")
Back to Archived Journals » Reports in Organic Chemistry » Volume 5
Recent advances in the discovery and design of multicomponent reactions for the generation of small-molecule libraries
Authors Malinakova H
Received 4 May 2015
Accepted for publication 11 June 2015
Published 4 September 2015 Volume 2015:5 Pages 75—90
DOI https://doi.org/10.2147/ROC.S65115
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Sean Kerwin
Helena C Malinakova
Department of Chemistry, The University of Kansas, Lawrence, KS, USA
Abstract: The current drug discovery process is dependent on the ability of synthetic chemistry to deliver libraries of small molecules with ever increasing levels of structural complexity and diversity. Sequencing multicomponent reactions (MCRs) with elaboration or cyclization steps represents an effective approach toward generating libraries of complex and diverse small molecules. This review highlights the most recent (2010–2015) examples of the application of reaction sequences including MCRs to the synthesis of diverse assemblies of small heterocycles. Sequences performed in multiple separate steps, or as one-pot operations, and including MCRs based on 1) carbonyl condensations; 2) isocyanide-based reactions; 3) cycloadditions; and 4) transition metal-mediated reactions are discussed.
Keywords: multicomponent reactions, sequential reactions, tandem reactions, divergent reactions, heterocycles, diversity
Introduction
The term multicomponent reactions (MCRs) describes processes that construct bonds between three or more substrates in one synthetic operation yielding structurally complex organic compounds.1 At the onset of their evolution, the majority of MCRs was based on classical condensations between carbonyl derivatives and various nucleophiles, the illustrative example being the first known MCR, the Strecker synthesis of amino acids from aldehydes, potassium cyanide, and ammonium chloride reported in 1850.2 Others, like the Mannich reaction,3 and a host of transformations designed for the synthesis of nitrogen-containing heterocycles including Biginelli,4 Hantzsch,5 or Asinger6 reactions, also rely on the classical condensation processes. The introduction of the isocyanide-based Ugi7 and Passerini reaction8 MCRs expanded the range of accessible products to compounds rich in biologically relevant and reactive amide and ester functionalities. Applications of Ugi-type MCRs dominated the field for an extended period of time, as indicated by the numbers of published reviews9 and monographs.10 Currently, MCRs exploiting virtually all the mechanistic pathways known to modern synthetic organic chemistry have been reported, including organocatalytic transformations, cycloadditions, and transition metal-catalyzed or radical-mediated protocols.11 The fundamental advantages of MCRs stem from the rapid increase of molecular complexity in the products that notably enhances the economy of synthesis. The modularity of MCRs permits one to independently vary any one of the multiple starting materials to efficiently generate assemblies of compounds possessing a common structural core decorated with diverse substituents. MCRs were warmly embraced by medicinal chemists seeking to generate large combinatorial libraries of compounds for biological screening and drug discovery.12 More recently, the advent of diversity-oriented synthesis (DOS)13 spearheaded further advances in the design of modern MCRs. At present, the development and application of MCRs reached a fully mature state, as indicated by the countless recent reviews focusing on roles played by MCRs in specialized areas, including the synthesis of bioactive molecules,14 in DOS,15 in the synthesis of heterocycles,16 natural product-like polycyclic structures,17 and others.
In 2011, Ruijter et al18 highlighted the need to maximize scaffold complexity and diversity in libraries of small organic products of MCRs, as well as the discovery of stereo-controlled MCRs as two major challenges for the future of the design of novel MCRs. Ruijter et al’s review also outlined the design strategies that promise to effectively address these challenges.18
Herein, examples from the recent literature (2010–2015) that showcase the use of “modular reaction sequences” (MRS) and “combination of MCRs” (MCR2)18 as novel strategies for maximizing the diversity of the resulting small-molecule libraries are reviewed.
The general representation of the design concepts discussed herein is shown in Figure 1. The common premise rests on the notion that performing several chemical reactions, including at least one MCR, as an uninterrupted one-pot process (tandem reactions), or a sequence that may require delayed additions of a component or a catalyst, ideally also performed in one reaction vessel (sequential reactions), will result in a notable increase in the structural complexity and diversity in the final products. Furthermore, the presence of properly matched reactive functionalities (FGn) in all the components of the initial MCR sets the stage for the subsequent tandem or sequential transformations (Figure 1).
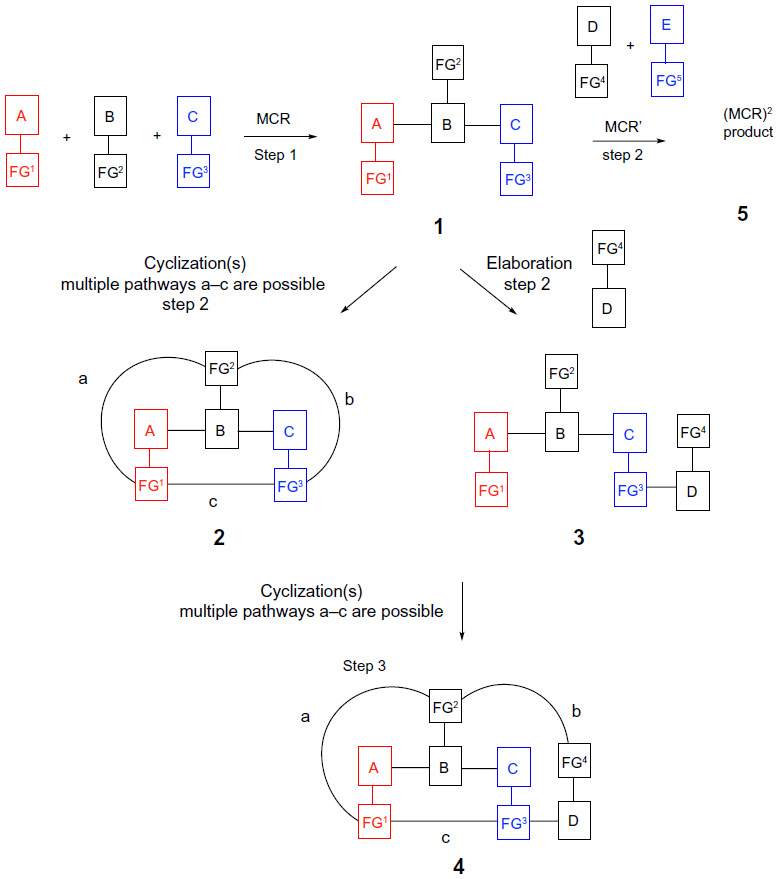 | Figure 1 Overview of sequential MCR/elaboration, tandem MCR/cyclization, and MCR2 strategies for the generation of diversity. |
Specifically, an MCR with properly functionalized components A, B, and C in step one delivers MCR product 1 that may undergo an intramolecular cyclization in situ via either pathway (a), (b), or (c), depending on the selected functional groups FG1–FG3, to deliver product 2 via a tandem (uninterrupted) one-pot two-step process (Figure 1). Alternatively, the functionalized product 1 can be further elaborated by the reaction with an additional component D to afford product 3 via a sequential (interrupted) one- or two-pot process. Functional groups in product 3 may be poised to initiate a subsequent in situ tandem cyclization via one of the multiple possible pathways A–C to yield product(s) 4. All these pathways fulfill the definition of a “modular reaction sequence”, as described in the review by Ruijter et al.18 Alternatively, product 1 of the initial MCR could become a substrate for a different MCR reaction that can be performed in step two following the addition of components D and E to deliver product 5 of MCR2. Ideally, the MCR2 protocol would be performed as a one-pot sequential reaction.
Aiming to highlight the diversity of mechanistic pathways employed by modern MCRs, protocols featured herein are organized according to the fundamental nature of the MCR step. Thus, reaction sequences involving MCRs based on 1) classical carbonyl condensations; 2) reactions with an isocyanide component; 3) cycloadditions; and 4) transition metal-catalyzed MCRs will be discussed in succession.
Most frequently, the development of novel MCRs has been aimed at the generation of libraries of molecules with potential drug-like properties, and thus naturally led to the prevalence of methodologies for the synthesis of medium-size heterocycles. This trend is reflected in all the transformations discussed herein. In Figure 2, examples of bioactive natural products, pharmaceutical drugs, and synthetic compounds with significant biological activities featuring core structures (highlighted in red) identical, or closely related to the structures of heterocycles prepared by the protocols described in Figures 3–7, are summarized. The data in Figure 2 underscore the relevance of the novel protocols for the drug discovery process.
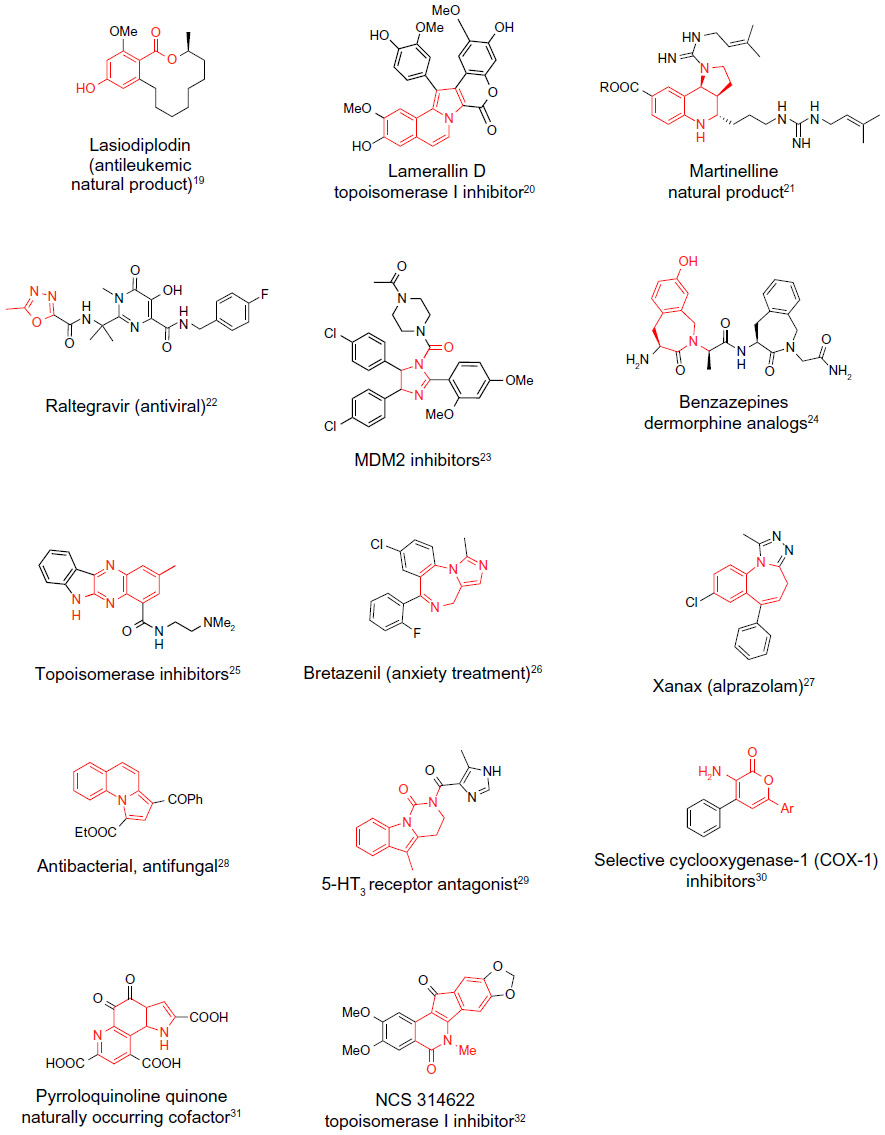 | Figure 2 Naturally occurring compounds, synthetic drugs, or bioactive compounds with core structures identical or closely related to the products of the MCRs described herein. |
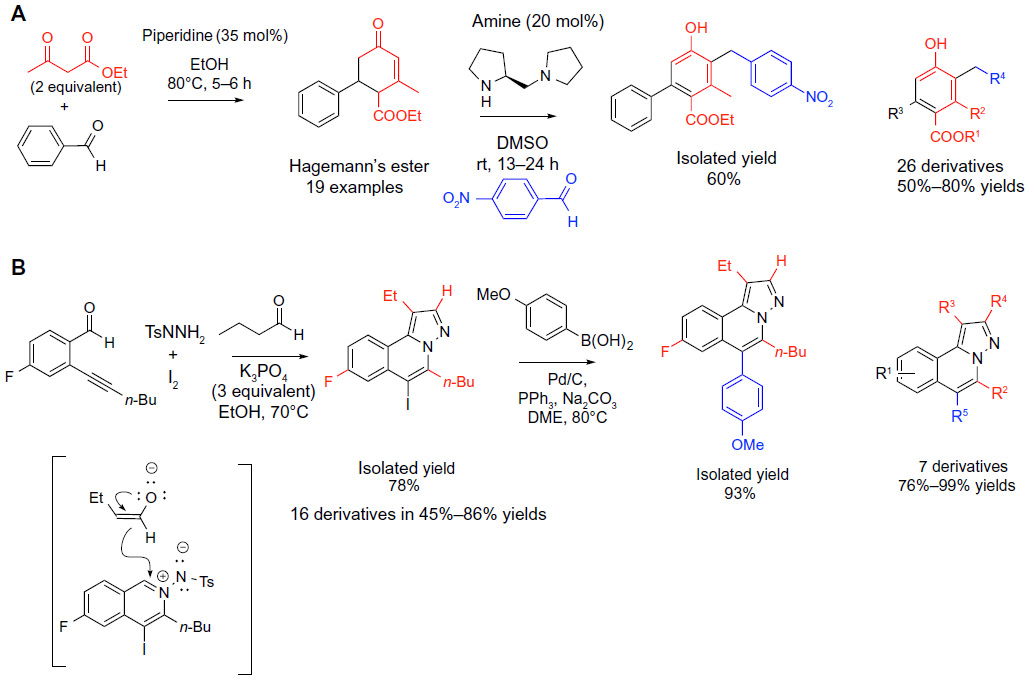 | Figure 3 Classical carbonyl condensations-based MCRs in sequential protocols (part 1). |
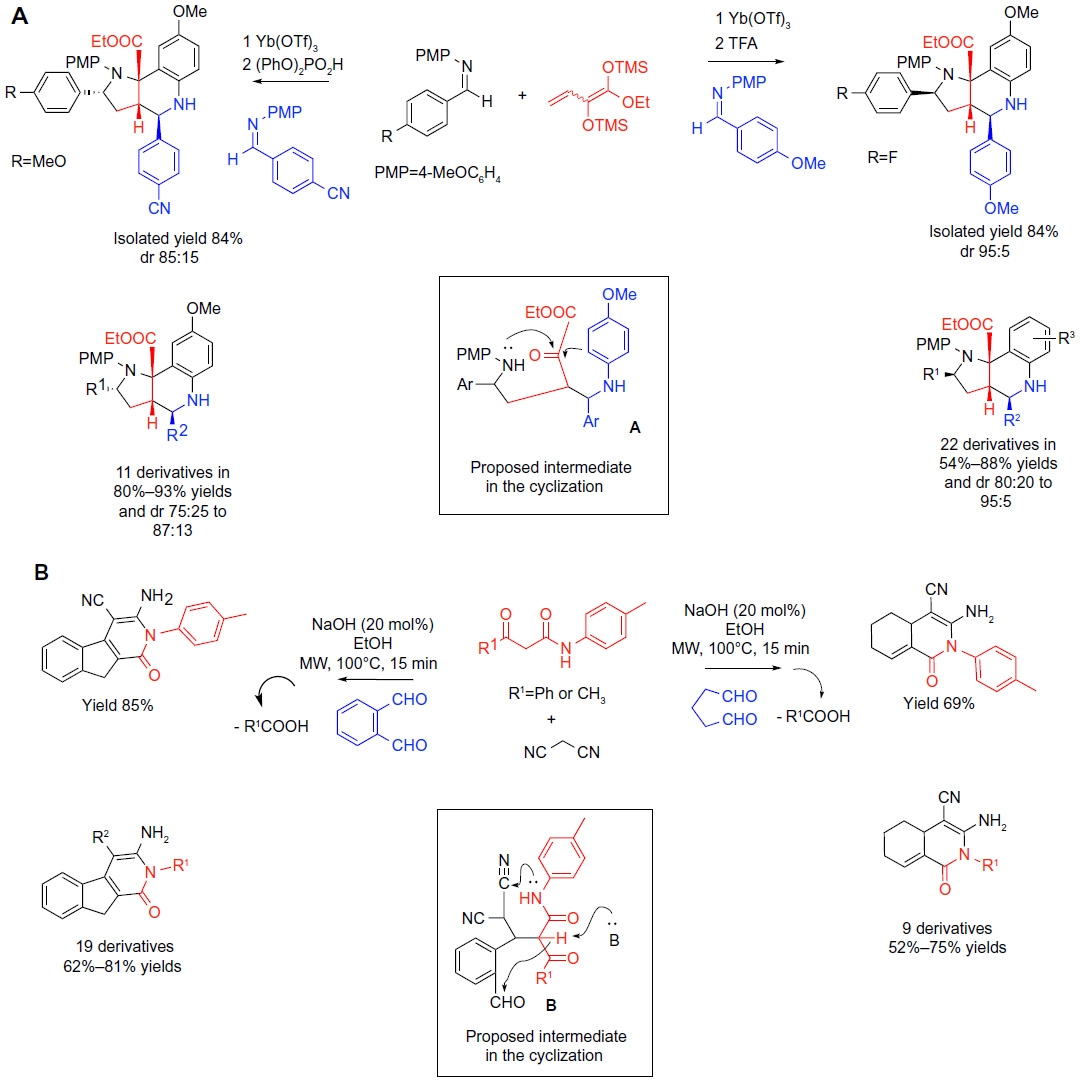 | Figure 4 Classical carbonyl condensations-based MCRs in sequential protocols (part 2). |
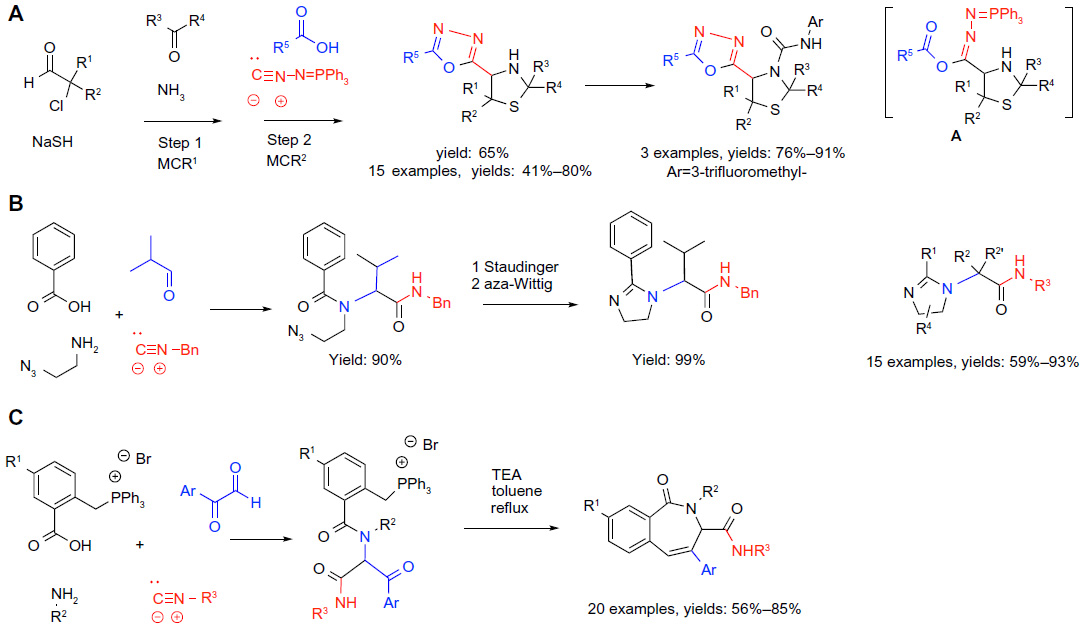 | Figure 5 Isocyanide-based MCRs in sequential protocols (part 1). |
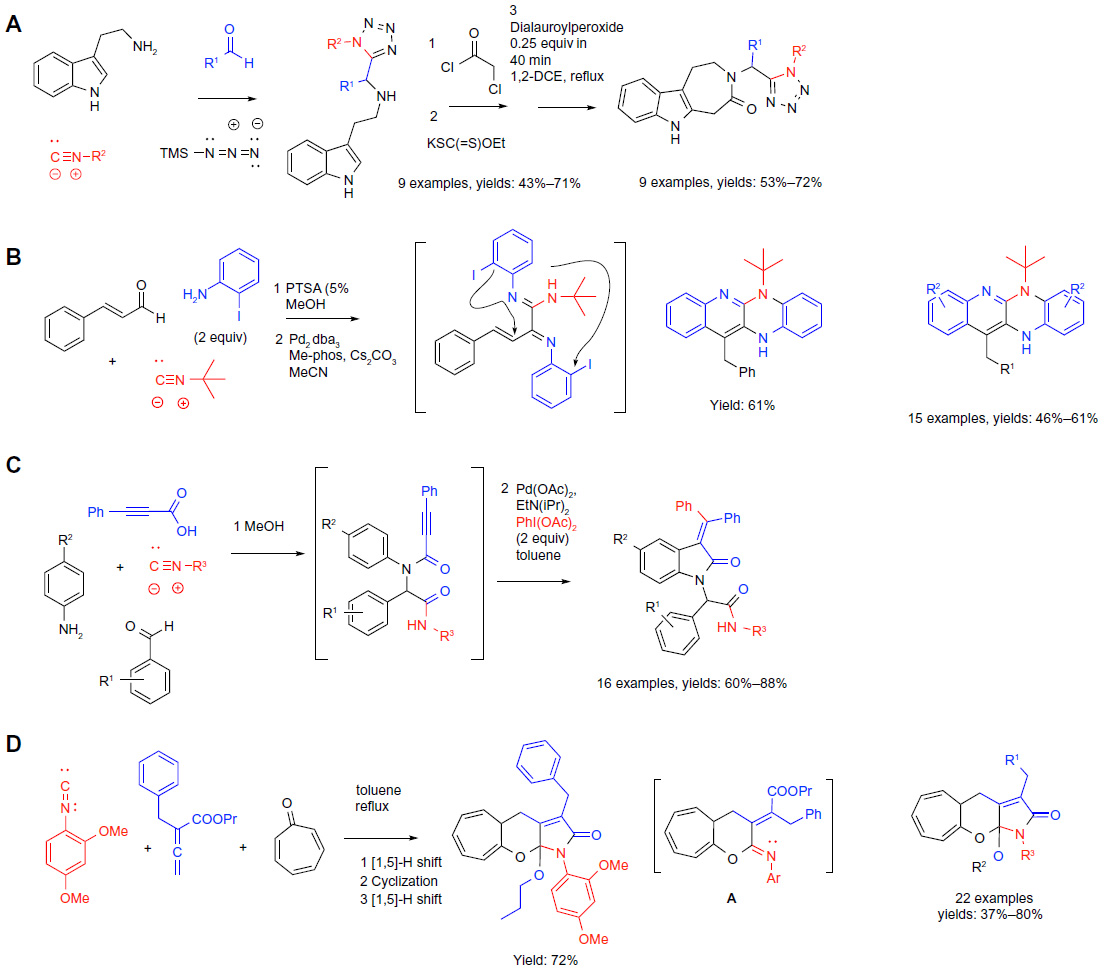 | Figure 6 Isocyanide-based MCRs in sequential protocols (part 2). |
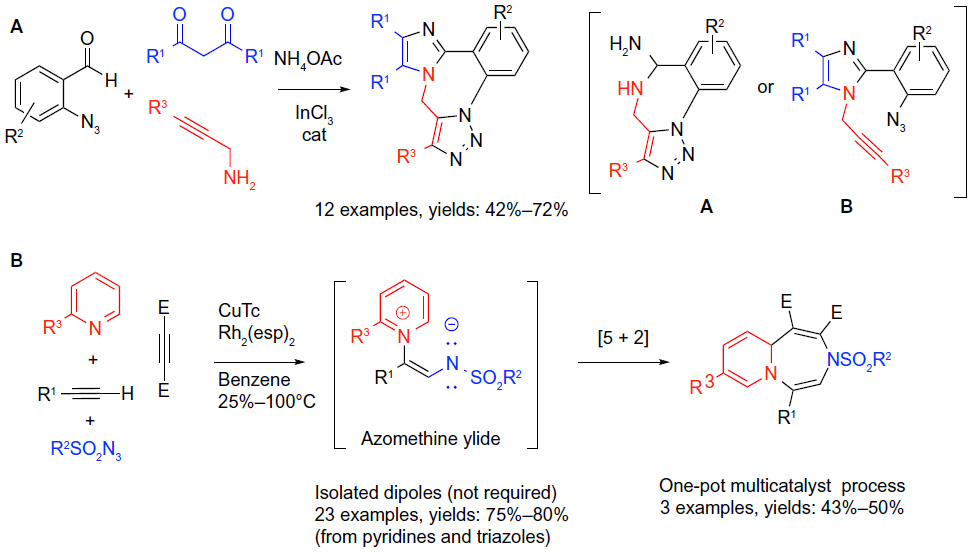 | Figure 7 Cycloaddition-based MCRs in sequential protocols. |
Classical carbonyl condensations-based MCRs in sequential protocols
Organocatalysis is a synthetic field rapidly growing in popularity. Ramachary et al33 reported an interesting application of amine-catalyzed classical condensation reactions for the preparation of diverse libraries of “push–pulls” olefins, phenols, and 2-methyl-2H-chromenes. A three-component coupling reaction of two equivalents of ethyl acetoacetate with an aldehyde catalyzed by piperidine afforded Hagemann’s esters via a tandem cascade of Knoevenagel/Michal/aldol/condensation/decarboxylation events (reaction A; Figure 3). A subsequent replacement of ethanol solvent with dimethyl sulfoxide and the addition of a distinct diamine catalyst ([S]-1-[2-pyrrolidinylmethyl]pyrrolidine) and various aromatic aldehydes into the same reaction vessel afforded the highly functionalized phenols in good yields (50%–80%) via Claisen–Schmidt/iso-aromatization events (Figure 3). It is notable that the choice of the diamine organocatalyst was critical for the success of the Claisen–Schmidt/iso-aromatization, favoring the fully aromatized condensation product. Overall, the protocol was used to produce a total of 26 of functionalized phenols in 50%–80% yields for the second step of the sequential process.33
Another example of the powers of the MCR/elaboration strategy was reported by Yu et al,34 who was seeking synthetic approaches to highly diversified H-pyrazolo(5,1-a) isoquinolines. For the initial MCR, Yu et al34 devised a tandem one-pot process involving inter- and intramolecular condensations and electrophilic cyclization between 2-alkynyl benzaldehyde, sulfonylhydrazide, bromine, or iodine as electrophiles, and ketones or aldehydes to afford H-pyrazolo(5,1-a)isoquinolines. The halogen atom was used in the second step as a functional handle for diversification of the resulting scaffolds via a Pd-catalyzed cross-coupling to various boronic acids (reaction B; Figure 3). The process is notable for the rapid assembly of the complex and highly substituted heterocyclic core in one step under mild conditions, as well as dual function of the halogens as activating reagents for the electrophilic cyclization and the handle for the subsequent elaboration. This strategy represents an insightful application of the reactivity demonstrated in the earlier work by Yue and Larock.35 Substituents highlighted in red and blue colors in Figure 3, reaction B indicate the points of diversification that were exploited to produce seven derivatives in 76%–99% yields for the final elaboration step (reaction B; Figure 3).
One approach toward producing molecular libraries with diverse core structures consists of substituting one of the components of the MCR with a structurally or electronically differentiated building block, which subsequently causes a divergence in the reaction pathways of the MCR. Transformations showcased in Figure 4 are examples of such methodologies termed the “single reactant replacement” by Ruijter et al.18
In the method shown as reaction A in Figure 4, the addition of 1,2-bis-trimethylsilyl enol ether into two moles of an N-arylimine served as the key MCR to deliver a multifunctional acyclic product A (Figure 4) capable of a subsequent in situ cyclization via imminium ion formation and its intramolecular trapping by an electron-rich aromatic ring to yield highly substituted hexahydropyrrolo(3,2-c)-quinolines (reaction A; Figure 4).36 The nucleophilic attack on the second mole of the imine was slower and required a time-resolved addition of a Bronsted acid. When instead of using two moles of the same arylimine, a different imine was added along with the Bronsted acid in a time-resolved manner, a large and substantially diverse set of pyrroloquinolines was produced (22 derivatives in 54%–88% yields; ratio of diastereomers (dr) 80: 20–95:5) (reaction A; Figure 4). Although aliphatic imines failed to participate in the same sequence, the transformation showed an interesting stereodivergency. When an electron-deficient imine was used in the delayed addition step (CNC6H4C[=NPMP]H instead of MeOC6H4C[=NPMP]H), the reaction conditions could be optimized to favor a distinct diastereomer by a range of 75:25–87:13 in diastereomeric ratios for eleven reported examples (80%–93% yields) (reaction A; Figure 4). In one example, the imine building blocks were replaced with the corresponding aldehydes (benzaldehyde and p-anisylaldehyde) and the amine (PMP-NH2), and the process was optimized to allow the entire sequence to be performed as a one-pot sequential five-component process on a nearly 800 mg scale of the produced pyrroloisoquinoline, demonstrating the practicality of the synthetic protocol.
The fundamentally analogous design of an MCR/cyclization sequence was employed in a recent report on the synthesis of indenopyridines and hydroisoquinolines.37 The structural divergence between these two core structures was controlled by the choice of either an aliphatic bisaldehyde or an aromatic 1,2-bisaldehyde delivering the hydroisoquinolines and indenopyridines, respectively. The bisaldehydes were reacted with 1,3-ketoamides and 1,3-biscyanides or 1,3-cyanoesters under basic conditions in a one-pot reaction (reaction B; Figure 4). Knoevenagel-type condensation followed by Michael addition generates a possible acyclic condensation product (structure B; Figure 4) that undergoes a rapid in situ cyclization due to the high density of compatible functional groups to yield the heterocyclic products. The methodology is attractive due to its operational simplicity and the demonstrated potential to obtain structurally diverse cores. The presence of orthogonal functionalities (CN, COOEt, and NH2) in the products of the MCR/cyclization sequences sets the stage for potential future diversification of the heterocyclic scaffolds.
Isocyanide-based MCRs in sequential protocols
Sequencing MCRs with other transformations may involve the preparation of one of the building blocks via a prior MCR yielding the so-called “MCR2” sequence.18 Alternatively, one of the building blocks for the MCR may contain a reactive functionality that is stable under the coupling conditions, but initiates a cyclization following the completion of the original MCR. Martens combined both these approaches in his preparation of thiazolidin-4-yl-1, 3,4-oxadiazoles (reaction A; Figure 5).38 First, five 3-thiazolines were prepared by the modified Asinger reaction.6 Subsequently, 3-thiazolines acting as the imine components of an Ugi-type process were reacted with carboxylic acids and isocyanides bearing an additional orthogonal functionality ([isocyanoimino]triphenylphosphorane). The three-component coupling (step 2) generated the presumed product A (Figure 5) which, instead of the traditional acyl-transfer step terminating the Ugi reaction, underwent an intramolecular aza-Wittig event yielding the substituted 1,3,4-oxadiazoles (reaction A; Figure 5). Finally, the possibility to further diversify the products via the reaction of the secondary amine group in the thiazolidine ring with a series of isocyanates giving urea derivatives was established (reaction A; Figure 5). Thus, this study exemplifies the powers of the sequential MCR2 strategy, combined with tandem cyclization and sequential elaboration for creating complexity and diversity in the targets.
Sequencing the Ugi-type MCRs with an intramolecular olefin formation via Staudinger/aza-Wittig39 or Wittig reaction40 was shown to be a popular synthetic tool. Two selected applications for the preparation of libraries of biologically relevant heterocycles are shown in reactions B and C in Figure 5.
The versatility of the Ugi reaction has been significantly expanded by the development of its variants.41 The Ugi-azide modification41 followed by a few “post MCR” functional group manipulations and terminated by a radical cyclization was applied to the synthesis of complex 3-tetrazolylmethyl-azepino(4,5-b)indol-4-ones relevant to drug discovery (reaction A; Figure 6).42 The MCR and two subsequent functional group manipulations yielding xanthates were performed in one pot without purifications. In the final operation, the treatment of xanthates with dilauroyl peroxide under heating or microwave irradiation afforded the azepine derivatives. Thus, although several steps are needed, the overall protocol is operationally simple and delivered nine examples of azepinoindolones diversified at two positions in synthetically practical yields (reaction A; Figure 6).
Recent advances in transition metal-mediated reactions of substrates possessing multiple heteroatoms set the stage for applying such transformation to the elaboration or cyclization of the Ugi-type products (reactions B and C; Figure 6).
Thus, the use of an amine component possessing an orthogonal aryl iodide functionality in a modified Ugi condensation afforded acyclic highly functionalized amino imides that were shown to effectively undergo double intramolecular Pd-catalyzed Heck/Hartwig–Buchwald cyclizations to afford dihydropyridopyrazines via a one-pot two-step sequential protocol (reaction B; Figure 6).43 Not surprisingly, the choice of the ligand (Me-phos) and solvent (acetonitrile) proved critical for the success of the double Pd-catalyzed cyclization, as well as the use of Pd2dba3 as the palladium source. The one-pot process was realized via the removal of methanol solvent after the first reaction step, followed by the addition of acetonitrile and the components of the Pd-catalytic system (Pd2dba3, Me-phos, Cs2CO3) into the crude reaction mixture to afford the target heterocycles in good yields.
Pd-catalyzed C–C bond-forming reactions under oxidative conditions have received much attention44 due to their practical significance and remaining intriguing mechanistic questions. Seeking modular methodology for a selective construction of diverse 3-(diphenylmethylidene)-2,3- dihydro-1H-indol-2-ones free of (3-phenylmethylidene)-2, 3-dihydro-1H-indol-2-one byproducts, a protocol consisting of a sequential Ugi four-component coupling and inter-/intramolecular coupling/cyclization induced by PhI(OAc)2 under Pd(OAc)2 catalysis has been developed (reaction C; Figure 6).45 The reaction sequence was performed without the full isolation and purification of the Ugi coupling products. The removal of the methanol solvent was followed by the addition of toluene along with the reagents for the Pd-catalyzed coupling/cyclization delivering the final indolone products in good yields (16 examples; 60%–88% yields). Mechanistically, a pathway alternative to the one proposed by the authors can be envisioned, involving the C–H aromatic palladation via the palladium in oxidation state Pd(II), and termination by the coupling of the heterocyclic organopalladium(II) intermediate with PhI(OAc)2 reagent through a Pd(II)/Pd(IV) cycle.
The methodologies utilizing sequences of Ugi-type MCRs with cutting edge Pd-catalyzed transformations described herein (reactions B and C; Figure 6) highlight the synthetic potential arising from combination of the strengths of two very distinct transformations.
Besides the “traditional” condensations, new types of reactivities of isocyanides have been exploited for the design of MCRs. A synthetic process based on an initial condensation of isocyanides with 1,2-disubstituted allenoates and tropolone was recently described (reaction D; Figure 6).46 The product A (Figure 6) of the three-component coupling engaged in a series of cyclization/H-shifts/cyclization/H-shift cascade events in situ, yielding the terminal tricyclic heterocycles. The substitution patterns of the heterocycles could be diversified at three distinct positions. The protocol demonstrates the utility of a one-pot tandem (uninterrupted) MCR/rearrangement/cyclization strategy made possible by the presence of properly positioned reactive functionalities in the building blocks of the initial MCR process (reaction D; Figure 6).
Cycloaddition-based MCRs in sequential protocols
The popular “click chemistry” involving a Cu-catalyzed cycloaddition of azides to alkynes provided the basis for the design of novel synthetic protocols and their impressive applications in bioorganic chemistry.47
The reactive functionalities requisite for the “click reaction” were incorporated into building blocks for the classical carbonyl condensation MCRs.48 An in situ intramolecular cycloaddition between the matched functionalities (azide and alkyne) was expected to deliver complex heterocycles featuring a diazepine fused to imidazole and triazole rings (reaction A; Figure 7). Thus, aldehydes possessing the azide group were condensed with 1,3-dicarbonyls, NH4OAc and propargyl amines under Lewis acid catalysis in a protocol anticipated to construct 1,3-imidazoles. However, in the presence of the “matched” azide and alkyne groups, the “click” (3 + 2) cycloaddition-forming triazole ring can occur in situ at any point in time when the two functionalities are brought into close proximity. Thus, either the intermediate A or B shown in Figure 7 was deemed possible, depending on the occurrence of the cycloaddition either prior to the completion of the traditional four-component coupling (A), or subsequent to the full assembly of the imidazole (B) (reaction A; Figure 7). The overall transformation was performed as a tandem one-pot protocol delivering 12 examples of heterocycles with three points of diversification.
Building on the powers of the “click reaction”, Lee et al49 reported an intriguing sequence that took advantage of the unique reactivity of 1-sulfonyl-1,2,3-triazoles. The process incorporates two distinct cycloadditions. A Rh-catalyzed reaction of the initial (3 + 2) cycloadducts with pyridines gave rise to isolable azomethine ylide dipoles that underwent an uncatalyzed (5 + 2) cycloaddition to afford highly substituted 1,4-diazepines (reaction B; Figure 7). A large number of diazepines (23 examples in 75%–80% yields) were prepared via a one-step Rh-catalyzed three-component coupling between triazoles synthesized separately via the Cu-catalyzed click process, pyridines, and alkynes. Ultimately, a tandem four-component multicatalytic (Cu/Rh) protocol was designed, in which the Cu-catalyzed click reaction is followed by the Rh-catalyzed in situ formation of the yilide and is terminated by the (5 + 2) process. This methodology was applied to a somewhat limited number of cases (three examples; 43%–50% yields) with encouraging results (reaction B; Figure 7). The transformation represents the first example of a metal-catalyzed multicomponent (5 + 2) cycloaddition. The novel protocol furthermore delivers 1,4-diazepine scaffolds fused to the pyridine ring that are closely related to medicinal psychoactive drugs.27
Transition metal-catalyzed MCRs in sequential protocols
The introduction of transition metal-catalyzed reactions to organic synthesis opened up the possibility to functionalize traditionally unreactive C–H bonds, as well as to realize other unprecedented transformations. Furthermore, fine distinctions between closely related transition metals, or other components of the catalytic systems (ligands, additives), were shown to give rise to divergent reaction pathways. The unique features of transition metal-catalyzed reactions contributed to the rising popularity of transition metal-catalyzed MCRs.50 Transition metal-catalyzed MCRs have been incorporated into various reaction sequences achieving impressive diversity in the resulting molecular ensembles. The effort to perform the sequential reactions as one-pot procedures brought attention to an emerging challenge of identifying reaction conditions that would avoid undesired interactions between the components of the multicatalytic systems. Such interactions may involve reactions between the two different transition metal complexes, and/or the additives that comprise the distinct catalytic systems, and may lead to a significant loss of activity (poisoning) of either one, or both, the catalysts present.51
A recent report on the preparation of pyrrolo(1,2-a)quinolines via a sequential iron(III)-catalyzed three-component coupling followed by a gold(III)-catalyzed intramolecular hydroarylation reaction illustrates a number of these features (reaction A; Figure 8).52 Mechanistically, the process is notable based on the importance of a proper choice of the two distinct transition metal catalysts. In the initial MCR, the FeCl3 catalyst selectively mediated the construction of the pyrrole ring in the presence of the alkyne functionality, instead of a possible undesired formation of indoles via a premature engagement of the alkyne. Presumably, the preferential coordination of the FeCl3 to the carbonyl group in the 1,3-dicarbonyls, as well as the nitro group, triggered the enamine formation with the aniline component, followed by the Michael addition of the enamine to the unsaturated nitro derivative yielding a substrate for an intramolecular cyclization via a Neff-like step53 to deliver the pyrrole. Lewis acids with stronger affinities for the alkyne group (for example, indium or gold salts) would not be suitable for this step. In contrast, in the “post-MCR” intramolecular hydroarylation,54 the selectivity of the Au(III)Cl3 for activating the alkyne for the nucleophilic attack by the pyrrole ring is critical. Thus, the resulting formal 6-endo addition of a C–H bond from the pyrrole across the alkyne group completes the cyclization delivering the targeted heterocyclic products (reaction A; Figure 8). The sequence of the two transformations was performed in two separate steps with isolation of the pyrrole derivatives. A large variety of pyrrolo(1,2-a)quinolones with four points of diversity was obtained in synthetically practical yields (19 examples; 50%–90% yields).
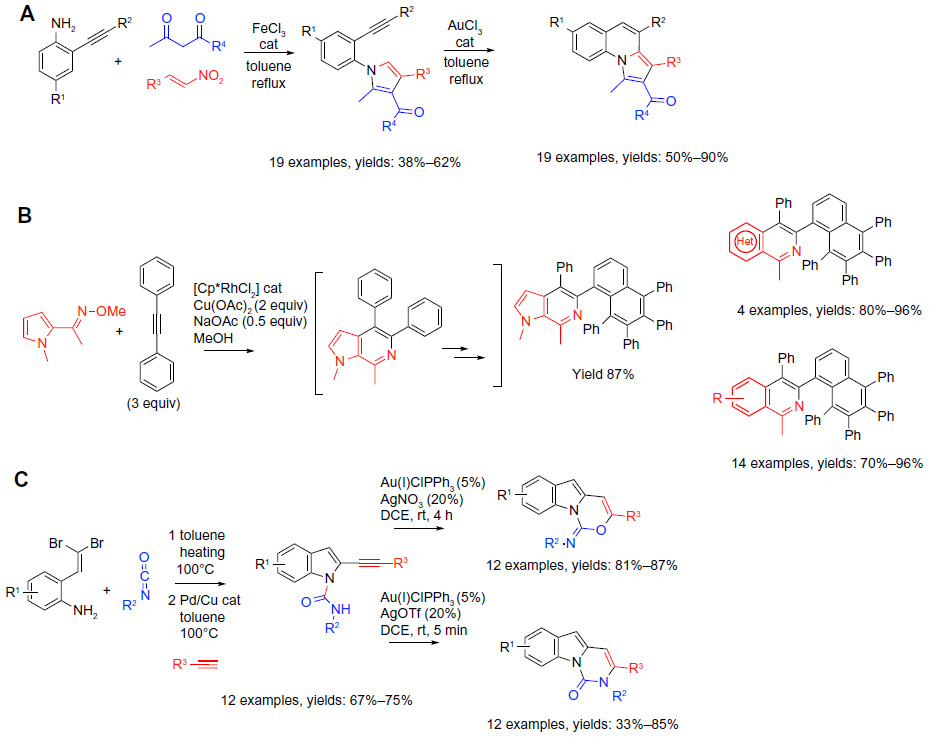 | Figure 8 Transition metal-catalyzed MCRs in sequential protocols (part 1). |
A one-pot tandem sequence of two MCR reactions (for example, the MCR2 strategy shown in reaction B; Figure 8) exemplifies the powers of multiple sequential metal-mediated C–H activations. The method55 exploits Rh(III)-catalyzed oxidative coupling between aromatic or heteroatomatic substrates, bearing a directing group and multiple molecules of diphenyl acetylene to afford polycyclic products with complex structures. The method was reported by Liu et al,55 and builds on the prior work by Too et al56 and Song et al.57 At the onset of the process, heteroatom-directed (N-methyl ketoximes) Rh(III)-catalyzed oxidative aromatic C–H activation generates a metalacycle intermediate that undergoes the insertion of the first diphenyl acetylene molecule to ultimately generate an isoquinoline ring. In a repeat of the same mechanistic sequence, the isoquinoline undergoes in situ sequential annulations with two additional moles of diphenyl acetylene, taking advantage of the directing abilities of the nitrogen in the isoquinoline ring. The contribution of this study consists in the successful optimization of the reaction conditions to favor the completion of the entire sequence of selective annulations with three molecules of diphenyl acetylene. Although electron-deficient aromatic ketoximes were the most favorable substrates for the rate determining C–H activation step, a broad range of substituted aromatic, as well as electron-rich (pyrrole, thiophene) heterocyclic ketoximes, reacted successfully in this multicomponent annulation process. A sample reaction was successfully realized on a gram scale. Overall, a number of diverse annulated heterocycles were obtained in good yields (15 examples; 70%–96%) (reaction B; Figure 8).
The potential to perform two distinct palladium-catalyzed reactions concomitantly using a properly optimized common catalytic system has been already showcased herein by the post-MCR elaboration of the functionalized Ugi product in reaction B, Figure 6.43 An analogous concept provided the basis for Pd-mediated MCR between 2-(2,2-dibromovinyl)anilines, isocyanates, and terminal alkynes (reaction C; Figure 8).58 The initial pretreatment of the anilines with the isocyanates followed by the addition of the Pd/Cu catalytic system along with diverse terminal alkynes led to two distinct cross-coupling reactions that formed Csp2–Csp and Csp2–N bonds of the indole core in a manner consistent with the prior work of Fayol et al.59 In the second step of the sequence, two divergent reaction pathways were uncovered and could be controlled by the choice of the reagent, an approach exemplifying the “conditions-based divergence (CBD) concept”, as defined by Ruijter et al.18 The Au/Ag-mediated cyclization proceeded selectively via the 6-endo rather than the 5-exo mode in both the pathways. However, depending on the specific choice of the counterions in the Ag(I)X component (AgOTf versus AgNO3), either the N- or O-atom of the amide group became involved in the cyclization (reaction C; Figure 8). Control experiments suggested that the presence of both the Ag(I) cation, as well as the corresponding counterions (eg, anions NO3– or OTf– along with the AuClPPPh3 catalyst) were required to cleanly realize the two regiochemically divergent pathways. The authors proposed that coordination of the oxygen in the amide group to the Ag(I) ion is critical for directing the attack of the O heteroatom on the harder electrophilic center arising from the activation of the alkyne via the in situ-formed (AuPPh3)NO3. In contrast, activation of the alkyne via the softer (AuPPh3)OTf complex favors the attack by the softer N-heteroatom from the amide group (reaction C; Figure 8). This work showcases how divergent post-MCR cyclizations could be used to deliver libraries of heterocycles with distinct structural cores.
The availability of transition metal-catalyzed additions of C–H bonds in relatively unreactive organic nucleophiles to in situ-generated imines and imminium ions expanded the range of components that can be employed in the corresponding MCRs.60 Furthermore, in contrast to the traditional carbonyl condensation MCRs, the transition metal-catalyzed analogs proceed under neutral conditions, and therefore permit a broad range of functionalities to be present in the building blocks. Consequently, a variety of cyclization pathways become available to the MCR products. Thus, the variation of specific structural features in just one of the building blocks has the potential to open up divergent cyclization pathways, giving rise to organic products with diverse core structures. The most recent examples of synthetic methodologies designed according to these principles are discussed in Figure 9.
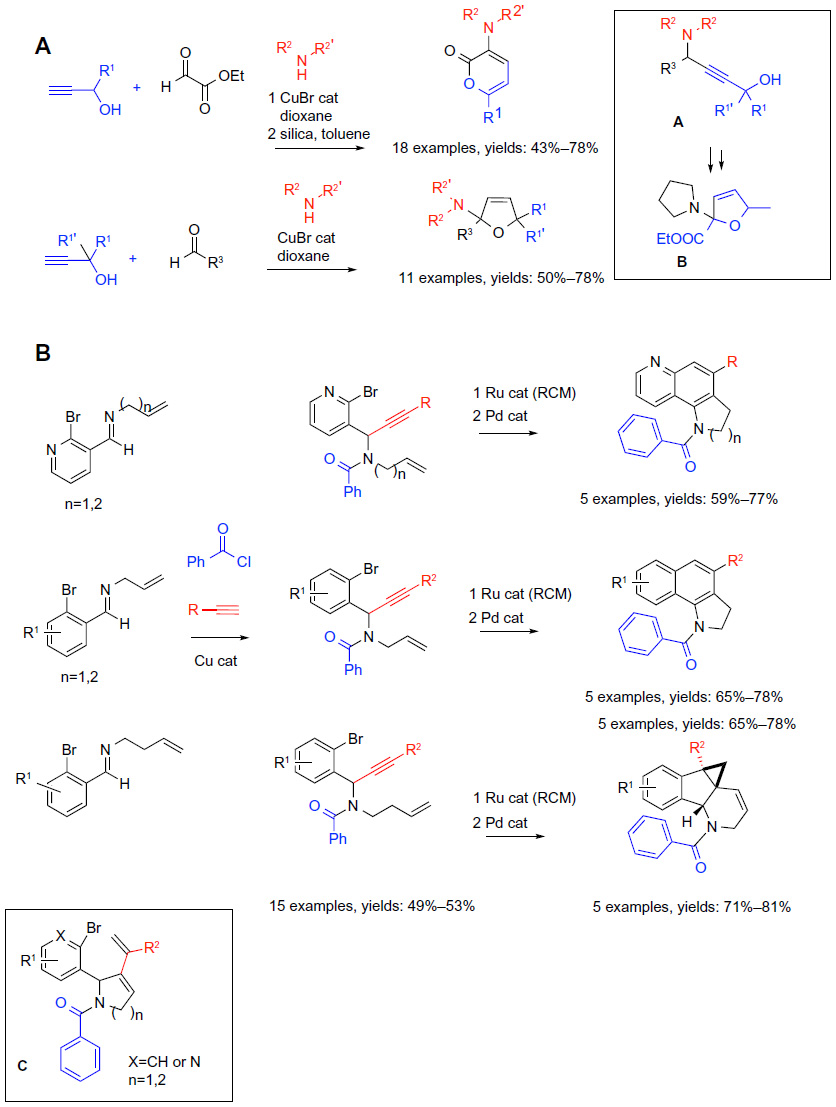 | Figure 9 Transition metal-catalyzed MCRs in sequential protocols (part 2). |
Exploring Cu-catalyzed three-component coupling of terminal alkynes, aldehydes, and amines, Fan and Ma61 observed the formation of an unexpected cyclized pyrone derivative as a minor product in reactions utilizing ethyl glyoxylate, along with a secondary amine and a terminal unprotected propargyl alcohol. An optimized protocol involving a CuBr-catalyzed coupling of terminal propargyl alcohols, ethyl glyoxylate, and secondary amines in 1,4-dioxane followed by the treatment of the crude product with silica in toluene afforded an extensive series of 3-amino-2- pyrone derivatives. The products were diversified at the C-6 positions by the variation of the substituent (R1) on the propargyl alcohol, and featured diverse substituents (R2) on the nitrogen, reflecting the choice of the secondary amine component (reaction A; Figure 9).61 A mechanistic study indicated that a 2,5-dihydrofurane derivative B could be isolated prior to treatment with silica. Presumably, the amino propargyl amine/alcohol A (Figure 9) that results from the initial MCR, undergoes an alkyne-to-allene isomerization prior to the cyclization to yield the pyrones. Based on these mechanistic considerations, the authors established that the replacement of the ethyl glyoxylate with diverse electron-deficient aldehydes (R3C[=O]H), and the elimination of the treatment with silica, provided a general protocol for the synthesis of 2-amino-2,5-dihydrofuranes with three points of diversity (reaction A; Figure 9). Both of these protocols were performed as sequential one-pot operations.
The author’s laboratory has been developing novel reaction sequences of Cu-catalyzed MCR originally reported by Black and Arndtsen,62 followed by other transition metal-catalyzed cyclizations with the aim of synthesizing structurally diverse N-heterocycles. In an earlier study,63 a sequence of the Cu-catalyzed MCR followed by an intramolecular two-step Pd-catalyzed annulation afforded a combinatorial library of dihydroindeno(1,2-c)isoquinolines featuring three points of diversity. The more recent work highlights the possibility to uncover divergent reaction pathways via structural variations in one of the substrates (reaction B; Figure 9).64 Thus, the CuCl-catalyzed coupling of N-allyl or N-homoallyl imines derived from 2-bromoarylcarbaldehydes or pyridine-2-bromo-3-carbaldehyde with terminal aromatic alkynes and benzoyl chloride afforded a total of 15 highly functionalized acyclic enynes, generally in practical 49%–53% yields. The products of the first step were isolated and characterized. In the second step of the sequence, the Ru-catalyzed enyne ring-closing metathesis was performed, giving rise to dienes C (Figure 9). The presence of the aryl or heteroaryl bromide group along with the diene set the stage for a subsequent Pd-catalyzed intramolecular cyclization. We observed that both the N-allyl and N-homoallyl imines of pyridine-2-bromo-3-carbaldehyde afforded either pyrroloquinolines (number [n]=1; N-allyl) or tetrahydrophenanthrolines (n=2; N-homoallyl) via Pd-catalyzed 6-endo cyclization/isomerization. In contrast, the outcomes of the Pd-catalyzed cyclizations of the N-allyl and N-homoallyl imines derived from 2-bromoaryl carbaldehydes proceeded via divergent pathways. The N-allyl imines underwent Pd-catalyzed cyclization in the 6-endo mode yielding benzoindolines, whereas the N-homoallyl imines afforded polycyclic indenopyridine derivatives via an apparent 5-exo cyclization followed by the 3-exo carbopalladation to form the three-membered ring (reaction B; Figure 9). Thus, heterocycles with a quite distinct three-dimensional shape, either the flat benzoindolines or the bowl-shaped indenopyridines, could be obtained via the same reaction sequence simply by utilizing homologous N-substituents in the imine components. The divergence between the 5-exo and 6-endo pathways of the Pd-catalyzed cyclization was proposed to originate from differences in the stability and reactivity of the different (n=1 or n=2) kinetic 5-exo cyclization products of the reversible carbopalladation. The kinetic product can either react further (n=2) or revert back (n=1) to the substrates, which then proceed to undergo the 6-endo carbopalladation.
As part of the study, conditions for performing the Ru-catalyzed enyne ring-closing metathesis and the Pd-catalyzed intramolecular Heck cyclization as an interrupted one-pot sequential operation without isolation of the diene intermediates were developed. The yields of the final products (calculated from the enynes) obtained from the one-pot protocols improved by 3%–22% in comparison to the combined yields of the two individual steps from the two-step two-pot protocols.
Conclusion
MCRs have the potential to generate libraries of small molecules with high levels of complexity and diversity. Recently, a powerful strategy for maximizing these features has been introduced. In the MCR reaction, building blocks possessing orthogonal functionalities are employed, and the MCR is sequenced with additional elaboration or cyclization processes that greatly increase the molecular complexity and diversity of the products. The most powerful synthetic methodologies arise when the practical aspects are properly optimized and the entire sequence can be performed as a one-pot operation. Furthermore, the most diverse products are produced when divergent reaction pathways could be opened up by variations in the structures of the substrates for the MCR, or by the choice of the reagents or reaction conditions.
Herein, recent studies aimed at designing such protocols and their applications to the synthesis of diverse assemblies of medicinally relevant heterocycles have been reviewed. Strategies exploiting a broad range of mechanistic types of MCRs, including those based on traditional carbonyl condensations, isocyanide-based MCRs, cycloadditions-based MCRs, and transition metal-catalyzed MCRs were described. Examples of protocols realized as one-pot sequential (interrupted) transformations have been provided (eg, by reactions B and C in Figure 6, and reaction A in Figure 9). The most accomplished protocols for one-pot tandem sequences could be found in reactions B in Figure 4, reaction D in Figure 6, reactions A and B in Figure 7, and reaction B in Figure 8.
Future outlook
The studies described herein serve as inspiration for the future development of novel MCRs to satisfy the growing demand for more diverse libraries of small molecules as probes for use in drug discovery. One of the challenges that need to be addressed consists of identifying more selective catalysts and optimum reaction conditions that would increase the functional group tolerance, and therefore diversity, in the products of the initial MCRs. In order to produce libraries with high structural diversity, further advances are needed in the development of divergent sequential processes, where divergent reaction pathways could arise either due to the variation in the reaction conditions, catalysts used, or a small structural variation in the building blocks. Finally, in order to make the production of the diverse libraries environmentally sustainable and economically viable, much effort needs to be invested into the design of sequential protocols that can be performed as one-pot operations. The current state-of-the-art indicates that all these challenges can be addressed, and one-pot sequential processes involving an MCR followed by subsequent elaborations are likely to become a critically important tool for drug discovery.
Disclosure
The author reports no conflicts of interest in this work.
References
Muller TJJ, editor. Science of Synthesis: Multicomponent Reactions. Georg Thieme Verlag KG: Stuttgart; 2014. | |
Strecker A. Ueber die künstliche Bildung der Milchsäure und einen neuen, dem Glycocoll homologen Körper. Justus Liebigs Annalen der Chemie. 1850;75(1):27–45. German. | |
Mannich C, Krösche W. Ueber ein Kondensationsprodukt aus Formaldehyd, Ammoniak und Antipyrin. Archiv der Pharmazie. 1912;250(1):647–667. German. | |
Biginelli P. Ueber Aldehyduramide des Acetessigäthers. II. Berchte der Deutschen Chemischen Gesellschaft. 1891;24(2):2962–2967. German. | |
Hantzsch A. Condensationprodukte aus Aldehydamonniak und Ketonartigen Verbindungen. Chem Ber. 1881;14:1637–1638. German. | |
Asinger F. Über die gemeinsame Einwirkung von Schwefel und Ammoniak auf Ketone I. Angew Chem. 1956;68(12):413. German. | |
Ugi I, Meyr R, Fetzer U, Steinbrüker C. Versuche mit Isonitrilen. Angew Chem. 1959;71(11):386. German. | |
Banafi L, Riva R. The Passerini reaction. Organic Reactions. 2005;65:1–140. | |
Dömling A, Ugi I. Multicomponent reactions with isocyanides. Angew Chem Int Ed Engl. 2000;39(18):3168–3210. | |
Nenajdenko V, editor. Isocyanide Chemistry: Applications in Synthesis and Material Science. Weinheim, Germany: Wiley-VCH; 2012. | |
Zhu J, Bienaymé H, editors. Multicomponent Reactions. Weinheim, Germany: Wiley-VCH; 2005. | |
Slobbe P, Ruijter E, Orru RVA. Recent applications of multicomponent reactions in medicinal chemistry. MedChemComm. 2013;3:1189–1218. | |
Knapp JM, Kurth MJ, Shaw JT, Younai A. Strategic applications of multicomponent reactions in diversity-oriented synthesis. In: Trabocchi A, editor. Diversity-Oriented Synthesis: Basics and Applications in Organic Synthesis, Drug Discovery, and Chemical Biology. Hoboken, NJ: John Wiley & Sons, Inc.; 2013:29–57. | |
Dömling A, Wang W, Wang K. Chemistry and biology of multicomponent reactions. Chem Rev. 2012;112(6):3083–3135. | |
Biggs-Houck JE, Younai A, Shaw JT. Recent advances in multicomponent reactions for diversity-oriented synthesis. Curr Opin Chem Biol. 2010;14(3):371–382. | |
Sunderhaus JD, Martin SF. Applications of multicomponent reactions to the synthesis of diverse heterocyclic scaffolds. Chemistry. 2009;15(6):1300–1308. | |
Ulaczyk-Lesanko A, Hall DG. Wanted: new multicomponent reactions for generating libraries of polycyclic natural products. Curr Opin Chem Biol. 2005;9(3):266–276. | |
Ruijter E, Scheffelaar R, Orru RV. Multicomponent reaction design in the quest for molecular complexity and diversity. Angew Chem Int Ed Engl. 2011;50(28):6234–6246. | |
Leet KH, Hayashi N, Okano M, et al. Lasiodiplodin, a potent antileukemic macrolide from Euphorbia splendens. Phytochemistry. 1982;21(5):1119–1121. | |
Reddy MV, Rao MR, Rhodes D, et al. Lamellarin alpha 20-sulfate, an inhibitor of HIV-1 integrase active against HIV-1 virus in cell culture. J Med Chem. 1999;42(11):1901–1907. | |
Witherup KM, Ransom RW, Graham AC, et al. Martinelline and martinellic acid, novel G-protein linked receptor antagonists from the tropical plant Martinella iquitosensis (Bignoniaceae). J Am Chem Soc. 1995;117(25):6682–6685. | |
Summa V, Petrocchi A, Bonelli F, et al. Discovery of raltegravir, a potent, selective orally bioavailable HIV-integrase inhibitor for the treatment of HIV-AIDS infection. J Med Chem. 2008;51(18):5843–5855. | |
Vassilev LT, Vu BT, Graves B, et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303 (5659):844–848. | |
Ballet S, Misicka A, Kosson P, et al. Blood–brain barrier penetration by two dermorphin tetrapeptide analogues: role of lipophilicity vs structural flexibility. J Med Chem. 2008;51(8):2571–2574. | |
Deady LW, Kaye AJ, Finlay GJ, Baguley BC, Denny WA. Synthesis and antitumor properties of N-[2-(dimethylamino)ethyl]carboxamide derivatives of fused tetracyclic quinolines and quinoxalines: a new class of putative topoisomerase inhibitors. J Med Chem. 1997;40(13):2040–2046. | |
Tashma Z, Raveh L, Liani H, et al. Bretazenil, a benzodiazepine receptor partial agonist, as an adjunct in the prophylactic treatment of OP poisoning. J Appl Toxicol. 2001;21 Suppl 1:S115–S119. | |
Hadjipavlou-Litina D, Hansch C. Quantitative structure-activity relationships of the benzodiazepines. A review and reevaluation. Chem Rev. 1994;94(6):1483–1505. | |
Hazra A, Mondal S, Maity A, et al. Amberlite-IRA-402 (OH) ion exchange resin mediated synthesis of indolizines, pyrrolo [1,2-a] quinolines and isoquinolines: antibacterial and antifungal evaluation of the products. Eur J Med Chem. 2011;46(6):2132–2140. | |
Kato M, Ito K, Nishino S, Yamakuni H, Takasugi H. New 5-HT3 (serotonin-3) receptor antagonists. I. Synthesis and structure-activity relationships of pyrido[1,2-a]indoles. Chem Pharm Bull (Tokyo). 1994;42(12):2546–2555. | |
Chu XP, Zhou QF, Zhao S, et al. Synthesis and biological evaluation of 3-amino-2-pyrones as selective cyclooxygenase-1 (COX-1) inhibitors. Chin Chem Lett. 2013;24(2):120–122. | |
Min Z, Wang L, Jin J, et al. Pyrroloquinoline quinone induces cancer cell apoptosis via mitochondrial-dependent pathway and down-regulating cellular Bcl-2 protein expression. J Cancer. 2014;5(7):609–624. | |
Kohlhagen G, Paull KD, Cushman M, Nagafuji P, Pommier Y. Protein-linked DNA strand breaks induced by NSC 314622, a novel noncamptothecin topoisomerase I poison. Mol Pharmacol. 1998;54(1):50–58. | |
Ramachary DB, Ramakumar K, Bharanishashank A, Narayana VV. Sequential one-pot combination of multireactions through multicatalysis: a general approach to rapid assembly of functionalized push-pull olefins, phenols, and 2-methyl-2H-chromenes. J Comb Chem. 2010; 12(6):855–876. | |
Yu X, Pan X, Wu J. An efficient route to diverse H-pyrazolo[5,1-a]oisoquinolines via sequential multi-component/cross-coupling reactions. Tetrahedron. 2011;67(6):1145–1149. | |
Yue D, Larock RC. Synthesis of 2,3-disubstituted benzo[b]thiophenes via palladium-catalyzed coupling and electrophilic cyclization of terminal acetylenes. J Org Chem. 2002;67(6):1905–1909. | |
Boomhoff M, Yadav AK, Appun J, Schneider C. Modular, flexible, and stereoselective synthesis of pyrroloquinolines: rapid assembly of complex heterocyclic scaffolds. Org Lett. 2014;16(23):6236–6239. | |
Feng X, Wang JJ, Xun Z, Huang ZB, Shi DQ. Multicomponent strategy to indeno[2,1-c]pyridine and hydroisoquinoline derivatives through cleavage of carbon–carbon bond. J Org Chem. 2015;80(2):1025–1033. | |
Brockmeyer F, van Gerven D, Saak W, Martens J. Two sequential multicomponent reactions: synthesis of thiazolidin-4-yl-1,3,4-oxadiazoles under mild conditions. Synthesis. 2014;46(12):1603–1612. | |
Welsch SJ, Umkehrer M, Kalinski C, et al. Synthesis of substituted imidazolines by an Ugi/Staudinger/aza-Wittig sequence. Tetrahedron Lett. 2015;56(8):1025–1029. | |
Wang L, Ren ZL, Ding MW. Synthesis of 2,3-Dihydro-1H-2- benzazepin-1-ones and 3H-2-benzoxepin-1-ones by isocyanide-based multicomponent reaction/Wittig sequence starting from phosphonium salt precursors. J Org Chem. 2015;80(1):641–646. | |
El Kaim L, Grimaud L. Beyond the Ugi reaction: less conventional interactions between isocyanides and iminium species. Tetrahedron. 2009;65(11):2153–2171. | |
Gordillo-Cruz RE, Rentería-Gómez A, Islas-Jácome A, et al. Synthesis of 3-tetrazolylmethyl-azepino[4,5-b]indol-4-ones in two reaction steps: (Ugi-azide/N-acylation/SN2)/free radical cyclization and docking studies to a 5-Ht(6) model. Org Biomol Chem. 2013;11(38):6470–6476. | |
Che C, Yang B, Jiang X, et al. Syntheses of fused tetracyclic quinolines via Ugi-variant MCR and Pd-catalyzed bis-annulation. J Org Chem. 2014;79(1):436–440. | |
Deprez NR, Sanford MS. Reactions of hypervalent iodine reagents with palladium: mechanisms and applications in organic synthesis. Inorg Chem. 2007;46(6):1924–1935. | |
Ghabraie E, Balalaie S. Sequential Ugi four-component reaction (4-CR)/C-H activation using (diacetoxyiodo)benzene for the synthesis of 3-(diphenylmethylidene)-2,3-dihydro-1H-indol-2-ones. Helvetica Chimica Acta. 2014;97(11):1555–1563. | |
Jia S, Su S, Li C, Jia X, Li J. Multicomponent cascade cycloaddition involving tropone, allenoate, and isocyanide: a rapid access to a 7,6,5-fused tricyclic skeleton. Org Lett. 2014;16(21):5604–5607. | |
Agard NJ, Baskin JM, Prescher JA, Lo A, Bertozzi CR. A comparative study of bioorthogonal reactions with azides. ACS Chem Biol. 2006; 1(10):644–648. | |
Nguyen HH, Palazzo TA, Kurth MJ. Facile one-pot assembly of imidazotriazolobenzodiazepines via indium(III)-catalyzed multicomponent reactions. Org Lett. 2013;15(17):4492–4495. | |
Lee DJ, Han HS, Shin J, Yoo EJ. Multicomponent [5 + 2] cycloaddition reaction for the synthesis of 1,4-diazepines: isolation and reactivity of azomethine ylides. J Am Chem Soc. 2014;136(33):11606–11609. | |
Balme G, Didier B, Monteiro N. Chapter 8. Metal-catalyzed multicomponent reactions. In: Zhu J, Bienaymé H. editors. Multicomponent Reactions. Weinheim, Germany: Wiley-VCH Verlag GmbH and Co; 2005:224–276. | |
Zhang L, Qureshi Z, Sonaglia L, Lautens M. Sequential rhodium/palladium catalysis: enantioselective formation of dihydroquinolinones in the presence of achiral and chiral ligands. Angew Chem Int Ed Engl. 2014;53(50):13850–13853. | |
Sarkar S, Bera K, Jalal S, Jana U. Synthesis of structurally diverse polyfunctional pyrrolo[1,2-a]quinolines by sequential iron-catalyzed three-component coupling and gold-catalyzed hydroarylation reactions. European J Org Chem. 2013;27:6055–6061. | |
Noland WE. The NEF reaction. Chem Rev. 1955;55(1):137–155. | |
Samala S, Mandadapu AK, Saifuddin M, Kundu B. Gold-catalyzed sequential alkyne activation: one-pot synthesis of NH-carbazoles via cascade hydroarylation of alkyne/6-endo-dig carbocyclization reactions. J Org Chem. 2013;78(13):6769–6774. | |
Liu B, Hu F, Shi BF. Synthesis of sterically congested polycyclic aromatic hydrocarbons: rhodium(III)-catalyzed cascade oxidative annulations of aryl ketoximes with diphenylacetylene by sequential cleavage of multiple C-H bonds. Adv Synth Catal. 2014;356(11–12):2688–2696. | |
Too PC, Chua SH, Wong SH, Chiba S. Synthesis of azaheterocycles from aryl ketone O-acetyl oximes and internal alkynes by Cu-Rh bimetallic relay catalysts. J Org Chem. 2011;76(15):6159–6168. | |
Song G, Chen D, Pan CL, Crabtree RH, Li X. Rh-catalyzed oxidative coupling between primary and secondary benzamides and alkynes: synthesis of polycyclic amides. J Org Chem. 2010;75(21):7487–7490. | |
Gupta S, Koley D, Ravikumar K, Kundu B. Counter ion effect in Au/Ag-catalyzed chemoselective 6-endo-dig N- and O-cyclizations of enyne-urea system: diversity-oriented synthesis of annulated indoles. J Org Chem. 2013;78(17):8624–8633. | |
Fayol A, Fang YQ, Lautens M. Synthesis of 2-vinylic indoles and derivatives via a Pd-catalyzed tandem coupling reaction. Org Lett. 2006;8(19):4203–4206. | |
Zhang XS, Chen K, Shi ZJ. Transition metal-catalyzed direct nucleophilic addition of C-H bonds to carbon-heteroatom double bonds. Chemical Science. 2014;5:2146–2159. | |
Fan W, Ma S. Copper(I)-catalyzed three-component reaction of terminal propargyl alcohols, aldehydes, and amines: synthesis of 3-amino-2-pyrones and 2,5-dihydrofurans. Angew Chem Int Ed Engl. 2014;53(52):14542–14545. | |
Black DA, Arndtsen BA. Copper-catalyzed coupling of imines, acid chlorides, and alkynes: a multicomponent route to propargylamides. Org Lett. 2004;6(7):1107–1110. | |
Kumar S, Painter TO, Pal BK, Neuenswander B, Malinakova HC. Application of sequential Cu(I)/Pd(0)-catalysis to solution-phase parallel synthesis of combinatorial libraries of dihydroindeno[1,2-c]isoquinolines. ACS Comb Sci. 2011;13(5):466–477. | |
Raikar SN, Malinakova HC. Divergent reaction pathways of homologous and isosteric propargyl amides in sequential Ru/Pd-catalyzed annulations for the synthesis of heterocycles. J Org Chem. 2013;78(8):3832–3846. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.