")
Back to Journals » International Journal of Nanomedicine » Volume 15
Pulmonary-Affinity Paclitaxel Polymer Micelles in Response to Biological Functions of Ambroxol Enhance Therapeutic Effect on Lung Cancer
Authors He W, Xiao W , Zhang X, Sun Y, Chen Y, Chen Q, Fang X, Du S, Sha X
Received 2 September 2019
Accepted for publication 14 January 2020
Published 4 February 2020 Volume 2020:15 Pages 779—793
DOI https://doi.org/10.2147/IJN.S229576
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 6
Editor who approved publication: Dr Linlin Sun
Wenxiu He,1,* Wenze Xiao,2,* Xiulei Zhang,1 Yali Sun,1 Yiting Chen,1 Qinyue Chen,1 Xiaoling Fang,1 Shilin Du,3 Xianyi Sha1,4
1Key Laboratory of Smart Drug Delivery, Ministry of Education, Center for Medical Research and Innovation, Shanghai Pudong Hospital, School of Pharmacy, Fudan University, Shanghai 201203, People’s Republic of China; 2Department of Rheumatology, Shanghai Pudong Hospital, Fudan University Pudong Medical Center, Shanghai 201399, People’s Republic of China; 3Department of Emergency Medicine, Zhongshan Hospital, Fudan University, Shanghai 200032, People’s Republic of China; 4The Institutes of Integrative Medicine of Fudan University, Shanghai 200040, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Xianyi Sha; Shilin Du Tel +86-21-51980072
Email [email protected]; [email protected]
Purpose: Cancer chemotherapy effect has been largely limited by cell autophagy and little drug accumulation at the action sites. Herein, we designed an intelligent strategy involving paclitaxel (PTX) polymer micelles in response to biological functions of ambroxol (Ax). The amphiphilic polymers polyethyleneglycol-polylactic acid (PEG-PLA) and Pluronic P105 were selected as nanocarriers to encapsulate PTX to form into lung affinity PEG-PLA/P105/PTX micelles. Ax which can up-regulate the secretion of pulmonary surfactant (PS) and inhibit autophagy was hired to change the microenvironment of the lung, thereby promoting the lung accumulation and increasing cell-killing sensitivity of the micelles.
Methods: The physical and chemical properties of the micelles were characterized including size, morphology, critical micellar concentration (CMC) and in vitro drug release behavior. The therapeutic effects of the combination regimen were characterized both in vitro and in vivo including study on Ax in promoting the secretion of pulmonary surfactant, in vitro cytotoxicity, cellular uptake, Western blotting, in vivo biodistribution, in vivo pharmacokinetics and in vivo antitumor efficacy.
Results: The PEG-PLA/P105/PTX micelles showed a particle size of 16.7 ± 0.5 nm, a nearly round shape, small CMC and sustained drug release property. Moreover, the in vitro results indicated that Ax could increase PS and LC3 protein secretion and enhance the cytotoxicity of PEG-PLA/P105/PTX micelles toward A549 cells. The in vivo results indicated that the combination therapeutic regimen could promote the micelles to distribute in lung and enhance the therapeutic effect on lung cancer.
Conclusion: This multifunctional approach of modulating the tumor microenvironment to enhance drug transportation and cell-killing sensitivity in the action sites might offer a new avenue for effective lung cancer treatment.
Keywords: lung cancer, pulmonary-affinity micelles, ambroxol, pulmonary microenvironment, combination therapy
Introduction
As population ages and air pollution increases, incidence and mortality of lung cancer have been rising.1,2 Despite the emergence of various therapies, chemotherapy, undeniably, still dominates lung cancer treatment clinically.3 Traditional first-line chemotherapy drugs often face the dilemma of drug resistance and adverse effects.4–7 For more effective treatments, various tumor-targeting nano-preparations have been developed.8–11 Unfortunately, few can meet the expectations of clinical treatment. On one hand, due to the complexity of tumor microenvironment,12–14 the general strategy of size adjustment and targeting modification for nanoparticles alone cannot make enough drugs reach the cancerous sites. On the other hand, chemotherapy drugs can induce tumor cell-protective autophagy,15–19 which can reduce the cell-killing sensitivity of drugs. Protective autophagy20,21 is the process of delivering damaged organelles or proteins to lysosomes for degradation after tumorigenesis, thereby providing nutrients to rapidly growing tumor cells. Autophagy is a double-edged sword in cell metabolism. On one hand, protective autophagy prevents cell apoptosis; on the other hand, excessive autophagy prevents cells from maintaining the basic structure and causes autophagy cell death. But when tumors occur, autophagy mainly plays a role in maintaining tumors' survival.22–24 Therefore, in view of the above-mentioned dilemma of lung cancer chemotherapy, we believe that actively modulating tumor microenvironment to attract more drugs in the cancerous sites, or (and) combining chemotherapeutics with autophagy inhibitors will improve the therapeutic effect.
Until now, several literatures have reported regulating tumor microenvironment to enhance the retention of drugs.25–29 For example, Ji et al25 designed an MMP-2-responsive liposome which could achieve tumor-targeting delivery and release of pirfenidone at the pancreatic stellate cells-enriched pancreatic tumors. The released pirfenidone could down-regulate the extracellular matrix expression of pancreatic stellate cells, which increase drug penetration in the tumor, thereby improving the therapeutic effect of gemcitabine. There have been also some reports of enhancing the effectiveness of chemotherapy drugs through combining with cell autophagy inhibitors.30–32 For instance, Zhang et al22 reported docetaxel-loaded dendritic copolymer nanoparticles by co-treatment with autophagy inhibitor to enhance the therapeutic effects of breast cancer. However, there is still no strategy that can not only regulate tumor microenvironment to promote the accumulation of drugs in tumor sites but also inhibit cell autophagy to increase the cytotoxicity of chemotherapy drugs.
Lung has unique characteristics that distinguish it from other organs, for example, it is rich in pulmonary surfactant (PS). PS is a kind of phospholipid and protein complex synthesized by alveolar type II epithelial cells. Its main function is to reduce alveolar surface tension, maintain pulmonary fluid balance and regulate local inflammation and immune response.33,34 Pluronic P105 can interact with PS through Van der Waals forces and hydrogen bonding,35 thereby increasing the distribution of Pluronic P105-containing polymer micelles in the lung.36,37 Ambroxol (Ax) is a kind of drug used to treat respiratory diseases in clinic.38,39 Literature have reported that Ax can promote alveolar type II epithelial cells to secrete PS and reduce the alveolar surface tension.34,35 In addition, our previous work found that Ax can inhibit cell autophagy through inducing autophagosome aggregation, thereby enhancing the cell-killing effect of chemotherapy drugs.40 Therefore, the combination of Ax and nano-drugs in lung cancer treatment has innate advantages and great potential for clinical transformation.
Based on the above research, in order to solve the aforementioned two obstacles faced by lung cancer chemotherapy, we employed amphiphilic polymers polyethyleneglycol-Polylactic acid (PEG-PLA) and Pluronic P105 as nanocarriers to encapsulate PTX to form into lung affinity PEG-PLA/P105/PTX micelles (Figure 1A). When the micelles are combined with Ax, the micelles will fully respond to the biological functions of Ax. First, the biological function of Ax to up-regulate the secretion of pulmonary surfactant promotes the PEG-PLA/P105/PTX micelles to distribute in the lung. Second, the biological function of Ax to inhibit cell autophagy enhances the cell-killing sensitivity of PTX (Figure 1B). The in vitro experiment results demonstrated that Ax could increase the cytotoxicity of PEG-PLA/P105/PTX micelles. The in vivo experiment results showed that Ax could facilitate the micelles to distribute in the lung and remarkably improve the lung cancer therapeutic effect. We can conclude that by employing biologically functional “molecular chaperones” to actively modulate the tumor microenvironment, thereby enhancing the nano-drug transportation and cell-killing sensitivity in the action sites, might be a promising strategy for lung cancer treatment.
 |
Figure 1 Schematic illustration of pulmonary-tendency paclitaxel polymer micelles in response to biological functions of ambroxol for synergistic lung cancer therapy. (A) Preparation of PEG-PLA/P105/PTX micelles. (B) Ax can up-regulate the secretion of pulmonary surfactant and inhibit tumor cell autophagy; subsequently, the PEG-PLA/P105/PTX micelles respond to the biological functions of Ax with stronger lung tendency and cytotoxicity. Abbreviations: Ax, ambroxol; PTX, paclitaxel; PEG-PLA, polyethyleneglycol-polylactic acid; PS, pulmonary surfactant. |
Materials and Methods
Materials
Ax was purchased from Boehringer-Ingelheim Co., Ltd (Ingelheim, Germany). PTX was purchased from Dalian Meilun Biotech Co., Ltd (Dalian, China). PEG2000-PLA2000 was provided by East China University of Science and Technology. Pluronic P105 was brought from BASF, Ludwigshafen (Germany). DiR was obtained from Sigma-Aldrich (Saint Louis, USA), A549 lung cancer cells and A549-luciferase lung cancer cells were purchased from Shanghai cell bank, Chinese Academy of Sciences (China). ICR mice, BALB/c nude mice and SD rats were purchased from the animal experiment center of Fudan University.
Study on Ax in Promoting the Secretion of Pulmonary Surfactant
All animal experiments were conducted in accordance with the ethical requirements of Fudan University Institutional Animal Care and Use Committee (IACUC). To determine the optimal administration strategy of Ax in up-regulating pulmonary surfactant secretion, different doses (20, 50, 100 mg/kg) of Ax were intravenously injected into the male ICR mice (18–22 g, 6–8 weeks) every 4 days for 4 times (3 mice in each group). Mice injected with normal saline were regarded as the control group. Mice were decapitated the day after the last administration and lung tissues of each group were excised to measure the pulmonary surfactant associated protein-A41 (SP-A) by immunohistochemical staining to determine the optimal dose.
Afterward, to further screen the optimal treatment time, the optimal dose of Ax was intravenously injected into the ICR mice every 4 days for different times (1, 2, 3, 4, 5 times, 3 mice per group). The SP-A level of each group was measured as mentioned above.
Pulmonary Affinity and Optimal Ratio Screening of Pluronic P105
To study the pulmonary affinity of PEG-PLA/P105 micelles, we encapsulated DiR, a near-infrared fluorescence dye, into the PEG-PLA micelles and PEG-PLA/10% P105 micelles by thin-film hydration method. Briefly, DiR (2 mg), PEG-PLA (90 mg) and P105 (10 mg) were dissolved in 5 mL acetonitrile, the mixture was evaporated under 37°C with a rotary evaporator for 30 mins to form a thin film. Then, the thin film was hydrated with 10 mL deionized water for 10 mins and the resulting solution was filtered through a 0.22 μm filter to remove the adhesion debris and unencapsulated DiR. ICR mice were randomly divided into 3 groups (12 mice per group) and intravenously injected with free DiR (1 mg/kg), PEG-PLA/DiR micelles and PEG-PLA/10% P105/DiR micelles (each equivalent to 1 mg/kg DiR), respectively. Afterward, mice were decapitated at predetermined time points postinjection (2, 6, 12, 24 h) and the main organs were harvested to measure the fluorescence intensity using the In Vivo Imaging System (IVIS, Xenogen, USA).
Next, we screened the optimal ratio of Pluronic P105 in PEG-PLA/P105 micelles. DiR was encapsulated into the PEG-PLA/5% P105 micelles, PEG-PLA/10% P105 micelles and PEG-PLA/20% P105 micelles, respectively. Mice were randomly divided into 3 groups (12 mice per group) and intravenously injected with PEG-PLA/5% P105/DiR micelles, PEG-PLA/10% P105/DiR micelles and PEG-PLA/20% P105/DiR micelles (each equivalent to 1 mg/kg DiR), respectively. The fluorescence intensity of each group was measured as mentioned above.
Fabrication and Characterization of PEG-PLA/P105/PTX micelles
The PEG-PLA/P105/PTX micelles of the optimal prescription were prepared by thin-film hydration method mentioned above. Briefly, PTX (10 mg), PEG-PLA (80 mg) and P105 (10 mg) were dissolved in 5 mL acetonitrile, and the mixture was evaporated under 37°C with a rotary evaporator for 30 mins to form a thin film. Then, the thin film was hydrated with 10 mL deionized water for 10 mins, and the resulting solution was filtered through a 0.22 μm filter to remove the adhesion debris and unencapsulated PTX. The drug-loading content of the micelles was characterized by HPLC (Agilent 1260 Infinity, USA) and the retention time of PTX was 4.45 mins (Supporting Information, Figure S1). We also prepared the PEG-PLA/P105 micelles in the same way. The hydration particle sizes, Zeta potentials and the morphology of the micelles were characterized by dynamic light scattering (DLS, Zetasizer Nano ZS, Malvern, England) and transmission electron microscopy (TEM, JEM-1400 plus, Japan Electronics Corporation), respectively. To prepare the TEM samples, a drop of the micelle solution was dropped on a carbon film copper and allowed to dry at room temperature, and a drop of phosphotungstic acid solution was then added to stain for 3 mins. The CMC of the micelles was tested by iodine spectroscopy method,42 (Supporting Information). In addition, to investigate the stability of the micelle solution, we centrifuged the micelle solution at 10,000 rpm for 10 mins and took the supernatant to determine the PTX relative percentage at different time point within 48 hrs.
The in vitro drug release behavior of the micelles was tested through dialysis method. Briefly, phosphate buffer solution (0.01 mol/L, pH 7.4) containing 0.5% Tween 80 was used as the release medium. 1 mL free PTX (0.2 mg/mL) and PEG-PLA/P105/PTX micelle solution (equivalent to 0.2 mg PTX) were added in dialysis bags (MWCO = 3.5 KDa) respectively. And then the dialysis bags were put into centrifuge tubes containing 49-mL release medium to ensure drug release under sink condition and shaken at 37°C, 100 rpm. 0.5 mL release medium was taken out at 0.5, 1, 2, 4, 6, 8, 10, 12 and 24 hrs, and at the same time the same volume of fresh release medium was added. The concentration of PTX was measured by HPLC.
In vitro Cytotoxicity
In vitro cytotoxicity was determined by methyl thiazolyl tetrazolium (MTT) assay. A549 lung cancer cells were cultured with RPMI 1640 culture medium containing 10% fetal bovine serum and 1% penicillin-streptomycin under an atmosphere containing 5% CO2 at 37°C. cells in the logarithmic growth phase were seeded in 96-well plates at a density of 8×103 cells/well and incubated for 24 hrs to allow adherence. Next, cells were co-treated with different concentrations of PEG-PLA/P105/PTX micelles and Ax (equivalent to 0.01–0.1 μg/mL PTX and 0–100 μM of Ax, n = 3) for 24, 48 and 72 hrs, respectively. Thereafter, the culture medium was exchanged by 0.5 mg/mL MTT solution and incubated for 4 hrs at 37°C. The absorbance of each well was measured at 570 nm with a Microplate Reader (Synergy 2, Bio Tek, USA) after the MTT solution was replaced by 200 μL DMSO. The control well was cells cultured with normal culture medium and the blank well was well cultured without cells. The cell viability of each well was calculated according to the following formula:

Cellular Uptake
Cellular uptake experiment was used to determine whether Ax could affect the cellular uptake behavior of the micelles. Since PTX cannot be fluorescent, we employed Coumadin−6 (Cou−6), a hydrophobic fluorescent probe, to encapsulate into the PEG-PLA/P105 micelles to study the cellular uptake behavior of PEG-PLA/P105 micelles. The PEG-PLA/P105/Cou−6 micelles were prepared in the same way as the PEG-PLA/P105/PTX micelles except that PTX was replaced by Cou−6. Although there have limitations to replace drugs by fluorescent probes, since they have different chemical structures and half-lives. The fate of nanodrugs mainly depends on the material properties and particle size.43 The PEG-PLA/P105/Cou-6 micelles have similar particle size as the PEG-PLA/P105/PTX micelles, it is reasonable to replace PTX with Cou-6. A549 cells were seeded at a density of 2×105 cells per well in 6-well plates for overnight incubation and pretreated with or without 100 μM Ax for 24 hrs. Afterward, cells were treated with PEG-PLA/P105/Cou-6 micelles (equivalent to 0.1 μg/mL Cou-6, n = 3) at 37°C for 0.5, 1, 2 and 4 hrs, respectively. The supernatant of each well was removed and rinsed with cold PBS 3 times. Fluorescence intensity of each well was photographed under an inverted fluorescence microscope (Leica DMI4000B, Germany).
Western Blotting
In our previous study, we have proved that Ax can block autophagic flux through promoting autophagosome accumulation. The microtubule-associated protein 1 light chain 3 (LC3) was the marker protein of autophagosomes.15 Western blotting could be used to determine the lung cancer cell autophagy regulated by PEG-PLA/P105/PTX micelles combined with Ax by measuring the LC3 proteins. Specifically, A549 cells in the logarithmic growth phase were seeded in 6-well plates at a density of 5×105 cells/well. After cells were attached, PEG-PLA/P105/PTX micelles, free Ax and PEG-PLA/P105/PTX micelles + Ax (equivalent to 0.5 μg/mL PTX and 50 μM Ax) diluted with culture medium were added into each well and incubated at 37°C for 24 hrs, respectively. Cells incubated with normal culture medium were regarded as the control group. Next, cells were collected and the LC3 proteins, which indirectly reflect the number of autophagosomes, were determined by Western blotting.
In vivo Biodistribution
Male ICR mice (18–22 g, 6–8 weeks) were randomly divided into 5 groups (12 mice per group). Group A was injected with normal saline, group B was injected with free DiR, group C was injected with PEG-PLA/P105/DiR micelles, group D was injected with PEG-PLA/DiR micelles after injecting with 100 mg/kg of Ax every 4 days for 4 times, group E was injected with PEG-PLA/P105/DiR micelles after injecting with 100 mg/kg of Ax every 4 days for 4 times (each equivalent to 1 mg/kg of DiR), respectively. Mice were then sacrificed and the main organs were harvested to test the fluorescence intensity at 2, 6, 12 and 24 hrs postinjection.
In vivo Pharmacokinetics
Male SD rats (180–220 g, 6–8 weeks) were randomly divided into 4 groups (6 mice per group). Group A was injected with PTX, group B was injected with PTX after injecting with 100 mg/kg Ax every 4 days for 4 times, group C was injected with PEG-PLA/P105/PTX micelles, group D was injected with PEG-PLA/P105/PTX micelles after injecting with 100 mg/kg Ax every 4 days for 4 times (each equivalent to 10 mg/kg PTX), respectively. The PTX concentration in the blood of each group was measured at different time points.
In vivo Antitumor Efficacy
To establish the orthotopic lung tumor model, 200 μL A549-luciferase cells (2 × 107/mL) was intravenously injected into each male BALB/c nude mouse (18–22 g, 6–8 weeks). The lung tumor growth was observed using the IVIS. When the fluorescence intensity of the lung tissues reached about 1 × 106 (p/s/cm2/sr)/(μW/cm2), the tumor-bearing mice were randomly divided into 6 groups (13 mice per group). Each group was injected with normal saline, Ax (100 mg/kg), PTX (10 mg/kg), PEG-PLA/P105/PTX micelles (equivalent to 10 mg/kg PTX), PTX (3 mg/kg) + Ax (100 mg/kg), and PEG-PLA/P105/PTX micelles (equivalent to 3 mg/kg PTX) + Ax (100 mg/kg) at 0, 4, 8, 12 days, respectively. The lung tumor growth of each group was observed using the IVIS once every 4 days for two consecutive weeks. 3 mice in each group were sacrificed on the 15th day, and lung tissues were harvested to measure the LC3 protein expression. The death time of the remaining mice in each group was recorded to plot the survival curves (10 mice per group).
Statistical Analysis
All the results were reported as the mean ± standard deviation of at least three independent experiments. Data comparisons were performed by Student’s t-test or one-way analysis of variance (ANOVA). Log-rank test was used to compare the median survival values. Data analyses were conducted using GraphPad Prism 5 software (GraphPad Software Inc., La Jolla, CA, USA) or SPSS 19.0 (Statistical Product and Service Solutions, IBM, USA). P < 0.05 was regarded as a statistically significant difference.
Results and Discussion
Study on Ax in Promoting the Secretion of Pulmonary Surfactant
To determine the optimal administration strategy for Ax in up-regulating the secretion of pulmonary surfactant. ICR mice were injected with various doses of Ax for four times and SP-A (yellow-stained protein) was measured by immunohistochemical stain. Compared with normal saline, 20 mg/kg Ax does not up-regulate the SP-A secretion, and 50 mg/kg Ax can significantly up-regulate the SP-A secretion, while 100 mg/kg Ax has the most significant effect on up-regulating SP-A secretion (Figure 2A). Since a dose of 100 mg/kg in mice is equivalent to the maximum clinical dose in humans, we did not further test the higher dose.
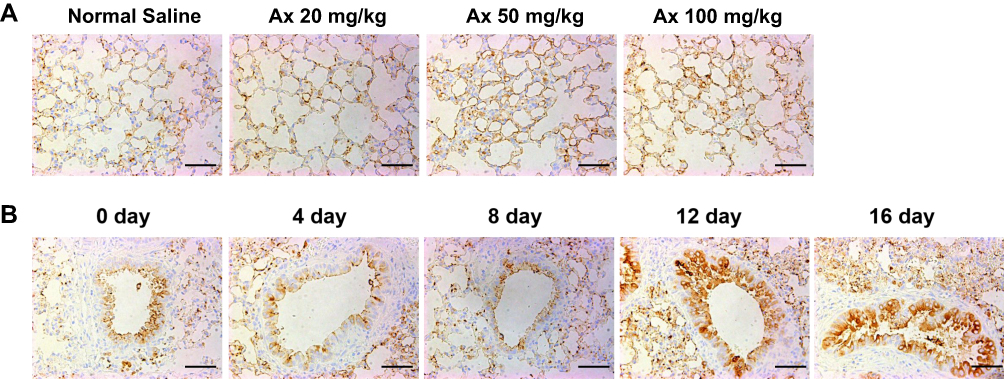 |
Figure 2 Study on Ax in promoting the secretion of pulmonary surfactant. (A) SP-A expression of each group treated with different concentrations of Ax. (B) SP-A expression of each group treated with Ax (100 mg/kg) for different times. Note: Scale bar = 25 μm.Abbreviation: Ax, ambroxol. |
Furthermore, ICR mice were injected with 100 mg/kg Ax for various times. With the increase of administration times, the expression of SP-A increased. However, after four doses, the maximum SP-A expression was obtained. There was no further improved progress shown on the fifth doses (Figure 2B). Therefore, the maximal effect of SP-A expression could be achieved by 100 mg/kg Ax with four consecutive doses.
Pulmonary Affinity and Optimal Ratio Screening of P105
To demonstrate the pulmonary affinity of P105, DiR containing micelles were prepared to trace their distribution in vivo. At different time points, the fluorescence intensity of the lung tissues in the PEG-PLA/DiR micelles group was significantly higher than that in the DiR group. Compared with the PEG-PLA/DiR micelles group, the PEG-PLA/10% P105/DiR micelles group showed significantly stronger fluorescence intensity (Figure 3A). The fluorescence intensity AUC (area under the curve) of the lung tissues in each group was 2.142 ± 0.107 (DiR), 2.796 ± 0.094 (PEG-PLA/DiR micelles) and 4.139 ± 0.620 (PEG-PLA/10% P105/DiR micelles) (×106 p/s/cm2/sr)/(μW/cm2), respectively (Figure 3B). These results indicate that the micelles themselves have favorable lung affinity and the P105-containing micelles have intrinsic lung affinity.
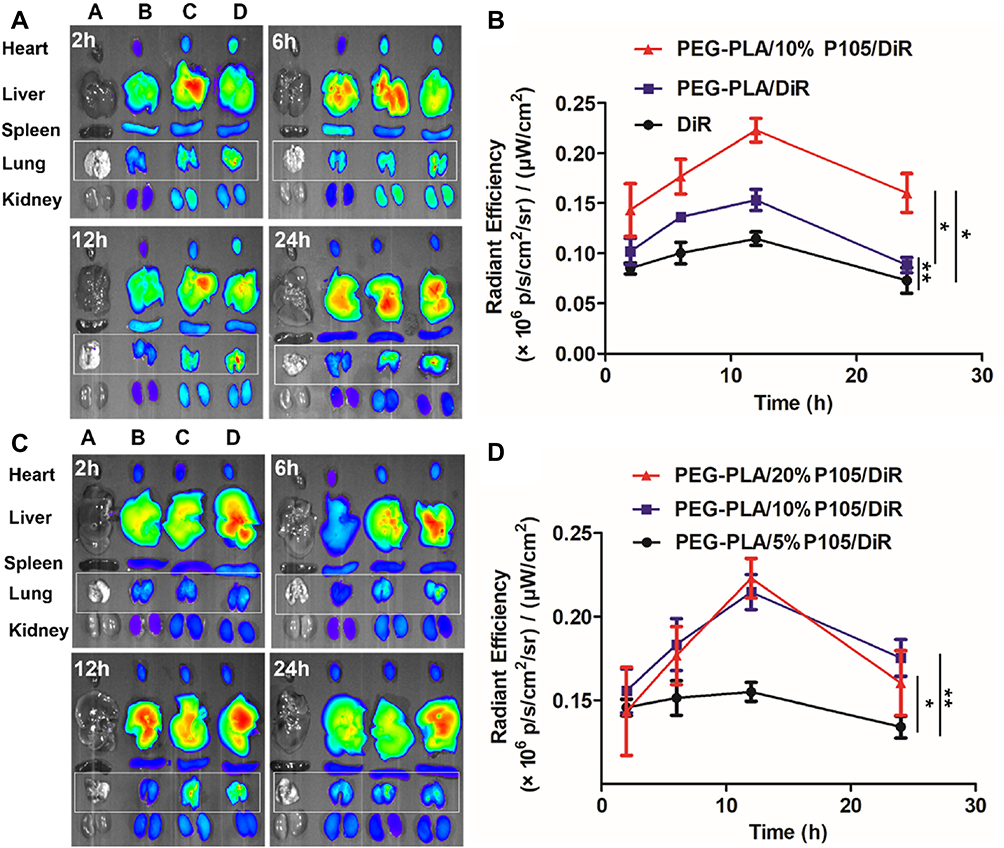 |
Figure 3 Study on the pulmonary affinity of P105. (A) Fluorescence photographs of the main organs (A): normal saline, (B) DiR, (C) PEG-PLA/DiR micelles and (D) PEG-PLA/P105/DiR micelles and (B) fluorescence intensity of the lung tissues of each group at different time points. (C) Fluorescence photographs of the main organs (A): normal saline, (B) PEG-PLA/5% P105/DiR micelles, (C) PEG-PLA/10% P105/DiR micelles and (D) PEG-PLA/20% P105/DiR micelles and (d) lung fluorescence intensity of each group at different points. *P < 0.05, **P < 0.01. Note: Data are represented as mean ± SD, n = 3Abbreviations: PEG-PLA/5% P105/DiR, PEG-PLA/5% P105/DiR micelles; PEG-PLA/10% P105/DiR, PEG-PLA/10% P105/DiR micelles; PEG-PLA/20% P105/DiR, PEG-PLA/20% P105/DiR micelles. |
To further screen out the optimal ratio of P105 in PEG-PLA/P105 micelles, DiR was encapsulated into PEG-PLA/P105 micelles containing different ratios of P105 to trace their distribution in vivo. At different time points, the fluorescence intensity of the lung tissues in PEG-PLA/10% P105/DiR micelles group was significantly higher than that in PEG-PLA/5% P105/DiR micelles group. However, there was no significant difference between the PEG-PLA/10% P105/DiR micelles group and PEG-PLA/20% P105/DiR micelles group (Figure 3C). The fluorescence intensity AUC of the lung tissues in each group was 3.307 ± 0.319 (PEG-PLA/5% P105/DiR micelles), 4.139 ± 0.620 (PEG-PLA/10% P105/DiR micelles), 4.212 ± 0.452 (PEG-PLA/20% P105/DiR micelles) (×106 p/s/cm2/sr)/(μW/cm2), respectively (Figure 3D). These results indicate that 10% P105 had achieved satisfying pulmonary affinity. Considering that a high ratio of P105 decreased the drug loading and stability of the micelles (the PTX relative percentage of the PEG-PLA/20% P105/PTX micelle solution decreased to 80% at 48 h), 10% P105 was selected for subsequence prescription research.
Fabrication and Characterization of PEG-PLA/P105/PTX micelles
Referring to the experimental results of 3.2, we finally determined that the optimal ratio of P105 was 10%. We prepared and characterized the PEG-PLA/10% P105 micelles (hereinafter referred to as PEG-PLA/P105 micelles) and PEG-PLA/10% P105/PTX micelles (hereinafter referred to as PEG-PLA/P105/PTX micelles). The drug encapsulation efficiency was 99.3%. The average hydration diameters were 16.8 ± 0.5 nm for PEG-PLA/P105 micelles and 16.7 ± 0.5 nm for PEG-PLA/P105/PTX micelles, indicating that the encapsulated drug does not affect the micelle size (Figure 4A and C). The Zeta potentials were −4.2 ± 0.6 mV for PEG-PLA/P105 micelles and −4.1 ± 0.4 mV for PEG-PLA/P105/PTX micelles. The TEM image indicated that the micelles had nearly round shapes and uniform particle sizes which were in accord with the DLS results (Figure 4B and D). The CMC values of Pluronic P105, PEG-PLA and PEG-PLA/P105 were 0.0068% (w/v), 0.0141% (w/v) and 0.0132% (w/v), respectively (Supporting Information, Figure S2). The small CMC of PEG-PLA/P105 indicates the strong dilution resistance of the micelles. The PTX relative percentage of the micelle solution at different time points was between 95% and 105% (Figure 4E), indicating the micelles remain stable within 48 hrs.
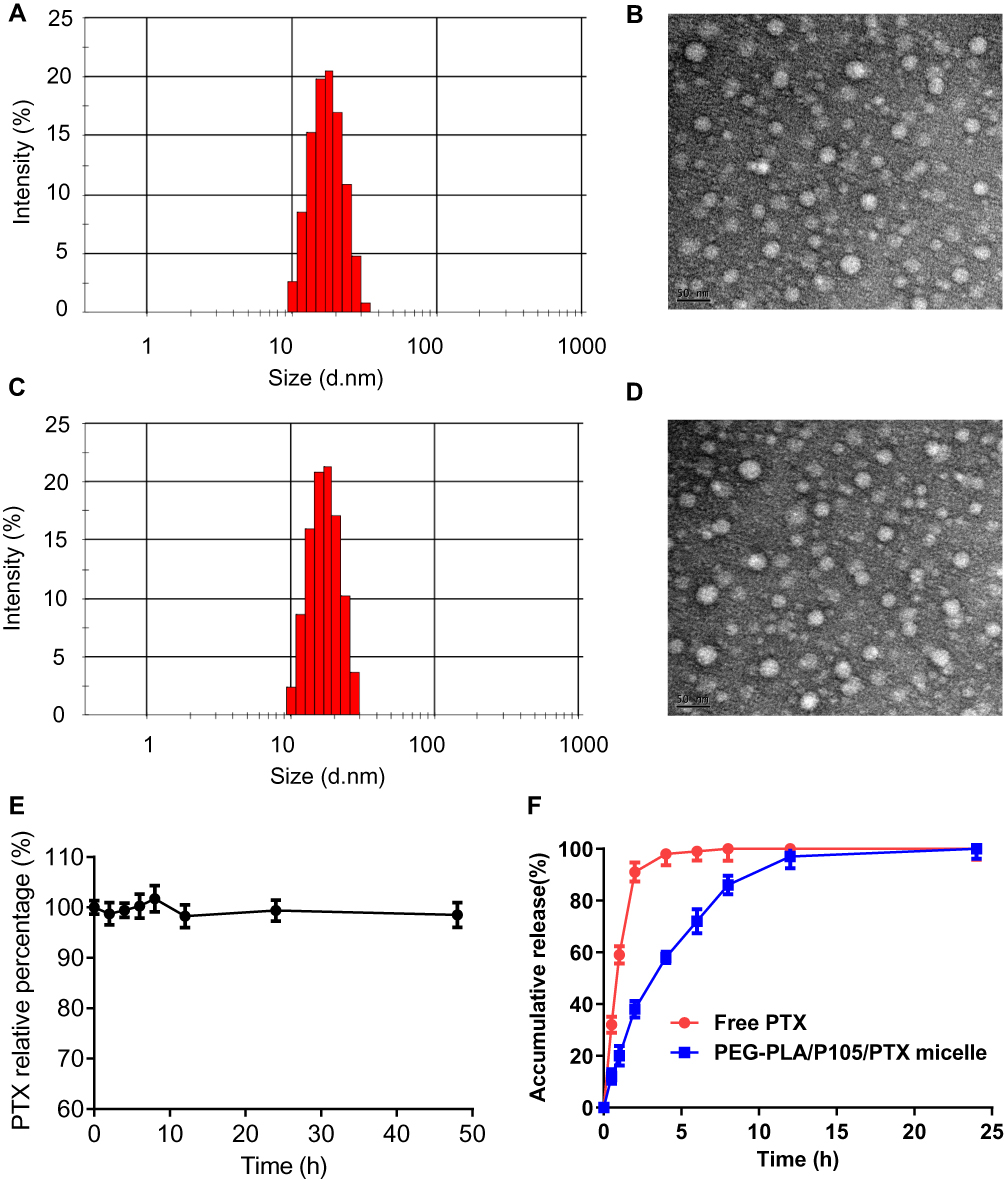 |
Figure 4 Characterization of PEG-PLA/P105/PTX micelles. (A) Particle size distribution and (B) TEM image of PEG-PLA/P105 micelles, (C) particle size distribution and (D) TEM image of PEG-PLA/P105/PTX micelles, (E) PTX relative percentage of PEG-PLA/P105/PTX micelles within 48 hrs and (F) in vitro drug release behavior of PEG-PLA/P105/PTX micelles. Note: (D) Data are represented as mean ± SD, n = 3.Abbreviations: PTX, paclitaxel. |
As shown in Figure 4D, free PTX released more than 90% at 2 hrs, PEG-PLA/P105/PTX micelles released approximately 60% at 6 hrs and nearly 100% at 12 hrs. The in vitro drug release behavior demonstrated the sustained-release property of the micelles.
In vitro Cytotoxicity
To determine whether PEG-PLA/P105/PTX micelles combined with Ax can enhance the killing effect on A549 lung cancer cells. Cells were co-treated with different concentrations of micelles and Ax. The results showed that Ax significantly increased the cytotoxicity of the micelles at all concentrations and incubation times. The higher Ax concentration, the stronger cell-killing ability of the micelles (Figure 5). The IC50 values of the PEG-PLA/P105/PTX micelles in combination with 20, 50 and 100 μM Ax on A549 cells following 24, 48 and 72 hr treatment were calculated (Table 1). The IC50 values of the PEG-PLA/P105/PTX micelles at 24 hrs with no Ax or combined with 100 μM Ax were 87.09 ± 4.12 ng/mL and 1.14 ± 0.08 ng/mL, respectively. This means that 100 μM Ax could enhance the therapeutic effects of PEG-PLA/P105/PTX micelles by 76-folds. The IC50 values of 48 hrs and 72 hrs also indicate that Ax could significantly increase the cytotoxicity of PEG-PLA/P105/PTX micelles.
 |
Table 1 IC50 Values of PEG-PLA/P105/PTX micelles in Combination with 20, 50 and 100 μM of Ax on A549 Cells Following 24, 48 and 72 hrs Treatment, Respectively. |
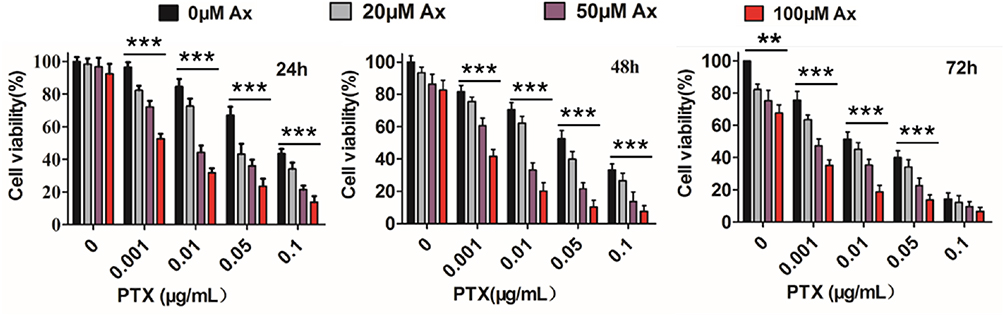 |
Figure 5 Viability of A549 cells co-treated with 0.001–0.1 μg/mL PEG-PLA/P105/PTX micelles and 20–100 μM Ax for 24, 48 and 72 hrs, respectively. **P < 0.01, ***P < 0.001. Note: Data are represented as mean ± SD, n = 6.Abbreviations: Ax, ambroxol; PTX, paclitaxel. |
Cellular Uptake and Western Blotting
To verify whether Ax increases the cytotoxicity of PEG-PLA/P105/PTX micelles because Ax increases the cellular uptake of the micelles. We measured the cellular uptake of the micelles with or without Ax co-incubation. Interestingly, cells showed similar green fluorescence intensity after incubating with PEG-PLA/P105/cou-6 micelles or PEG-PLA/P105/cou-6 micelles + Ax at different time points (Figure 6A), which means Ax does not affect the cellular uptake of the micelles. Therefore, Ax does not enhance the cytotoxicity of the micelles by increasing the cellular uptake.
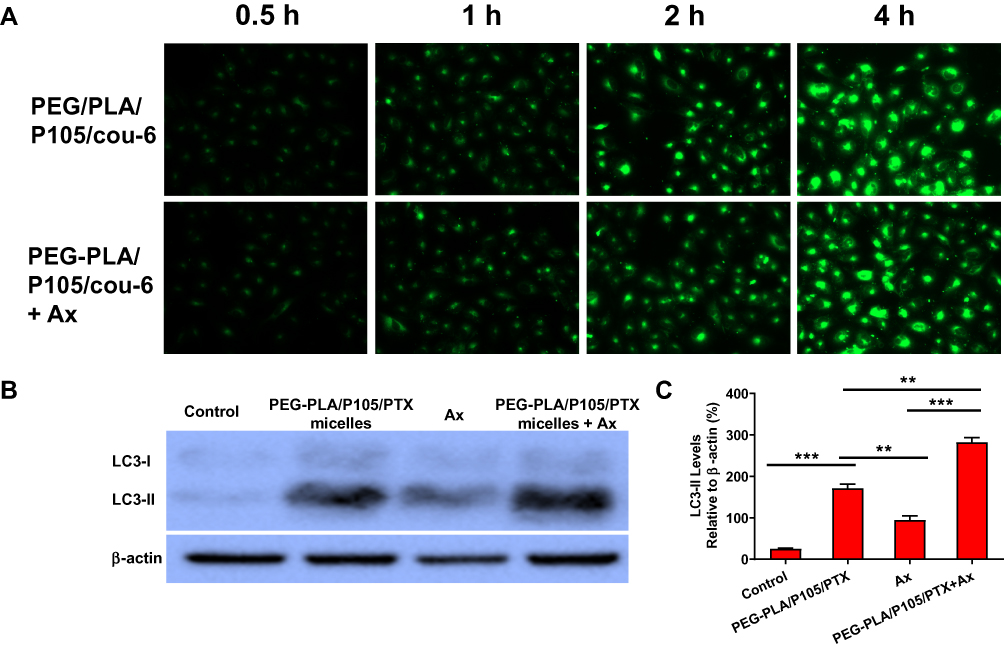 |
Figure 6 (A) Cellular uptake of PEG-PLA/P105 micelles regulated by Ax. (B) LC3 proteins measured by Western blotting and (C) quantitative analysis of LC3-II protein of PEG-PLA/P105/PTX micelles regulated by Ax. **P < 0.01, ***P < 0.001. Note: Data are represented as mean ± SD, n = 3.Abbreviations: cou-6, coumarin 6; PEG-PLA/P105/cou-6, PEG-PLA/P105/cou-6 micelles; PEG-PLA/P105/PTX, PEG-PLA/P105/PTX micelles; Ax, ambroxol; PTX, paclitaxel. |
To determine whether Ax increases the cytotoxicity of PEG-PLA/P105/PTX micelles because Ax inhibits cell autophagy. We next examined the LC3 proteins of A549 cells incubated with PEG-PLA/P105/PTX micelles + Ax. Compared with the control group, both PEG-PLA/P105/PTX micelles and Ax alone could increase the LC3 proteins to a certain degree, and the combination of PEG-PLA/P105/PTX micelles and Ax could further enhance the LC3 proteins (Figure 6B and C). The above experimental results indicated that Ax increased the cytotoxicity of PEG-PLA/P105/PTX micelles by inhibiting cell autophagy.
In vivo Biodistribution
In order to investigate the biological effect of Ax on the biodistribution of PEG-PLA/P105 micelles, we conducted an in vivo biodistribution experiment. Compared with the PEG-PLA/P105/DiR micelles group, the lung fluorescence intensity in Ax + PEG-PLA/P105/DiR micelles group was significantly stronger, indicating that Ax could facilitate the micelles to distribute in lung through increasing PS secretion. In addition, the lung fluorescence intensity in Ax + PEG-PLA/P105/DiR micelles group was stronger than that in Ax + PEG-PLA/DiR micelles group, further demonstrating the lung affinity of P105-containing micelles (Figure 7A). The lung fluorescence intensity AUC of each group was 3.919 ± 0.331 (PEG-PLA/P105/DiR micelles), 4.149 ± 0.400 (Ax + PEG-PLA/DiR micelles) and 5.535 ± 0.515 (Ax + PEG-PLA/P105/DiR micelles) (×106 p/s/cm2/sr)/(μW/cm2), respectively (Figure 7B). There are two reasons for us not incorporating Ax and P105 in a single micelle system. First, Ax is hydrophilic and it is not easy to be encapsulated into the micelles. Second, Ax itself has favorable lung affinity and can rapidly distribute in the lung (Figure S3 and Table S1).
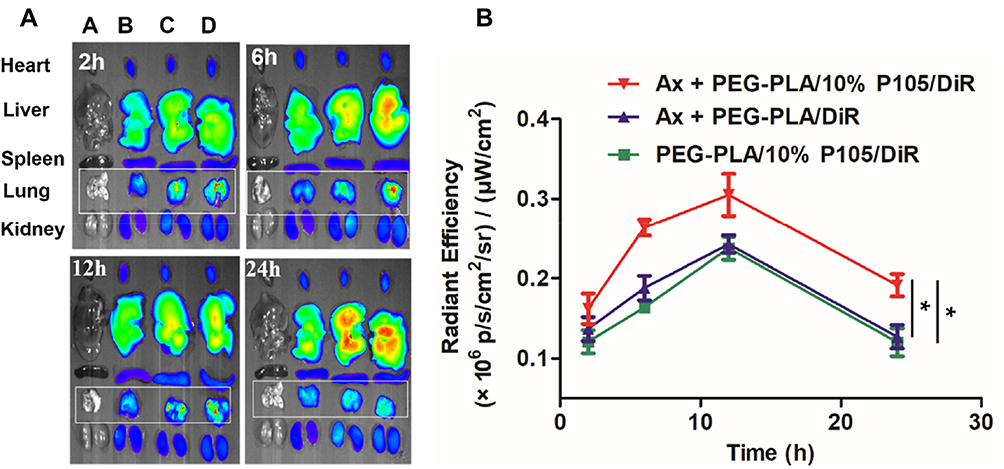 |
Figure 7 In vivo biodistribution of PEG-PLA/P105 micelles regulated with Ax. (A) Fluorescence photographs of the main organs (A): normal saline, (B) PEG-PLA/P105/DiR micelles, (C) Ax + PEG-PLA/DiR micelles, and (D) Ax + PEG-PLA/P105/DiR micelles and (B) lung fluorescence intensity of each group at different time points. *P < 0.05.Note: Data are represented as mean ± SD, n = 3.Abbreviations: PEG-PLA/10% P105/DiR, PEG-PLA/10% P105/DiR micelles; PEG-PLA/DiR, PEG-PLA/DiR micelles; Ax, ambroxol. |
In vivo Pharmacokinetics
Subsequently, we performed a pharmacokinetic study of PEG-PLA/P105/PTX micelles combined with Ax in SD rats. Group C (PEG-PLA/P105/PTX micelles) had a longer elimination half-life, a larger AUC and a slower clearance rate than group A (free PTX), suggesting the long-circulation effect of the micelles (Figure 8A). The longer the circulation time of nanoparticles in the body, the better the accumulation of nanoparticles in tumor sites.44,45 In addition, group D (Ax + PEG-PLA/P105/PTX micelles) displayed the longest t1/2β (elimination half-life, 3.96 h), the largest AUC (13,525 µg/L/h) and the slowest clearance rate (0.74 L/h/kg) (Table 2). Moreover, the PTX concentration in lung tissue of group D (Ax + PEG-PLA/P105/PTX micelles) at 24 hrs postinjection is 4 times higher than the group B (Ax + PTX) and 2 times higher than the Group C (PEG-PLA/P105/PTX micelles) (Figure 8B). These results, on the one hand, confirmed the long-circulation of PEG-PLA/P105/PTX micelles, and on the other hand, proved that Ax could slow down the plasma clearance and prolong the elimination half-life of the micelles.
 |
Table 2 Pharmacokinetic Parameters of PTX, Ax + PTX, PEG-PLA/P105/PTX micelles and Ax + PEG-PLA/P105/PTX micelles (n = 6) |
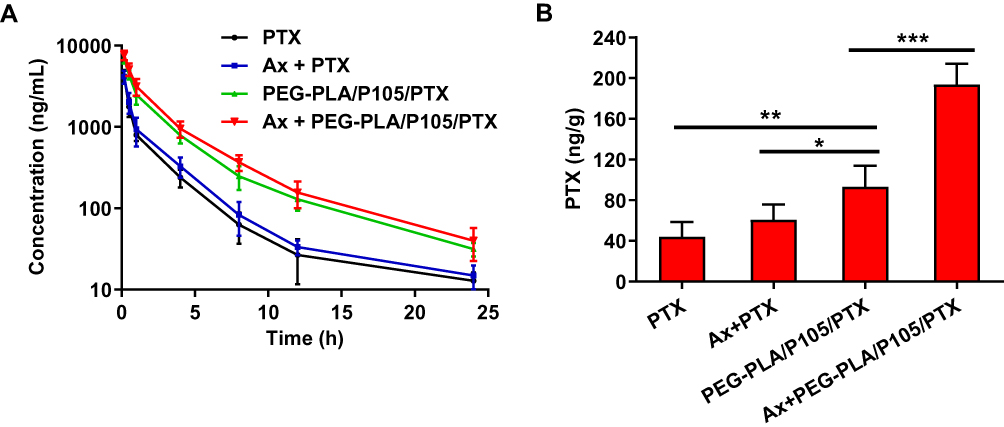 |
Figure 8 (A) PTX plasma concentration–time curves of free PTX, Ax + PTX, PEG-PLA/P105/PTX micelles and Ax + PEG-PLA/P105/PTX micelles. (B) The concentrations of PTX in lung tissues at 24 hrs postinjection. *P < 0.05, **P < 0.01, ***P < 0.001. Notes: Data are represented as mean ± SD, n = 6.Abbreviations: PEG-PLA/P105/PTX, PEG-PLA/P105/PTX micelles; Ax, ambroxol; PTX, paclitaxel. |
In vivo Antitumor Efficacy
Encouraged by the superior in vitro cytotoxicity effect of the combination regimen. The in vivo therapeutic effect of PEG-PLA/P105/PTX micelles combining with Ax was determined on an orthotopic A549 lung tumor model. As shown in the IVIS, Ax alone had almost no therapeutic effect compared to normal saline. PTX (3 mg/kg) + Ax (100 mg/kg) showed comparable tumor inhibition effect to PEG-PLA/P105/PTX micelles (equivalent to 10 mg/kg of PTX), but better than PTX (10 mg/kg). While PEG-PLA/P105/PTX micelles (3 mg/kg) + Ax (100 mg/kg) showed the strongest tumor suppression effect with the longest median survival time (MST) for 52.5 days (Figure 9A, D and E). In addition, LC-3 proteins were determined by Western blotting. The superior tumor inhibition effect, the higher LC3 protein expression in lung tumor tissues (Figure 9B and C). It might indicate that Ax enhanced the anti-tumor effect of PEG-PLA/P105/PTX micelles by interfering cell autophagy. Above all, these results indicated that PEG-PLA/P105/PTX micelles combined with Ax could remarkably improve the lung tumor therapeutic effect.
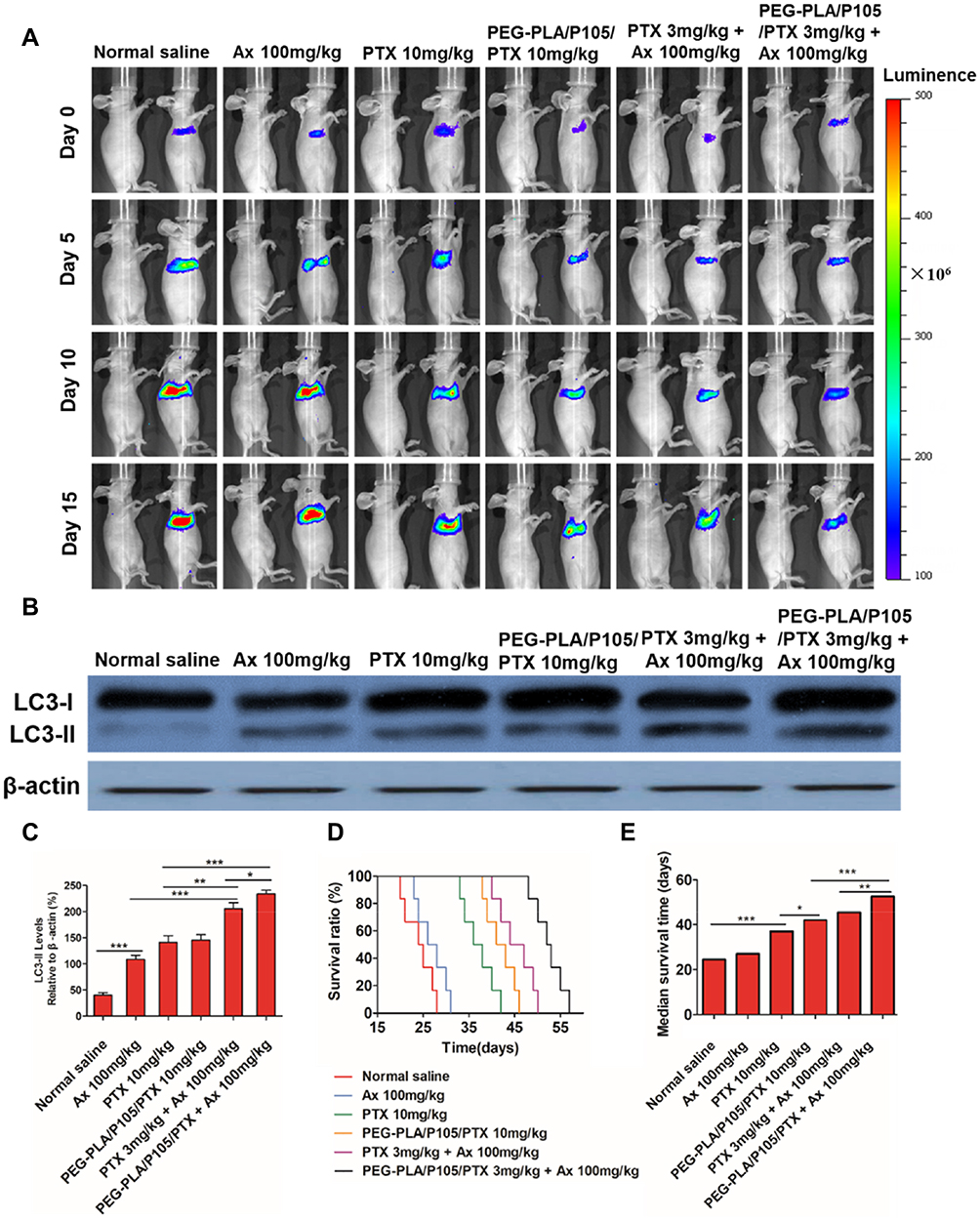 |
Figure 9 In vivo anticancer effects of PEG-PLA/P105/PTX micelles combined with Ax. (A) The changes in pulmonary tumor size detected at 0, 5, 10, 15 days by IVIS. The left-side mice in each group represent the bright-field picture. (B) LC3 proteins of the lung tissues measured by Western blotting and (C) quantitative analysis of LC3-II protein of the lung tissues. (D) Survival curves and (E) median survival times of the pulmonary tumor-bearing mice at different treatment groups. *P < 0.05, **P < 0.01, ***P < 0.001. Notes: (C) Data are represented as mean ± SD, n=3; (D) and (E) Data are represented as mean ± SD, n = 10.Abbreviations: PEG-PLA/P105/PTX, PEG-PLA/P105/PTX micelles; Ax, ambroxol; PTX, paclitaxel. |
Conclusion
In this work, considering two major obstacles to cancer chemotherapy – little drug accumulation at the tumor sites and the decreased cytotoxicity induced by cell autophagy – we have developed a lung cancer chemotherapy regimen. Ax can increase PS secretion and suppress tumor cell autophagy. The pulmonary-affinity PEG-PLA/P105/PTX micelles could adequately respond to the biological functions of Ax. Compared with the general strategies that only focus on the functional modification of nano-formulations, we employed a “molecular chaperone” Ax to change the microenvironment of cancerous lesion, thereby promoting the accumulation and cell-killing sensitivity of the micelles. Both the in vitro and in vivo experiment results demonstrated that the rational designed PEG-PLA/P105/PTX micelles combine with Ax could produce an effective anticancer effect. Our versatile approach of modulating tumor microenvironment to enhance the drug transportation and cell-killing sensitivity in the action sites may bring new enlightenment to the clinical treatment of lung cancer.
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (81773201), Fudan-SIMM Joint Research Fund (FU-SIMM20182005), PDH-SPFDU Joint Research Fund (RHJJ2018-02), Development Project of Shanghai Peak Disciplines-Integrative Medicine (20180101).
Disclosure
The authors declare no conflicts of interest in this work.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69(1):7–34. doi:10.3322/caac.21551
2. Bray F, Ferlay J, Soerjomataram I, et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. doi:10.3322/caac.21492
3. Tsuchiya N, Doai M, Usuda K, Uramoto H, Tonami H. Non-small cell lung cancer: whole-lesion histogram analysis of the apparent diffusion coefficient for assessment of tumor grade, lymphovascular invasion and pleural invasion. PLoS One. 2017;12(2):e0172433. doi:10.1371/journal.pone.0172433
4. Li X, Lewis MT, Huang J, et al. Intrinsic resistance of tumorigenic breast cancer cells to chemotherapy. J Natl Cancer Inst. 2008;100(9):672–679. doi:10.1093/jnci/djn123
5. Shi J, Kantoff PW, Wooster R, Farokhzad OC. Cancer nanomedicine: progress, challenges and opportunities. Nat Rev Cancer. 2017;17(1):20–37. doi:10.1038/nrc.2016.108
6. Shi J, Xiao Z, Kamaly N, Farokhzad OC. Self-assembled targeted nanoparticles: evolution of technologies and bench to bedside translation. Acc Chem Res. 2011;44(10):1123–1134. doi:10.1021/ar200054n
7. Yu MK, Park J, Jon S. Targeting strategies for multifunctional nanoparticles in cancer imaging and therapy. Theranostics. 2012;2(1):3–44. doi:10.7150/thno.3463
8. Jiang TY, Zhang ZH, Zhang YL, et al. Dual-functional liposomes based on pH-responsive cell-penetrating peptide and hyaluronic acid for tumor-targeted anticancer drug delivery. Biomaterials. 2012;33(36):9246–9258. doi:10.1016/j.biomaterials.2012.09.027
9. Cui YN, Xu QX, Chow PKH, Wang DP, Wang CH. Transferrin-conjugated magnetic silica PLGA nanoparticles loaded with doxorubicin and paclitaxel for brain glioma treatment. Biomaterials. 2013;34(33):8511–8520. doi:10.1016/j.biomaterials.2013.07.075
10. Xu XL, Lu KJ, Zhu ML, et al. Sialic acid-functionalized pH-triggered micelles for enhanced tumor tissue accumulation and active cellular internalization of orthotopic hepatocarcinoma. ACS Appl Mater Interfaces. 2018;10(38):31903–31914. doi:10.1021/acsami.8b09498
11. Wan XM, Min YZ, Bludau H, et al. Drug combination synergy in worm-like polymeric micelles improves treatment outcome for small cell and non-small cell lung cancer. ACS Nano. 2018;12(3):2426–2439. doi:10.1007/s12272-001-1189-2
12. de Palma M, Biziato D, Petrova TV. Microenvironmental regulation of tumour angiogenesis. Nat Rev Cancer. 2017;17(8):457–474. doi:10.1038/nrc.2017.51
13. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. doi:10.1016/j.cell.2011.02.013
14. Kessenbrock K, Plaks V, Werb Z. Matrix metalloproteinases: regulators of the tumor microenvironment. Cell. 2010;141(1):52–67. doi:10.1016/j.cell.2010.03.015
15. Yao CW, Kang KA, Piao MJ, et al. Reduced autophagy in 5-fluorouracil resistant colon cancer cells. Biomol Ther. 2017;25(3):315–320. doi:10.4062/biomolther.2016.069
16. Zhang Q, Si SH, Schoen S, et al. Suppression of autophagy enhances preferential toxicity of paclitaxel to folliculin-deficient renal cancer cells. J Exp Clin Cancer Res. 2013;32:99–109. doi:10.1186/1756-9966-32-99
17. Zhang SF, Wang XY, Fu ZQ, et al. TXNDC17 promotes paclitaxel resistance via inducing autophagy in ovarian cancer. Autophagy. 2015;11(2):225–238. doi:10.1080/15548627.2014.998931
18. Yang ZNJ, Chee CE, Huang SB, Sinicrope FA. The role of autophagy in cancer: therapeutic implications. Mol Cancer Ther. 2011;10(9):1533–1541. doi:10.1158/1535-7163.Mct-11-0047
19. Zhang YJ, Sha R, Zhang L, et al. Harnessing copper-palladium alloy tetrapod nanoparticle-induced pro-survival autophagy for optimized photothermal therapy of drug-resistant cancer. Nat Commun. 2018;9:4236–4248. doi:10.1038/s41467-018-06529-y
20. Peynshaert K, Manshian BB, Joris F, et al. Exploiting intrinsic nanoparticle toxicity: the pros and cons of nanoparticle-induced autophagy in biomedical research. Chem Rev. 2014;114(15):7581–7609. doi:10.1016/j.biocel.2016.11.003
21. Yang S, Wang X, Contino G, et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev. 2011;25(7):717–729. doi:10.1101/gad.2016111
22. Zhang XD, Yang Y, Liang X, et al. Enhancing therapeutic effects of docetaxel-loaded dendritic copolymer nanoparticles by co-treatment with autophagy inhibitor on breast cancer. Theranostics. 2014;4(11):1085–1095. doi:10.7150/thno.9933
23. Mathew R, Karantza-Wadsworth V, White E. Role of autophagy in cancer. Nat Rev Cancer. 2007;7(12):961–967. doi:10.1038/nrc2254
24. White E. Deconvoluting the context-dependent role for autophagy in cancer. Nat Rev Cancer. 2012;12(6):401–410. doi:10.1038/nrc3262
25. Ji TJ, Li SP, Zhang YL, et al. An MMP-2 responsive liposome integrating antifibrosis and chemotherapeutic drugs for enhanced drug perfusion and efficacy in pancreatic cancer. ACS Appl Mater Interfaces. 2016;8(5):3438–3445. doi:10.1021/acsami.5b11619
26. Ji TJ, Lang JY, Wang J, et al. Designing liposomes to suppress extracellular matrix expression to enhance drug penetration and pancreatic tumor therapy. ACS Nano. 2017;11(9):8668–8678. doi:10.1021/acsnano.7b01026
27. Prasad P, Gordijo CR, Abbasi AZ, et al. Multifunctional albumin-MnO2 nanoparticles modulate solid tumor microenvironment by attenuating hypoxia, acidosis, vascular endothelial growth factor and enhance radiation response. ACS Nano. 2014;8(4):3202–3212. doi:10.1021/nn405773r
28. Kong M, Tang JM, Qiao Q, et al. Biodegradable hollow mesoporous silica nanoparticles for regulating tumor microenvironment and enhancing antitumor efficiency. Theranostics. 2017;7(13):3276–3292. doi:10.7150/thno.19987
29. Ikeda Y, Hisano H, Nishikawa Y, Nagasaki Y. Targeting and treatment of tumor hypoxia by newly designed prodrug possessing high permeability in solid tumors. Mol Pharm. 2016;13(7):2283–2289. doi:10.1021/acs.molpharmaceut.6b00011
30. Zhang XD, Dong YC, Zeng XW, et al. The effect of autophagy inhibitors on drug delivery using biodegradable polymer nanoparticles in cancer treatment. Biomaterials. 2014;35(6):1932–1943. doi:10.1016/j.biomaterials.2013.10.034
31. Ma XW, Wu YY, Jin SB, et al. Gold nanoparticles induce autophagosome accumulation through size-dependent nanoparticle uptake and lysosome impairment. ACS Nano. 2011;5(11):8629–8639. doi:10.1021/nn202155y
32. Adiseshaiah PP, Clogston JD, McLeland CB, et al. Synergistic combination therapy with nanoliposomal C6-ceramide and vinblastine is associated with autophagy dysfunction in hepatocarcinoma and colorectal cancer models. Cancer Lett. 2013;337(2):254–265. doi:10.1016/j.canlet.2013.04.034
33. Nathan N, Taytard J, Duquesnoy P, et al. Surfactant protein A: a key player in lung homeostasis. Int J Biochem Cell Biol. 2016;81:151–155. doi:10.1016/j.biocel.2016.11.003
34. Mitsuhashi A, Goto H, Kuramoto T, et al. Surfactant protein A suppresses lung cancer progression by regulating the polarization of tumor-associated macrophages. Am J Pathol. 2013;182(5):1843–1853. doi:10.1016/j.ajpath.2013.01.030
35. Chang LC, Chang YY, Gau CS. Interfacial properties of Pluronics and the interactions between Pluronics and cholesterol/DPPC mixed monolayers. J Colloid Interface Sci. 2008;322(1):263–273. doi:10.1016/j.jcis.2008.02.051
36. Han LM, Guo J, Zhang LJ, Wang QS, Fang XL. Pharmacokinetics and biodistribution of polymeric micelles of paclitaxel with pluronic P123. Acta Pharmacol Sin. 2006;27(6):747–753. doi:10.1016/j.jcis.2008.02.051
37. Wang YZ, Li YJ, Zhang LJ, Fang XL. Pharmacokinetics and biodistribution of paclitaxel-loaded pluronic P105 polymeric micelles. Arch Pharmacal Res. 2008;31(4):530–538. doi:10.1007/s12272-001-1189-2
38. Baranwal AK, Murthy AS, Singhi SC. High-dose Oral Ambroxol for early treatment of Pulmonary acute respiratory distress syndrome: an exploratory, randomized, controlled pilot trial. J Trop Pediatr. 2015;61(5):339–350. doi:10.1093/tropej/fmv033
39. Li Q, Yao GQ, Zhu X. High-dose ambroxol reduces pulmonary complications in patients with acute cervical spinal cord injury after surgery. Neurocrit Care. 2012;16(2):267–272. doi:10.1007/s12028-011-9642-4
40. Zhang XL, Chen QY, Chen MY, et al. Ambroxol enhances anti-cancer effect of microtubule-stabilizing drug to lung carcinoma through blocking autophagic flux in lysosome-dependent way. Am J Cancer Res. 2017;7(12):2406–2421.
41. Hasegawa Y, Takahashi M, Ariki S, et al. Surfactant protein A down-regulates epidermal growth factor receptor by mechanisms different from those of surfactant protein D. J Biol Chem. 2017;292(45):18565–18576. doi:10.1074/jbc.M117.800771
42. Ridder KB, Davies-Cutting CJ, Kellaway IW. Surfactant solubility and aggregate orientation in hydrofluoroalkanes. Int J Pharm. 2005;295:57–65. doi:10.1016/j.ijpharm.2005.01.027
43. Moore TL, Rodriguez-Lorenzo L, Hirsch V, et al. Nanoparticle colloidal stability in cell culture media and impact on cellular interactions. Chem Soc Rev. 2015;44:6287–6305. doi:10.1039/c4cs00487f
44. Jain RK, Stylianopoulos T. Delivering nanomedicine to solid tumors. Nat Rev Clin Oncol. 2010;7(11):653–664. doi:10.1038/nrclinonc.2010.139
45. Kim SC, Kim DW, Shim YH, et al. In vivo evaluation of polymeric micellar paclitaxel formulation: toxicity and efficacy. J Control Release. 2001;72(1–3):191–202. doi:10.1016/s0168-3659(01)00275-9
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.