")
Back to Journals » Drug Design, Development and Therapy » Volume 9
Rationale and clinical utility of the darunavir–cobicistat combination in the treatment of HIV/AIDS
Authors Putcharoen O, Do T, Avihingsanon A, Ruxrungtham K
Received 17 April 2015
Accepted for publication 26 June 2015
Published 23 October 2015 Volume 2015:9 Pages 5763—5769
DOI https://doi.org/10.2147/DDDT.S63989
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Shu-Feng Zhou
Opass Putcharoen,1 Tanya Do,2 Anchalee Avihingsanon,2 Kiat Ruxrungtham1,2
1Department of Medicine, Faculty of Medicine, Chulalongkorn University, 2The HIV Netherlands Australia Thailand (HIV-NAT) Research Collaboration, The Thai Red Cross AIDS Research Center, Bangkok, Thailand
Abstract: This article is to provide an update overview of cobicistat (COBI)-boosted darunavir in response to its recent approval by the US Food and Drug Administration, and inclusion as an alternative first-line regime in the 2015 treatment guidelines in the US. COBI is a relatively new non-antiretroviral cytochrome P450 3A inhibitor or pharmacoenhancer. The rationale behind COBI development was to provide an alternative to ritonavir (RTV) as a protease inhibitor pharmacoenhancer, due to associated adverse events with short- and long-term RTV use, such as gastrointestinal intolerability, drug–drug interactions, insulin resistance, lipodystrophy, and hyperlipidemia. Although in vitro studies suggest that COBI may result in a lower incidence of undesired drug–drug interactions and lipid-associated disorders than RTV, not all Phase III studies have well addressed these issues, and the data are limited. However, Phase III studies have demonstrated tolerability, noninferiority, and bioequivalence of COBI compared to RTV. Two main advantages of COBI over RTV-containing regimes have been noted as follows: 1) COBI has no anti-HIV activity; therefore, resistance to COBI as a booster in addition to protease inhibitor resistance is of little concern, allowing for COBI-containing regimes in future. 2) COBI’s solubility and dissolution rate allow for co-formulated/fixed-dose combination products. Nonetheless, prior to initiating COBI-containing treatment regimens, the following should be considered: 1) COBI may increase serum creatinine levels and reduce estimated glomerular filtration rate (GFR) without affecting actual GFR; 2) potential drug–drug interaction data are insufficient, warranting caution when initiating COBI in conjunction with concomitant medication or in individuals with multiple comorbidities; 3) food plays a pivotal role in boosting darunavir exposure, warranting caution and patient education on the importance of taking COBI-containing regimens with appropriate amounts of food; and 4) data on the success of COBI-containing regimens in treatment-experienced patients are limited.
Keywords: first-line regime, pharmacoenhancer, adverse events, ritonavir, drug–drug interactions, tolerability
Introduction
HIV therapy has evolved to better serve HIV patients. With effective treatment utilizing a combination of drug classes, the disease has changed from being considered deadly to one that simply necessitates chronic care management.1 Currently, there are six classes of antiretroviral agents approved for therapy in treatment-naïve and treatment-experienced patients. These six classes are the nucleoside/nucleotide reverse transcriptase inhibitors (NRTIs) that are the backbone of therapy, non-nucleoside reverse transcriptase inhibitors (NNRTIs), protease inhibitors (PIs), fusion inhibitor, a CCR5 antagonist and integrase strand transfer inhibitors (INSTIs).2,3 The recommended antiretroviral regimens comprise two NRTIs plus the third agent being an NNRTI, PI, or INSTI. These antiretroviral drug classes target the virus at different phases of the replication cycle, thereby achieving viral suppression in the majority of patients during the last 20 years1,3–5 and supporting the need for global scale-up of antiretroviral therapy.6 Factors that guide clinician in antiretroviral initiation and maintenance practices are favorable efficacy, low adverse events, ease of administration, and minimal food or drug interactions. In conjunction, individualized patient preferences and past medical history render specific regimens to be more favorable than others.2,3 However, treatment is currently lifelong, short- and long-term adverse events and resistance are not uncommon, and the development of new HIV medications promising superiority over what is currently available is lesser in comparison to years past, underscoring the need to appropriately address awareness and knowledge of treatment options and consequences thereof among clinicians and patients alike.1,3–5,7 Cobicistat (COBI) is a relatively new non-antiretroviral cytochrome P450 3A inhibitor (booster/pharmacoenhancer) that can be used as an alternative to ritonavir (RTV) with potentially fewer drug–drug interactions, metabolic adverse effects, and fixed-dose combinations (FDCs), reducing pill burden and thus improving the likelihood of adherence.8–12 The aim of this article is to provide an overview of the history and rationale behind darunavir (DRV)-boosted COBI (DRV/c) and to outline efficacy and tolerability findings, in order to inform clinician’s prescriptive practices in response to its recent approval by the US Food and Drug Administration (FDA) and inclusion as an alternative pharmacoenhancer PI within first-line regime 2015 treatment guidelines of the Department of Health and Human Services.
HIV-1 protease enzyme, mechanisms of HIV-1 PI, and limitations in un-boosted PI use
The protease enzyme crystal structure was first discovered in 1988,13 and the first PI was available for HIV treatment in 1995.14 An HIV-1 protease is an aspartyl protease, and one of the essential components in the HIV virion. This enzyme is a homodimer composed of two identical subunits within a 99-amino acid chain, connected by antiparallel beta-sheets. The site of enzyme activity is located at the center of the dimer. Protease enzymes have two Asp residues at the site to catalyze peptide bonding of the polyprotein chain to viral core proteins and functional proteins. The catalyzing event takes place in the virion right after budding.15,16
The PI drug class is a potent antiviral agent, achieving viral suppression in treatment-naïve and treatment-experienced patients. PIs are peptidic-like compounds that bind to the cleavage site of the enzyme, preventing viral polyprotein binding. This mechanism thereby interferes with processing of the viral structural and nonstructural proteins, inhibiting viral maturation and preventing infection into new cells.17 However, due to the evolution and high genetic heterogeneity of HIV, development of resistance to PIs is inevitable after HIV exposure. The resistant mutations modify amino acids at the binding sites of the PI and lower binding affinity. Cross resistance from one PI to another can occur because some PIs share binding sites or are located in close proximity.7
PIs are extensively metabolized by cytochrome P450 3A4 (CYP3A) at the liver and intestines. Most PIs have a short plasma half-life when administered as an un-boosted, mono-PI. In the infancy of PI-containing regimens, they had a high pill burden, short dosing intervals, and restricted administration in conjunction with other medications or food. These factors created barriers to initiation and maintenance. In addition, metabolic complications such as insulin resistance and gastrointestinal disturbances were noted in these regimes.18 This changed, however, with the introduction of RTV as a PI booster approximately 15 years ago.19
The PI DRV is a nonpeptidic compound developed for HIV treatment. This compound uses the Asp-29 and Asp-30 position of the protease enzyme.20 DRV has a high affinity for binding to wild-type (WT) and mutant protease. This property is the key to its high genetic barrier to resistance. DRV demonstrates a fast association (Kon) but a very slow dissociation (Koff) from WT HIV-1 protease compared with other PIs.21 The slower rate of dissociation explains its efficacy in long-term use.22–24 However, as an un-boosted PI, DRV is quickly metabolized and has a short plasma half-life similar to other PIs. Therefore, low-dose RTV was introduced in combination with DRV as a means to prolong DRV’s half-life. This combination has demonstrated long-term efficacy and tolerability in treatment-naïve and treatment-experienced patients.23,24
RTV mechanisms of action
RTV is a PI developed by Abbott Laboratories, initially approved for HIV treatment at high dose levels as a component of an antiretroviral regimen. Similar to other PIs, high-dose RTV has a very short plasma half-life and significant metabolic side effects. However, in lower doses (100–200 mg per day), this agent was found to be an excellent booster for other PIs through two mechanisms. RTV inhibits metabolism of other PIs via the CYP3A enzyme, thereby increasing their half-life. The CYP3A enzyme is a major contributor to the elimination of a number of therapeutic agents.25,26 In addition, RTV inhibits the p-glycoprotein that pumps drugs out of intestinal cells. Together, this results in higher and more prolonged concentrations of drugs that are metabolized via the P450 pathway. At higher plasma concentrations, the now boosted PI can overcome replication of WT and potentially PI-resistant strains. This strategy contributed to the reduction in pill burden and dosing frequency encouraging treatment adherence.27
When co-administered with DRV, RTV prolongs the terminal half-life of DRV to 15 hours.28 The currently recommended dose of DRV/RTV (DRV/r) is 800/100 mg once daily in treatment-naïve patients and 600/100 mg twice daily in treatment-experienced patients.2 The pharmacokinetic (PK) enhancement properties of low-dose RTV have expended its utility to other anti-HIV agents such as elvitegravir and maraviroc.29 However, although RTV is administered as a booster, it still induces some side effects such as dyslipidemia and gastrointestinal intolerance. Furthermore, low-dose RTV has the potential for drug resistance, as it retains activity against HIV replication during co-administration. Due to the challenges associated with low-dose RTV, the search began for new booster alternatives with fewer adverse effects and a decreased susceptibility to resistance.
Development of COBI
The new booster COBI (GS-9350) was approved in 2012 for use in HIV therapy in the form of a FDC: elvitegravir/COBI/tenofovir disoproxil fumarate (TDF)/emtricitabine (FTC) with the trade name Stribild. COBI was originally developed by Gilead Laboratory. The chemical structure of COBI is related to RTV in that it maintains potent inhibition of CYP3A but without activity against HIV replication. COBI inhibits human CYP3A via direct interaction and mechanism-based inhibition at the heme group of CYP3A. The kinetics of COBI’s inactivation of human hepatic microsomal CYP3A, measured by kinact and K1, is comparable to RTV. The inactivation of CYP3A is possibly sustained until new CYP3A is synthesized by hepatocytes. The spectrum of COBI in the inhibition of human microsomal hepatic CYP3A is as broad as in RTV.9 In addition, lipid accumulation assays in vitro and ex vivo demonstrated a lower effect on lipid accumulation and insulin-mediated glucose metabolism in adipocytes.9 The PKs and pharmacodynamics of single- and multi-dose COBI were studied in healthy volunteers for clinical validation, and it was found that COBI increased systemic exposure of CYP3A substrates equivalent to that of RTV with no dose-limiting toxicities between 50 mg per day and 300 mg per day dosing.30 Drug–drug interactions of COBI were not shown to be as extensive as RTV, although it is speculated that drug–drug interactions with COBI are most likely similar to RTV. Therefore, caution must be exercised when administering COBI with drugs known to interact with RTV; contraindications or a need for dose adjustment should be considered and anticipated.
The recommended first-line PI in treatment-naïve patients, in accordance with the Department of Health and Human Services 2015 treatment guidelines, is boosted DVR, in combination with two NRTIs. DRV can be boosted with either RTV or COBI. Elion et al and Gallant et al compared the efficacy of ATV/RTV (ATV/r) vs ATV/COBI (ATV/c) in treatment-naïve patients with a TDF/FTC backbone and found similar results.31,32 The Phase III trial conducted by Gallant et al was a randomized, partially placebo-controlled, and double-blind multicenter study. At week 48, no significant difference in treatment response between COBI (85%) and RTV (87%) was detected. Rates of CD4 recovery were also comparable between the two arms. Furthermore, the difference in the number of adverse events such as hyperbilirubinemia and jaundice was not significant. Reduction in the mean estimated glomerular filtration rate (eGFR, Cockcroft–Gault, mL/min) occurred in both treatment arms but was slightly more prominent in the COBI arm (nine vs four participants). This study concluded that COBI was a noninferior pharmacoenhancer with a comparable efficacy and safety profile to RTV, when combined with a PI.
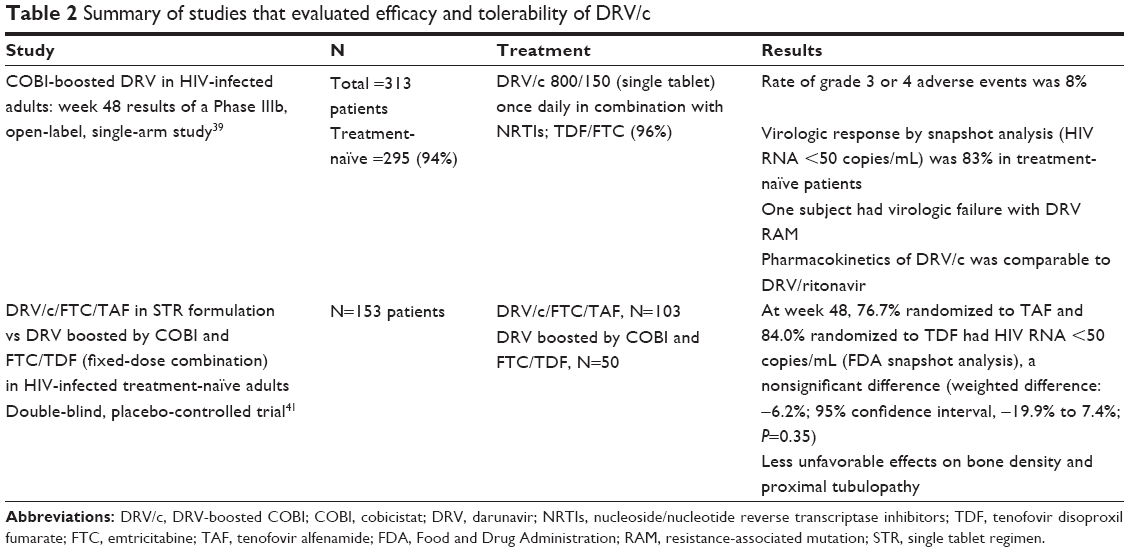
Thus, on January 29, 2015, the FDA approved DRV/c (Prezcobix®) based on the bioequivalence data (TMC114IFD1003 study, Table 1) and a clinical study evaluating the safety and efficacy of DRV/c for the treatment of HIV-1 in adults with no DRV resistance-associated mutations (GS-US-216-0130 study, Table 2), making it the first COBI FDC approved by the FDA.33
 | Table 1 Comparison of the pharmacokinetics of darunavir/ritonavir vs darunavir/cobicistat in HIV-infected subjects |
 | Table 2 Summary of studies that evaluated efficacy and tolerability of DRV/c |
PKs of DRV/c and DRV/r
One of the first PK studies of DRV/c and DRV/r co-formulations studied 33 healthy subjects.30 The geometric mean ratio (GMR) of DRV/c vs DRV/r was equivalent for area under the curve GMR with COBI at 102% and RTV at 90% (confidence interval [CI], 97.4–106), Cmax GMR COBI at 103% and RTV at 90% (CI, 100–106), and trough GMR COBI at 69.4% and RTV at 90% (CI, 59.0–81.7). When analyzing pre-dose DRV concentrations after participants received multiple doses, equivalent-level GMRs of COBI at 90% and RTV at 89.4% were detected (CI, 80.4–99.4). DRV/c trough concentrations remained more than 18 times above the protein-binding-adjusted 50% effective concentration (EC50) for WT (nonresistant) virus (55 ng/mL). Pre-dose DRV levels were more than 37 times above the EC50 for WT virus. DRV/c concentrations were equivalent to the data observed in ARTEMIS study.34
Renal safety of DRV/c
COBI inhibits creatinine excretion in detectable amounts at the proximal tubules via the human multidrug and toxin extrusion protein 1, the transporter system responsible for excreting creatinine. After administration of COBI, rising of serum creatinine in some patients resulted in a reduction of eGFR but without actual changes to kidney function.35 Studies have noted that in HIV-positive participants receiving COBI, an elevation of serum creatinine of approximately 15% from baseline could be observed, reaching maximal levels at week 2. Therefore, renal monitoring is essential in patients receiving COBI. It is recommended that COBI-containing regimens should not be initiated in patients with a creatinine clearance of less than 70 cc/mL2. However, recent data from a clinical study demonstrated that switching from ATV/r to ATV/c and DRV/r to DRV/c was safe up to 96 weeks in patients with creatinine clearances between 50 and 89 cc/min.38,39
Drug–drug interactions with COBI administration
COBI potentially may have fewer drug–drug interactions due to its more selective inhibition of CYP3A than RTV. In a study assessing the median half-life and mean apparent clearance of midazolam, COBI increased the median half-life 2.1- and 3.7-fold with a reduced mean clearance of 93% and 95%, respectively, whereas RTV increased the half-life 4.9-fold and reduced the mean clearance by 96%.30 At present, the recommendations regarding potential drug–drug interactions are based on predicted interactions in addition to clinical trial findings.33 Thus, caution should be exercised when initiating COBI in conjunction with concomitant medication or in individuals with multiple comorbidities.
Food effect on DRV/c
In a study where participants were administered DRV/c under fasting or fed conditions (standardized intake and high fat/high calorie intake), bioequivalence of DRV/c 800/150 mg as an FDC found that food increased DRV exposure, and recommended that DRV/c be administered with food.36 Furthermore, participants given higher fat/higher calorie food demonstrated a significant increase in DRVs maximum plasma concentrations of 2.27-fold and the area under the curve of 1.63- to 1.70-fold in comparison to fasting conditions, but no increase in adverse events was noted, and no recommendations on the optimal amount of food intake when taking DRC/c were provided.
Clinical studies of DRV/c in a FDC use
In treatment-experienced participants
Limited data are available on the use of DRV/c in treatment-experienced participants. And, in the three studies including treatment-experienced participants, the participant numbers were small. McDonald et al and Fisher et al studied the effects of switching 73 participants to either DRV/c- or ATV/c-containing regimes with a creatinine clearance of 50–89 mL/min.37,38 Findings demonstrated efficacy and tolerability with a similar renal profile as seen in other studies. The study by Tashima et al included 18 treatment-experienced and 295 treatment-naïve participants but did not address any specific findings in the treatment-experienced group nor made any recommendations regarding DRV/c use in treatment-experienced participants.39
In treatment-naïve patients
DRV/c at 800/150 mg as a single FDC tablet, in combination with two active nucleoside analogs (one being tenofovir, 99%), was evaluated in a 48-week Phase IIIb, open-label, single-arm, and multicenter study.43 This study included 313 subjects, mostly treatment-naïve patients (94%) with 86% of the patients completing evaluation at week 48. Eighty-three percent of treatment-naïve patients had plasma HIV RNA lower than 50 copies/mL by FDA snapshot analysis. There were no differences in response rates between patients with baseline HIV RNA >100,000 copies/mL vs patients with <100,000 copies/mL. The median increase in CD4 count at 48 weeks was 169 cells/mm3, and the discontinuation rate due to adverse effects was 5%. Measured parameters for PKs from 59 patients demonstrated that mean (standard deviation) Cmax, Ctau, and C0 h of DRV were 7,663 (1,920) ng/mL, 1,311 (969) ng/mL, and 1,560 (1,328) ng/mL, respectively. The population-based DRV/c PKs was consistent with the PKs of DRV/r, and the mean DRV C0 h was optimal for suppression of WT HIV-1. This study demonstrated that a fixed-dose of DRV/c is effective and safe.40
A quadruple single-tablet regimen of DRV/c/tenofovir alfenamide/FTC
One study has evaluated the efficacy of DRV/c in combination with tenofovir alfenamide (TAF) and FTC in a single-tablet regimen, in comparison to DRV/c plus TDF and FTC.41 This study enrolled 153 treatment-naïve participants with an eGFR at or greater than 70 cc/min. The investigators randomized 103 participants to receive DRV/c/TAF/FTC and 50 to receive DRV/c/TDF/FTC once daily. At week 48, 76.7% of the participants administered DRV/c/TAF/FTC and 84.0% of the participants administered DRV/c/TDF/FTC had plasma HIV RNA less than 50 copies/mL, weighted difference: −6.2% (CI, −19.9% to 7.4%; P=0.35). Those who experienced adverse events were graded as mild/moderate in severity. However, two patients had adverse events that led to discontinuation of the treatment regime in each arm. With respect to eGFR, participants in the DRV/c/TDF/FTC exhibited a greater reduction in eGFR and a greater change in proximal tubular proteinuria with a mean reduction in eGFR of −10.6 (TDF arm) vs −2.9 (TAF arm) (P=0.017). This study demonstrated the superiority of TAF to TDF in combination with DVR/c/FTC, possibly mitigating renal impairment concerns in COBI-containing regimens.
Summary
DRV/c as FDC has recently been approved by the FDA in treatment-naïve and treatment-experienced HIV-infected patients. This is based on comparable PK, efficacy, and safety profiles. The studied DRV/c/TAF/FTC single pill has shown to be well-tolerated and efficacious in a Phase II 48-week clinic trial, and is likely to be approved for clinical use in the near future. Familiarity with COBI-containing regimes is of importance to clinicians globally, as Gilead has signed a license agreement with Medicine Patent Pool reducing the cost and improving access in low- to middle-income countries.42 The studies completed to date address the concerns surrounding increased serum creatinine levels and reduced eGFR in COBI-containing regimes and should be considered prior to initiation. Furthermore, patient education should underline the importance of taking COBI-containing regimes with food. With increased use and study data, potential drug–drug interactions will become more evident, and until that time, caution when initiating COBI in conjunction with concomitant medication or in individuals with multiple comorbidities should be exercised. Likewise, data regarding the effects of COBI-containing regimens in treatment-experienced patients will expand to better inform clinician practices.
Acknowledgments
KR is supported by the Senior Research Scholar from the Thailand Research Fund (TRF). OP is supported by the Research Chair Grant, National Science and Technology Development Agency, Thailand.
Disclosure
KR has served as a consultant for Merck, Tibotec, and Mylan and has had paid speaking engagements with Bristol-Meyers Squibb, Merck, Roche, Jensen-Cilag, GlaxoSmithKline, Gilead, Abbvie, Thai GPO, and Mylan Lab Limited. OP has had paid speaking engagements with Bristol-Meyers Squibb, Merck, Jensen-Cilag, Gilead, Abbvie, and Mylan Lab Limited. The other authors report no conflicts of interest in this work.
References
Hirsch MS, Kuritzkes DR. The future of HIV treatment. J Acquir Immune Defic Syndr. 2012;60(suppl 2):S39–S40. | ||
Panel on Antiretroviral Guidelines for Adults and Adolescents. Guidelines for the use of antiretroviral agents in HIV-1-infected adults and adolescents. Department of Health and Human Services. Available at http://www.aidsinfo.nih.gov/ContentFiles/AdultandAdolescentGL.pdf. Accessed April 12, 2015. | ||
Este JA, Cihlar T. Current status and challenges of antiretroviral research and therapy. Antiviral Res. 2010;85(1):25–33. | ||
Boone LR, Koszalka GW. Antiretroviral drug development for HIV: challenges for the future. Curr Opin Investig Drugs. 2010;11(8):863–867. | ||
Deeks SG. HIV infection, inflammation, immunosenescence, and aging. Annu Rev Med. 2011;62:141–155. | ||
Vitoria M, Vella S, Ford N. Scaling up antiretroviral therapy in resource-limited settings: adapting guidance to meet the challenges. Curr Opin HIV AIDS. 2013;8(1):12–18. | ||
Wainberg MA, Zaharatos GJ, Brenner BG. Development of antiretroviral drug resistance. N Engl J Med. 2011;365(7):637–646. | ||
Deeks ED. Cobicistat: a review of its use as a pharmacokinetic enhancer of atazanavir and darunavir in patients with HIV-1 infection. Drugs. 2014;74(2):195–206. | ||
Xu L, Liu H, Murray BP, et al. Cobicistat (GS-9350): a potent and selective inhibitor of human CYP3A as a novel pharmacoenhancer. ACS medicinal chemistry letters. 2010;1(5):209–213. | ||
DeJesus E, Rockstroh JK, Henry K, GS-236-0103 Study Team, et al. Co-formulated elvitegravir, cobicistat, emtricitabine, and tenofovir disoproxil fumarate versus ritonavir-boosted atazanavir plus co-formulated emtricitabine and tenofovir disoproxil fumarate for initial treatment of HIV-1 infection: a randomised, double-blind, phase 3, non-inferiority trial. Lancet. 2012. 379(9835):2429–2438. | ||
Mitra A, Wu Y. Challenges and opportunities in achieving bioequivalence for fixed-dose combination products. AAPS J. 2012;14(3):646–655. | ||
Pourkavoos N. Unique risks, benefits, and challenges of developing drug–drug combination products in a pharmaceutical industrial setting. Comb Prod Ther. 2012;2:2. | ||
Seelmeier S, Schmidt H, Turk V, von der Helm K. Human immunodeficiency virus has an aspartic-type protease that can be inhibited by pepstatin A. Proc Natl Acad Sci USA. 1988;85(18):6612–6616. | ||
Babe LM, Rose J, Craik CS. Trans-dominant inhibitory human immunodeficiency virus type 1 protease monomers prevent protease activation and virion maturation. Proc Natl Acad Sci U S A. 1995;92(22):10069–10073. | ||
Davies DR. The structure and function of the aspartic proteinases. Annu Rev Biophys Biophys Chem. 1990;19:189–215. | ||
Kohl NE, Emini EA, Schleif WA, et al. Active human immunodeficiency virus protease is required for viral infectivity. Proc Natl Acad Sci U S A. 1988;85(13):4686–4690. | ||
Miller M, Schneider J, Sathyanarayana BK, et al. Structure of complex of synthetic HIV-1 protease with a substrate-based inhibitor at 2.3 A resolution. Science. 1989;246(4934):1149–1152. | ||
Currier JS. Cardiovascular risk associated with HIV therapy: review by Judith Currier, MD. J. Acquir. Immune Defic. Syndr. 2002;31:S16–S23. | ||
Cahn P, Sued O. Boosting HIV treatment options: good news, new challenges. J Infect Dis. 2013;208(1):4–6. | ||
King NM, Prabu-Jeyabalan M, Nalivaika EA, Wigerinck P, de Bethune MP, Schiffer CA. Structural and thermodynamic basis for the binding of TMC114, a next-generation human immunodeficiency virus type 1 protease inhibitor. J Virol. 2004;78(21):12012–12021. | ||
Dierynck I, De Wit M, Gustin E, et al. Binding kinetics of darunavir to human immunodeficiency virus type 1 protease explain the potent antiviral activity and high genetic barrier. J Virol. 2007;81(24):13845–13851. | ||
Ortiz R, Dejesus E, Khanlou H, et al. Efficacy and safety of once-daily darunavir/ritonavir versus lopinavir/ritonavir in treatment-naive HIV-1-infected patients at week 48. AIDS. 2008;22(12):1389–1397. | ||
Orkin C, DeJesus E, Khanlou H, et al. Final 192-week efficacy and safety of once-daily darunavir/ritonavir compared with lopinavir/ritonavir in HIV-1-infected treatment-naive patients in the ARTEMIS trial. HIV Med. 2013;14(1):49–59. | ||
Deeks ED. Darunavir: a review of its use in the management of HIV-1 infection. Drugs. 2014;74(1):99–125. | ||
Zhou S, Yung Chan S, Cher Goh B, et al. Mechanism-based inhibition of cytochrome P450 3A4 by therapeutic drugs. Clin Pharmacokinet. 2005;44(3):279–304. | ||
Kempf DJ, Marsh KC, Kumar G, et al. Pharmacokinetic enhancement of inhibitors of the human immunodeficiency virus protease by coadministration with ritonavir. Antimicrob Agents Chemother. 1997;41(3):654–660. | ||
Hull MW, Montaner JS. Ritonavir-boosted protease inhibitors in HIV therapy. Ann Med. 2011;43(5):375–388. | ||
Boffito M, Jackson A, Amara A, et al. Pharmacokinetics of once-daily darunavir-ritonavir and ata zanavir-ritonavir over 72 hours following drug cessation. Antimicrob Agents Chemother. 2011;55(9): 4218–4223. | ||
Macías J, Recio E, Márquez M, et al. Efficacy and safety of once-daily maraviroc plus ritonavir-boosted darunavir in pretreated HIV-infected patients in a real-life setting. HIV Med. 2014;15(7):417–424. | ||
Mathias AA, German P, Murray BP, et al. Pharmacokinetics and pharmacodynamics of GS-9350: a novel pharmacokinetic enhancer without anti-HIV activity. Clin Pharmacol Ther. 2010;87(3):322–329. | ||
Elion R, Cohen C, Gathe J, et al. Phase 2 study of cobicistat versus ritonavir each with once-daily atazanavir and fixed-dose emtricitabine/tenofovir df in the initial treatment of HIV infection. AIDS. 2011;25(15):1881–1886. | ||
Gallant JE, Koenig E, Andrade-Villanueva J, et al. Cobicistat versus ritonavir as a pharmacoenhancer of atazanavir plus emtricitabine/tenofovir disoproxil fumarate in treatment-naive HIV type 1-infected patients: week 48 results. J Infect Dis. 2013;208(1):32–39. | ||
Therapeutics J. PREZCOBIX™ (darunavir and cobicistat) [prescribing information]; Janssen Therapeutics, Titusville NJ, USA. 2015. | ||
Boffito M, Miralles D, Hill A. Pharmacokinetics, efficacy, and safety of darunavir/ritonavir 800/100 mg once-daily in treatment-naive and -experienced patients. HIV Clin Trials. 2008;9(6):418–427. | ||
German P, Liu HC, Szwarcberg J, et al. Effect of cobicistat on glomerular filtration rate in subjects with normal and impaired renal function. J Acquir Immune Defic Syndr. 2012;61(1):32–40. | ||
Kakuda TN, Van De Casteele T, Petrovic R, et al. Bioequivalence of a darunavir/cobicistat fixed-dose combination tablet versus single agents and food effect in healthy volunteers. Antivir Ther. 2014;19(6):597–606. | ||
McDonald CK, Martorell C, Ramgopal M, et al. Cobicistat-boosted protease inhibitors in HIV-infected patients with mild to moderate renal impairment. HIV Clin Trials. 2014;15(6):269–273. | ||
Fisher M, McDonald C, Moyle G, et al. Switching from ritonavir to cobicistat in HIV patients with renal impairment who are virologically suppressed on a protease inhibitor. J Int AIDS Soc. 2014;17(4 suppl 3):19824. | ||
Tashima K, Crofoot G, Tomaka FL, et al. Cobicistat-boosted darunavir in HIV-1-infected adults: week 48 results of a phase IIIb, open-label single-arm trial. AIDS Res Ther. 2014;11:39. | ||
Kakuda TN, Opsomer M, Timmers M, et al. Pharmacokinetics of darunavir in fixed-dose combination with cobicistat compared with coadministration of darunavir and ritonavir as single agents in healthy volunteers. J Clin Pharmacol. 2014;54(8):949–957. | ||
Mills A, Crofoot G Jr, McDonald C, et al. Tenofovir alafenamide vs tenofovir disoproxil fumarate in the first protease inhibitor-based single tablet regimen for initial HIV-1 therapy: a randomized phase 2 study. J Acquir Immune Defic Syndr. 2015;69(4):439–445. | ||
Medicines Patent Pool Signs Licence Agreement with Gilead to Increase Access to HIV/AIDS Medicines. Available at http://www.medicinespatentpool.org/. Accessed May 27, 2015. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.