")
Back to Archived Journals » Antibody Technology Journal » Volume 4
Rapid detection of different human anti-HCV immunoglobulins on electrical biochips
Authors Blohm L, Püttmann C, Holz S, Piechotta G, Albers J, Dammers C, Kleines M, Krüttgen A, Melmer G, Nähring J, Barth S, Nebling E
Received 2 January 2014
Accepted for publication 18 March 2014
Published 26 June 2014 Volume 2014:4 Pages 23—32
DOI https://doi.org/10.2147/ANTI.S54763
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Lars Blohm,1,2,§ Christiane Püttmann,3,§ Simone Holz,1 Gundula Piechotta,1,2 Jörg Albers,1,2 Christina Dammers,4–6 Michael Kleines,7,8 Alexander Krüttgen,8,9 Georg Melmer,2,10 Jörg Nähring,2,5 Stefan Barth,3,5,§ Eric Nebling1,2,§
1Department of Biotechnical Microsystems, Fraunhofer Institute for Silicon Technology, Itzehoe, Germany; 2POCDIA GmbH, Itzehoe, Germany; 3Department of Experimental Medicine and Immunotherapy, Institute for Applied Medical Engineering, Uniklinik RWTH Aachen, Germany; 4Institute of Biology VII, Molecular Biotechnology, RWTH Aachen University, Aachen, Germany; 5Department of Pharmaceutical Product Development, Fraunhofer Institute for Molecular Biology and Applied Ecology, Aachen, Germany; 6Institute of Complex Systems (ICS-6), Forschungszentrum Jülich, Jülich, Germany; 7Department of Hygiene, Microbiology, Social Medicine, Division of Virology, Medical University IBK, Innsbruck, Austria; 8Medizinisches Versorgungszentrum Dr Stein und Kollegen, Mönchengladbach, Germany; 9Department of Medical Microbiology, Division of Virology, Uniklinik RWTH Aachen, Germany; 10Pharmedartis GmbH, Aachen, Germany
§These authors contributed equally
Abstract: The detection of hepatitis C virus (HCV) in the blood of patients is currently based on immunological assays (enzyme-linked immunosorbent assay [ELISA] and recombinant immunoblot assay) that use different HCV epitopes to detect anti-HCV antibodies, and these tests usually require laboratories and trained personnel. The ELISA-based systems are also time consuming. Portable diagnostic devices offering rapid test results would therefore be advantageous in the field of medical care. To facilitate the fast and reliable diagnosis of HCV, we used a miniaturized automated system based on a cartridge with an integrated electrical biochip for the decentralized detection of anti-HCV antibodies against the Core, NS3, and NS4A proteins. This system allows the detection of virus-specific antibodies in 2 µL of serum or whole blood within 15 minutes using an ELISA directly on a gold electrode array containing HCV proteins as the capture antigen. The sensitivity of this system is comparable with standard microtiter plate ELISAs, but the duration of the novel assay is 5%–6% that of standard ELISAs.
Keywords: ELISA, point-of-care, cartridge, lab-on-chip
Introduction
More than 150 million people are chronically infected with hepatitis C virus (HCV) and thus have an increased risk of liver cirrhosis and liver cancer.1 The virus causes 3–4 million new infections per year worldwide, but 40% of patients achieve full recovery,2 and recent advances in antiviral therapy ensure that the mortality rate is approximately 0.2% (350,000 people per year).1 Unlike hepatitis A virus and hepatitis B virus, there is no vaccine available for HCV.
The diagnosis of HCV infection is important because most infections are caused by blood-to-blood contact with undiagnosed and symptom-free carriers, or by contaminated blood transfusions or transplants.3,4 Intravenous or nasal drug use and tattooing or acupuncture with unsafe materials and equipment are currently the main routes of transmission.5 An easy-to-use and inexpensive detection system would therefore be advantageous and valuable for routine HCV testing in all blood banks around the world. The determination of HCV viral load and genotype is also important because it helps to identify the most efficient therapeutic approach, but this currently requires different polymerase chain reaction-based diagnostic systems.6
Current state-of-the-art antibody (Ab)-based methods can identify patients carrying HCV. First, an enzyme immunoassay (EIA) or chemiluminescence immunoassay is carried out to detect anti-HCV Abs in the blood. To confirm this first diagnosis, Centers for Disease Control and Prevention guidelines initially recommended an additional recombinant immunoblot assay (RIBA), but more recent guidelines recommend the detection of viral ribonucleic acid (RNA).7 Furthermore, the Chiron RIBA HCV 3.0 Strip Immunoblot Assay is no longer available for HCV testing.8
Since the discovery of HCV in 1989, four generations of serological test systems have improved the sensitivity and specificity of EIA-based HCV diagnosis.9 Current third-generation assays include different HCV proteins, such as Core (c22-3), NS3 (c33p), NS4 (c100-3), and NS5, thus achieving an average sensitivity of 98%.10 One major drawback of these test systems is the number of false-negative results, reflecting late Ab responses, and false-positive results caused by other issues.11 The error rate has been reduced in the most recent fourth-generation assays, which detect immunoglobulin (Ig) G as well as IgM Ab responses.10,11 These improvements allow EIA-based systems to be used reliably in low-risk environments such as blood banks, but will not entirely remove the need for RNA-based test systems because RNA can be used as a direct marker of viral replication and the discrimination between active and cured infections.12
An alternative to standard colorimetric enzyme-linked immunosorbent assays (ELISAs) is the cyclovoltammetric detection of HCV antigens13 or the electrical biochip platform that uses “single electrode redox cycling” and detects anti-Core antibodies in the blood.14 In combination with advanced lab-on-chip functionality, this technology has now been improved by the addition of two additional antigens to the biochip. Here we describe a demonstrator that integrates the HCV proteins Core, NS3, and NS4A, and measures an anti-HCV Ab response in diluted serum or whole blood samples in less than 15 minutes. A special biochip cartridge allows direct sample collection and its dilution in a fully automated analysis system. The portability of the system makes it suitable as a candidate to improve diagnostics in small hospitals or medical practices, especially in the field of point-of-care testing.
Material and methods
HCV serum samples
Tests were carried out on 71 anonymized patient serum samples with a defined HCV status, according to the regulations of the University Hospital Aachen, Aachen, Germany. All samples were previously characterized using the ARCHITECT® anti-HCV assay (Abbott Laboratories, Abbott Park, IL, USA), identifying 39 sera as anti-HCV-negative and 32 as anti-HCV-positive. The samples were inactivated with 1% Tween-20 at 37°C for 4 hours by diluting 1:2 with phosphate-buffered saline with Tween-20 (PBS-T). For ELISA, the blood samples of HCV-negative healthy individuals (C1–3) were analyzed and the C2 sample was used as a reference.
Reagents
All chemicals for antigen preparation and ELISA measurements were purchased from Carl Roth GmbH + Co, KG (Karlsruhe, Germany). 2,2′-azino-bis(3-ethylbenzthiazoline-6-sulphonic acid) was purchased from Roche Applied Science (Mannheim, Germany), and the rabbit anti-human IgG peroxidase conjugate (Fab specific) was purchased from Sigma-Aldrich GmbH (Taufkirchen, Germany).
PBS pH 7.4, Tween-20, trehalose, MgCl2, 4-aminophenyl-β-D-galactopyranoside (p-APG), and bovine serum albumin (BSA) were purchased from Sigma-Aldrich GmbH. Human IgG was obtained from Fitzgerald Industries (Concord, MA, USA), and goat anti-human IgG β-galactosidase conjugate was purchased from Southern Biotech (Birmingham, AL, USA).
Preparation of antigens
The Core antigen was produced as previously described.14 For NS3 and NS4A, a consensus sequence was generated by amino acid alignment (see supplementary material) and synthesized by Mr. Gene GmbH (Regensburg, Germany). The sequences were transferred to vector pET32a+ (Merck Chemicals, Nottingham, UK) using the enzymes NcoI-HF and NotI-HF (New England Biolabs GmbH, Frankfurt am Main, Germany) and expressed as thioredoxin (Trx)-fusion proteins in Escherichia coli strain Rosetta2 DE3 pLysS (Merck Chemicals). The cells were cultivated in a BioFlo 110 fermenter (7 l, New Brunswick Scientific, Enfield, CT, USA) using synthetic medium, and were lysed by sonication in 8 M urea buffer, pH 8.0. After overnight incubation, the cellular debris was removed by centrifugation (40,000 g, 90 minutes), the pellet was processed again as described previously, and the protein was purified from the pooled supernatants using a ÄKTA fast protein liquid chromatography system and a 4 mL Ni-TED column (Macherey-Nagel GmbH and Co, KG, Düren, Germany). The protein concentration was determined using the BCA Protein Assay Kit (Thermo Fisher Scientific, Pierce, Rockford, IL, USA) with a BSA or lysozyme standard for Trx-NS3 and Trx-NS4A, respectively. The integrity of the proteins was confirmed by mass spectrometry.
Microtiter plate ELISA
The ELISA was carried out as previously described14 after coating the plates with 50 ng/well of the full-length recombinant HCV Core protein, 100 ng/well of Trx-NS3, or 100 ng/well of Trx-NS4A. After blocking with 1% (w/v) BSA, putative HCV-positive sera were diluted 1:200 in PBS and applied to the plate. Bound anti-HCV antibodies were detected using a rabbit anti-human IgG peroxidase conjugate (Fab specific, diluted 1:5,000). After incubation for 45 minutes, 2,2′-azino-bis (3-ethylbenzthiazoline-6-sulphonic acid) was added and the reaction was monitored by spectrophotometry at 405 nm. The total test time was 4 hours.
Biochip fabrication and integration
The disposable silicon-based chips were manufactured on the industrial semiconductor production line at the Fraunhofer Institute for Silicon Technology, Itzehoe, Germany.14–16 The new layout was based on chip dimensions of 8×10 mm and the production was modified to process 8-inch silicon wafers. The chips carried 16 unstructured gold electrode positions, each with a diameter of 350 μm. The electrical interface of the chips was modified to form one row of contact pads with a pitch of 0.5 mm, allowing the use of commercial connectors.
The wafers were thermally oxidized to a depth of 650 nm for insulation. The gold electrodes comprised vacuum-deposited stacks of 20 nm tantalum as a bonding agent and a final 150 nm upper gold layer. A gold counter-electrode and an iridium oxide reference electrode were also integrated into the chip, structured by photolithography and lift-off technology. The conducting paths were passivated against the wet samples using a combination of 400 nm silicon nitride and a 2 μm structured photoresist. Only the electrodes and the contact pad areas were opened by dry etching. This combination of the hydrophilic electrode surface and surrounding hydrophobic passivation optimized the dispensing and immobilizing of the capture molecules.
The antigens were applied to the gold electrodes using a piezo-driven microdispensing device from GeSiM GmbH (Großerkmannsdorf, Germany). Human IgG was used as a positive control (300 μg/mL in PBS) and BSA as a negative control (100 μg/mL in PBS). The antigens were spotted on to the chip at three positions each, each deposit comprising 26 droplets representing 10 nL per array electrode. The HCV Core antigen (10 μg/mL in PBS with 3 M urea) was deposited at positions 4 and 7, the NS3 antigen (280 μg/mL in PBS with 3 M urea) at positions 5 and 8, and the NS4A antigen (80 μg/mL in PBS with 3 M urea) at positions 6 and 9. The antigens were initially diluted 1:2.5 in PBS. Positions 1–3 were designated the “positive control” and positions 10–12 the “negative control”, because they were used for mathematical standardization. The immobilization of these capture molecules was achieved by thiol–gold interaction and hydrophobic adsorption. The spotted chips were incubated for 2.5 hours at room temperature in a humidity chamber. After washing with PBS-T, the chips were blocked with BSA (0.5 mg/mL in PBS) for 20 minutes, washed with deionized water, and dried under a vacuum. The chips were designed to be disposable and were discarded after a single assay.
The spotted biochips were housed in a polycarbonate microfluidic cartridge. A flow-through cell allowed the samples and reagents to be passed successively over the biochips in the cartridge. The cartridge also contained a sample collector and a dilution reservoir together with fluid channels and five ports connecting the biochips to the measurement device. The latter was designed to incorporate a disposable reagent container (for buffer, enzyme conjugate, substrate, and waste) and a biochip cartridge adapter to facilitate automated analysis via two miniaturized peristaltic pumps (Prolatec GmbH, Radebeul, Germany) and six pinch solenoid valves (Sirai®, Bussero, Italy). The total size of the measurement device was 144×250×244 mm. An integrated multichannel potentiostat was used to change the reaction potential of each electrode between 200 mV and –350 mV and to measure the resulting currents at a 5 pA resolution with a range of ±200 nA. The electronic components controlled the assay procedure and also the pump, valves, and temperature regulation unit. The biochip cartridge adapter allowed the biochip to be connected simultaneously to the multichannel potentiostat and the controlled heating unit, as well as connecting the cartridge to the fluidic system of the measurement device.
The assay was automatically controlled by the proprietary software “MCDDE” (Fraunhofer Institute of Silicon Technology, Itzehoe, Germany), which regulated the action of valves, pumps, and heating. The resulting data were analyzed using OriginPro 8G (OriginLab Corporation, Northampton, MA, USA).
Analysis on electrical biochips
The 15-minute HCV test on the electrical biochip was carried out by pipetting 2 μL of serum into the cartridge sample collector. Once inserted into the measurement device, the sample was diluted automatically 1:250 with buffer. Whole blood samples were taken from a droplet on the fingertip into the cartridge sample collector by capillary transfer. Spotted biochips were already integrated into the cartridge, which was connected to the fluidic, electrical, and thermal systems. All steps were carried out with the chip temperature set to 38°C.
Goat anti-human β-galactosidase was diluted 1:400 in PBS-T containing MgCl2, trehalose, and BSA, and the substrate p-APG was provided at a concentration of 1 mg/mL. The diluted samples were passed across the chip and incubated for 8 minutes to allow anti-HCV antibodies to bind the HCV target antigens on the electrodes. The bound antibodies (and human IgG control) were then enzyme-labeled for a further 3 minutes, before the position-specific generation of the electrochemically active substrate 4-hydroxyaniline from p-APG was used to provide the readout by single electrode redox cycling in a stop flow mode.14 A further 4 minutes were necessary for all dilution, reaction, and washing steps.
The production of 4-hydroxyaniline was measured by switching the gold electrodes on the biochip between +200 mV and –350 mV with a frequency of 1 Hz. Current measurements were taken 120 milliseconds after switching, resulting in position-specific increasing oxidation and reduction currents. The magnitude of these oxidation and reduction currents was summarized to create a current slope for each chip position, beginning 1 second after stop flow mode. Finally, linear regression analysis of a 6-second time period yielded target concentration-dependent slopes in the nA/min range.
A mean value was generated for each triplicate (positive and negative control) or duplicate (target antigens) on each individual chip. A standardized procedure was carried out by setting the negative control values (chip position 10–12) to 0, then adjusting the positive control and HCV values accordingly. The positive control was set to 1,000 nA/min and the HCV signals were normalized relative to this value.
Results and discussion
Design of proteins
The efficacy of the current anti-Core Ab detection system14 was enhanced by integrating two new proteins on to the biochip. All published sequences for NS3 and NS4A were aligned, and a genotype-independent protein sequence was designed and synthesized. The location of these proteins in the HCV polyprotein is shown in Figure 1. To enhance the production of these proteins in E. coli, they were expressed as Trx–tag fusions.17 This was important because both proteins contain N-terminal hydrophobic stretches that may reduce expression levels, as shown for the HCV glycoprotein E2.18 We found that the Trx component of the fusion protein does not interfere with the test system, because the background level does not increase when compared with the results for the Core protein alone. As discussed for the HCV Core protein, both NS proteins were stored in 8 M urea buffer after purification and were used for the analysis of patient sera by standard ELISA and biochip measurements.
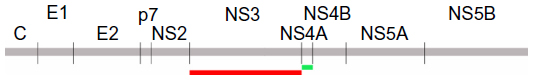 | Figure 1 The location of NS3 and NS4A proteins within the hepatitis C virus polyprotein. The complete NS3 (marked in red, residues 1027–1658) and NS4A (marked in green, residues 1658–1712) proteins were expressed as thioredoxin fusions to enhance the recombinant protein yields. |
Microtiter plate ELISA
Standard ELISAs were used to confirm the suitability and optimize the binding conditions before integrating them with the biochip. The two NS proteins were coated at twice the concentration used for the Core protein, but this still fell within the normal range for coating ELISA plates, because concentrations of 0.2 ng to 1 μg have been reported for non-Ab proteins.19–21 In addition to 71 patient samples that had tested HCV-positive or HCV-negative, three serum samples from healthy, HCV-negative individuals (C1–3) were analyzed and served as a negative control group for normalization. Because all the patient samples were tested in triplicate, a maximum of 20 different measurements could be taken on one ELISA plate. In order to allow the comparison of all results, samples C1–3 were analyzed on each plate. The serum with the medium extinction (C2) was used to normalize the patient data. The cutoff to discriminate HCV-positive and HCV-negative patients was set to 2 (Figure 2).
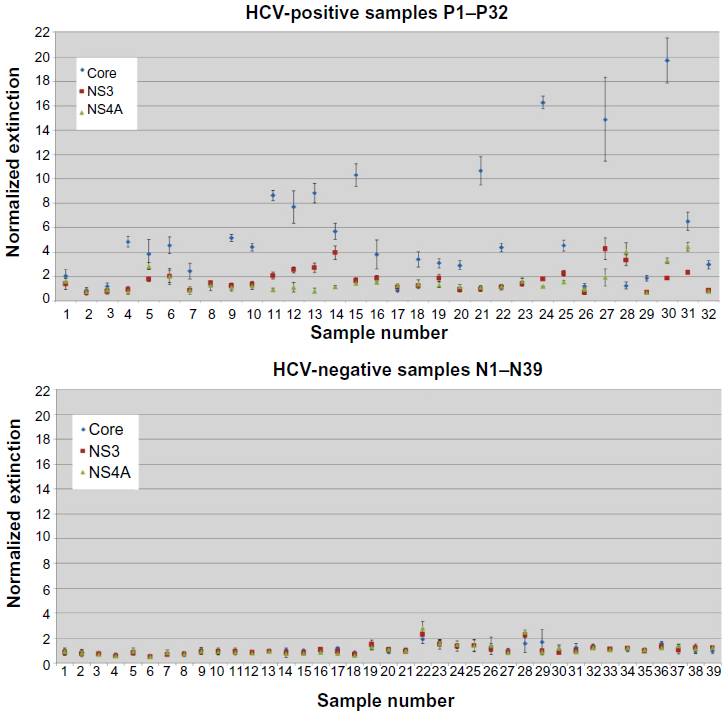 | Figure 2 All 71 patient serum samples were measured by standard enzyme-linked immunosorbent assay. The upper graph shows the normalized data for the 32 hepatitis C virus (HCV)-positive samples, and the lower graph shows the equivalent for the 39 HCV-negative samples. All measurements were taken twice in triplicates. The cutoff value was set to 2. Standardized target signals below this value were set as negative, and above this value as positive. Borderline signals are defined as those samples that span the cutoff value. |
Previously, we achieved the efficient detection of HCV-positive serum samples using the Core protein.14 The addition of two HCV-NS proteins should increase the sensitivity of the detection system. Among the 32 samples that previously tested HCV-positive, 78.1% (25/32) gave a positive ELISA result based only on anti-Core antibodies. The incorporation of data from NS3 and NS4A increased the positive test results from the infected patients to 81.3% (26/32) by eliminating the formerly false-negative result from sample P28. The borderline signal of sample P29 could not be improved by including data from the two NS proteins. The number of false-positive results did not increase. Two patients (N22 and N28) previously identified as HCV-negative now tested positive when the NS proteins were included, and two further patients (N26 and N29) showed a borderline signal. Therefore, the standard ELISA achieved an overall accuracy of 88.7% (63/71). The reproducibility, based on samples producing one positive and one negative result, was 93.0% (five inconclusive samples among 71 tested).
Analysis on electrical biochips
The HCV Core, NS3 and NS4A antigens, together with the positive and negative controls, were spotted at specific positions on the biochip using a piezo-driven spotting device. Figure 3A shows an individual unspotted biochip, as produced in the Fraunhofer Institute for Silicon Technology cleanroom environment, whereas Figure 3B shows a biochip spotted with 10 nL sample droplets at each position. The spotted proteins were immobilized by thiol–gold interaction and hydrophobic adsorption. After incubation and protein binding, the blocked and washed chips were housed in the biochip cartridge, where they remained stable for more than 12 months at 4°C and for several weeks at room temperature. This allows the housed chips to be distributed to different laboratories without a cold chain.
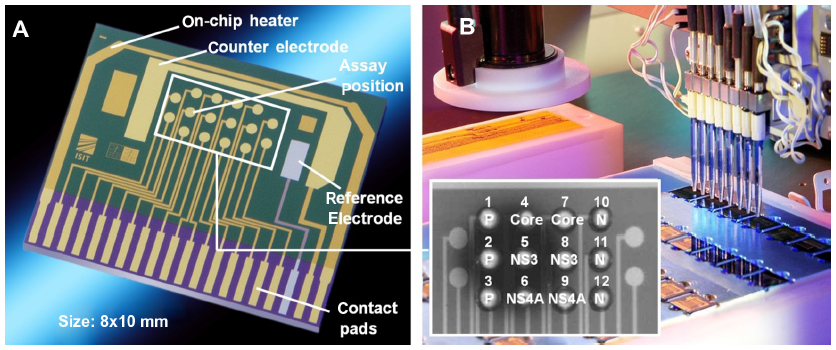 | Figure 3 (A) The Fraunhofer Institute for Silicon Technology biochip is equipped with an iridium oxide reference electrode, a gold counter-electrode, and 16 gold array working electrodes. The chip size is 8×10 mm2, and each working electrode is 350 μm in diameter. (B) The piezo-driven microdispenser is shown in the background, and spotted working electrodes on the chip are shown at the front. The positive control (P, positions 1–3) and the negative control (N, positions 10–12) were spotted in triplicate, whereas Core (positions 4, 7), NS3 (positions 5, 8), and NS4A (positions 6, 9) were spotted in duplicate. |
The portable, fully automated biochip system was developed for point-of-care diagnostics. It allows the straightforward collection of microliter-sized blood samples combined with automated direct sample dilution within the disposable biochip cartridge. The latter is equipped with the inserted biochip, a sample collector, and a reservoir for automated sample dilution. In our comparability measurements, 2 μL of each serum sample was pipetted into the collector, whereas other commercially available EIA-based systems require a sample volume of 20–100 μL.9 In practical use, pricking the fingertip with a sterile needle allows a 2 μL sample to be collected by capillary transfer.
The collector was sealed with a female Luer taper and the cartridge inserted into the measurement device. Pushing the cartridge into the cartridge adapter resulted in the electrical contacts on the biochip, connecting to the heater and the five fluidic ports of the cartridge in parallel. A separate pump diluted the 2 μL sample by pumping 500 μL of buffer through the collector within the sample dilution reservoir. Because the serum samples are prediluted 1:2, the final concentration of the sample was therefore 1:500. Pumping the diluted sample, enzyme conjugate, substrate, and all intermediate washing buffers was achieved automatically by the microfluidic components of the device. The incubation time and pump rates were modified to determine the optimized assay program with duration less than 15 minutes, which is faster than any other comparable commercial test system.9
The electrochemical detection method single electrode redox cycling and the related signal evaluation achieved sensitive and reproducible results (Figure 4). The microtiter plate ELISA described previously, based on 39 HCV-negative and 32 HCV-positive serum samples, was repeated with the point-of-care biochip system for comparability tests. Each serum sample was measured in duplicate to test the reproducibility of automated sample dilution. In Figure 4A, five measurements are picked out for detailed explanation. The positive control positions 1–3 showed homogeneous signals in each triplicate, as well as comparisons from chip to chip. The average signal was ~2,600 nA/min regardless of the sample. The negative control positions 10–12 resulted in signals marginally below 0 in each triplicate, regardless of the individual chip, indicating the absence of nonspecific binding. The negative control serum N4 yielded signals below 0 at the Core, NS3, and NS4A positions, demonstrating the absence of nonspecific binding at the target positions and a negative classification in the biochip assay. The weak positive samples P3 and P28 showed signals at the Core positions in the case of P3 and at the NS3 and NS4A positions in the case of P28, whereas the negative positions remained below 0. Serum P4 was recorded as positive, reflecting the 600 nA/min signal at the Core positions. The average signal values for serum P27 were 220 nA/min for NS4A, 900 nA/min for NS3, and 2,150 nA/min for Core, thus identifying this sample as a strong positive. The semiquantitative presentation of results is therefore practicable using this system.
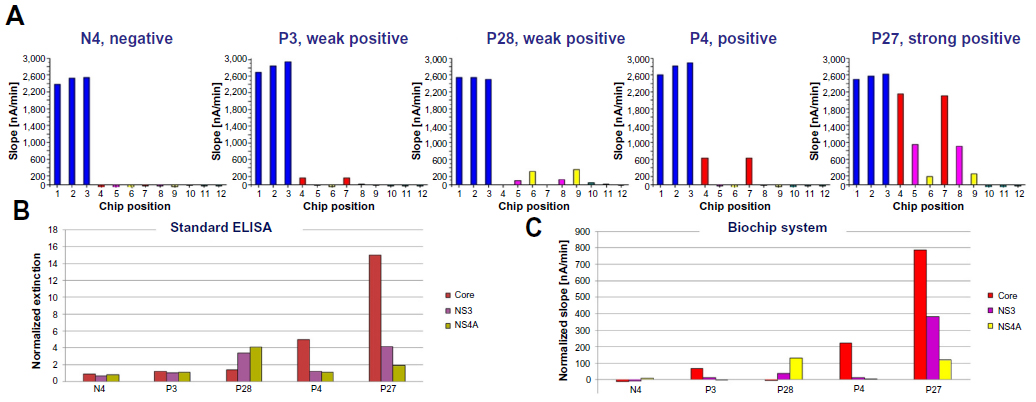 | Figure 4 Measurements of five individual serum samples. (A) Signals obtained at the different chip positions for each patient, as well as the control measurements: positive control positions 1–3 (blue), Core positions 4 and 7 (red), NS3 positions 5 and 8 (pink), NS4A positions 6 and 9 (yellow), negative control positions 10–12 (green). The assay time was 15 minutes. To compare the overall measurement of the enzyme-linked immunosorbent assay (ELISA) and biochip results, the data for the five selected patients are summarized in (B) and (C), respectively. |
Comparing the data for the same patients obtained by ELISA (Figure 4B) and biochip analysis (Figure 4C) revealed similar outcomes. Nevertheless, the biochip assay is much easier to interpret because there is a greater difference between positive and negative signals compared with the ELISA. In particular, the weak positive signal for sample P3 can be identified more clearly as an HCV-infected sample in the biochip assay.
The standardization of target signals relative to positive control signals also results in a better interassay reproducibility. The 71 samples were measured and normalized using the biochip system (Figure 5). The cutoff value was set to 20 nA/min. Standardized target signals below this value were set as negative and values above were set as positive. Using the point-of-care biochip system, three HCV-positive samples were identified as false negatives against all targets (P8, P17, and P23) – sample P8 was false negative in one measurement, whereas samples P17 and P23 were false negative in both measurements (Figure 5A). P17 and P23 were false negatives in the microtiter plate ELISA too, indicating the absence of antibodies against the three tested HCV proteins. In contrast, the Abbott ARCHITECT system identified these sera as anti-HCV positive, possibly because the Abbott system detects not only IgG antibodies directed against HCV but also IgM antibodies that are generated earlier in the infection. The incorporation of NS3 and NS4A data allowed sample P28 to test positive in the microtiter plate ELISA and the biochip assay. Two of the HCV-negative samples were detected as false positives in one of two measurements (N22 and N27), and N27 recorded an NS4A value of 20 nA/min (Figure 5B). Overall, we recorded four false results among 71 samples, which is equivalent to a test correctness of 94.4%. In comparison, the accuracy of the microtiter plate ELISA was 88.7% (8/71). The biochip therefore achieved greater correctness than the microtiter plate ELISA, in addition to a 16-fold saving in the duration of the assay. The automated microfluidic assay procedure also uses low sample and reagent volumes. In most cases, the double measurement of a serum sample resulted in comparable signals, indicating that the automated sample dilution was highly repeatable (Figure 5A).
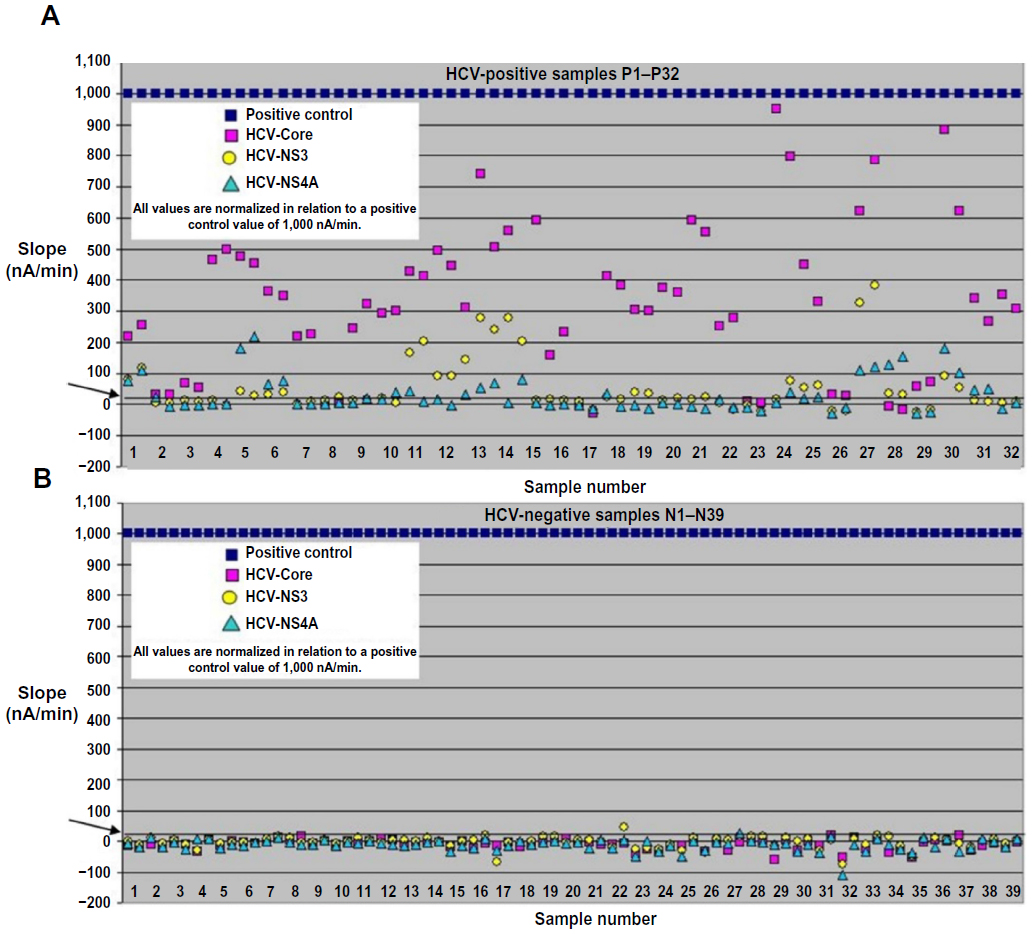 | Figure 5 All 71 serum samples measured with the biochip system. (A) Normalized data for the 32 hepatitis C virus (HCV)-positive samples shown with each measurement in duplicate. (B) Normalized data for the 39 negative samples shown with each measurement in duplicate. The line at 20 nA/min represents the cutoff value (arrow). Standardized target signals below this value are set as negative, and above this value as positive. |
The Abbott ARCHITECT system achieved a reproducibility of 99.9% and an assay correctness of 95.0% among more than 1,000 samples by detecting anti-HCV IgM and IgG. The assay time for a single sample is ~30 minutes, thus our biochip system was much faster.
Measuring whole blood samples
For point-of-care applications it is essential that no further sample preparation is needed after collection. Therefore, we tested our cartridge-based biochip system with realistic whole blood samples. HCV-negative blood was sampled by drawing 2 μL into the cartridge from a droplet on a fingertip (Figure 6A). After closing the collector, the cartridge was inserted into the measurement device (Figure 6B). The test was carried out as described previously for serum samples, using HCV-negative serum as a negative control (Figure 6C) and 2 μL of HCV-negative whole blood spiked with 1 μL of positive serum P24 as a positive serum control (Figure 6C). The presence of red blood cells did not affect the performance of the electrical biochips or the automated analysis in the cartridge.
 | Figure 6 (A) Acquisition of a 2 μL whole blood sample (negative or spiked) directly using the cartridge sample collector. (B) Point-of-care biochip device with inserted cartridge and open reagent container. (C) Measurements for signal comparison between serum and spiked whole blood. Positive control positions 1–3 (blue), Core positions 4 and 7 (red), NS3 positions 5 and 8 (pink), NS4A positions 6 and 9 (yellow), negative control positions 10–12 (green). The assay time was 15 minutes. |
Conclusion
We have developed a point-of-care diagnostic system based on electrical biochips. The system allows the fully automated and sensitive detection of anti-HCV antibodies in serum or whole blood samples within 15 minutes. A robust assay design is combined with safe and direct sample collection into the cartridge, thus avoiding syringes, cannulas, and further pipetting steps. Red blood cells do not affect the signal intensity or the background level compared with serum tests. Only 2 μL of whole blood is needed to detect a putative HCV infection, and automated dilution by the system achieves reproducible results. The signal-to-noise ratio is better than in the microtiter plate ELISA, and the assay correctness of 94% fits within the given standard. The introduction of the NS3 and NS4A proteins improves the assay correctness by 3%, by eliminating a false-negative result. Our system therefore appears suitable for a point-of-care approach where only lancets and the biochip cartridge are required for taking whole blood samples. The robust platform saves time and reagent costs, and thus offers a safe, inexpensive, and portable alternative to current laboratory assays.
Acknowledgments
We thank Michael Küpper (IMB, RWTH Aachen University) for assistance with mass spectrometry and its evaluation, Thorsten Sieben (Uniklinik RWTH Aachen) and Severin Schmies (Fraunhofer IME, Aachen) for helping with the fermentation and purification of both NS proteins, and Dr Richard Twyman for critical reading of the manuscript.
Disclosure
The authors report no conflicts of interest in this work.
References
World Health Organization. Hepatitis C. Fact sheet No 164. Available from: http://www.who.int/mediacentre/factsheets/fs164/en/. Accessed March 27, 2014. | |
Burguete-Garcia AI, Conde-Gonzalez CJ, Jimenez-Mendez R, et al. Hepatitis C seroprevalence and correlation between viral load and viral genotype among primary care clients in Mexico. Salud Publica de Mexico. 2011;53 Suppl 1:S7–S12. | |
Attaullah S, Khan S, Khan J. Trend of transfusion transmitted infections frequency in blood donors: provide a road map for its prevention and control. J Transl Med. 2012;10:20. | |
Dominguez-Gil B, Andres A, Campistol JM, Morales JM. Should we be using kidneys from hepatitis C virus-infected donors? Curr Opin Nephrol Hypertens. 2011;20(6):599–604. | |
Calvaruso V, Craxi A. 2011 European Association of the Study of the Liver hepatitis C virus clinical practice guidelines. Liver Int. 2012; 32 Suppl 1:2–8. | |
Chevaliez S, Pawlotsky JM. Diagnosis and management of chronic viral hepatitis: antigens, antibodies and viral genomes. Best Pract Res Clin Gastroenterol. 2008;22(6):1031–1048. | |
Centers for Disease Control and Prevention. Testing for HCV infection: an update of guidance for clinicians and laboratorians. MMWR. 2013; 62(18):362–365. | |
Center for Biologics Evaluation and Research. Information for Blood Establishments: Discontinuation of CHIRON® RIBA® HCV 3.0 SIA (RIBA). Online: US Food and Drug Administration (FDA). 2013. | |
Cabezas-Fernandez M, Cabeza-Barrera M. Introduction of an automated system for the diagnosis and quantification of hepatitis B and hepatitis C viruses. Open Virol J. 2012;6:122–134. | |
Cao J, Chen Q, Zhang H, et al. Novel evolved immunoglobulin (Ig)-binding molecules enhance the detection of IgM against hepatitis C virus. PloS One. 2011;6(4):e18477. | |
Yuksel P, Caliskan R, Ergin S, et al. New approaches to in vitro diagnosis of hepatitis C infection a reason for post transfusion hepatitis: diagnostic value of determination of hepatitis C virus core antigen. Transfus Apher Sci. 2011;45(3):247–250. | |
Ember SW, Schulze H, Ross AJ, et al. Fast DNA and protein microarray tests for the diagnosis of hepatitis C virus infection on a single platform. Anal Bioanal Chem. 2011;401(8):2549–2559. | |
Dai X-F, Bai Y, Liu Z-M, Li M-J, Cai P-X. A HCV electrochemical immunosensor for the detection of serum sample. Journal of Instrumental Analysis. 2006;26(1):49–51. | |
Kraus S, Kleines M, Albers J, et al. Quantitative measurement of human anti-HCV Core immunoglobulins on an electrical biochip platform. Biosens Bioelectron. 2011;26(5):1895–1901. | |
Albers J, Grunwald T, Nebling E, Piechotta G, Hintsche R. Electrical biochip technology – a tool for microarrays and continuous monitoring. Anal Bioanal Chem. 2003;377(3):521–527. | |
Elsholz B, Worl R, Blohm L, et al. Automated detection and quantitation of bacterial RNA by using electrical microarrays. Anal Chem. 2006;78(14):4794–4802. | |
LaVallie ER, DiBlasio EA, Kovacic S, Grant KL, Schendel PF, McCoy JM. A thioredoxin gene fusion expression system that circumvents inclusion body formation in the E. coli cytoplasm. Biotechnology (N Y). 1993;11(2):187–193. | |
Liu J, Kong Y, Zhu L, Wang Y, Li G. High-level expression of the C-terminal hydrophobic region of HCV E2 protein ectodomain in E. coli. Virus Genes. 2002;25(1):5–13. | |
Jorissen H, Bektas N, Dahl E, et al. Production and characterisation of monoclonal antibodies against RAI3 and its expression in human breast cancer. BMC Cancer. 2009;9:200. | |
Kah O, Pontet A, Nunez Rodriguez J, Calas A, Breton B. Development of an enzyme-linked immunosorbent assay for goldfish gonadotropin. Biol Reprod. 1989;41(1):68–73. | |
Sreedevi C, Hafeez M, Subramanyam KV, Anand Kumar P, Chengalva Rayulu V. Development and evaluation of flow through assay for detection of antibodies against porcine cysticercosis. Trop Biomed. 2011;28(1):160–170. |
Supplementary materials
Consensus sequence NS3
N- -> C-terminal direction
APITAYAQQTRGLLGTIVTSLTGRDKNEVEGEVQVLSTATQTFLGTTINGVMWTVYHGAGSKTLAGPK GPVIQMYTNVDQDLVGWPAPPGAKSLTPCTCGSSDLYLVTRHADVIPARRRGDTRGSLLSPRPISTLK GSSGGPVLCPSGHAVGIFRAAVCTRGVAKALDFVPVESLETTMRSPVFTDNSTPPAVPQTYQVGHLH APTGSGKSTKVPAAYAAQGYKVLVLNPSVAATLGFGAYMSKAHGIDPNIRTGVRTITTGAPITYSTYGK FLADGGCSGGAYDIIICDECHSTDATTILGIGTVLDQAETAGARLVVLATATPPGSVTVPHPNIEEVALP TTGEIPFYGKAIPLELIKGGRHLIFCHSKKKCDELAKQLTSLGLNAVAYYRGLDVSVIPTSGDVVVCATD ALMTGFTGDFDSVIDCNVAVTQTVDFSLDPTFTIETTTVPQDAVSRSQRRGRTGRGRLGIYRYVSPGE RPSGMFDSVVLCECYDAGCAWYELTPAETTVRLRAYLNTPGLPVCQDHLEFWEGVFTGLTHIDAHFL SQTKQAGENFPYLVAYQATVCARAKAPPPSWDTMWKCLIRLKPTLHGPTPLLYRLGAVQNEVTLTHPI TKYIMTCMSADLEVVT
Consensus sequence NS4A
N- -> C-terminal direction
STWVLVGGVLAALAAYCLSVGSVVIVGRIILSGKPAVIPDREVLYQQFDEMEEC
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.