectivity in comparison to those bearing electron-withdrawing groups at the same position. Furthermore, the variation in the substituents on the ester moiety of the substrate did not result in any significant change in yield or EE value. With regard to reactivity of oxazolinones, the substrate bearing a bulkier substituent (t-butyl) at the C4 position gave both poor yield and enantioselectivity and had longer reaction time. Considering the exclusive diastereoselectivity of the reaction, the authors proposed the inverse electron-demand hetero-Diels–Alder mechanism in an endo-manner and proposed the transition state 119.
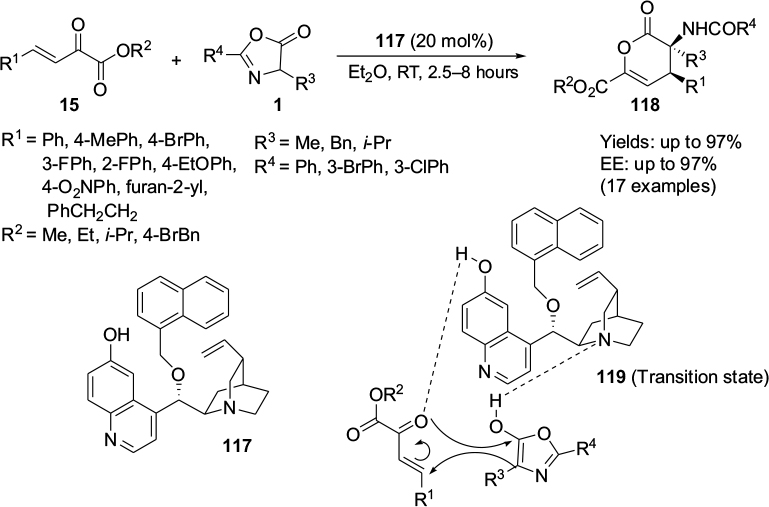 | Figure 38 Application of Cinchona alkaloid-based catalyst with a hydroxyl group on C6′ and an alkoxy group on C9 in [4+2]-cycloaddition. Abbreviations: RT, room temperature; EE, enantiomeric excess. |
Chen et al developed an asymmetric dearomatic Diels–Alder protocol for the reactions of heteroarene-embedded dienes 120 with maleimides 86 using a Cinchona-based primary amine 7a catalyst.73 The heteroarene moieties included benzofuran, thiophene, furan, and N-methylindole. The protocol, which leads to an array of chiral-fused frameworks with high molecular complexity, involves activation of diene systems by in situ trienamine formation. A representative example from 3-substituted heteroarenes is shown in Figure 39. The synthetic utility of a cycloadduct 121 was shown by its catalytic reduction to a multifunctional hexahydrobenzofuran with five continuous stereocenters.
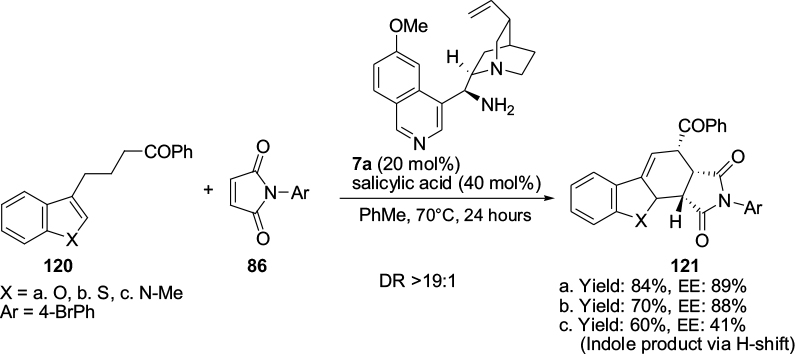 | Figure 39 Application of a Cinchona-based primary amine catalyst in Diels–Alder protocol. Abbreviations: DR, diastereomeric ratio; EE, enantiomeric excess. |
As mentioned earlier, isatin and its derivatives have emerged as powerful substrates for the synthesis of several biologically important spirooxindoles in recent years.14 The reactions of isatins, 3-isothiocyanato-2-oxindoles, and isatylidene malononitriles have been investigated for asymmetric synthesis of enantioenriched spirooxindoles employing modified Cinchona alkaloids as chiral organocatalysts. Wang et al have undertaken several studies on the asymmetric synthesis of spirooxindoles by cycloadditions catalyzed by Cinchona-alkaloid derivatives. An asymmetric [4+2]-annulation of isatins 9 with but-3-yn-2-one 122 used a dimeric Cinchona-alkaloid organocatalyst (DHQD)2-phthalazine 123 in the presence of 3.0 equivalents of D-diethyl tartrate in a mixed solvent containing 1:1 ratio of diphenyl ether and diethyl ether to afford the substituted spiro[indoline-3,2′-pyran]-2,4′-(3′H)-diones 124 (Figure 40) with good-to-excellent yields and high enantioselectivity under mild conditions.74 The authors proposed that the H-bonding interaction between isatin and d-diethyl tartrate may increase the steric hindrance between the organocatalyst and d-diethyl tartrate. Subsequently, nucleophilic attack of the enolate on the Re face of the imine may take place.
 | Figure 40 Application of dimeric Cinchona-alkaloid organocatalyst (DHQD)2PHAL in [4+2]-annulation of isatins with but-3-yn-2-one. Abbreviations: DHQD, dihydroquinidine; PHAL, phthalazine; EE, enantiomeric excess. |
Application of Cinchona-derived catalysts in [3+2]-cycloaddition reactions
Arróniz et al employed a Cinchona alkaloid-derived catalyst in a formal asymmetric [3+2]-cycloaddition of isocyanoacetates 43 to α,β-unsaturated ketones 125, leading to the formation of dihydropyrroles 126 (Figure 41).75 The reaction, however, required application of a silver salt as well, which formed a complex with isocyanate and increased the acidity of its α-proton, making it easier for the quinuclidine nitrogen to abstract this proton from isocyanates. After deprotonation, a chiral ion pair 127 may form by hydrogen bonding of the C9 hydroxyl group with nucleophile. This follows a 1,4-addition of nucleophile to vinyl ketone, generating a new stereocenter 128. The hydroxyl group at the C6′ position plays a key role in biasing the approach of the electrophile by forming a hydrogen bond with the ketone group.
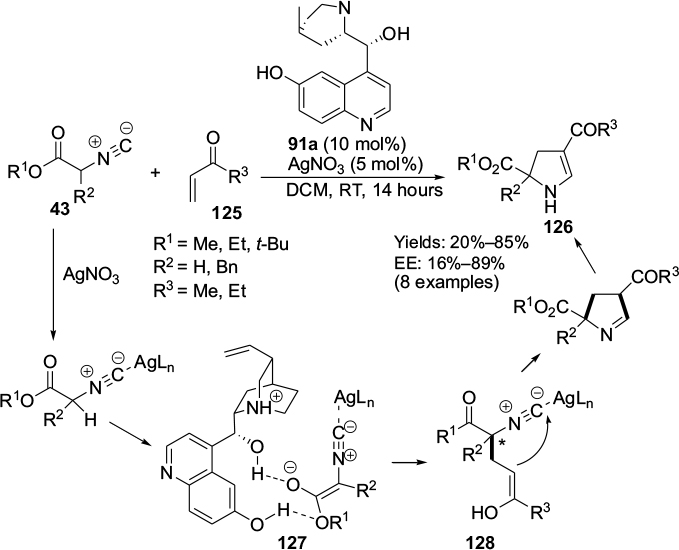 | Figure 41 Application of Cinchona-based catalyst with C6′ and C9 hydroxyl groups in [3+2]-cycloaddition. Abbreviations: DCM, dichloromethane; RT, room temperature; EE, enantiomeric excess. |
Fei et al investigated the reactions of isatylidene malononitriles 129 with Seyferth–Gilbert reagent 130 for the asymmetric synthesis of spiro-phosphonylpyrazoline-oxindoles 132 in the presence of several modified Cinchona alkaloids.76 The reactions in the presence of Cinchona alkaloid 131 with a bulky 2,2,3-trimethylbutoxy substituent at the C6′ position of the quinoline ring in a mixture of solvents containing cyclopentyl methyl ether (CPME) and dichloromethane led to the synthesis of several chiral spiro-phosphonylpyrazoline-oxindoles bearing two adjacent quaternary carbons in yields and enantioselectivity up to 99% (Figure 42). The nature of substituents on the phenyl ring had no effect on enantioselectivity, but the electron-donating substituents reduced the yield. The position of substituents on the ring, however, affected enantioselectivity: a substituent at the C7 position decreased enantioselectivity. It is interesting to see that the N–H of the isatin was tolerated in this reaction. However, the reaction requires a minimum of 4 days for completion. The authors suggested that this [3+2]-cycloaddition reaction proceeded via a Michael-type 1,4-addition and cyclization sequence. The Cinchona-alkaloid derivative could work as a bifunctional catalyst: the hydroxyl group as a Brønsted acid might have activated the isatylidene malononitrile by hydrogen bonding, and the amine moiety might have activated the C3 position of isatylidene malononitrile from its Si face.
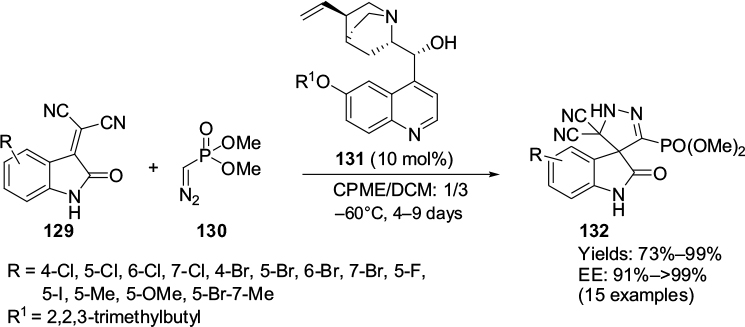 | Figure 42 Application of modified Cinchona-alkaloid catalyst with C9 hydroxyl and C6′ alkoxy groups in synthesis of chiral spiro-phosphonylpyrazoline-oxindoles. Abbreviations: CPME, cyclopentyl methyl ether; DCM, dichloromethane; EE, enantiomeric excess. |
Asano and Matsubara reported an enantioselective formal [3+2]-cycloaddition that occurred through hemiacetal intermediates, formed from the reactions of aldehydes 134 with γ-hydroxy-α,β-unsaturated ketones 133, leading to asymmetric synthesis of 1,3-dioxolanes 135.77 Cinchona alkaloid-based thiourea 44a was observed to be the most efficient catalyst, leading to the formation of products in excellent yields (71%–99%) and with high enantioselectivity (70%–98% EE) from both electron-rich and electron-poor enones at room temperature in cyclopentyl methyl ether (Figure 43). The use of an electron-deficient ketone instead of an aldehyde in the reaction afforded 1,3-dioxolane with 99% yield and moderate stereoselectivity (70% EE). The reaction, however, offered moderate diastereoselectivity (up to 4.7:1 DR). The authors attributed the enantioselectivity of this reaction largely to the step comprising oxy-Michael addition from the hemiacetal intermediates. The synthetic utility of the 1,3-dioxolanes was demonstrated by obtaining products from their ring-opening or reduction of their ketone group.
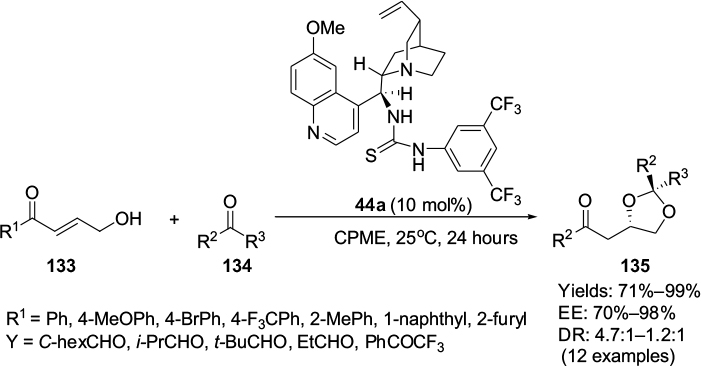 | Figure 43 Application of Cinchona alkaloid-based thiourea in asymmetric synthesis of 1,3-dioxolane by a formal [3+2]-cycloaddition. Abbreviations: CPME, cyclopentyl methyl ether; EE, enantiomeric excess; DR, diastereomeric ratio. |
Zhao et al reported the use of a quinine-derived bifunctional amine-thiourea-bearing sulfonamide (Figure 44) as a multiple hydrogen-bonding donor catalyst in the reactions of isatins with α-substituted isocyanoacetates.78 Several isatin derivatives with diverse electronic and steric environments underwent formal [3+2]-cycloaddition, affording optically active spirooxindole oxazolines. The reactions occurred most efficiently in diethyl ether, affording products with good yields (51%–95%), high diastereoselectivity (up to >20:1 DR), and good-to-excellent enantioselectivity (80%–97% EE). The reaction of 5-chloroisatin with ethyl 2-isocyanoacetate (with no substituent at the α-position), however, offered poor diastereoselectivity (2:1 DR) and enantioselectivity (36% EE). Also, the α-isopropyl- and α-(2-methylphenyl)isocyanoacetates did not react with 5-chloroisatin under the standard conditions.
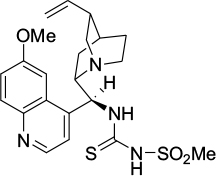 | Figure 44 A quinine-derived bifunctional amine-thiourea catalyst bearing sulfonamide. |
The [3+2]-cycloaddition reactions of 3-isothiocyanato-2-oxindoles 136 with allenic esters 108 or 2-butynedioic acid diester in the presence of squaramide catalyst 94a with stronger hydrogen-bonding donors furnished spirooxindoles 137 (Figure 45) with good yields and high enantioselectivity.79
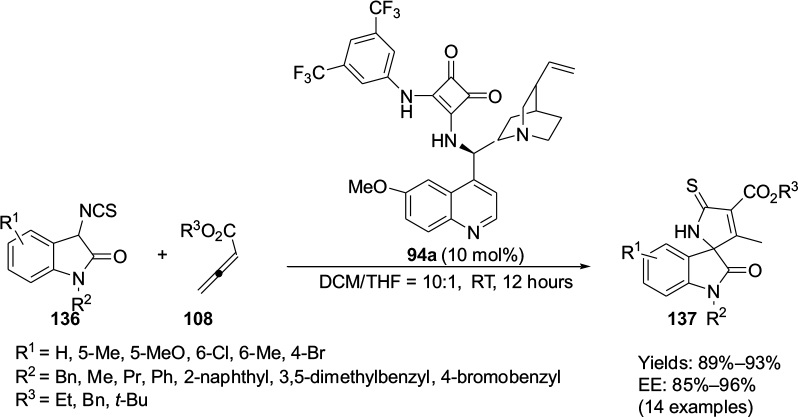 | Figure 45 Application of Cinchona-alkaloid squaramide catalyst in [3+2]-cycloaddition reactions of 3-isothiocyanato-2-oxindoles with allenic esters. Abbreviations: DCM, dichloromethane; THF, tetrahydrofuran; RT, room temperature; EE, enantiomeric excess. |
Cooperative catalysis by a Cinchona alkaloid-derived squaramide and AgSbF6 has been reported in the reaction of α-aryl isocyanoacetates with N-aryl maleimides.80 A formal [3+2]-cycloaddition reaction led to the formation of a wide range of optically active, substituted 1,3a,4,5,6,6a-hexahydropyrrolo[3,4-c]pyrrole derivatives with yields up to 98%, diastereoselectivity up to >20:1, and enantioselectivity up to 92% under mild reaction conditions. The two NH groups in the catalyst interacted with the esters by H-bonding with the carbonyl group, whereas the quinuclidine abstracted the proton from the 3-isothiocyanato-2-oxindoles. The final product was thus obtained through Michael addition/cyclization, which was followed by a series of transformations of the intermediate obtained.
A critical review of the cycloaddition reactions involving Cinchona alkaloid as catalysts suggests that tremendous progress has been made in this area. Isatins appear to be celebrity substrates. Cycloaddition reactions of appropriately designed imines and allenic esters have also been investigated. The C9 hydroxyl, C9 thiourea, C9 squaramide, dimeric Cinchona alkaloid, C9 amine, and C9 benzyloxy derivatives have been employed to achieve enantioselective synthesis of cycloaddition product via [2+2]-, [3+2]-, and [4+2]-cycloaddition reactions. In the only example of [2+2]-cycloaddition reported, a catalyst bearing a benzyloxy group at C9 and a side chain with two amide groups at C6′ was used. The two amide groups probably activate the imine by hydrogen bonding. The hydroxyl group at C6′/C9 and amido group at C-6′ also activate the imine and C3 position of 2-oxindole by H-bonding in the formation of [4+2]-cycloaddition products. In [3+2]-cycloaddition of isocyanoacetates to α,β-unsaturated ketones, the hydroxyl group at the C6′ position plays a key role in biasing the approach of the electrophile by forming a hydrogen bond with the ketone group.
Conclusion
Cinchona alkaloid-derived catalysts are highly desirable in achieving diastereo- and enantioselectivity in nucleophilic addition reactions. This class of catalysts has contributed immensely to the development of stereoselective synthetic methods. Several new catalysts have been designed and developed as optimized catalysts for aldol addition, Mannich addition, Strecker reaction, and Michael additions/cyclization. Most of these catalysts are developed by modifying the C6′ and C9 positions of the Cinchona alkaloids. These positions usually bear amine, urea, thiourea, alkoxy, hydroxyl, and amide functionality. Some dimeric Cinchona-alkaloid catalysts have also been employed in addition reactions.
Asymmetric aldol reactions of isatins and β,γ-unsaturated α-ketoesters using Cinchona-derived catalysts have led to the enantioselective formation of synthetically useful products. These reactions are mainly catalyzed by Cinchona alkaloid-based catalysts with NH2, OH, and alkoxy groups at the C9 position. Enamine catalysis by C9 Cinchona-based amine is the most common approach in these reactions. The application of such catalysts in Mannich and Strecker reactions of imines with nitroalkanes, α-nitro esters, oxazolinones, butenolides, and α-aryl isocyanoacetates has led to the synthesis of the corresponding adducts with very good yields and very high enantioselectivity. The C9 thiourea catalysts are more commonly used in such reactions. The thiourea moiety activates the azomethine carbon via H-bonding, and the quinuclidine nitrogen activates the nucleophile. Some novel Cinchona squaramides and BINOL–quinine–squaramide catalysts have been developed. Cinchona-alkaloid derivatives bearing a thiourea moiety have been employed in Michael-addition reactions of diverse types of substrates. Isatins and their derivatives have occupied a prominent place among different classes of heterocyclic compounds as a substrate in different types of addition reactions. The addition reactions of isatins and their derivatives, in the presence of Cinchona alkaloid-based catalysts, have furnished biologically demanding chiral 3-substituted 2-oxindoles and spirooxindoles. Based on this development, it is anticipated that the reactions of many other substrates suitable for these reactions will be investigated by employing the catalysts from the available domains. There is also scope for further development of new classes of Cinchona alkaloid-based catalysts.
Acknowledgment
The authors thank the Chemistry Department, University of Botswana, for providing the necessary facilities.
Disclosure
The authors report no conflicts of interest in this work.
References
Pellisier H. Asymmetric organocatalysis. Tetrahedron. 2007;63:9267–9331. | ||
Kacprzak K, Gawronski J. Cinchona alkaloids and their derivatives: versatile catalysts and ligands in asymmetric synthesis. Synthesis (Stuttg). 2001;2001:961–998. | ||
Marcelli T, Hiemstra H. Cinchona alkaloids in asymmetric organocatalysis. Synthesis (Stuttg). 2010;2010:1229–1279. | ||
Yeboah EM, Yeboah SO, Singh GS. Recent applications of Cinchona alkaloids and their derivatives as catalysts in metal-free asymmetric synthesis. Tetrahedron. 2011;67:1725–1762. | ||
Stegbauer L, Sladojevich F, Dixon DJ. Bifunctional organo/metal cooperative catalysis with Cinchona alkaloid scaffolds. Chem Sci. 2012;3:942–958. | ||
Duan JD, Li PF. Asymmetric catalysis mediated by primary amines derived from Cinchona alkaloids: recent advances. Catal Sci Technol. 2014;4:311–320. | ||
Bisai V, Bisai A, Singh VK. Enantioselective organocatalytic aldol reactions using small organic molecules. Tetrahedron. 2012;68:4541–4580. | ||
Heravi MM, Asadi S. Recent applications of organocatalysts in asymmetric aldol reactions. Tetrahedron Asymmetry. 2012;23:1431–1465. | ||
Franchino A, Jakubec P, Dixon DJ. Enantioselective synthesis of (-)-chloramphenicol via silver-catalyzed asymmetric isocyanoacetate aldol reaction. Org Biomol Chem. 2016;14:93–96. | ||
Zheng Y, Deng L. Catalytic asymmetric direct aldol reaction of α-alkyl azlactones and aliphatic aldehydes. Chem Sci. 2015;6:6510–6514. | ||
Wang W, Ma X, Wan J, Cao J, Tang Q. Preparation and confinement effect of a heterogeneous 9-amino-9-dexoy-epi-cinchonidine organocatalyst for asymmetric aldol addition in aqueous medium. Dalton Trans. 2012;41:5715–5726. | ||
Wan J, Zhao Z, Wang F, Ma X. An acid/base-regulated recyclable strategy for homogeneous Cinchona alkaloid-derived primary amine organocatalysts in aldol, vinylogous Michael and double Michael cascade reactions. European J Org Chem. 2015;2015:5755–5763. | ||
Perera S, Naganaboina VK, Wang L, et al. Organocatalytic highly enantioselective synthesis of β-formyl-α-hydroxyphosphonates. Adv Synth Catal. 2011;353:1729–1734. | ||
Singh GS, Desta ZY. Isatins as privileged molecules in design and synthesis of spiro-fused cyclic frameworks. Chem Rev. 2012;112:6104–6155. | ||
Guo Q, Zhao CG. Primary amine catalyzed aldol reaction of isatins with acetaldehyde. Tetrahedron Lett. 2012;13:1768–1771. | ||
Kumar A, Chimni SS. Organocatalytic asymmetric direct aldol reaction of pyruvic aldehyde dimethyl acetal with isatin derivatives. European J Org Chem. 2013;2013:4780–4786. | ||
Li P, Zhao J, Li F, Chan AS, Kwong FY. Highly enantioselective and efficient organocatalytic aldol reaction of acetone with β,γ-unsaturated α-keto ester. Org Lett. 2010;12:5616–5619. | ||
Liu C, Dou X, Lu Y. Organocatalytic asymmetric aldol reaction of hydroxyacetone with β,γ-unsaturated α-keto esters: facile access to chiral tertiary alcohols. Org Lett. 2011;13:5248-5251. | ||
Lin N, Deng YQ, Zhang ZW, Wang Q, Lu G. Asymmetric synthesis of β-hydroxy-α-amino acid derivatives by organocatalytic aldol reactions of isocyanoesters with β,γ-unsaturated α-keto esters. Tetrahedron Asymmetry. 2014;25:650–657. | ||
El-Sepelgy O, Mlynarski J. Biomimetic direct aldol reaction of pyruvate esters with chiral aldehydes. Adv Synth Catal. 2013;355:281–286. | ||
Peng L, Wang LL, Bai JF, et al. Highly effective and enantioselective phosphoaldol reaction of diphenyl phosphite with N-alkylated isatins catalyzed by quinine. Tetrahedron Lett. 2011;52:1157–1160. | ||
Guo Q, Bhanushali M, Zhao CG. Quinidine thiourea catalyzed aldol reaction of unactivated ketones: highly enantioselective synthesis of 3-hydroxy-3-alkylindolin-2-ones. Angew Chem Int Ed Engl. 2010;49:9460–9464. | ||
Liu GG, Zhao H, Lan YB, et al. Asymmetric cross aldol addition of isatins with α,β-unsaturated ketones catalyzed by a bifunctional Brønsted acid Brønsted base organocatalyst. Tetrahedron. 2012;68:3843–3850. | ||
Allu S, Molleti N, Panem R, Singh VK. Enantioselective organocatalytic aldol reaction of unactivated ketones with isatins. Tetrahedron Lett. 2011;52:4080–4083. | ||
Okumus S, Tanyeli C, Demir AS. Asymmetric aldol addition of α-azido ketones to ethyl pyruvate mediated by a Cinchona-based bifunctional urea catalyst. Tetrahedron Lett. 2014;55:4302–4305. | ||
Han Z, Yang W, Tan CH, Jiang Z. Organocatalytic asymmetric Mannich reactions of 5H-oxazol-4-ones: highly enantio- and diastereoselective synthesis of chiral α-alkylisoserine derivatives. Adv Synth Catal. 2013;355:1505–1511. | ||
Fan W, Kong S, Cai Y, Wu G, Miao Z. Diastereo- and enantioselective nitro-Mannich reaction of α-substituted nitroacetates to N-phosphoryl imines catalyzed by Cinchona alkaloid thiourea organocatalysts. Org Biomol Chem. 2013;11:3223–3229. | ||
Chauhan P, Chimni SS. Organocatalytic asymmetric synthesis of 3-amino-2-oxindole derivatives bearing a tetrasubstituted stereocenter. Tetrahedron Asymmetry. 2013;24:343–356. | ||
Zhao MX, Zing L, Zhou H, Shi M. Cinchona alkaloid mediated asymmetric Mannich reaction of isocyanoacetates with isatin-derived ketimines and subsequent cyclization: enantioselective synthesis of spirooxindole imidazolines. RSC Adv. 2015;5:75648–75652. | ||
Liu YL, Zhou J. Organocatalytic asymmetric cyanation of isatin derived N-Boc ketoimines. Chem Commun. 2013;49:4421–4423. | ||
Wang D, Liang JY, Feng JC, et al. The quinine thiourea catalyzed asymmetric Strecker reaction: an approach for the synthesis of 3-aminooxindoles. Adv Synth Catal. 2013;355:548–558. | ||
Yan WJ, Wang D, Feng JC, Li P, Zhao DP, Wang R. Synthesis of N-alkoxycarbonyl imines derived from isatins and their application in enantioselective synthesis of 3-aminooxindoles. Org Lett. 2012;14:2512–2515. | ||
Guo YL, Zhang Y, Qi LW, Tian F, Wang LX. Organocatalytic direct asymmetric vinylogous Mannich reaction of γ-butenolides with isatin-derived ketimines. RSC Adv. 2014;4:27286–27289. | ||
Kumar A, Kaur J, Chimni SS, Jassal AK. Organocatalytic enantioselective aza-Henry reaction of ketimines derived from isatins: access to optically active 3-amino-2-oxindoles. RSC Adv. 2014;4:24816–24819. | ||
Hara N, Nakamura S, Sano M, Tamura R, Funahashi Y, Shibata N. Enantioselective synthesis of AG-041R by using N-heteroarenesulphonyl Cinchona alkaloid amides as organocatalysts. Chemistry. 2012;18:9276–9280. | ||
Barrulas PC, Genoni A, Benaglia M, Burke AJ. Cinchona-derived picolinamides: effective organocatalysts for stereoselective imine hydrosilylation. European J Org Chem. 2014;2014:7339–7342. | ||
Csákÿ AG, de la Herrán G, Murcia MC. Conjugate addition reactions of carbon nucleophiles to electron-deficient dienes. Chem Soc Rev. 2010;39:4080–4102. | ||
Zhang Y, Wang W. Recent advances in organocatalytic asymmetric Michael reactions. Catal Sci Technol. 2012;2:42–53. | ||
Chen WY, Li P, Xie JW, Li XS. Organocatalytic and enantioselective Michael reaction of malonates to 3-nitro-2H-chromenes. Catal Commun. 2011;12:502–504. | ||
Ramireddy N, Abbaraju S, Zhao CG. Organocatalyzed enantioselective synthesis of 2-amino-5-oxo-5,6,7,8-tetrahydro-4H-chromene-3-carboxylates. Tetrahedron Lett. 2011;52:6792–6795. | ||
Rana NK, Singh VK. Enantioselective enolate protonation in sulfa-Michael addition to α-substituted N-acryloyloxazolidin-2-ones with bifunctional organocatalyst. Org Lett. 2011;13:6520–6523. | ||
Li XJ, Peng FZ, Li X, et al. Enantioselective and regioselective organocatalytic conjugate addition of malonates to nitroenynes. Chem Asian J. 2011;6:220–225. | ||
Luo J, Jiang C, Wang H, Xu LW, Lu Y. Direct asymmetric Michael addition of phthalide derivatives to chalcones. Tetrahedron Lett. 2013;54:5261–5265. | ||
Yu L, Xie X, Wu S, et al. Organocatalytic asymmetric Michael addition of 2-naphthols to alkylideneindolenines generated in situ from arenesulfonylalkylindoles. Tetrahedron Lett. 2013;54:3675–3678. | ||
Zhao MX, Dai TL, Liu R, et al. Enantioselective Michael addition of 3-aryloxindoles to a vinyl bisphosphonate ester catalyzed by a Cinchona alkaloid-derived thiourea catalyst. Org Biomol Chem. 2012;10:7970–7979. | ||
Cai CW, Zhu XL, Wu S, et al. Organocatalytic asymmetric Michael addition of oxazolones to arylsulfonyl indoles: facile access to syn-configured α,β-disubstituted tryptophan derivatives. European J Org Chem. 2013;2013:456–459. | ||
Wang JJ, Lao J, Hu ZP, et al. Organocatalytic asymmetric conjugate addition of cyclic 1,3-dicarbonyl compounds to β,γ-unsaturated α-ketoesters. Arch Org Chem. 2010;9:229–243. | ||
Chen JR, Cao YJ, Zou YQ, et al. Novel thiourea-amine bifunctional catalysts for asymmetric conjugate addition of ketones/aldehydes to nitroalkenes: rational structural combination for high catalytic efficiency. Org Biomol Chem. 2010;8:1275–1279. | ||
Chen JR, Zou YQ, Fu L, Ren F, Tan F, Xiao WJ. Highly enantioselective Michael addition of aldehydes to nitroolefins catalyzed by primary amine thiourea organocatalysts. Tetrahedron. 2010;66:5367–5372. | ||
Shi X, He W, Li H, Zhang X, Zhang S. Highly efficient and enantioselective Michael addition of acetylacetone to nitroolefins catalyzed by chiral bifunctional organocatalyst bearing multiple hydrogen-bonding donors. Tetrahedron Lett. 2011;52:3204–3207. | ||
Liu B, Han X, Dong Z, Lv H, Zhou HB, Dong C. Highly enantioselective Michael addition of 1,3-dicarbonyl compounds to nitroalkenes catalyzed by designer chiral BINOL-quinine-squaramide: efficient access to optically active nitroalkanes and their oxazole derivatives. Tetrahedron Asymmetry. 2013;24:1276–1280. | ||
da Silva RC, da Silva GP, Sangi DP, et al. 1,1-Diamino-2-nitroethylenes as excellent hydrogen bond donor organocatalysts in the Michael addition of carbon-based nucleophiles to β-nitrostyrenes. Tetrahedron. 2013;69:9007–9012. | ||
Fang X, Dong XQ, Wang CJ. Highly efficient organocatalytic asymmetric Michael addition of homoserine lactone derived cyclic imino esters to nitroolefins. Tetrahedron Lett. 2014;55:5660–5662. | ||
Ashokkumar V, Siva A. Cinchona alkaloid-based chiral catalysts act as highly efficient multifunctional organocatalysts for the asymmetric conjugate addition of malonates to nitroolefins. Org Biomol Chem. 2015;13:10216–10225. | ||
Campbell NR, Sun B, Singh RP, Deng L. Cinchona alkaloid-catalyzed enantioselective amination of α,β-unsaturated ketones: an asymmetric approach to Δ2-pyrazolines. Adv Synth Catal. 2011;353:3123–3128. | ||
Liu W, Mei D, Wang W, Duan W. Highly enantioselective conjugate addition of nitroalkanes to enones catalyzed by Cinchona alkaloid derived primary amine. Tetrahedron Lett. 2013;54:3791–3793. | ||
Molleti N, Allu, Ray SK, Singh VK. Bifunctional chiral urea catalyzed highly enantioselective Michael addition of 1,3-dicarbonyl compounds to 2-enoylpyridines. Tetrahedron Lett. 2013;54:3241–3244. | ||
Huang X, Yi WB, Ahad D, Zhang W. Recyclable cinchona alkaloid catalyzed asymmetric Michael addition reaction. Tetrahedron Lett. 2013;54:6064–6066. | ||
Das U, Chen YR, Tsai YL, Lin W. Organocatalytic enantioselective direct vinylogous Michael addition of γ-substituted butenolides to 3-aroyl acrylates and 1,2-diaroylethylenes. Chemistry. 2013;19:7713–7717. | ||
He Y, Bao X, Qu J, Wang B. Asymmetric tandem Michael addition/oxidation of pyrazolones with p-benzoquinone catalyzed by cinchona alkaloids. Tetrahedron Asymmetry. 2015;26:1382–1387. | ||
Cabanillas A, Davies CD, Male L, Simpkins NS. Highly enantioselective access to diketopiperazines via cinchona alkaloid catalyzed Michael additions. Chem Sci. 2015;6:1350–1354. | ||
Zhao MX, Zhu HK, Dai TL, Shi M. Cinchona alkaloid squaramide-catalyzed asymmetric Michael addition of α-aryl isocyanoacetates to β-trifluoromethylated enones and its application in the synthesis of chiral β-trifluoromethylated pyrrolines. J Org Chem. 2015;80:11330–11338. | ||
Miyaji R, Asano K, Matsubara S. Asymmetric chroman synthesis via an intramolecular oxy-Michael addition by bifunctional organocatalysts. Org Biomol Chem. 2014;12:119–122. | ||
Dai L, Wang SX, Chen FE. A bifunctional Cinchona alkaloid-squaramide catalyst for the highly enantioselective conjugate addition of thiols to trans-chalcones. Adv Synth Catal. 2010;352:2137–2141. | ||
Malerich JP, Hagithara, Rawal VH. Chiral squaramide derivatives are excellent hydrogen bond donor catalysts. J Am Chem Soc. 2008;130:14416–14417. | ||
Rana K, Unhale R, Singh VK. Enantioselective sulfa-Michael addition of thioacids to α,β-unsaturated ketones with bifunctional organocatalyst. Tetrahedron Lett. 2012;53:2121–2124. | ||
Breman AC, Telderman SE, van Santem RP, Scott JI, van Marseeveen JH. Cinchona alkaloid catalyzed sulfa-Michael addition reactions leading to enantiopure β-functionalized cysteines. J Org Chem. 2015;80:10561–10574. | ||
Paquette LA. Asymmetric cycloaddition reactions. In: Morrison JD, editor. Asymmetric Synthesis. Vol 3. New York: Academic Press; 2012:455–501. | ||
Denis JB, Masson J, Retailleau P, Zhu J. Cinchona alkaloid catalyzed formal enantioselective [2+2]-cycloadditions of allenoates and imines: synthesis of 2,4-disubstituted imines. Angew Chem Int Ed Engl. 2011;50:5356–5360. | ||
Pei CK, Shi M. Quinidine derived organocatalysts for the nucleophile promoted asymmetric [4+2]-cycloaddition reaction of salicyl N-tosylimine with allenic esters. Tetrahedron Asymmetry. 2011;22:1239–1248. | ||
Pei CK, Jiang Y, Wei Y, Shi M. Enantioselective synthesis of highly functionalized phosphonate-substituted pyrans or dihydropyrans through asymmetric [4+2]-cycloaddition of β,γ-unsaturated α-ketophosphonates with allenic esters. Angew Chem Int Ed Engl. 2012;51:11328–11332. | ||
Ying Y, Chai Z, Wang HF, et al. Bifunctional Cinchona alkaloids-catalyzed asymmetric [4+2] cycloaddition reaction of β,γ-unsaturated α-keto esters with oxazolones. Tetrahedron. 2011;67:3337–3342. | ||
Xiao YC, Yue CZ, Chen PQ, Chen YC. Asymmetric dearomatic Diels-Alder reactions of diverse heteroarenes via π-system activation. Org Lett. 2014;16:3208–3211. | ||
Wang Q, Lian Z, Xu Q, Shi M. Asymmetric [4+2] annulations of isatins with but-3-yn-2-one. Adv Synth Catal. 2013;355:3344–3350. | ||
Arróniz C, Gil-González A, Semak V, Escolano C, Bosch J, Amat M. Cooperative catalysis for the first asymmetric formal [3+2] cycloaddition reactions of isocyanoacetates to α,β-unsaturated ketones. European J Org Chem. 2011;2011:3755–3760. | ||
Fei TD, Du F, Ning Y, Peng Y. Organocatalytic enantioselective 1,3-dipolar cycloadditions between Seyferth-Gilbert reagent and isatylidene malononitriles: synthesis of chiral spiro phosphonylpyrazoline oxindoles. Org Lett. 2015;17:1308–1311. | ||
Asano K, Matsubara S. Asymmetric synthesis of 1,3-dioxolanes by organocatalytic formal [3+2] cycloaddition via hemiacetal intermediates. Org Lett. 2012;14:1620–1623. | ||
Zhao MX, Zhou H, Tang WH, Qu WS, Shi M. Cinchona alkaloid-derived thiourea-catalyzed diastereoselective and enantioselective [3+2] cycloaddition reaction of isocyanoacetates to isatins: a facile access to optically active spirooxindole oxazolines. Adv Synth Catal. 2013;355:1277–1283. | ||
Du D, Ziang Y, Xu Q, Shi M. Enantioselective construction of spirooxindole derivatives: asymmetric [3+2] cyclization of isothiocyanatooxindoles with allenic esters or 2-butynedioic acid diesters. Adv Synth Catal. 2013;355:2249–2256. | ||
Zhao MX, Wei DK, Ji FH, Zhao XL, Shi M. Asymmetric formal [3+2] cycloaddition reaction of α-aryl isocyanoesters with N-aryl maleimides by bifunctional cinchona alkaloids-based squaramide/cooperative catalysis. Chem Asian J. 2012;7:2777–2781. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.