")
Back to Journals » Drug Design, Development and Therapy » Volume 10
Rational design and synthesis of novel diphenyl ether derivatives as antitubercular agents
Authors Sankar Kar S, Bhat V, Rao P N P, P Shenoy V, Bairy I, Gautham G
Received 11 January 2016
Accepted for publication 16 March 2016
Published 18 July 2016 Volume 2016:10 Pages 2299—2310
DOI https://doi.org/10.2147/DDDT.S104037
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Wei Duan
Sidhartha S Kar,1 Varadaraj Bhat G,1 Praveen PN Rao,2 Vishnu P Shenoy,3 Indira Bairy,4 G Gautham Shenoy1
1Department of Pharmaceutical Chemistry, Manipal College of Pharmaceutical Sciences, Manipal University, Manipal, India; 2School of Pharmacy, Health Sciences Campus, University of Waterloo, Waterloo, ON, Canada; 3Department of Microbiology, Kasturba Medical College, Manipal, 4Melaka-Manipal Medical College, Manipal University, Manipal, India
Abstract: A series of triclosan mimic diphenyl ether derivatives have been synthesized and evaluated for their in vitro antitubercular activity against Mycobacterium tuberculosis H37Rv. The binding mode of the compounds at the active site of enoyl-acyl carrier protein reductase of M. tuberculosis has been explored. Among them, compound 10b was found to possess antitubercular activity (minimum inhibitory concentration =12.5 µg/mL) comparable to triclosan. All the synthesized compounds exhibited low levels of cytotoxicity against Vero and HepG2 cell lines, and three compounds 10a, 10b, and 10c had a selectivity index more than 10. Compound 10b was also evaluated for log P, pKa, human liver microsomal stability, and % protein binding, in order to probe its druglikeness. Based on the antitubercular activity and druglikeness profile, it may be concluded that compound 10b could be a lead for future development of antitubercular drugs.
Keywords: dihenyl ether, tuberculosis, cytotoxicity, druglikeness
Introduction
Tuberculosis (TB), caused by Mycobacterium tuberculosis, is the leading cause of mortality worldwide, as indicated by the World Health Organization report.1 Currently, there is a need for antitubercular (anti-TB) agents effective against the drug-resistant strains of M. tuberculosis.2,3
Synthesis of mycolic acid, which is a central constituent of the mycobacterial cell wall, is carried out by fatty acid synthase (FAS) systems, namely, FAS-I and -II. Inhibition of type II fatty acid biosynthesis (FAS-II) pathway is a promising strategy in devising novel anti-TB agents.4,5
The final reaction in the FAS-II pathway of M. tuberculosis is catalyzed by the enoyl-acyl carrier protein reductase (ENR), which mediates the nicotinamide adenine dinucleotide (reduced form)-dependent reduction of trans-2-enoyl-acyl carrier protein to acyl-acyl carrier protein, and this biosynthetic pathway is an excellent target for antimicrobial agents.
Triclosan (1), a diphenyl ether derivative (Figure 1), exhibits a minimum inhibitory concentration (MIC) value of 12.5 μg/mL against M. tuberculosis H37Rv. Triclosan is an inhibitor of mycobacterial ENR enzyme.6 Unlike isoniazid, triclosan does not require prior activation to bind with ENR.7 Several investigators have attempted to develop diphenyl ether-based anti-TB agents, which chemically resemble triclosan (1).8–12 Triclosan has drawbacks including poor solubility13 and suboptimal bioavailability.14
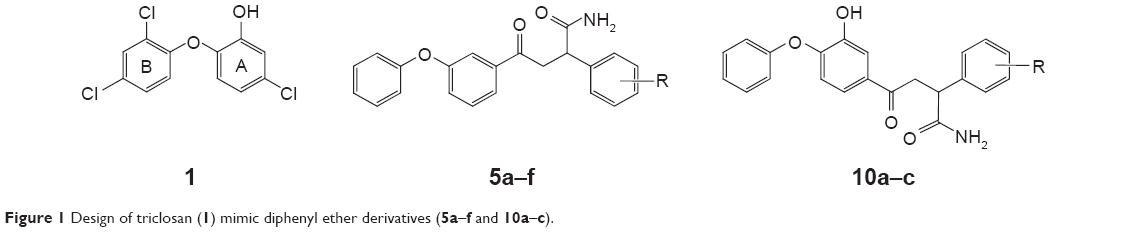 | Figure 1 Design of triclosan (1) mimic diphenyl ether derivatives (5a–f and 10a–c). |
Encouraged with the outcome of our previous work on the anti-TB activity of diphenyl ethers,15,16 we decided to further explore the diphenyl ether scaffold. In the present study, we report the design of triclosan mimic diphenyl ether derivatives as anti-TB agents using computational techniques simultaneously applying Lipinski’s criteria. The aim of the present study was to develop triclosan mimic diphenyl ether derivatives as potential anti-TB agents with improved druglikeness.
Materials and methods
Materials
Column chromatography was carried out on 100–200 mesh silica gel. Progress of the reactions was monitored by thin layer chromatography (TLC) using aluminum-backed sheets of silica gel 60 F24 (EMD Millipore, Billerica, MA, USA). Melting points were recorded with a laboratory melting point apparatus as uncorrected values. 1H NMR and 13C NMR spectra were recorded on an NMR spectrometer (AV400 – 400 MHz High-Resolution Multinuclear FT-NMR Spectrometer; Bruker India Scientific Pvt. Ltd., Bangalore, India) using dimethyl sulfoxide (DMSO)-d6 as the solvent. Mass spectrometry (MS) data were obtained on liquid chromatography (LC)–MS (Agilent 6520 series, Q-TOF LC/MS; Agilent Technologies, Santa Clara, CA, USA) and gas chromatography (GC)–MS (Shimadzu GC-17A, GCMS-QP5050A; Shimadzu Analytical (India) Pvt. Ltd., Mumbai, India). The purity of the final compounds was checked by reverse-phase high-performance liquid chromatography (HPLC) (ultra fast liquid chromatograph [UFLC], Shimadzu) using C-18 column in isocratic mode solvent systems (methanol and buffer, pH =7.4) and was found to be ≥95%.
Synthesis of 1-(3-phenoxy-phenyl)-ethanone (2)
To the stirred solution of 3-hydroxy acetophenone (3 g, 22.02 mmol) in anhydrous dichloromethane (120 mL), activated molecular sieves (4 Å, 3 g), phenylboronic acid (4.02 g, 33.18 mmol), copper (II) acetate (7.98 g, 44.04 mmol), and anhydrous pyridine (3.48 g, 44.04 mmol, 3.51 mL) were added successively. The resulting suspension was stirred at 25°C–27°C. Progress of the reaction was monitored by TLC using hexane:ethyl acetate (9:1). After the completion of reaction (72 hours), the reaction mixture was diluted with dichloromethane (100 mL) and filtered under reduced pressure. The filtrate was washed with dilute hydrochloric acid (2 M, 75 mL), followed by water (75 mL), dried over anhydrous MgSO4, and evaporated under reduced pressure. The crude compound was purified by column chromatography over silica 100–200 mesh with hexane:ethyl acetate (9:1) as the mobile phase to obtain the target compound.
Yield =2.25 g (48%); Rf=0.95 (hexane:ethyl acetate = 9:1); λmax=301 nm (MeOH); 1H NMR (400 MHz, (DMSO)-d6) δ ppm: 7.77–7.72 (m, 1H), 7.55–7.51 (m, 1H), 7.47–7.46 (m, 1H), 7.43–7.39 (m, 2H), 7.28–7.26 (ddd, J=8.0, 2.8, and 0.8 Hz, 1H), 7.20–7.16 (m, 1H), 7.06–7.03 (m, 2H), 2.55 (s, 3H); calculated for C14H12O2 [M+]: 212.24, found GC–MS (EI, m/z): 212 (M)+, 197 (M-CH3)+, 169 (M-COCH3)+.
General method for the synthesis of 3-(3-phenoxyphenyl)-1-aryl prop-2-en-1-one (3a–f)
To a solution of compound 2 (1 g, 4.711 mmol) and aryl acetophenones (4.711 mmol) in absolute alcohol (25 mL), ethanolic solution of KOH (0.527 g, 9.423 mmol) was added at 25°C–27°C. The reaction mixture was stirred at ambient temperature. Progress of the reaction was monitored by TLC using hexane:ethyl acetate (8:2). After the completion of reaction (14 hours), the reaction mixture was poured into ice-cold water (100 mL) and the residue was extracted with ethyl acetate (3×50 mL). The combined organic layers were washed with water, brine, dried over anhydrous MgSO4, and evaporated under reduced pressure. The crude compound was purified by column chromatography over silica 100–200 mesh with hexane:ethyl acetate (8:2) as the mobile phase to obtain the target compound.
1-(3-Phenoxyphenyl)-3-phenylprop-2-en-1-one (3a)
Yield =1.1 g (78%); mp =72°C–74°C; Rf=0.66 (hexane:ethyl acetate =8:2); λmax=309.80 nm (MeOH); 1H NMR (400 MHz, (DMSO)-d6) δ ppm: 7.96–7.94 (dd, J=8.0 and 1.2 Hz, 2H), 7.83 (d, J=15.6 Hz, 1H), 7.77–7.72 (m, 3H), 7.65 (d, J=8.0 Hz, 2H), 7.48–7.45 (m, 2H), 7.37 (d, J=7.8 Hz, 1H), 7.32–7.25 (m, 1H), 7.23 (t, J=7.6 Hz, 1H), 7.16–7.14 (m, 2H), 7.12–7.08 (m, 1H); calculated for C21H16O2 [M+]: 300.35, found LC–MS (+ESI, m/z): 301.1069 (M+H)+.
1-(3-Phenoxyphenyl)-3-p-tolylprop-2-en-1-one (3b)
Yield =1.0 g (68%); mp =58°C–60°C; Rf=0.8 (hexane:ethyl acetate =8:2); λmax=321.40 nm (MeOH); 1H NMR (400 MHz, (DMSO)-d6) δ ppm: 7.96–7.93 (td, J=8.0, 1.2, and 1.2 Hz, 1H), 7.85 (d, J=15.6 Hz, 1H), 7.77 (d, J=8.4 Hz, 2H), 7.72 (s, 1H), 7.69–7.68 (m, 1H), 7.57 (t, J=8.0 Hz, 1H), 7.44–7.39 (m, 2H), 7.307–7.301 (dd, J=2.4 and 0.8 Hz, 1H), 7.28–7.25 (m, 2H), 7.20–7.16 (m, 1H), 7.08–7.06 (m, 2H), 2.34 (s, 3H); calculated for C22H18O2 [M+]: 314.38, found LC–MS (+ESI, m/z): 315.1473 (M+H)+.
3-(4-Methoxyphenyl)-1-(3-phenoxyphenyl)prop-2-en-1-one (3c)
Yield =1.2 g (77%); mp =80°C–82°C; Rf=0.6 (hexane:ethyl acetate =8:2); λmax=343.20 nm (MeOH); 1H NMR (400 MHz, (DMSO)-d6) δ ppm: 7.87 (d, J=15.6 Hz, 1H), 7.76–7.73 (m, 2H), 7.54–7.50 (dt, J=8.0 and 1.2 Hz, 1H), 7.47–7.46 (m, 1H), 7.43–7.39 (m, 2H), 7.27–7.25 (ddd, J=8.4, 2.8, and 1.2 Hz, 1H), 7.20–7.14 (m, 3H), 7.05–7.02 (m, 2H), 6.82–6.80 (dd, J=8.4 and 1.2 Hz, 2H), 3.70 (s, 3H); calculated for C22H18O3 [M+]: 330.38, found LC–MS (+ESI, m/z): 331.2903 (M+H)+.
3-(2-Methoxyphenyl)-1-(3-phenoxyphenyl)prop-2-en-1-one (3d)
Yield =1.2 g (77%); mp =55°C–57°C; Rf=0.71 (hexane:ethyl acetate =8:2); λmax=316.40 nm (MeOH); 1H NMR (400 MHz, (DMSO)-d6) δ ppm: 7.76–7.74 (m, 2H), 7.52 (t, J=7.8 Hz, 1H), 7.48 (t, J=2.0 Hz, 1H), 7.43–7.39 (m, 2H), 7.28–7.25 (ddd, J=8.4, 2.8, and 1.2 Hz, 2H), 7.19–7.14 (m, 2H), 7.05–7.02 (m, 2H), 6.81 (d, J=7.2 Hz, 2H), 6.74–6.71 (m, 1H), 3.70 (s, 3H); calculated for C22H18O3 [M+]: 330.38, found LC–MS (+ESI, m/z): 331.2903 (M+H)+.
3-(4-Fluorophenyl)-1-(3-phenoxyphenyl)prop-2-en-1-one (3e)
Yield =1.1 g (67%); mp =68°C–70°C; Rf=0.71 (hexane:ethyl acetate =8:2); λmax=310.20 nm (MeOH); 1H NMR (400 MHz, (DMSO)-d6) δ ppm: 7.94 (d, J=15.6 Hz, 1H), 7.76–7.74 (td, J=7.6 and 1.2 Hz, 2H), 7.52 (t, J=8.0 Hz, 1H), 7.48 (t, J=2.0 Hz, 1H), 7.43–7.38 (m, 3H), 7.30–7.25 (m, 4H), 7.11–6.99 (m, 3H), calculated for C21H15FO2 [M+]: 318.34, found LC–MS (+ESI, m/z): 319.1206 (M+H)+.
3-(4-Chlorophenyl)-1-(3-phenoxyphenyl)prop-2-en-1-one (3f)
Yield =1.12 g (72%); mp =77°C–79°C; Rf=0.84 (hexane:ethyl acetate =8:2); λmax=305.40 nm (MeOH); 1H NMR (400 MHz, (DMSO)-d6) δ ppm: 7.89 (d, J=15.6 Hz, 1H), 7.76–7.73 (m, 2H), 7.52 (t, J=8.0 Hz, 1H), 7.49–7.48 (m, 2H), 7.43–7.39 (m, 3H), 7.27–7.26 (dd, J=2.8 and 1.2 Hz, 1H), 7.20–7.15 (m, 2H), 7.05–7.02 (m, 3H); calculated for C21H15ClO2 [M+]: 334.80, found LC–MS (+ESI, m/z): 335.1613 (M+H)+.
General method for the synthesis of 4-oxo-4-(3-phenoxyphenyl)-2-arylbutanenitrile (4a–f)
Cyanohydrin acetone (2.32 mmol), tributyl methyl ammonium hydroxide (1.16 mmol), and K2CO3 (2.32 mmol) were added successively to a solution of chalcone (3a–f, 1.16 mmol) in acetone (15 mL) and water (1 mL). The resulting reaction mixture was refluxed at 57°C–58°C for 14 hours. Progress of the reaction was monitored by TLC using hexane:ethyl acetate (8:2) as the mobile phase. After the completion of reaction (14 hours), the solvent was evaporated under reduced pressure. The residue was treated with ice-cold water and extracted with ethyl acetate (3×25 mL). The organic layers were pooled, washed with water, brine, dried over anhydrous MgSO4, and evaporated under reduced pressure. The crude compound was purified by column chromatography over silica 100–200 mesh with hexane:ethyl acetate (8:2) as the mobile phase to obtain the target compound.
4-Oxo-4-(3-phenoxyphenyl)-2-phenylbutanenitrile (4a)
Yield =0.32 g (84%); mp =62°C–64°C; Rf=0.47 (hexane:ethyl acetate =8:2); λmax=224.4 nm (MeOH); 1H NMR (400 MHz, (DMSO)-d6) δ ppm: 8.02–7.98 (td, J=9.2, 2.2, and 2.4 Hz, 2H), 7.61–7.57 (td, J=9.2, 2.2, and 2.4 Hz, 2H), 7.42–7.36 (m, 3H), 7.28 (d, J=7.6 Hz, 1H), 7.16 (t, J=1.0 Hz, 1H), 7.17–7.12 (m, 2H), 7.02–6.94 (m, 2H), 6.92–6.91 (dd, J=2.4 and 1.2 Hz, 1H), 4.62–4.59 (dd, J=8.8 and 5.2 Hz, 1H), 4.01–3.95 (dd, J=18.4 and 8.8 Hz, 1H), 3.74–3.68 (dd, J=18.4 and 4.8 Hz, 1H); calculated for C22H17NO2 [M+]: 327.38, found LC–MS (+ESI, m/z): 328.1341 (M)+.
4-Oxo-4-(3-phenoxyphenyl)-2-p-tolylbutanenitrile (4b)
Yield =0.365 g (93%); Rf=0.62 (hexane:ethyl acetate =8:2); λmax=300 nm (MeOH); 1H NMR (400 MHz, (DMSO)-d6) δ ppm: 7.39–7.35 (m, 2H), 7.31 (t, J=8.0 Hz, 1H), 7.13 (d, J=7.2 Hz, 1H), 7.08–7.06 (m, 1H), 7.02–7.01 (m, 4H), 6.99 (t, J=1.6 Hz, 2H), 6.98–6.95 (m, 1H), 6.86–6.83 (ddd, J=8.0, 2.4, and 0.8 Hz, 1H), 4.51–4.46 (dd, J=8.0 and 5.4 Hz, 1H), 4.10–4.05 (dd, J=14.4 and 8.0 Hz, 1H), 3.51–3.46 (dd, J=14.6 and 8.8 Hz, 1H), 2.23 (s, 3H); calculated for C23H19NO2 [M+]: 341.40, found LC–MS (+ESI, m/z): 342.1509 (M+H)+.
2-(4-Methoxyphenyl)-4-oxo-4-(3-phenoxyphenyl)butanenitrile (4c)
Yield =0.348 g (84%); mp =52°C–54°C; Rf=0.70 (hexane:ethyl acetate =8:2); λmax=254.4 nm (MeOH); 1H NMR (400 MHz, (DMSO)-d6) δ ppm: 7.42–7.40 (dd, J=7.6 and 2 Hz, 2H), 7.38 (t, J=4.4 Hz, 1H), 7.16 (t, J=1.2 Hz, 1H), 7.14–7.07 (m, 3H), 7.02–6.98 (m, 3H), 6.88–6.88 (dd, J=2.4 and 0.8 Hz, 1H), 6.86–6.81 (m, 2H), 4.53–4.48 (dd, J=10.8 and 6.0 Hz, 1H), 4.13–4.07 (dd, J=18.8 and 10 Hz, 1H), 3.71 (s, 3H), 3.60–3.55 (dd, J=18.4 and 6.0 Hz, 1H); calculated for C23H19NO3 [M+]: 357.40, found LC–MS (+ESI, m/z): 358.1012 (M+H))+.
2-(2-Methoxyphenyl)-4-oxo-4-(3-phenoxyphenyl)butanenitrile (4d)
Yield =0.33 g (80%); mp =58°C–60°C; Rf=0.71 (hexane:ethyl acetate =8:2); λmax=271 nm (MeOH); 1H NMR (400 MHz, (DMSO)-d6) δ ppm: 7.39–7.34 (m, 2H), 7.31 (t, J=7.8 Hz, 1H), 7.16–7.11 (m, 2H), 7.10–7.07 (m, 2H), 7.00–6.95 (m, 3H), 6.86–6.83 (ddd, J=8, 2.4, and 0.8 Hz, 1H), 6.73–6.69 (m, 3H), 4.52–4.48 (dd, J=11.2 and 6.0 Hz, 1H), 4.12–4.07 (dd, J=14.2 and 6 Hz, 1H), 3.70 (s, 3H), 3.56–3.50 (dd, J=14.2 and 5.6 Hz, 1H); calculated for C23H19NO3 [M+]: 357.40, found LC–MS (+ESI, m/z): 358.1012 (M)+.
2-(4-Fluorophenyl)-4-oxo-4-(3-phenoxyphenyl)butanenitrile (4e)
Yield =0.342 g (85%); mp =60°C–62°C; Rf=0.84 (hexane:ethyl acetate =8:2); λmax=303 nm (MeOH); 1H NMR (400 MHz, (DMSO)-d6) δ ppm: 7.41–7.37 (m, 2H), 7.32–7.29 (m, 1H), 7.26 (t, J=7.8 Hz, 1H), 7.20–7.15 (m, 2H), 7.10–7.08 (m, 2H), 7.05–7.04 (m, 3H), 7.00–6.96 (dd, J=2.8 and 1.2 Hz, 1H), 6.88–6.84 (m, 1H), 4.56–4.50 (m, 1H), 3.98–3.93 (m, 1H), 3.38–3.33 (m, 1H); calculated for C22H16FNO2 [M+]: 345.37, found LC–MS (+ESI, m/z): 345.1254 (M)+.
2-(4-Chlorophenyl)-4-oxo-4-(3-phenoxyphenyl)butanenitrile (4f)
Yield =0.35 g (83%); mp =69°C–71°C; Rf=0.86 (hexane:ethyl acetate =8:2); λmax=295.4 nm (MeOH); 1H NMR (400 MHz, (DMSO)-d6) δ ppm: 7.39–7.35 (m, 2H), 7.34–7.29 (m, 1H), 7.25–7.21 (m, 2H), 7.13–7.08 (m, 2H), 7.08–7.05 (m, 2H), 7.00–6.96 (m, 3H), 6.88–6.84 (m, 1H), 4.56–4.5 (m, 1H), 3.98–3.93 (m, 1H), 3.38–3.33 (m, 1H); calculated for C22H16ClNO2 [M+]: 361.82, found LC–MS (+ESI, m/z): 362.1623 (M+H)+.
General method for the synthesis of 4-oxo-2-(3-phenoxyphenyl)-4-arylbutanamide (5a–f)
To the solution of 4-oxo-4-(3-phenoxy-phenyl)-2-aryl-butyronitrile (4a–f) (0.76 mmol) in DMSO (8 mL), anhydrous K2CO3 (0.209 g, 1.52 mmol) was added. Hydrogen peroxide solution (28%, 1.52 mmol) was added drop wise at 5°C–10°C. The reaction mixture was stirred at 25°C–27°C. Progress of the reaction was monitored by TLC using hexane:ethyl acetate (8:2) as the mobile phase. After the completion of reaction (2 hours), the reaction mixture was poured into ice-cold water and the precipitate obtained was extracted with ethyl acetate (3×25 mL). The organic layers were separated, pooled, washed with water, brine, dried over anhydrous MgSO4, and evaporated under reduced pressure. The crude compound was purified by column chromatography over silica 100–200 mesh with hexane:ethyl acetate (5:5) as the mobile phase to obtain the target compound with good yield.
4-Oxo-4-(3-phenoxyphenyl)-2-phenylbutanamide (5a)
Yield =0.165 g (70%); mp =165°C–167°C; Rf=0.21 (hexane:ethyl acetate =6:4); λmax=295.8 nm (MeOH); 1H NMR (400 MHz, (DMSO)-d6) δ ppm: 7.78 (d, J=7.6 Hz, 1H), 7.52 (t, J=8.0 Hz, 1H), 7.48–7.47 (m, 2H), 7.43–7.39 (m, 2H), 7.38–7.36 (m, 2H), 7.30–7.25 (m, 3H), 7.21–7.19 (m, 2H), 7.17–7.15 (m, 2H), 6.79 (s, 1H), 4.06–4.02 (dd, J=10.0 and 4.4 Hz, 1H), 3.86–3.79 (dd, J=17.6 and 10.0 Hz, 1H), 3.18–3.13 (dd, J=17.6 and 4.0 Hz, 1H); 13C NMR (100.64 MHz, (DMSO)-d6) δ ppm: 198.08, 174.34, 157.61, 156.61, 140.96, 138.85, 131.02, 130.70, 128.76, 128.24, 127.16, 124.45, 123.70, 119.48, 117.36, 42.18; calculated for C22H19NO3 [M+]: 345.39, found LC–MS (+ESI, m/z): 346.1485 (M+H)+.
4-Oxo-4-(3-phenoxyphenyl)-2-p-tolylbutanamide (5b)
Yield =0.2 g (73%); mp =139°C–141°C; Rf=0.25 (hexane:ethyl acetate =6:4); λmax=299 nm (MeOH); 1H NMR (400 MHz, (DMSO)-d6) δ ppm: 7.78–7.76 (dd, J=6.4 and 1.2 Hz, 1H), 7.52 (t, J=8.0 Hz, 1H), 7.46 (t, J=2.0 Hz, 1H), 7.43–7.39 (m, 3H), 7.28–7.27 (dd, J=2.4 and 0.8 Hz, 1H), 7.26–7.23 (m, 2H), 7.19–7.15 (m, 1H), 7.09 (d, J=8.0 Hz, 2H), 7.05–7.03 (m, 2H), 6.75 (s, 1H), 4.01–3.97 (dd, J=10.0 and 4.4 Hz, 1H), 3.83–3.76 (dd, J=18.0 and 10.0 Hz, 1H), 3.14–3.09 (dd, J=18.0 and 4.4 Hz, 1H), 2.24 (s, 3H); 13C NMR (100.64 MHz, (DMSO)-d6) δ ppm: 198.12, 174.49, 157.62, 156.60, 137.94, 131.02, 130.70, 129.29, 128.08, 124.46, 123.67, 119.49, 117.32, 46.29, 42.57, 42.19, 21.08; calculated for C23H21NO3 [M+]: 359.42, found LC–MS (+ESI, m/z): 360.1588 (M+H)+.
2-(4-Methoxyphenyl)-4-oxo-4-(3-phenoxyphenyl)butanamide (5c)
Yield =0.2 g (77%); mp =102°C–104°C; Rf=0.21 (hexane:ethyl acetate =6:4); λmax=273.8 nm (MeOH); 1H NMR (400 MHz, (DMSO)-d6) δ ppm: 7.77 (d, J=7.6 Hz, 1H), 7.52 (t, J=8.0 Hz, 1H), 7.46 (s, 1H), 7.41 (t, J=8.0 Hz, 3H), 7.27 (d, J=8.4 Hz, 3H), 7.17 (t, J=7.4 Hz, 1H), 7.04 (d, J=8.0 Hz, 2H), 6.85 (d, J=8.8 Hz, 2H), 6.74 (s, 1H), 3.98 (d, J=6.0 Hz, 1H), 3.82–3.75 (dd, J=17.6 and 9.2 Hz, 1H), 3.70 (s, 3H), 3.16–3.12 (dd, J=18.0 and 4.0 Hz, 1H); 13C NMR (100.64 MHz, (DMSO)-d6) δ ppm: 198.17, 174.69, 158.56, 157.61, 156.59, 138.89, 132.91, 131.03, 130.70, 129.21, 124.47, 123.67, 119.49, 117.31, 114.16, 55.52, 42.57; calculated for C23H21NO4 [M+]: 375.42, found LC–MS (+ESI, m/z): 376.1043 (M+H)+.
2-(2-Methoxyphenyl)-4-oxo-4-(3-phenoxyphenyl)butanamide (5d)
Yield =0.18 g (70%); mp =153°C–155°C; Rf=0.24 (hexane:ethyl acetate =6:4); λmax=272 nm (MeOH); 1H NMR (400 MHz, (DMSO)-d6) δ ppm: 7.76 (d, J=8.0 Hz, 1H), 7.51 (t, J=7.8 Hz, 1H), 7.46 (s, 1H), 7.39 (t, J=8.8 Hz, 2H), 7.28–7.25 (dd, J=8.4 and 1.6 Hz, 1H), 7.23–7.17 (m, 3H), 7.16–7.12 (m, 1H), 7.04 (d, J=7.6 Hz, 2H), 6.98 (d, J=8.0 Hz, 1H), 6.89 (t, J=7.6 Hz, 2H), 6.82 (s, 1H), 4.43–4.39 (dd, J=10.0 and 3.6 Hz, 1H), 3.78 (s, 3H), 3.76–3.66 (m, 1H), 3.01–2.95 (m, 1H); 13C NMR (100.64 MHz, (DMSO)-d6) δ ppm: 198.00, 174.69, 158.50, 157.61, 156.59, 135.00, 132.32, 131.05, 130.00, 129.2, 124.5, 124.11, 119.95, 118.12, 114.34, 56.12, 40.5; calculated for C23H21NO4 [M+]: 375.42, found LC–MS (+ESI, m/z): 376.1043 (M+H)+.
2-(4-Fluorophenyl)-4-oxo-4-(3-phenoxyphenyl)butanamide (5e)
Yield =0.19 g (70%); mp =130°C–132°C; Rf=0.27 (hexane:ethyl acetate =6:4); λmax=271 nm (MeOH); 1H NMR (400 MHz, (DMSO)-d6) δ ppm: 7.78 (d, J=7.6 Hz, 1H), 7.53 (d, J=8 Hz, 1H), 7.49 (d, J=8.8 Hz, 2H), 7.43–7.39 (m, 4H), 7.28–7.26 (dd, J=8.0 and 2.0 Hz, 1H), 7.17 (t, J=7.2 Hz, 2H), 7.11 (t, J=7.6 Hz, 1H), 7.04 (d, J=7.6 Hz, 2H), 6.81 (s, 1H), 4.07–4.03 (dd, J=10.8 and 4.0 Hz, 1H), 3.85–3.78 (dd, J=18.0 and 10.0 Hz, 1H), 3.19–3.14 (dd, J=17.6 and 4.4 Hz, 1H); 13C NMR (100.64 MHz, (DMSO)-d6) δ ppm: 196.80, 174.9, 159.00, 157.66, 156.5, 133.60, 132.2, 131.00, 130.5, 128.87, 124.5, 123.16, 118.5, 117.2, 114.00, 43.83, 41.25; calculated for C22H18FNO3 [M+]: 363.38, found LC–MS (+ESI, m/z): 364.134 (M+H)+.
2-(4-Chlorophenyl)-4-oxo-4-(3-phenoxyphenyl)butanamide (5f)
Yield =0.205 g (71%); mp =105°C–107°C; Rf=0.29 (hexane:ethyl acetate =6:4); λmax=274.5 nm (MeOH); 1H NMR (400 MHz, (DMSO)-d6) δ ppm: 7.75 (d, J=8.0 Hz, 1H), 7.64 (d, J=8.0 Hz, 1H), 7.49–7.46 (m, 2H), 7.43–7.39 (m, 4H), 7.24–7.21 (dd, J=8.4 and 2.0 Hz, 1H), 7.14–7.11 (m, 2H), 7.07 (t, J=7.6 Hz, 1H), 7.04–7.00 (m, 2H), 6.87 (s, 1H), 4.10–4.06 (dd, J=10.4 and 5.6 Hz, 1H), 3.92–3.86 (m, 1H), 3.18–3.14 (dd, J=15.6 and 5.6 Hz, 1H); 13C NMR (100.64 MHz, (DMSO)-d6) δ ppm: 199.20, 177.65, 159.69, 158.24, 156.5, 134.00, 132.43, 131.65, 130.15, 129.8, 124.5, 121.44, 118.54, 116.82, 115.12, 42.5, 41.2; calculated for C22H18ClNO3 [M+]: 379.84, found LC–MS (+ESI, m/z): 380.1416 (M+H)+.
Synthesis of 1-(3-methoxy-4-phenoxyphenyl)ethanone (6)
To the stirred solution of 4-hydroxy-3-methoxy-acetophenone (3 g, 18.05 mmol) in anhydrous dichloromethane (90 mL), activated molecular sieves (4 Å, 3 g), phenylboronic acid (4.49 g, 36.82 mmol), copper (II) acetate (7.36 g, 40.58 mmol), and anhydrous pyridine (5.70 g, 72.15 mmol, 5.82 mL) were added successively. The resulting suspension was stirred at 25°C–27°C. Progress of the reaction was monitored by TLC using hexane:ethyl acetate (8:2) as the mobile phase. After the completion of reaction (72 hours), the reaction mixture was diluted with dichloromethane (40 mL) and filtered under reduced pressure. The filtrate was washed with dilute aqueous hydrochloric acid solution (2 M, 50 mL), followed by water (50 mL), dried over anhydrous MgSO4, and evaporated under reduced pressure. The crude compound was purified by column chromatography over silica 100–200 mesh with hexane:ethyl acetate (8:2) as the mobile phase to obtain the target compound as a white crystalline solid.
Yield =4.12 g (94%); mp =82°C–84°C; Rf=0.9 (hexane:ethyl acetate =8:2); λmax=269.6 nm (MeOH); 1H NMR (400 MHz, (DMSO)-d6) δ ppm: 7.60 (d, J=8.0 Hz, 2H), 7.36 (t, J=7.8 Hz, 2H), 7.12 (t, J=7.2 Hz, 1H), 7.00 (d, J=8.0 Hz, 1H), 6.94 (d, J=8.0 Hz, 2H), 3.83 (s, 3H), 2.56 (s, 3H); calculated for C15H14O3 [M+]: 242.27, found LC–MS (+ESI, m/z): 243.1032 (M+H)+.
Synthesis of 1-(3-hydroxy-4-phenoxyphenyl)ethanone (7)
To a solution of 1-(3-methoxy-4-phenoxyphenyl) ethanone (6) (4 g, 16.52 mmol) in anhydrous dichloromethane (60 mL) maintained at −78°C under nitrogen atmosphere, BBr3 (1 M, 8.28 g, 33.04 mmol) was added. The reaction mixture was stirred for 2 hours at −78°C; then its temperature was allowed to increase up to 10°C and it was stirred continuously. Progress of the reaction was monitored by TLC using hexane:ethyl acetate (6:4). After the completion of reaction (6 hours), the reaction was quenched by pouring the reaction mixture into ice-cold aqueous sodium bicarbonate with continuous stirring. The organic layer was separated, washed with water, brine, dried over anhydrous MgSO4; and evaporated under reduced pressure. The crude compound was purified by column chromatography over silica 100–200 mesh with hexane:ethyl acetate (6:4) as the mobile phase to obtain the target compound.
Yield =3.6 g (96%); mp =87°C–89°C; Rf=0.63 (hexane:ethyl acetate =6:4); λmax=269.4 nm (MeOH); 1H NMR (400 MHz, (DMSO)-d6) δ ppm: 9.89 (s, 1H), 7.51 (d, J=2.0 Hz, 1H), 7.45–7.43 (dd, J=8.4 and 2.4 Hz, 1H), 7.37–7.33 (m, 2H), 7.11–7.07 (m, 1H), 6.96 (s, 1H), 6.94–6.92 (m, 2H), 2.50 (s, 3H); calculated for C14H12O3 [M+]: 228.24, found LC–MS (+ESI, m/z): 229.085 (M+H)+.
General method for the synthesis of 1-(3-Hydroxy-4-phenoxyaryl)ethanone (8a–c)
Compounds 8a–c were synthesized by following the method adopted for the synthesis of compounds 3a–f.
1-(3-Hydroxy-4-phenoxyphenyl)-3-phenylprop-2-en-1-one (8a)
Yield =1 g (72%); mp =94°C–96°C; Rf=0.61 (hexane:ethyl acetate =6:4); λmax=322.2 nm (MeOH); 1H NMR (400 MHz, (DMSO)-d6) δ ppm: 9.98 (s, 1H), 7.87–7.84 (m, 3H), 7.71 (d, J=7.6 Hz, 1H), 7.68 (t, J=2.4 Hz, 2H), 7.44 (t, J=2.6 Hz, 3H), 7.36 (t, J=7.8 Hz, 2H), 7.10 (t, J=7.4 Hz, 1H), 7.01–6.94 (m, 3H); calculated for C21H16O3 [M+]: 316.35, found LC–MS (+ESI, m/z): 317.1157 (M+H)+.
1-(3-Hydroxy-4-phenoxyphenyl)-3-p-tolylprop-2-en-1-one (8b)
Yield =0.8 g (55%); mp =152°C–153°C; Rf=0.82 (hexane:ethyl acetate =6:4); λmax=321.8 nm (MeOH); 1H NMR (400 MHz, (DMSO)-d6) δ ppm: 9.91 (s, 1H), 7.81 (s, 1H), 7.76 (t, J=7.4 Hz, 2H), 7.70–7.67 (dd, J=8.4 and 2.4 Hz, 2H), 7.66 (d, J=2.4 Hz, 1H), 7.38–7.34 (td, J=7.0 and 2.0 Hz, 2H), 7.26 (d, J=8.0 Hz, 2H), 7.12–7.08 (m, 1H), 7.00–6.95 (m, 3H), 2.34 (s, 3H); calculated for C22H18O3 [M+]: 330.38, found LC–MS (+ESI, m/z): 331.1348 (M+H)+.
1-(3-Hydroxy-4-phenoxyphenyl)-3-(4-methoxyphenyl) prop-2-en-1-one (8c)
Yield =0.94 g (62%); mp =138°C–140°C; Rf=0.70 (hexane:ethyl acetate =6:4); λmax=352.4 nm (MeOH); 1H NMR (400 MHz, (DMSO)-d6) δ ppm: 9.89 (s, 1H), 7.83–7.80 (dd, J=8.8 and 1.6 Hz, 2H), 7.70 (d, J=3.6 Hz, 2H), 7.66–7.65 (m, 2H), 7.38–7.34 (m, 2H), 7.12–7.08 (m, 1H), 7.01–6.97 (m, 3H), 6.96–6.94 (m, 2H), 3.81 (s, 3H); calculated for C22H18O4 [M+]: 346.38, found LC–MS (+ESI, m/z): 347.1256 (M+H)+.
General method for the synthesis of 4-(3-hydroxy-4-phenoxyphenyl)-4-oxo-2-aryl butanenitrile (9a–c)
Compounds 9a–c were synthesized by following the method adopted for synthesis of compounds 4a–f.
4-(3-Hydroxy-4-phenoxyphenyl)-4-oxo-2-phenyl butanenitrile (9a)
Yield =0.3 g (80%); mp =81°C–82°C; Rf=0.28 (hexane:ethyl acetate =6:4); λmax=275.4 nm (MeOH); 1H NMR (400 MHz, (DMSO)-d6) δ ppm: 9.94 (s, 1H), 7.54–7.47 (m, 4H), 7.42–7.33 (m, 5H), 7.20 (d, J=8.0 Hz, 1H), 7.10 (t, J=7.4 Hz, 1H), 6.94 (d, J=8.0 Hz, 2H), 4.59–4.49 (m, 1H), 3.92–3.81 (m, 1H), 3.65–3.56 (m, 1H); calculated for C22H17NO3 [M+]: 343.38, found LC–MS (+ESI, m/z): 344.1294 (M+H)+.
4-(3-Hydroxy-4-phenoxyphenyl)-4-oxo-2-p-tolylbutanenitrile (9b)
Yield =0.33 g (83%); mp =60°C–62°C; Rf=0.32 (hexane:ethyl acetate =6:4); λmax=274.8 nm (MeOH); 1H NMR (400 MHz, (DMSO)-d6) δ ppm: 9.97 (s, 1H), 7.52 (d, J=2.4 Hz, 1H), 7.49–7.46 (dd, J=8.4 and 2.4 Hz, 1H), 7.38–7.33 (m, 4H), 7.19 (d, J=7.6 Hz, 2H), 7.12–7.07 (dt, J=7.4 and 1.0 Hz, 1H), 6.95–6.92 (m, 3H), 4.52–4.49 (dd, J=8.8 and 5.2 Hz, 1H), 3.87–3.80 (dd, J=18.8 and 9.2 Hz, 1H), 3.61–3.55 (dd, J=18.0 and 5.6 Hz, 1H), 2.28 (s, 3H); calculated for C23H19NO3 [M+]: 357.40, found LC–MS (+ESI, m/z): 358.1448 (M+H)+.
4-(3-Hydroxy-4-phenoxyphenyl)-2-(4-methoxyphenyl)-4-oxobutanenitrile (9c)
Yield =0.305 g (81%); mp =52°C–54°C; Rf=0.31 (hexane:ethyl acetate =6:4); λmax=274.8 nm (MeOH); 1H NMR (400 MHz, (DMSO)-d6) δ ppm: 9.95 (s, 1H), 7.76 (d, J=2.4 Hz, 1H), 7.50–7.45 (m, 2H), 7.41–7.38 (m, 3H), 7.26 (d, J=8.0 Hz, 2H), 7.17–7.13 (m, 2H), 7.08–7.02 (m, 2H), 4.57–4.54 (dd, J=8.4 and 5.4 Hz, 1H), 3.89–3.80 (m, 1H), 3.67–3.58 (m, 1H), 2.28 (s, 3H); calculated for C23H19NO4 [M+]: 373.40, found LC–MS (+ESI, m/z): 374.1396 (M+H)+.
General method for the synthesis of 4-(3-hydroxy-4-phenoxyphenyl)-4-oxo-2-aryl butanamide (10a–c)
Compounds 10a–c were synthesized by following the method adopted for synthesis of compounds 5a–f.
4-(3-Hydroxy-4-phenoxyphenyl)-4-oxo-2-phenylbutanamide (10a)
Yield =0.215 g (82%); mp =143°C–145°C; Rf=0.09 (hexane:ethyl acetate =5:5); λmax=269.4 nm (MeOH); 1H NMR (400 MHz, (DMSO)-d6) δ ppm: 9.87 (s, 1H), 7.51 (t, J=5.0 Hz, 1H), 7.50–7.46 (m, 2H), 7.43 (s, 1H), 7.38–7.34 (m, 3H), 7.33–7.30 (m, 2H), 7.28–7.20 (m, 1H), 7.09 (t, J=7.2 Hz, 1H), 6.95–6.92 (dd, J=8.4 and 5.2 Hz, 3H), 6.80–6.76 (m, 1H), 4.07–4.04 (dd, J=10.0 and 4.0 Hz, 1H), 3.81–3.74 (m, 1H), 3.13–3.07 (dd, J=18.0 and 4.4 Hz, 1H); 13C NMR (100.64 MHz, (DMSO)-d6) δ ppm: 197.28, 174.41, 157.20, 149.25, 148.08, 141.13, 133.75, 130.33, 129.30, 128.78, 128.20, 128.05, 127.14, 123.49, 120.90, 120.62, 117.88, 116.65, 46.73, 41.89; calculated for C22H19NO4 [M+]: 361.39, found LC–MS (+ESI, m/z): 362.1562 (M+H)+.
4-(3-Hydroxy-4-phenoxyphenyl)-4-oxo-2-p-tolylbutanamide (10b)
Yield =0.235 g (86%); mp =135°C–137°C; Rf=0.12 (hexane:ethyl acetate =5:5); λmax=269.6 nm (MeOH); 1H NMR (400 MHz, (DMSO)-d6) δ ppm: 9.87 (s, 1H), 7.52 (d, J=2.0 Hz, 1H), 7.48–7.46 (dd, J=8.4 and 2.0 Hz, 1H), 7.43 (s, 1H), 7.36–7.32 (m, 2H), 7.24 (d, J=8 Hz, 2H), 7.10 (d, J=7.2 Hz, 3H), 6.95–6.92 (m, 3H), 6.76 (s, 1H), 4.02–3.99 (dd, J=10.0 and 4.4 Hz, 1H), 3.78–3.71 (dd, J=17.6 and 11.2 Hz, 1H), 3.09–3.03 (dd, J=17.6 and 4.8 Hz, 1H), 2.25 (s, 3H); 13C NMR (100.64 MHz, (DMSO)-d6) δ ppm: 197.34, 174.59, 157.19, 149.24, 148.07, 138.11, 136.17, 133.77, 130.33, 129.31, 128.05, 123.49, 120.89, 120.63, 117.88, 116.64, 46.32, 42.57, 41.89, 21.09; calculated for C23H21NO4 [M+]: 375.42, found LC–MS (+ESI, m/z): 376.1511 (M+H)+.
4-(3-Hydroxy-4-phenoxyphenyl)-2-(4-methoxyphenyl)-4-oxobutanamide (10c)
Yield =0.23 g (81%); mp =123°C–125°C; Rf=0.11 (hexane:ethyl acetate =6:4); λmax=270.6 nm (MeOH); 1H NMR (400 MHz, (DMSO)-d6) δ ppm: 9.92 (s, 1H), 7.62 (d, J=2.4 Hz, 1H), 7.54–7.51 (m, 2H), 7.48 (s, 1H), 7.38–7.35 (m, 2H), 7.24 (d, J=8 Hz, 2H), 7.18 (t, J=7.6 Hz, 1H), 7.05–6.99 (m, 2H), 6.84 (s, 1H), 4.00–3.96 (dd, J=10.2 and 5.4 Hz, 1H), 3.91–3.86 (dd, J=16.6 and 10.2 Hz, 1H), 3.71 (s, 3H), 3.14–3.09 (m, 1H); 13C NMR (100.64 MHz, (DMSO)-d6) δ ppm: 197.39, 174.78, 158.56, 157.20, 149.24, 148.07, 133.79, 133.09, 130.32, 129.17, 123.49, 120.89, 120.63, 117.88, 116.66, 114.18, 55.52, 45.86, 42.58, 41.99; calculated for C23H21NO5 [M+]: 391.42, found LC–MS (+ESI, m/z): 392.135 (M+H)+.
Results and discussion
The detailed methodology for anti-TB screening, pKa determination, log P determination, cytotoxicity studies, and liver microsomal stability studies is given in Supplementary material.
Triclosan mimic novel diphenyl ether derivatives were designed (Figure 1) with functional diversifications at the C-3 position of ring-A. With the intention of improving the hydrogen bond interactions at the catalytic sites of InhA enzyme and decreasing the lipophilicity of diphenyl ether moiety, hydrogen bond donors/acceptors were introduced in the substituted alkyl chain. Sivaraman et al17 reported that the chloro substitutions in ring-B of triclosan (1) are involved in unfavorable steric interactions with the enzyme, and their deletion from the scaffold increases the affinity sevenfold. Hence, diphenyl ether derivatives were designed without chloro substitutions in ring-B. Removal of halogens also helps in decreasing the lipophilicity of the designed molecules.
Compounds 5a–f were prepared to probe the effect of presence of electron-withdrawing carbonyl group adjacent to ring-A of diphenyl ether ring (Figure 2). To begin with, 3-phenoxy acetophenone (2) was prepared through the Chan–Lam coupling reaction using Cu(OAc)2.18 Chalcones (3a–f) (Figure 2) were synthesized by reacting 3-phenoxy acetophenone (2) with different aryl aldehydes.19 Primary amide functionality was incorporated at the β-carbon of the chalcones through base-catalyzed selective hydration of the hydrocyanated chalcones20 in the presence of hydrogen peroxide to obtain the corresponding amides (5a–f).21
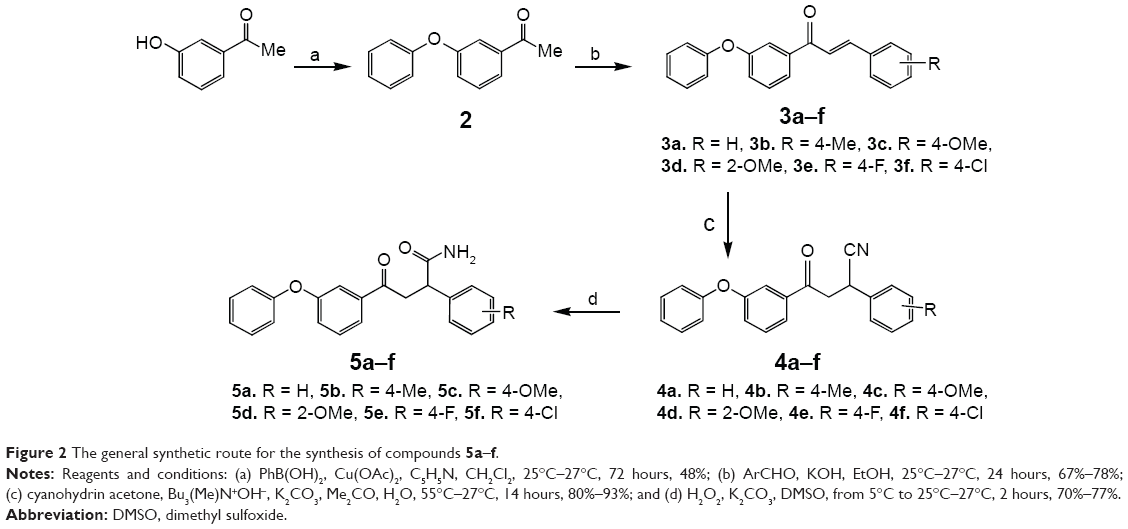 | Figure 2 The general synthetic route for the synthesis of compounds 5a–f. |
Phenolic –OH of the A-ring of triclosan plays a crucial role in binding to the catalytic site of enoyl-acyl carrier protein reductase of M. tuberculosis. In addition to this, diphenyl ethers substituted with long alkyl chains at the C-4 position of ring-A have been reported to display better affinity than triclosan toward enoyl-acyl carrier protein reductase of M. tuberculosis.6 However, the lipophilicity of the reported diphenyl ethers is high. Therefore, it was decided to synthesize diphenyl ether derivatives with hydrophilic substituents at the fourth position and –OH group at the C-2 position of ring-A (Figure 1). These changes were made to decrease the lipophilicity of the diphenyl ether derivatives. At the same time, it was ensured that the newly designed molecules achieved similar orientation and interactions as those of triclosan at the target binding site. Synthesis of compounds 10a–c is described in Figure 3. Acetovanillone was condensed with phenyl boronic acid by Chan–Lam O-arylation reaction. Compound 6 that was obtained was subjected to BBr3-assisted demethylation reaction as described by Gillmore et al22 to obtain compound 7. Compounds 8a–c, 9a–c, and 10a–c were prepared by using the same chemistry shown in Figure 2.
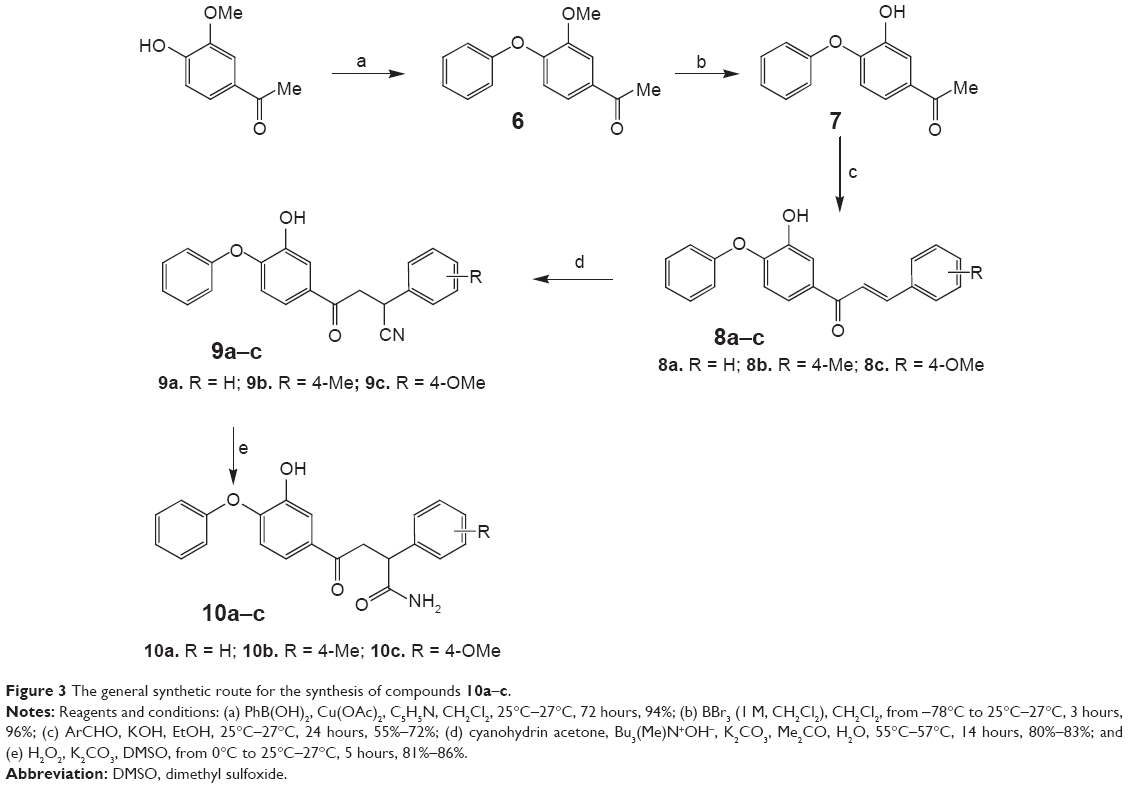 | Figure 3 The general synthetic route for the synthesis of compounds 10a–c. |
The synthesized diphenyl ether derivatives were screened in vitro for anti-TB activity against M. tuberculosis H37Rv strain using microplate Alamar Blue assay method.23,24 Known InhA inhibitors, triclosan and isoniazid, were used as standard drugs for comparison.
Compounds 5a–f were screened for their anti-TB potential to study the effect of the presence carbonyl group at the proximity of ring-A on the anti-TB activity. This series of compounds possessed very weak activity (MIC ≥50 μg/mL). This indicates that structural modifications at C-3 of ring-A in diphenyl ether are unfavorable for anti-TB activity. Potential toxicity of the synthesized diphenyl ether derivatives was evaluated on mammalian Vero cell lines and HepG2 cell lines using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay technique. Data showed that compounds 5a–f are not cytotoxic against (half-maximal cytotoxicity concentration >300 μg/mL) against Vero and HepG2 cells (Table 1).25–27
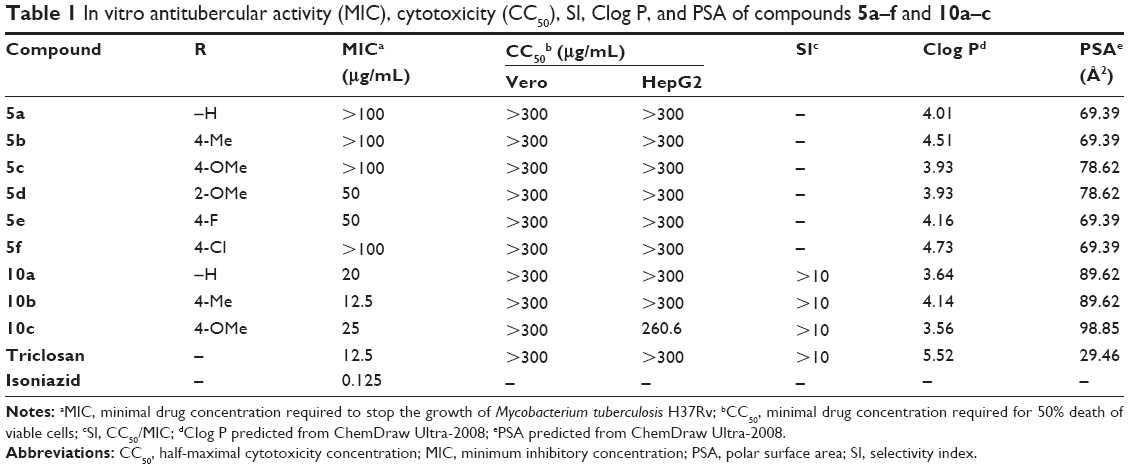 | Table 1 In vitro antitubercular activity (MIC), cytotoxicity (CC50), SI, Clog P, and PSA of compounds 5a–f and 10a–c |
Compounds 10a–c were screened for their anti-TB activity. Compound 10b showed MIC value of 12.5 μg/mL, which is equal to the MIC of triclosan. The Clog P and polar surface area of compound 10b (Table 1) showed that it is comparatively more polar than triclosan and obeys the Lipinski’s rule for druglikeness.28 All the compounds of this series possessed acceptable safety profile against Vero as well as HepG2 cells.
To understand the interaction of the proposed diphenyl ether derivatives with mycobacterial ENR, molecular docking study was conducted in Schrodinger-201029 using PDB 1P45 as the target protein.30 The molecular docking study of compounds 5a–f showed that the amide group at β-carbon was oriented toward the pyrophosphate part of nicotinamide adenine dinucleotide (NAD) forming hydrogen bond with it. Although this series of compounds showed apparently good docking scores, their interactions with the pyrophosphate moiety of NAD proved unfavorable for anti-TB activity. Molecular docking study of compound 10b revealed that the prochiral carbonyl group was oriented opposite to the plane of NAD, and lost hydrogen bonding interactions with both 2′ OH of NAD and Tyr-158 (Figure 4). However, the flexible amide group of 10b formed hydrogen bond with the carbonyl oxygen of nicotinamide of NAD. Compound 10c oriented in a different manner and displayed hydrogen bonding interaction with the pyrophosphate region of NAD. This hydrogen bonding interaction was noticed to be unfavorable for anti-TB activity. Although these three compounds lost hydrogen bonding interactions at the catalytic site of ENR, they displayed strong van der Waals interaction with NAD. Although the most promising compound 10b did not possess the hydrogen bonding interaction pattern of triclosan, it fitted better inside the active site of the ENR to show superior docking score (Table 2).
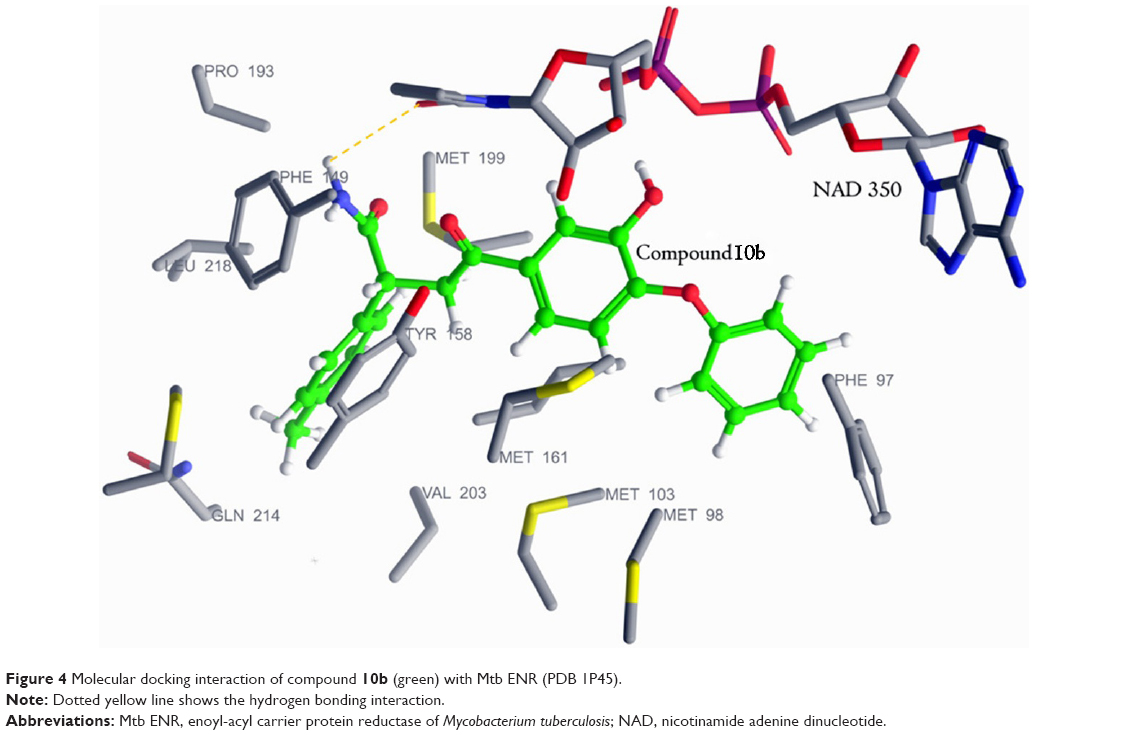 | Figure 4 Molecular docking interaction of compound 10b (green) with Mtb ENR (PDB 1P45). |
 | Table 2 Molecular docking results of compounds 5a–f and 10a–c |
In-depth analysis of docking results and their correlation with the anti-TB activity of these series of compounds showed that docking orientations, van der Waals interactions, and hydrogen bonding interactions with NAD play a decisive role in improving the anti-TB activity.
In order to understand the effect of lipophilicity on anti-TB activity, experimental log P of the most promising compound 10b was determined by reverse-phase HPLC method31,32 and is shown in Table 3. The log P obtained was compared with its calculated Clog P using ChemDraw Ultra-2008 software (Cambridge, MA, USA) (Table 1). Compound 10b demonstrated moderate log P and was within the limit of Lipinski’s28 suggested parameters for oral bioavailability. In-depth analysis of predicted Clog P of compounds indicated that there is no relationship between lipophilicity and the observed anti-TB activity.
 | Table 3 Evaluation of druglikeness of compound 10b |
pKa of the representative compound 10b was determined using the reverse-phase HPLC method (Table 3).33 Compound 10b was found to be weakly acidic (pKa =7.59). The weak acidity of the molecule may be due to the effect of the carbonyl group on the acidity of ionizable phenolic OH group at C-2 position of ring-A. pKa of compound 10b showed that it would have remained in the ionized form to a certain extent at pH 7.4 during the in vitro whole-cell assay (microplate Alamar Blue assay). Hence, insignificant penetration into the mycobacterial cell wall may be the sole reason for its moderate anti-TB activity.
The protein binding of compound 10b was evaluated by reverse-phase HPLC method using human serum albumin column (Table 3).34,35 Compound 10b was observed to be highly susceptible to protein binding (90.5%), whereas plasma protein binding of triclosan is known to be even higher than compound 10b.36 Human liver microsomal stability (%) of compound 10b was evaluated at different time points (15 minutes, 30 minutes, 1 hour, and 2 hours).37,38 After 2 hours of incubation, 50.68% of 10b was found to be intact (Table 4).
 | Table 4 Human microsome stability study of compound 10b |
Conclusion
A series of triclosan mimic diphenyl ether derivatives were prepared and evaluated for their anti-TB activity against M. tuberculosis H37Rv strain. The synthesized compounds were found to be less lipophilic than triclosan. Among the synthesized molecules, compound 10b had anti-TB activity equal to that of triclosan and was less lipophilic than triclosan. 10b had good microsomal stability and low cytotoxicity. Docking studies indicate that there is scope for further optimization of diphenyl ether derivatives in order to gain access to new lead molecules as anti-TB agents.
Acknowledgments
This research was funded by the Indian Council of Medical Research, New Delhi. The authors thank Schrodinger Inc. for providing software to carry out molecular modeling studies. The authors are grateful to AICTE, New Delhi, DST, New Delhi, and Manipal University, Manipal, for providing necessary facilities to carry out the research work.
Disclosure
The authors report no conflicts of interest in this work.
References
Global Tuberculosis Report. Geneva: World Health Organization; 2014. | ||
Green KD, Garneau-Tsodikova S. Resistance in tuberculosis: what do we know and where can we go? Front Microbiol. 2013;4:208. | ||
Riccardi G, Pasca MR. Trends in discovery of new drugs for tuberculosis therapy. J Antibiot. 2014:67:655–659. | ||
Payne DJ, Warren PV, Holmes DJ, Ji Y, Lonsdale JT. Bacterial fatty-acid biosynthesis: a genomics-driven target for antibacterial drug discovery. Drug Discov Today. 2001;6:537–544. | ||
Lamichhane G. Novel targets in M. tuberculosis: search for new drugs. Trends Mol Med. 2001;17:25–33. | ||
Sullivan TJ, Truglio JJ, Boyne ME, et al. High affinity InhA inhibitors with activity against drug-resistant strains of Mycobacterium tuberculosis. ACS Chem Biol. 2006;1:43–53. | ||
Heath RJ, Yu YT, Shapiro MA, Olson E, Rock CO. Broad spectrum antimicrobial biocides target the FabI component of fatty acid synthesis. J Biol Chem. 1998;273:30316–30320. | ||
Tipparaju SK, Mulhearn DC, Yang S, et al. Design and synthesis of novel aryl ether inhibitors of the Bacillus anthracis enoyl-ACP reductase. ChemMedChem. 2008;3:1260–1268. | ||
Tipparaju SK, Joyasawal S, Forrester S, et al. Design and synthesis of 2-pyridones as novel inhibitors of the Bacillus anthracis enoyl–ACP reductase. Bioorg Med Chem Lett. 2008;18:3565–3569. | ||
Takahata S, Iida M, Osaki Y, et al. AG205, a novel agent directed against FabK of Streptococcus pneumoniae. Antimicrob Agents Chemother. 2006;50:2869–2871. | ||
Takahata S, Iida M, Yoshida T, et al. Discovery of 4-Pyridone derivatives as specific inhibitors of enoyl-acyl carrier protein reductase (FabI) with antibacterial activity against Staphylococcus aureus. J Antibiot. 2007;60:123–128. | ||
Kitagawa H, Ozawa T, Takahata S, Iida M, Saito J, Yamada M. Phenyl imidazole derivatives of 4-pyridone as dual inhibitors of bacterial enoyl-acyl carrier protein reductases FabI and FabK. J Med Chem. 2007;50:4710–4720. | ||
McLeod R, Muench SP, Rafferty JB, et al. Triclosan inhibits the growth of Plasmodium falciparum and Toxoplasma gondii by inhibition of apicomplexan Fab I. Int J Parasitol. 2001;31:109–113. | ||
Wang LQ, Falany CN, James MO. Triclosan as a substrate and inhibitor of 3′-phosphoadenosine-5′-phosphosulfate-sulfotransferase and UDP-glucuronosyl transferase in human liver fractions. Drug Metab Dispos. 2004;32:1162–1169. | ||
Thomas CA, Kar SS, Bairy I, Bhat VG, Shenoy VP, Shenoy GG. Synthesis and evaluation of antitubercular activity of novel diphenyl ether derivatives. Ind Glob J Pharm Sci. 2015;5:19–25. | ||
Thomas CA, Kar SS, Bairy I, Bhat VG, Shenoy VP, Shenoy GG. Design, synthesis and evaluation of antitubercular activity of triclosan analogues. Arabian J Chem. 2015 (accepted). Available from: http://dx.doi.org/10.1016/j.arabjc.2015.09.003 | ||
Sivaraman S, Sullivan TJ, Johnson F, et al. Inhibition of the bacterial enoyl reductase FabI by triclosan: a structure-reactivity analysis of FabI inhibition by triclosan analogues. J Med Chem. 2004;47:509–518. | ||
Evans DA, Katz JL, West TR. Synthesis of diaryl ethers through the copper-promoted arylation of phenols with arylboronic acids. An expedient synthesis of thyroxine. Tetrahedron Lett. 1998;39:2937–2940. | ||
Abdel Hafez OM, Ahmed MK, Haggag EE. Synthesis of some potentially bioactive compounds from visnaginone. Molecules. 2001;6:396–405. | ||
Ellis JE, Davis EM, Brower PL. A novel and practical method for hydrocyanation of chalcones. Org Pro Res Dev. 1997;1:250–252. | ||
Katritzky AR, Pilarski B, Urogdi L. Efficient conversion of nitriles to amides with basic hydrogen peroxide in dimethyl sulfoxide. Synthesis. 1989;12:949–952. | ||
Gillmore A, Lauret C, Roberts SM. A route to the structure proposed for puetuberosanol and approaches to the natural products marshrin and phebalosin. Tetrahedron. 2003;59:4363–4375. | ||
Leonard B, Coronel J, Siedner M. Inter- and intra-assay reproducibility of microplate alamar blue assay results for isoniazid, rifampicin, ethambutol, streptomycin, ciprofloxacin, and capreomycin drug susceptibility testing of Mycobacterium tuberculosis. J Clin Microbiol. 2008;46:3526–3529. | ||
Collins L, Franzblau SG. Microplate alamar blue assay versus BACTEC 460 System for high-throughput screening of compounds against Mycobacterium tuberculosis and Mycobacterium avium. Antimicrob Agents Chemother. 1997;41:1004–1009. | ||
Morgan DML. Tetrazolium (MTT) assay methods in molecular biology for cellular viability and activity. Polyamine Protocols. 1998;79:179–182. | ||
Singh M, Sasi P, Rai G, Gupta VH, Amarapurkar D, Wangikar PP. Studies on toxicity of antitubercular drugs namely isoniazid, rifampicin, and pyrazinamide in an in vitro model of HepG2 cell line. Med Chem Res. 2011;20:1611–1615. | ||
Sundermann VM, Knasmuller S, Wu XJ, Darroudi F, Kassie F. Use of a human- derived liver cell line for the detection of cytoprotective, antigenotoxic and cogenotoxic agents. Toxicology. 2004;198:329–340. | ||
Lipinski CA. Drug-like properties and the causes of poor solubility and permeability. J Pharmacol Toxicol Methods. 2000;44:235–249. | ||
Suite 2010: Maestro, version 9.2, Schrödinger, LLC, New York, NY, 2010. | ||
Kuo MR, Morbidoni HR, Alland D. Targeting tuberculosis and malaria through inhibition of enoyl reductase: compound activity and structural data. J Biol Chem. 2003;278:20851–20859. | ||
Lombardo F, Shalaeva MY, Tupper KA, Gao F, Abraham MH. ElogPoct: a tool for lipophilicity determination in drug discovery. J Med Chem. 2000;43:2922–2928. | ||
Wiczling P, Markuszewski MJ, Kaliszan R. Determination of pKa by pH gradient reversed-phase HPLC. Anal Chem. 2004;76:3069–3077. | ||
Kaliszan R, Haber P, Baczek T, Siluk D, Valko K. Lipophilicity and pKa estimates from gradient high-performance liquid chromatography. J Chromatogr A. 2002;965:117–127. | ||
Hollósy F, Valkó K, Hersey A, Nunhuck S, Kéri G, Bevan C. Estimation of volume of distribution in humans from high throughput HPLC-based measurements of human serum albumin binding and immobilized artificial membrane partitioning. J Med Chem. 2006;49:6958–6971. | ||
Rowley M, Kulagowski JJ, Watt AP, et al. Effect of plasma protein binding on in vivo activity and brain penetration of Glycine/NMDA receptor antagonists. J Med Chem. 1997;40:4053–4068. | ||
Rotroff DM, Wetmore BA, Dix DJ, et al. Incorporating human dosimetry and exposure into high-throughput in vitro toxicity screening. Toxicol Sci. 2010;117(2):348–358. | ||
Hill JR. In vitro drug metabolism using liver microsomes. Curr Prot Pharmacol. 2003;7.8.1–7.8.11. | ||
Mammalian Liver Microsomes Guidelines for Use. TF000017 Rev 1.0, 1, BD Biosciences. Available from: http://www.industrycortex.com/datasheets/profile/759758262/manual-tf000017-rev-1.0-mammalian-liver-microsomes-guidelines-for. Accessed April 12, 2014. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.