")
Back to Journals » Drug Design, Development and Therapy » Volume 8
Protective effect of picroside II on myocardial ischemia reperfusion injury in rats
Authors Wu N, Li W, Shu W, Jia D
Received 13 February 2014
Accepted for publication 18 March 2014
Published 14 May 2014 Volume 2014:8 Pages 545—554
DOI https://doi.org/10.2147/DDDT.S62355
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Nan Wu, Wenna Li, Wenqi Shu, Dalin Jia
Department of Cardiology, The First Affiliated Hospital of China Medical University, Shenyang, People's Republic of China
Abstract: The aim of this study was to determine the effect of picroside II on myocardial ischemia reperfusion injury in rats and to explore its underlying mechanism. Isolated rat hearts underwent 30 minutes of global ischemia followed by 120 minutes of reperfusion. Different doses of picroside II (1 µM, 10 µM, and 100 µM) were given 20 minutes before ischemia. Phosphoinositide 3-kinase inhibitor (wortmannin) and nitric oxide synthase (NOS) inhibitor (L-NG-nitroarginine methyl ester) were given 10 minutes before picroside II treatment. The cardiac function, myocardial infarct size, apoptosis, myocardial nitric oxide content, the expressions of Bcl-2 and Bax, and the activation of the phosphoinositide 3-kinase/Akt/endothelial NOS pathway were evaluated. Treatment with 10 µM and 100 µM picroside II significantly improved postischemic myocardial function, reduced myocardial infarct size, inhibited apoptosis, increased myocardial NO content, upregulated Bcl-2, downregulated Bax, and increased the phosphorylation of Akt and endothelial NOS, but cardioprotection was not shown in the 1 µM picroside II treatment group and was abrogated by wortmannin and L-NG-nitroarginine methyl ester. Furthermore, cardioprotection in the 100 µM picroside II treatment group was superior to that in the 10 µM picroside II treatment group. In conclusion, the data reveals that picroside II has a significant protective effect on myocardial ischemia reperfusion injury in a dose-dependent manner, which was mediated by upregulating the phosphoinositide 3-kinase/Akt/endothelial NOS pathway to increase nitric oxide production and regulating the expressions of Bcl-2 and Bax to inhibit apoptosis.
Keywords: picroside II, ischemia reperfusion injury, apoptosis, nitric oxide, endothelial nitric oxide synthase
Introduction
Ischemic heart disease is a leading cause of morbidity and mortality worldwide.1 Restoration of blood supply to the heart immediately is a key step in avoiding further damage of the heart.2 However, the abrupt reperfusion of an ischemic myocardium can itself further damage the ischemic tissue, which is termed as myocardial ischemia reperfusion injury (MIRI).3 Although ischemic preconditioning or ischemic postconditioning – a short series of repetitive cycles of brief reperfusion and ischemia performed immediately before or after sustained ischemia – increases the resistance against sustained ischemia of longer duration, they cannot really be widely used in the clinic treatment due to their traumatic manipulation.4–7 Therefore, much greater attention has been paid to exploring new drugs to prevent ischemia reperfusion injury that are free from the trauma resulting from ischemic preconditioning and ischemic postconditioning.8
Picroside II, an iridoid glycoside found in the root of Picrorhiza scrophulariiflora Pennell (Scrophulariaceae), has been demonstrated to have multiple pharmacologic actions, including decreasing oxidative stress, inhibition of apoptosis, and downregulating the expression of related inflammatory factors.9–13 However, the cardioprotection of picroside II has still not been determined in the animal model and the mechanism of picroside II’s cardioprotection on MIRI has not been clearly elucidated.
Therefore, the present study tried to determine the cardioprotection of picroside II on isolated rat hearts subjected to ischemia reperfusion injury and to further explore whether the protective effect of picroside II on MIRI was mediated by upregulating phosphoinositide 3-kinase (PI3K)/Akt/endothelial nitric oxide synthase (eNOS) pathway to increase nitric oxide (NO) production and regulating the expressions of Bcl-2 and Bax proteins to inhibit apoptosis.
Materials and methods
Animals
One-hundred and sixty male Wistar rats purchased from the Center of Experimental Animals (China Medical University, Shenyang, People’s Republic of China), weighing 300±10 g were used in this study. All animals used in this study were treated in accordance with the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health (Bethesda, MD, USA). The study protocol was approved by the institutional ethics committee.
Drugs
Picroside II was purchased from Meilun Biotech Co, Ltd (Dalian, People’s Republic of China). Wortmannin, L-NG-nitroarginine methyl ester (L-NAME), and 2,3,5-triphenyltetrazolium chloride were purchased from Sigma-Aldrich (St Louis, MO, USA).
Heart preparation
Rats were anesthetized with an intraperitoneal injection of pentobarbital sodium (100 mg/kg). Heparin (1,500 IU/kg) was administered intravenously to prevent intracoronary clot formation. The heart was rapidly excised and immediately immersed in ice-cold heparinized modified Krebs–Henseleit (KH) solution containing 127 mmol/L NaCl, 17.7 mmol/L NaHCO3, 5.1 mmol/L KCl, 1.5 mmol/L CaCl2, 1.26 mmol/L MgCl2, and 11 mmol/L D-glucose (pH =7.4). The heart was mounted on a Langendorff perfusion apparatus and retrogradely perfused through the aorta with recirculating KH solution saturated with 95% O2+ 5% CO2 at 37°C. The heart was maintained in a thermostatic chamber at 37°C. Perfusion was maintained at a constant pressure of 75 mmHg. The fluid-filled latex balloon was inserted in the left ventricle via the left atrium for pressure measurement. The balloon was connected to a pressure transducer and inflated to an initial left ventricle end-diastolic pressure between 8–10 mmHg.
Experimental protocol
The experimental protocol is shown in Figure 1. Rats were further divided into eight groups with 20 animals per group. In all groups, the isolated rat hearts were perfused with KH solution and allowed 10 minutes of stabilization. All hearts were subjected to 30 minutes of global ischemia followed by 120 minutes of reperfusion to induce the ischemia reperfusion model.
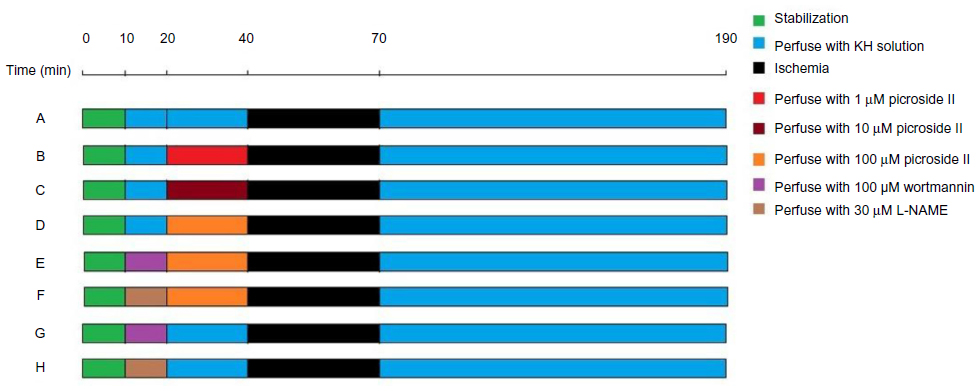 | Figure 1 Representation of experimental protocol. Rats were randomly divided into eight groups with 20 animals per group. (A) Control group; (B) 1 μM picroside II pretreatment group; (C) 10 μM picroside II pretreatment group; (D) 100 μM picroside II pretreatment group; (E) 100 μM picroside II pretreatment+wortmannin group; (F) 100 μM picroside II pretreatment+L-NAME group; (G) wortmannin control group; and (H) L-NAME control group. |
In the control group, the isolated rat heart preparations were perfused for 30 minutes with KH solution. Then, the preparations were subjected to ischemia reperfusion as described above. Three different concentrations of picroside II (1 μM, 10 μM, and 100 μM) were tested in the pretreatment groups (picroside II-1 group, picroside II-10 group, and picroside II-100 group, respectively). The isolated rat hearts were perfused for 10 minutes with KH solution, and then perfused with KH solution containing different concentrations of picroside II at the speed of 10 mL/minute for 20 minutes before ischemia.
To further determine whether the PI3K/Akt/eNOS pathway is involved in regulating the cardioprotection of picroside II on MIRI, four more groups treated with PI3K specific inhibitor (wortmannin) and NOS specific inhibitor (L-NAME) were added. In the picroside II-100+wortmannin group, the isolated rat hearts were perfused with KH solution containing 100 nM wortmannin at the speed of 10 mL/minute for 10 minutes; the rest of the procedure was the same as picroside II-100 group. In the picroside II-100+L-NAME group, the isolated rat hearts were perfused with 30 μM L-NAME at the speed of 10 mL/minute for 10 minutes; the rest of procedure was similar to the picroside II-100 group. In the wortmannin control group, wortmannin was given as described in the picroside II-100+wortmannin group, but 20 minutes of picroside II treatment was replaced by 20 minutes of perfusion with KH solution before ischemia. In the L-NAME control group, L-NAME was given as described in the picroside II-100+L-NAME group followed by perfusion with KH solution for 20 minutes instead of 20 minutes of picroside II treatment before ischemia.
Cardiac function monitoring
The assessment of cardiac function included heart rate (HR), left ventricular developed pressure (LVDP), positive first-order derivative of ventricular pressure (+dp/dt), negative first-order derivative of ventricular pressure (−dp/dt), and coronary flow (CF). These parameters were continuously monitored throughout the experimental protocol. The HR, LVDP, +dp/dt, −dp/dt, and CF were sampled and digitally processed via a homodynamic system (MP150; BIOPAC Systems, Inc., Goleta, CA, USA).
Measurement of infarct size
Infarct size was determined as previously described.14 Briefly, after 2 hours of reperfusion, the hearts were harvested, and the left ventricles were sectioned from apex to base into 2–3 mm sections following incubation for 20 minutes at 37°C in 1% 2,3,5-triphenyltetrazolium chloride; unstained tissue was carefully separated from stained tissue by an independent observer. In fact, while the unstained tissue represents the dead cells, the stained tissue represents the viable cells. The unstained mass was expressed as a percentage of total left ventricular mass. In fact, the total left ventricular mass also corresponds to the risk area because a global ischemia was induced.
Measurement of myocardial apoptosis
At the end of 120 minutes of reperfusion, the heart was removed as described earlier.14 Cardiomyocyte apoptosis was detected using an In Situ Cell Death Detection Kit (Hoffman-La Roche Ltd, Basel, Switzerland) according to the manufacturer’s instructions. Briefly, the tissue sections were washed with phosphate-buffered saline and then fixed in 4% paraformaldehyde solution before incubation in 20 μg/mL proteinase K for 10 minutes. After being washed with phosphate-buffered saline three times, the tissue sections were incubated with terminal deoxynucleotidyl transferase in a humidified chamber at 37°C for 60 minutes for incorporation of biotinylated nucleotides at the 3′-OH DNA ends. The reaction was terminated by transferring the slides to a 2× sodium citrate saline solution. Endogenous peroxidase activity was quenched by incubation in 0.3% hydrogen peroxide. Finally, streptavidin horseradish peroxidase was bound to the biotinylated nucleotides and peroxidase activity was demonstrated in each section by the application of a stable chromogen diaminobenzidine. In this technique, the apoptotic nuclei were stained dark brown. Normal nuclei were stained blue with hematoxylin. Six sections from each myocardial sample were randomly selected and ten microscopic fields (BX51 microscope; Olympus Corporation, Tokyo, Japan) per section were evaluated by two independent blind observers. In each field, the nuclei were counted and the percentage of terminal deoxynucleotidyl transferase deoxyuridine triphosphate nick end labeling-positive nuclei was calculated.
Measurement of NO content
After 30 minutes of global ischemia followed by 30 minutes of reperfusion, the hearts were removed quickly from the Langendorff apparatus and homogenized. The content of NO was measured using an NO assay kit (Jiancheng Bioengineering Institute, Nanjing, People’s Republic of China) according to the manufacturer’s instructions.
Western blotting
At the end of 30 minutes of reperfusion, the left ventricles were homogenized in a lysis buffer containing 10 mmol/L Tris-HCl, 20 orthophosphate, 1 mmol/L ethylene glycol tetraacetic acid, 1 mmol/L ethylenediaminetetraacetic acid, 2 mmol/L Na3VO4, and 1 mmol/L phenylmethylsulfonyl fluoride (pH =7.4). After sonication, the lysates were centrifuged, and the proteins were separated by electrophoresis on sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred onto polyvinylidene difluoride-plus membranes. The membranes were blocked with 5% skim milk followed by incubation overnight at 4°C with the antibodies Bcl-2 (1:1000) and Bax (1:1000; Abcam, Cambridge, UK) and Akt (1:500), β-actin (1:1000), phospho-Akt (at Ser473, 1:500), eNOS (1:500), and phospho-eNOS (at Ser1177, 1:500; Santa Cruz Biotechnology, Santa Cruz, CA, USA). After incubation, the membranes were washed three times with 0.1% Tween-20 for 15 minutes and incubated with horseradish peroxidase-conjugated goat anti-rabbit immunoglobulin G (1:2000; Santa Cruz Biotechnology) at 37°C for 2 hours. Relative densitometry was performed using a computerized software package (ImageJ 1.63; National Institutes of Health).
Statistical analysis
All results were expressed as mean ± standard deviation. Statistical analysis was performed by using SigmaStat software version 3.5 (Systat Software, Inc., San Jose, CA, USA). Differences between groups were evaluated using one-way analysis of variance, followed by Student–Newman–Keuls post hoc test. P<0.05 was considered statistically significant.
Results
Effect of picroside II on cardiac function
The values of HR, LVDP, +dp/dt, –dp/dt, and CF at baseline and different times of reperfusion are shown in Figure 2. There were no significant differences in the values of HR, LVDP, +dp/dt, –dp/dt, and CF at baseline among groups. In the picroside II-10 group and picroside II-100 group, the values of HR, LVDP, +dp/dt, –dp/dt, and CF were significantly increased compared with the control group (P<0.05). Furthermore, all values in the picroside II-100 group were higher than those in the picroside II-10 group (P<0.05). However, no significant differences in the values of cardiac function parameters were observed in the picroside II-1 group compared with the control group (P>0.05).
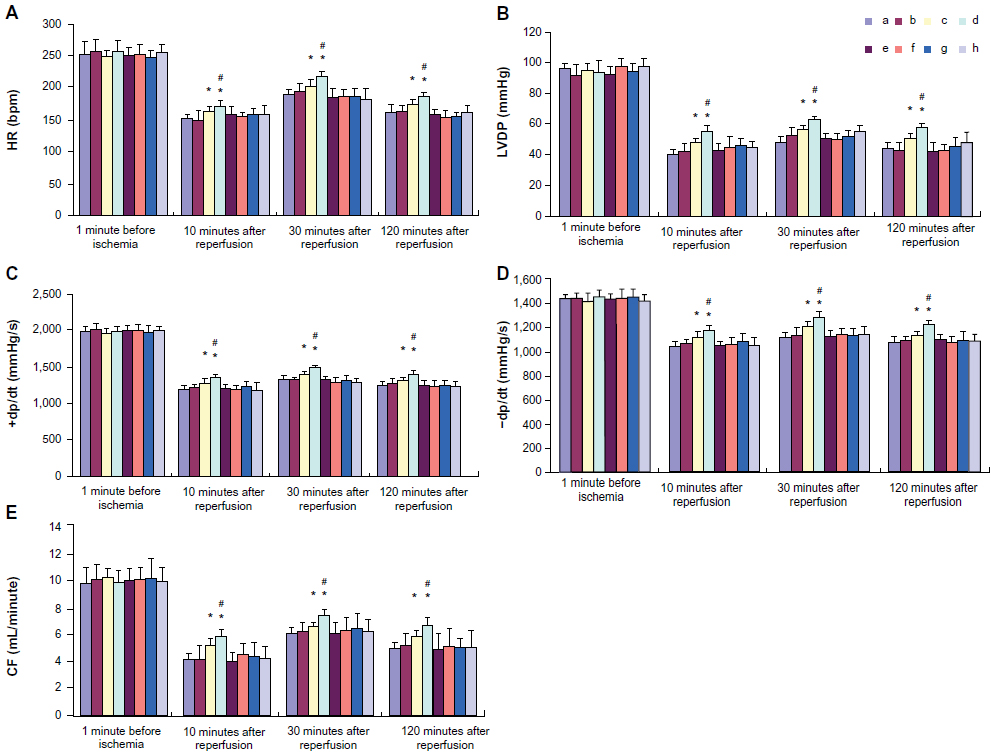 | Figure 2 Cardiac function parameters before and during reperfusion. (A) Control group; (B) 1 μM picroside II pretreatment group; (C) 10 μM picroside II pretreatment group; (D) 100 μM picroside II pretreatment group; (E) 100 μM picroside II pretreatment+wortmannin group; (F) 100 μM picroside II pretreatment+L-NAME group; (G) wortmannin control group; and (H) L-NAME control group. |
Effect of picroside II on infarct size
As shown in Figure 3, the infarct size was significantly reduced in the picroside II-10 group and picroside II-100 group as compared with that in the control group (36.3%±8.9% and 26.0%±6.4%, respectively, versus 46.5%±7.6%; P<0.05), but no significant reduction of infarct size was observed in the picroside II-1 group as compared with the control group (44.4%±7.9% versus 46.5%±7.6%; P>0.05). Meanwhile, the infarct size was further reduced in the picroside II-100 group compared with the picroside II-10 group (26.0%±6.4% versus 36.3%±8.9%; P<0.05). These results suggested that picroside II reduced infarct size in a dose-dependent manner.
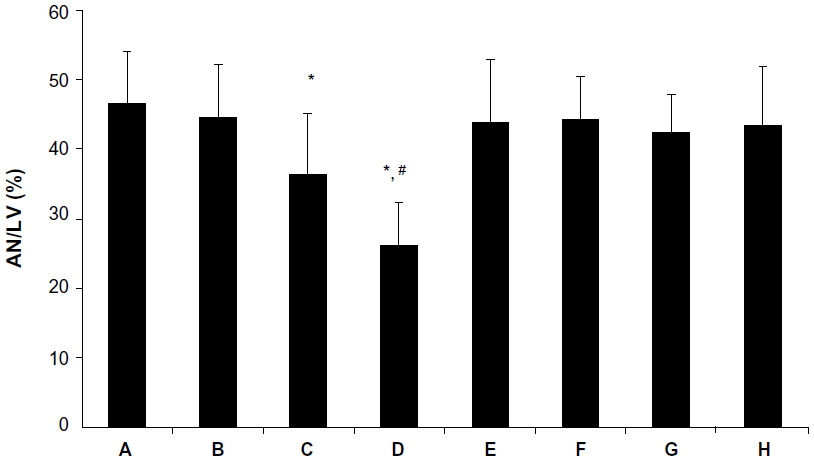 | Figure 3 AN expressed as a percentage of LV area. (A) Control group; (B) 1 μM picroside II pretreatment group; (C) 10 μM picroside II pretreatment group; (D) 100 μM picroside II pretreatment group; (E) 100 μM picroside II pretreatment+wortmannin group; (F) 100 μM picroside II pretreatment+L-NAME group; (G) wortmannin control group; and (H) L-NAME control group. |
Effect of picroside II on apoptosis
The percentage of cardiomyocyte apoptosis in all experimental groups is shown in Figure 4. As compared with the control group, the percentage of cardiomyocyte apoptosis was significantly reduced in the picroside II-10 group and picroside II-100 group (29.3%±5.9% and 21.5%±4.8%, respectively, versus 38.6%±3.7%; P<0.05), but no significant decrease was observed in the picroside II-1 group (37.5%±4.8% versus 38.6%±3.7%; P>0.05). Moreover, the anti-apoptosis effect of picroside II was also in a dose-dependent manner, as evidenced by a lower percentage of cardiomyocyte apoptosis in the picroside II-100 group compared with the picroside II-10 group (21.5%±4.8% versus 29.3%±5.9%; P<0.05).
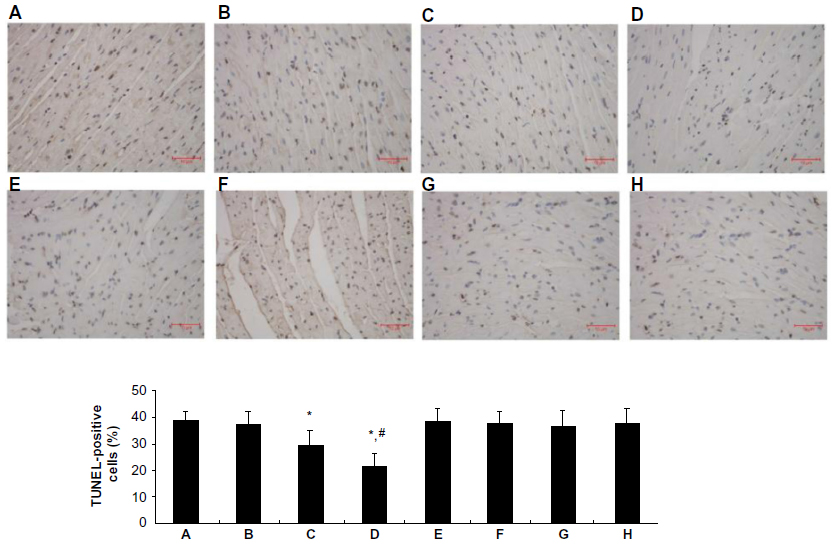 | Figure 4 Apoptosis after myocardial infarction. Photomicrographs were taken at 400× magnification. Apoptotic cardiomyocyte nuclei appear brown stained whereas TUNEL-negative nuclei appear blue. Histogram shows the percentage of TUNEL-positive cells (brown staining). (A) Control group; (B) 1 μM picroside II pretreatment group; (C) 10 μM picroside II pretreatment group; (D) 100 μM picroside II pretreatment group; (E) 100 μM picroside II pretreatment+wortmannin group; (F) 100 μM picroside II pretreatment+L-NAME group; (G) wortmannin control group; and (H) L-NAME control group. |
Effect of picroside II on NO content in rat myocardium
As shown in Figure 5, the content of NO in rat myocardium significantly increased in the picroside II-10 group and picroside II-100 group compared with the control group (4.40±1.08 μmol/g and 6.52±1.53 μmol/g, respectively, versus 2.62±0.64 μmol/g; P<0.05), however, the content of NO did not increase in the picroside II-1 group (2.75±1.19 μmol/g versus 2.62±0.64 μmol/g; P>0.05). Besides, the content of NO in the picroside II-100 group was remarkably higher compared with the picroside II-10 group (6.52±1.53 μmol/g versus 4.40±1.08 μmol/g; P<0.05), which suggests that picroside II could also increase the content of NO in ischemic myocardium in a dose-dependent manner.
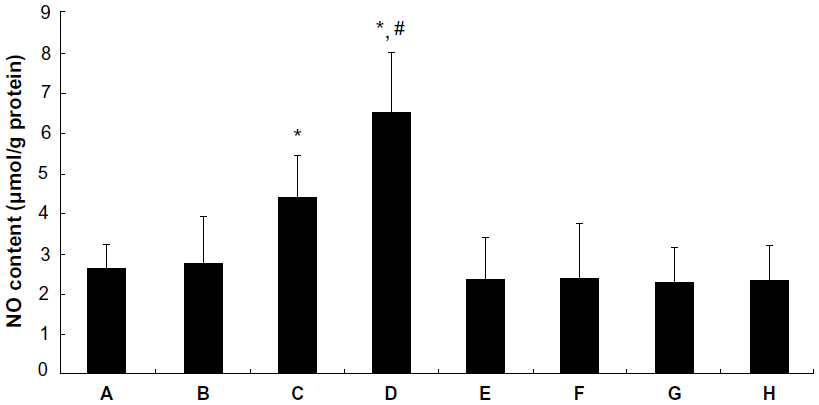 | Figure 5 The content of NO in myocardium. (A) Control group; (B) 1 μM picroside II pretreatment group; (C) 10 μM picroside II pretreatment group; (D) 100 μM picroside II pretreatment group; (E) 100 μM picroside II pretreatment+wortmannin group; (F) 100 μM picroside II pretreatment+L-NAME group; (G) wortmannin control group; and (H) L-NAME control group. |
Expression of Bcl-2 and Bax proteins
In order to elucidate the underlying mechanism of the anti-apoptosis effect of picroside II, the expression of apoptosis-related proteins Bcl-2 and Bax was further examined (Figure 6). As compared with the control group, there was a significant increase in the expression of Bcl-2 in the picroside II-10 group and picroside II-100 group (0.446±0.022 and 0.584±0.023, respectively, versus 0.140±0.015; P<0.05) (Figure 6B), whereas a remarkable decrease in the expression of Bax was shown in these two groups (0.399±0.019 and 0.193±0.034, respectively, versus 0.495±0.026; P<0.05) (Figure 6C) compared with the control group, which is consistent with previous data derived from the terminal deoxynucleotidyl transferase deoxyuridine triphosphate nick end labeling staining for apoptosis (Figure 4). These results reveal that picroside II could effectively upregulate the expression of Bcl-2 as well as downregulate the expression of Bax.
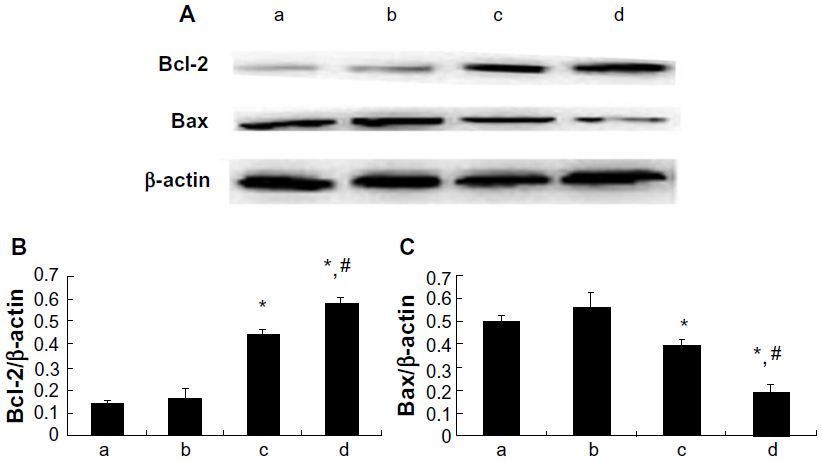 | Figure 6 Myocardial Bcl-2 and Bax expression. (A) Bcl-2 and Bax protein expressions detected by Western blotting; (B) Relative protein expression level of Bcl-2; (C) Relative protein expression level of Bax; (a) control group; (b) 1 μM picroside II pretreatment group; (c) 10 μM picroside II pretreatment group; and (d) 100 μM picroside II pretreatment group. |
Expression of Akt and eNOS proteins
As shown in Figure 7, treatment with 10 μM and 100 μM picroside II resulted in a significant increase in the phosphorylation of Akt compared with the control group (0.316±0.019 and 0.414±0.020, respectively, versus 0.186±0.011; P<0.05). Figure 8 shows a similar increase in the phosphorylation of eNOS (0.342±0.037 and 0.503±0.021, respectively, versus 0.253±0.021; P<0.05). However, there was no significant change in total expressions of Akt and eNOS in the picroside II-10 group and picroside II-100 group (Figures 7C and 8C). Moreover, the levels of phosphorylated Akt and eNOS were higher in the picroside II-100 group as compared with that in the picroside II-10 group. These results were in parallel with the results obtained with measurement of NO content in rat myocardium, indicating that picroside II increases NO production by upregulating the PI3K/Akt/eNOS pathway.
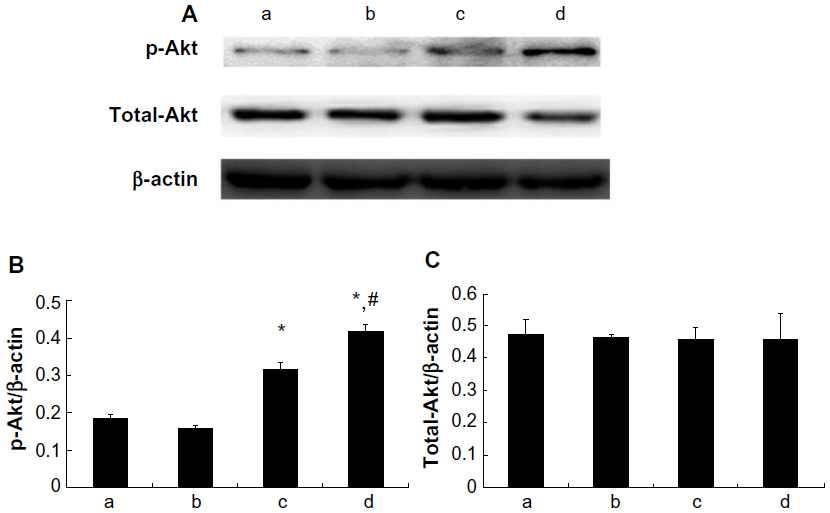 | Figure 7 Myocardial total-Akt and p-Akt expression. (A) p-Akt and total-Akt protein expressions detected by Western blotting; (B) Relative protein expression level of p-Akt; (C) Relative protein expression level of total-Akt; (a) control group; (b) 1 μM picroside II pretreatment group; (c) 10 μM picroside II pretreatment group; and (d) 100 μM picroside II pretreatment group. |
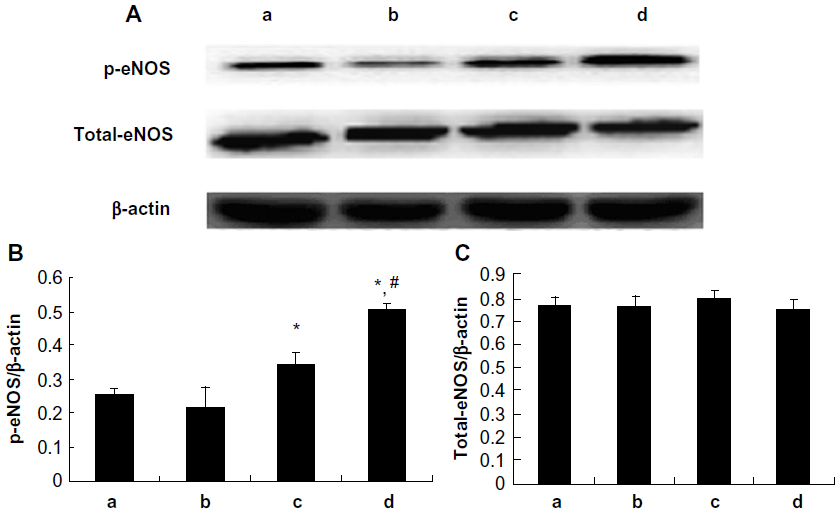 | Figure 8 Myocardial total-eNOS and p-eNOS expression. (A) p-eNOS and total-eNOS protein expressions detected by Western blotting; (B) Relative protein expression level of p-eNOS; (C) Relative protein expression level of total-eNOS; (a) control group; (b) 1 μM picroside II pretreatment group; (c) 10 μM picroside II pretreatment group; and (d) 100 μM picroside II pretreatment group. |
Effects of wortmannin and L-NAME on the cardioprotection of picroside II
To further assess the role of PI3K/Akt/eNOS pathway in the protective effect of picroside II, a PI3K specific inhibitor (wortmannin) and an NOS specific inhibitor (L-NAME) was given to the 100 μM picroside II treatment group. The results show that the cardioprotection of picroside II was abrogated by treatment with wortmannin or L-NAME alone, as evidenced by no significant differences in hemodynamic parameters (Figure 2), myocardial infarct size (Figure 3), the percentage of cardiomyocyte apoptosis (Figure 4), and the content of NO (Figure 5) in the picroside II-100+wortmannin group and picroside II-100+L-NAME group compared with the control group.
Discussion
Picroside II, one of the effective components extracted from the traditional Chinese medicine rhizoma coptidis hu, has already been demonstrated to have multiple protective effects, including alleviating hepatocyte oxidative stress damage, inhibiting cerebral ischemic reperfusion injury, and reducing the expression of related inflammatory factors.9–13 Moreover, a recent study has also shown that picroside II could protect cardiomyocytes against oxidative stress and apoptosis induced by hypoxia/reoxygenation injury.15 However, the cardioprotection of picroside II has still not been demonstrated in animal models. In the present study, it was found that picroside II pretreatment significantly increased the values of LVDP, +dp/dt, −dp/dt, and CF, and decreased the size of infarction, which suggests that picroside II could produce a protective effect on MIRI in rats.
One new finding of this study is that picroside II remarkably increased the content of NO in ischemic myocardium. Therefore, it is speculated that picroside II improves postischemic cardiac functional recovery as a result of an increase in NO production because NO can reinforce myocardial contractility.16,17 The main source of cardiac NO is generated through eNOS, which is expressed by coronary endothelial cells and cardiomyocytes and regulated by PI3K/Akt.18 Consequently, the influence of picroside II on the activation of the PI3K/Akt/eNOS signal pathway was further explored. As expected, the levels of Akt and eNOS phosphorylation in the picroside II treatment group were remarkably higher than in the picroside II untreated group. The results suggest that picroside II increased the synthesis of NO in an ischemic rat heart through upregulation of the PI3K/Akt/eNOS signal pathway. Meanwhile, this speculation was further confirmed as evidenced by the beneficial protection of picroside II being abolished by the administration of wortmannin and L-NAME. According to these results, it can be concluded that picroside II protects the heart against MIRI through activating the PI3K/Akt/eNOS signal pathway, leading to an increase in the synthesis of NO.
In addition, over-apoptosis is believed to be a key factor contributing to the process of MIRI, and one important molecular mechanism for cell apoptosis might be the modulation of the expression of the bcl-2 gene family, which plays a critical role for the common pathway of apoptosis.19,20 Thus, the rate of apoptosis and the expressions of Bcl-2 and Bax was detected in all experimental groups. The results show that picroside II significantly inhibited cardiomyocyte apoptosis induced by ischemia reperfusion injury, upregulated the expression of Bcl-2, and downregulated the expression of Bax, which agrees with the previous founding of Meng et al in a myocardium hypoxia/reoxygenation model.21 Therefore, it is speculated that Bcl-2 and Bax are involved in mediating the anti-apoptotic effects of picroside II on MIRI.
Consistent with most previous studies, the cardioprotection of picroside II is also in a dose-dependent manner.22 In the present study, the results revealed that the 1 μM picroside II treatment group did not show any beneficial effects in postischemic cardiac functional recovery, the size of infarction, the rate of apoptosis, and the content of NO, but the 10 μM and 100 μM picroside II treatment groups displayed a protective effect to a certain extent. Moreover, cardioprotection in the 100 μM picroside II treatment group is superior to the 10 μM picroside II treatment group as evidenced by the smaller size of infarction, less apoptosis, and greater NO production in the 100 μM picroside II treatment group compared with the 10 μM picroside II treatment group. Despite the fact that 100 μM picroside II achieved the maximum protective effect in the present study, the optimal dose of picroside II for preventing MIRI still needs to be further determined.
Conclusion
This study demonstrated that picroside II has a significant protective effect on MIRI in isolated rat hearts, which was mediated by upregulating the PI3K/Akt/eNOS pathway to increase NO production on the one hand and regulating the expression of Bcl-2 and Bax to inhibit apoptosis on the other.
Acknowledgment
This study is supported by Liaoning Provincial Science and Technology Projects, People’s Republic of China (2013021011).
Disclosure
The authors report no conflict of interests in this work.
References
Mathers CD, Loncar D. Projections of global mortality and burden of disease from 2002 to 2030. PLoS Med. 2006;3(11):e442. | |
Doorey AJ, Michelson EL, Topol EJ. Thrombolytic therapy of acute myocardial infarction. Keeping the unfulfilled promises. JAMA. 1992;268(21):3108–3114. | |
Jennings RB. Historical perspective on the pathology of myocardial ischemia/reperfusion injury. Circ Res. 2013;113(4):428–438. | |
Murry CE, Jennings RB, Reimer KA. Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation. 1986;74(5):1124–1136. | |
Zhao ZQ, Corvera JS, Halkos ME, et al. Inhibition of myocardial injury by ischemic postconditioning during reperfusion: comparison with ischemic preconditioning. Am J Physiol Heart Circ Physiol. 2003;285(2):H579–H588. | |
Ovize M, Thibault H, Przyklenk K. Myocardial conditioning: opportunities for clinical translation. Circ Res. 2013;113(4):439–450. | |
Ferdinandy P, Schulz R, Baxter GF. Interaction of cardiovascular risk factors with myocardial ischemia/reperfusion injury, preconditioning, and postconditioning. Pharmacol Rev. 2007;59(4):418–458. | |
Andreadou I, Iliodromitis EK, Koufaki M, Kremastinos DT. Pharmacological pre- and post-conditioning agents: reperfusion-injury of the heart revisited. Mini Rev Med Chem. 2008;8(9):952–959. | |
Li T, Liu JW, Zhang XD, Guo MC, Ji G. The neuroprotective effect of picroside II from hu-huang-lian against oxidative stress. Am J Chin Med. 2007;35(4):681–691. | |
Cao Y, Liu JW, Yu YJ, et al. Synergistic protective effect of picroside II and NGF on PC12 cells against oxidative stress induced by H2O2. Pharmacol Rep. 2007;59(5):573–579. | |
Gao H, Zhou YW. Inhibitory effect of picroside II on hepatocyte apoptosis. Acta Pharmacol Sin. 2005;26(6):729–736. | |
Li Q, Li Z, Xu XY, Guo YL, Du F. Neuroprotective properties of picroside II in a rat model of focal cerebral ischemia. Int J Mol Sci. 2010;11(11):4580–4590. | |
Guo Y, Xu X, Li Q, Li Z, Du F. Anti-inflammation effects of picroside II in cerebral ischemic injury rats. Behav Brain Funct. 2010;6:43. | |
Wu N, Zhang X, Guan Y, Shu W, Jia P, Jia D. Hypercholesterolemia abrogates the cardioprotection of ischemic postconditioning in isolated rat heart: roles of glycogen synthase kinase-3β and the mitochondrial permeability transition pore. Cell Biochem Biophys. Epub November 10, 2013. | |
Meng FJ, Hou ZW, Li Y, Yang Y, Yu B. The protective effect of picroside II against hypoxia/reoxygenation injury in neonatal rat cardiomyocytes. Pharm Biol. 2012;50(10):1226–1232. | |
Kitakaze M, Node K, Minamino T, et al. Role of nitric oxide in regulation of coronary blood flow during myocardial ischemia in dogs. J Am Coll Cardiol. 1996;27(7):1804–1812. | |
Beresewicz A, Karwatowska-Prokopczuk E, Lewartowski B, Cedro-Ceremuazynska K. A protective role of nitric oxide in isolated ischaemic/reperfused rat heart. Cardiovasc Res. 1995;30(6):1001–1008. | |
Dimmeler S, Fleming I, Fisslthaler B, Hermann C, Busse R, Zeilher AM. Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature. 1999;399(6736):601–605. | |
Gottlieb RA, Burleson KO, Kloner RA, Babior BM, Engler RL. Reperfusion injury induces apoptosis in rabbit cardiomyocytes. J Clin Invest. 1994;94(4):1621–1628. | |
Xie Z, Koyama T, Suzuki J, et al. Coronary reperfusion following ischemia: different expression of Bcl-2 and Bax proteins, and cardiomyocyte apoptosis. Jpn Heart J. 2001;42(6):759–770. | |
Meng FJ, Jiao SM, Yu B. Picroside II protects cardiomyocytes from hypoxia/reoxygenation-induced apoptosis by activating the PI3K/Akt and CREB pathways. Int J Mol Med. 2012;30(2):263–270. | |
Pei H, Su X, Zhao L, et al. Primary study for the therapeutic dose and time window of picroside II in treating cerebral ischemic injury in rats. Int J Mol Sci. 2012;13(3):2551–2562. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.