")
Back to Journals » Drug Design, Development and Therapy » Volume 10
Protective effect of fucoidan from Fucus vesiculosus on liver fibrosis via the TGF-β1/Smad pathway-mediated inhibition of extracellular matrix and autophagy
Authors Li J, Chen K , Li S, Feng J , Liu T, Wang F, Zhang R, Xu S, Zhou Y, Zhou S, Xia Y, Lu J, Zhou Y, Guo C
Received 20 October 2015
Accepted for publication 22 December 2015
Published 12 February 2016 Volume 2016:10 Pages 619—630
DOI https://doi.org/10.2147/DDDT.S98740
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Wei Duan
Jingjing Li,1 Kan Chen,1 Sainan Li,1 Jiao Feng,1 Tong Liu,1 Fan Wang,1 Rong Zhang,1,2 Shizan Xu,1,2 Yuqing Zhou,1,3 Shunfeng Zhou,1,3 Yujing Xia,1 Jie Lu,1 Yingqun Zhou,1 Chuanyong Guo1
1Department of Gastroenterology, Shanghai Tenth People’s Hospital, Tongji University School of Medicine, Shanghai, 2The First Clinical Medical College of Nanjing Medical University, Nanjing, 3Department of Gastroenterology, The First Affiliated Hospital of Soochow University, Suzhou, People’s Republic of China
Abstract: Liver fibrosis is a dynamic reversible pathological process in the development of chronic liver disease to cirrhosis. However, the current treatments are not administered for a long term due to their various side effects. Autophagy is initiated to decompose damaged or excess organelles, which had been found to alter the progression of liver fibrosis. In this article, we hypothesized that fucoidan from Fucus vesiculosus may attenuate liver fibrosis in mice by inhibition of the extracellular matrix and autophagy in carbon tetrachloride- and bile duct ligation-induced animal models of liver fibrosis. The results were determined using enzyme-linked immunosorbent assay, quantitative real-time polymerase chain reaction, Western blotting, and immunohistochemical staining. Fucoidan from F. vesiculosus could inhibit the activation of hepatic stellate cells and the formation of extracellular matrix and autophagosomes, and its effect may be associated with the downregulation of transforming growth factor beta 1/Smads pathways. Fucoidan, as an autophagy and transforming growth factor beta 1 inhibitor, could be a promising potential therapeutic agent for liver fibrosis.
Keywords: liver cirrhosis, hepatic stellate cells, bile duct ligation
Introduction
Liver fibrosis, a dynamic reversible pathological process that can cause chronic liver disease to develop into cirrhosis (mainly seen in intrahepatic fibrous dysplasia), results in the production and deposition of extracellular matrix (ECM) and an imbalance between synthesis and degradation.1 Approximately 70% of patients with chronic liver disease have associated liver fibrosis. If the process is not stopped, 25% will deteriorate further to decompensated cirrhosis accompanied by digestive tract hemorrhage, hepatic encephalopathy, and hepatorenal syndrome.2–4 The current treatments to tackle liver fibrosis include antiviral, antioxidant, and immune regulation therapies that are not administered in the long term due to their various side effects.5,6 Therefore, safe and effective screening of drugs used to control liver fibrosis plays an important role in cirrhosis and hepatoma prevention.
Hepatic ECM contains bioactive substances. Under normal physiological conditions, the ECM provides nutrition and is involved in the immune response. When stimulated by various cytokines, the ECM evolves to liver fibrosis due to synthesis of its components, including collagen I, fibronectin, laminin (LN), and hyaluronic acid, or reduced degradation induced by an imbalance between matrix metalloproteinases (MMPs) and tissue inhibitor of matrix metalloproteinases (TIMPs).7,8 A number of studies have demonstrated that the activation and proliferation of hepatic stellate cells (HSCs) are the key points in the production of ECM and includes the participation of other cells such as hepatic sinus endothelial cells, Kupffer cells, neutrophils, T-cells, and natural killer cells.1,7,9,10 HSCs differentiate into myofibroblasts (MFBs) with abundant production of α-smooth muscle actin (α-SMA) and ECM following liver damage, eventually leading to liver fibrosis.11 The related signaling pathways that influence each other in this process mainly include the transforming growth factor beta (TGF-β), platelet-derived growth factor, Wnt/β-catenin, Toll-like receptor, and other important pathways.7,12 The establishment of animal models that simulate the pathogenic mechanism has always been difficult, and commonly used drugs that induce liver fibrosis include carbon tetrachloride (CCl4) and dimethylnitrosamine. Another liver fibrosis model is induced by cholestasis via surgery with bile duct ligation (BDL).13–15 CCl4 can lead to the degeneration and necrosis of hepatic cells by directly dissolving liver cell membranes to initiate lipid peroxidation, which is similar to human liver fibrosis in morphology and pathophysiology.16 The CCl4-induced liver fibrosis model is widely used due to its stable pathological characteristics and good repeatability, while BDL-induced disease is akin to the biochemical and histological changes in human small nodular cirrhosis.17 These two animal models were jointly adopted in the present study to conduct drug screening for liver fibrosis.
Autophagy, defined by Ashford and Porter18 using an electron microscope when observing human liver cells, is a naturally occurring physiological process in which damaged or excess organelles are destroyed, to allow the synthesis of new organelles in the absence of nutrients and energy.18–21 Programmed cell death plays an essential role in acute liver damage, ischemia reperfusion, and other systemic diseases; however, less research has been carried out on liver fibrosis.22–27 In recent years, numerous studies have found that autophagy can alter the progression of liver fibrosis. Hernandez-Gea et al found that cells gained energy to supply materials for the activation of HSCs following lipid autophagic degradation.28 Liu et al showed that the autophagy inhibitor 3-methyladenine affected the proliferation and activation of HSCs to alleviate fibrosis.29 Therefore, autophagy could be regarded as a target in the prevention of liver fibrosis. Fucoidan, a polysaccharide extracted from brown seaweed, was reported to have many benefits, such as anti-inflammation and anticancer activities, and it affects oxidative stress and vascular physiology.30–32 In 2008, Hayashi et al33 demonstrated the protective effect of fucoidan on CCl4-induced liver fibrosis, while Hong et al15 reported that fucoidan could protect against liver fibrogenesis induced by dimethylnitrosamine through the inhibition of oxidative stress and inflammatory cytokine release.13,15 However, it is unclear whether fucoidan can ameliorate CCl4- and BDL-induced liver fibrosis as autophagy-targeting drugs, and its relationship with the TGF-β/Smad pathway is unknown.
In the present study, we hypothesized that fucoidan from Fucus vesiculosus may attenuate liver fibrosis in mice by inhibition of the ECM and autophagy in CCl4- and BDL-induced animal models of liver fibrosis. The potential mechanism of action of fucoidan was also investigated.
Materials and methods
Mice and fibrosis induction
Male C57 mice weighing 20–22 g were purchased from Shanghai Laboratory Animal Co., Ltd. (Shanghai, People’s Republic of China). All animals were housed in an air-conditioned room at a temperature of 25°C with a 12-hour alternating light and dark cycle and permitted free access to food and water. In the liver fibrosis models, mice were injected intraperitoneally with 1 mL/kg body weight of CCl4 (1:10 v/v; Sigma-Aldrich Co., St Louis, MO, USA) in olive oil twice a week for 8 weeks.33 Fucoidan (Sigma-Aldrich Co.) was diluted in saline and was administered orally at daily doses of 10 mg/kg, 25 mg/kg, and 50 mg/kg once a day. In BDL-induced liver fibrosis, all mice were anesthetized with 1.25% Nembutal (Sigma-Aldrich Co.) after a 24-hour fast. When the reflection and pain stimulation disappeared, the abdominal cavity was opened along the linea alba. A wet swab was used to expose the diaphragm and umbilical region and separate them from the bile duct, flank portal vein, and hepatic artery. The bile duct was ligated with two surgical knots using 5-0 suture and cut in between the two knots.
The abdominal cavity was washed with saline, and the two abdominal layers and skin were closed.17 The mice were fed in a warm and humid environment until fully awake and active and treated with and without drugs for 2 weeks. All animal experiments were performed in accordance with legal regulations and approved by the Animal Care and Use Committee of Shanghai Tongji University. Serum and liver tissues were obtained for the following determinations:
Mice with CCl4-induced liver fibrosis were randomly divided into six groups (eight mice per group) as follows:
- control (CCl4) group: saline by gavage;
- fucoidan (CCl4) group: fucoidan (50 mg/kg) daily by gavage;
- CCl4 group: CCl4 injected intraperitoneally;
- CCl4 + fucoidan (10) group: CCl4 injected intraperitoneally with fucoidan (10 mg/kg) daily by gavage;
- CCl4 + fucoidan (25) group: CCl4 injected intraperitoneally with fucoidan (25 mg/kg) daily by gavage; and
- CCl4 + fucoidan (50) group: CCl4 injected intraperitoneally with fucoidan (50 mg/kg) daily by gavage.
The BDL-induced liver fibrosis mice were randomly divided into groups as described earlier, including the control (BDL) group, fucoidan (BDL) group, BDL group, BDL + fucoidan (10) group, BDL + fucoidan (25) group, and BDL + fucoidan (50) group (eight mice per group).
Serum analysis
Serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST) were determined using an automated chemistry analyzer (Olympus AU1000; Olympus Corporation, Tokyo, Japan). Hydroxyproline and LN were determined using kits according to the manufacturers’ instructions.
Hematoxylin and eosin staining and Masson’s trichrome staining
Fresh liver samples were embedded in paraffin for histological examination using hematoxylin and eosin (H&E) and Masson’s trichrome staining. Sections (5 μm thick) were cut and prepared for further experiments. Collagen fiber stained blue, cytoplasm stained red, and nucleus stained blue-violet by Masson dye were observed under a light microscope with a digital camera (Leica Microsystems, Wetzlar, Germany).
Real-time polymerase chain reaction
Total RNA was extracted from frozen liver tissue by the pyrolysis of Trizol, chloroform, isopropyl alcohol, and ethanol (Thermo Fisher Scientific, Waltham, MA, USA) and transcribed into cDNA using the reverse transcription kit (TaKaRa Biotechnology, Dalian, People’s Republic of China). Real-time polymerase reaction (PCR) was carried out according to the protocols described by SYBR Premix EX Taq (TaKaRa Biotechnology) using a 7900HT Fast Real-Time PCR system (Thermo Fisher Scientific). The ratio of target genes and β-actin was calculated on the basis of the solubility curve. All primers used in the experiments are shown in Table 1.
 | Table 1 Nucleotide sequences of the primers used for qRT-PCR |
Western blotting analysis
The total tissue protein was extracted by radio immunoprecipitation assay lysis buffer with phenylmethanesulfonyl fluoride, and the concentration was calculated using the bicinchoninic acid protein assay (Kaiji, People’s Republic of China). Equal amounts of samples were added to 8%–12.5% sodium dodecyl sulfate-polyacrylamide gels and transferred onto polyvinylidene fluoride membranes that were sequentially blocked with defatted milk and incubated with specific primary and secondary antibodies. The primary antibodies used were as follows: α-SMA, TIMP1, MMP-9, Co-I, Beclin-1, P62 (1:500, all from Proteintech Group, Inc., Chicago, IL, USA); TGF-β1, smad2, p-smad2, LC3, β-actin (1:1,000, all from Cell Signaling Technology, Danvers, MA, USA); and smad3, p-smad3 (1:500, both from ABclonal, Cambridge, MA, USA). The Odyssey two-color infrared laser imaging system (LI-COR Biosciences, Lincoln, NE, USA) was used to scan the membranes for image acquisition.
Immunohistochemical staining
The prepared paraffin sections were dewaxed and rehydrated with different concentrations of alcohol. After washing with phosphate-buffered saline solution, the sections were pretreated using a microwave antigen retrieval technique with heating to 95°C and cooling to room temperature (four cycles). Antibodies, including α-SMA (1:100), TIMP1 (1:100), MMP-9 (1:100), Co-I (1:100), Beclin-1 (1:100), TGF-β1 (1:200), p-smad2 (1:100), LC3 (1:50), and p-smad3 (1:100), were added to the slices and then incubated at 4°C overnight. Following incubation with secondary antibody, a DAB kit was used and positive areas were observed by a microscope with a digital camera.
Electron microscopy
The liver tissues were prefixed with 3% glutaraldehyde buffered with 0.2 mmol/L cacodylate for 4 hours and postfixed in 1% osmium tetroxide for 1 hour. Autophagosomes were viewed by electron microscopy (JEM-1230; JEOL, Tokyo, Japan), and images were acquired.
Statistical analysis
The statistical analysis was performed using the SPSS 20.0 software package (IBM Corporation, Armonk, NY, USA). All data were compared by calculating the mean ± standard deviation using the Student–Newman–Keuls test or one-way analysis of variance. A P-value <0.05 was considered statistically significant. The positively stained areas were evaluated by integrated optical density and analyzed using Image-Pro Plus 6.0 (Media Cybernetics, Silver Spring, MD, USA).
Results
Fucoidan improved the liver function in liver fibrosis
ALT and AST are important indices for reflecting the status of liver function. We conducted a series of experiments to clarify the effect of fucoidan on liver enzyme levels. The serum levels of liver enzymes in the CCl4 and BDL groups were significantly elevated, which indicated serious liver damage. With increasing fucoidan dose (10 mg/kg, 25 mg/kg, and 50 mg/kg), the levels of ALT and AST declined gradually. However, 10 mg/kg of fucoidan had no obvious influence on the fibrosis caused by CCl4. Hydroxyproline, which can reflect the severity of liver fibrosis, is a characteristic amino acid in the synthesis of collagens. The hydroxyproline levels in liver tissue were higher in the model groups (both BDL and CCl4 groups) and decreased with increasing fucoidan concentration (Figure 1A). H&E and Masson staining were used to determine general morphology and collagen fibers in liver tissues. According to the results of H&E staining, the model groups showed inflammatory infiltrates, degeneration and necrosis in liver cells around the central vein, and connective tissue hyperplasia. The degree of damage in the fucoidan treatment groups was reduced. Consistent with the changes in liver enzymes and H&E staining, Masson staining of liver sections showed blue areas of collagen fiber and red areas of cytoplasm. Collagen fiber in the fucoidan groups showed a decreasing trend relative to the model groups (Figure 1B). These results indicated that different concentrations of fucoidan significantly alleviated liver fibrosis in a dose-dependent manner.
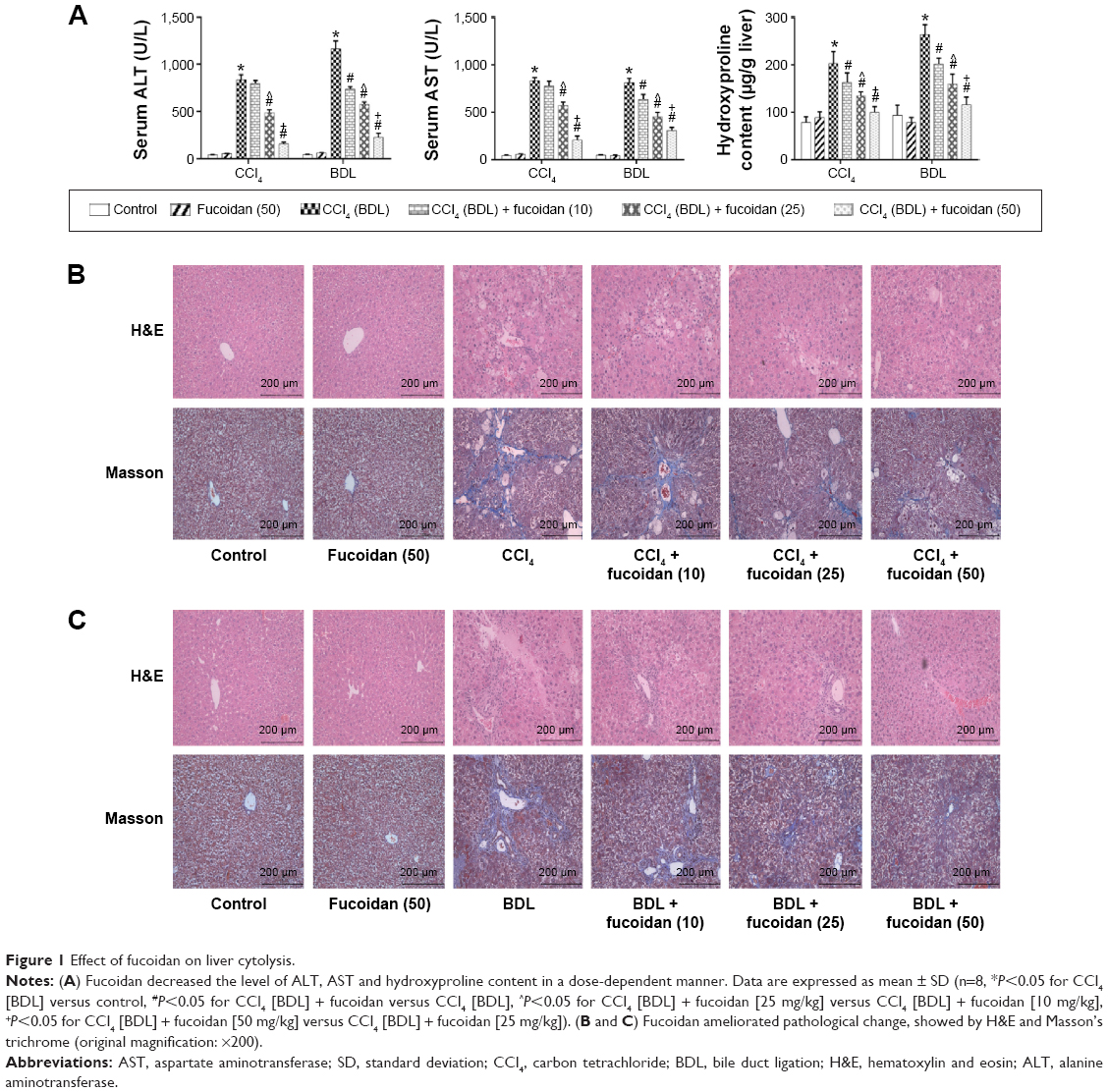 | Figure 1 Effect of fucoidan on liver cytolysis. |
Fucoidan inhibited the formation of ECM
Hyaluronic acid, fibronectin, LN, collagen I, α-SMA, MMPs, and TIMPs are involved in the synthesis of ECM. Enzyme-linked immunosorbent assay kits were used to detect serum hyaluronic acid and LN, and the results were consistent with those for ALT and AST. Increased serum levels in both model groups indicated severe fibrosis, and decreased levels in the fucoidan groups indicated attenuation of liver lesions (Figures 1A and 2A). PCR and Western blot were used to determine changes in the gene and protein levels of ECM components in liver tissues in the different treatment groups, respectively (Figure 2B and C). The results showed increased expression of collagen I, α-SMA, and TIMP1 in the model groups, and fucoidan inhibited this increase. Similarly, immunohistochemical staining of liver biopsies showed that the changes in these components in liver tissues were consistent with the results for gene and protein expression (Figure 2D). These results demonstrate that the production of ECM was inhibited by fucoidan to block the progression of liver fibrosis.
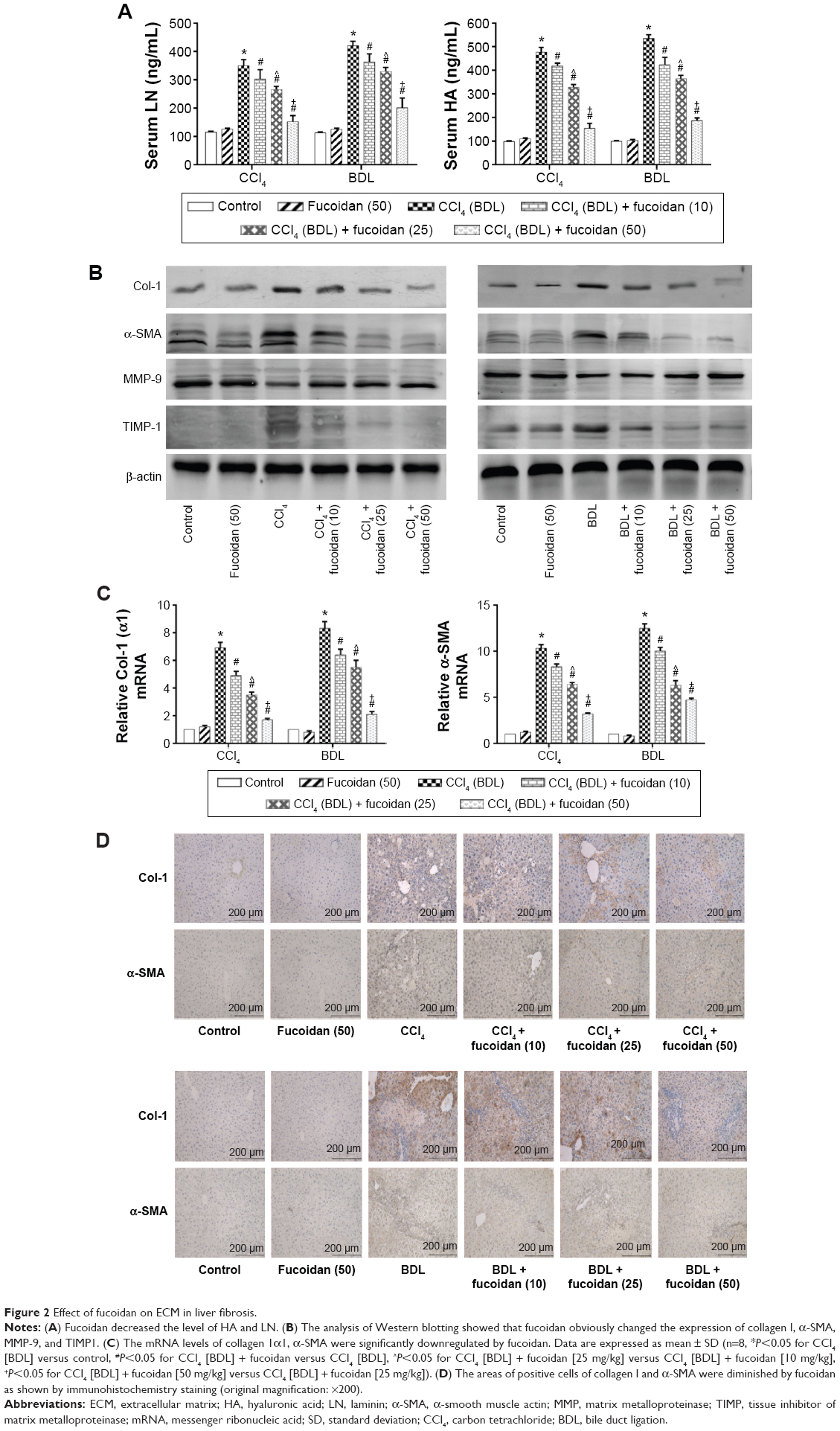 | Figure 2 Effect of fucoidan on ECM in liver fibrosis. |
Fucoidan decreased the formation of autophagosomes by downregulation of Beclin-1 and LC3
Autophagy can provide energy for the acceleration of liver fibrosis via the degradation of cell protein. Beclin-1, LC3, and P62 are important markers of autophagosome formation. To assess autophagy levels in liver tissues after fucoidan treatment, PCR, Western blot, and immunohistochemical staining were used to show the gene and protein tissue levels of these indices, respectively. The results showed that proteins, such as Beclin-1 and LC3, promoted autophagy and were significantly increased in the model groups with fibrosis (BDL and CCl4 groups). Fucoidan decreased cell damage induced by autophagy; thus, Beclin-1 and LC3 levels in the fucoidan-treated groups (10 mg/kg, 25 mg/kg, and 50 mg/kg) gradually decreased in a dose-dependent manner. P62 – an autophagy-related transporter – decreased in the model groups and increased following fucoidan treatment (Figure 3). All markers showed the formation of autophagosomes detected by electron microscopy, including the observation of microstructure. Compared with the control group, the number of autophagosomes in the BDL and CCl4 groups increased, and following fucoidan administration, the agglutinated chromatin in mitochondria and autophagy corpuscles was not easily seen. In summary, fucoidan mitigated the injury induced by autophagy via the inhibition of autophagosome formation by downregulating Beclin-1 and LC3.
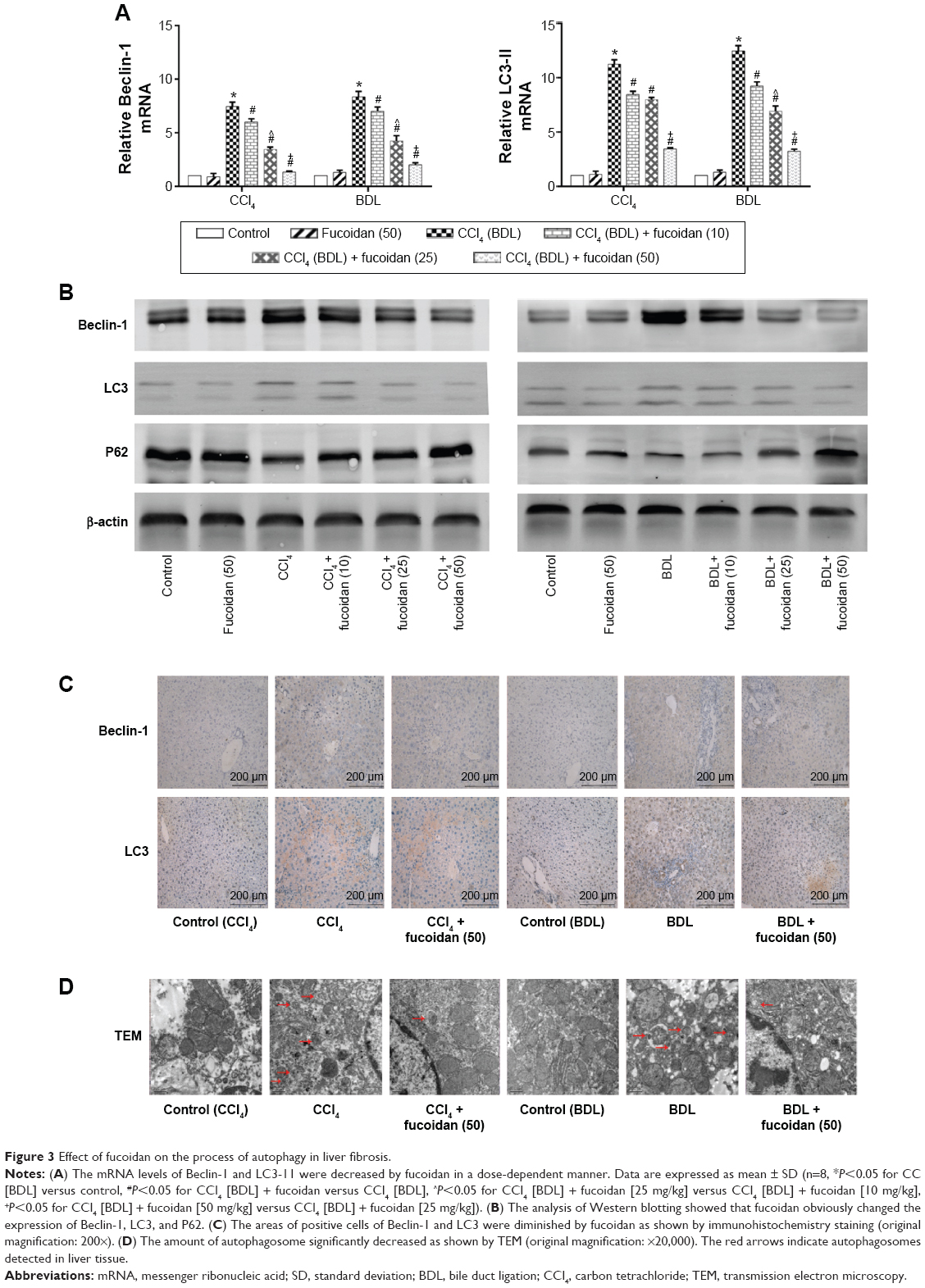 | Figure 3 Effect of fucoidan on the process of autophagy in liver fibrosis. |
Fucoidan prevented the activation of HSCs via the TGF-β1/Smads signaling pathway
TGF-β1 is mainly found in HSCs, hepatic sinus endothelial cells, and inflammatory cells during liver fibrosis and plays an important role in tissues and organs. As an important factor in improving liver fibrosis, TGF-β1 can mediate necrosis and autophagy by the activation of smads, which are phosphorylated in the nuclear region. In order to confirm the conduction pathway, we determined the levels of TGF-β1 in serum and tissues and its gene and protein expression (Figure 4A). The results showed that the levels of these factors were upregulated in the model groups with liver fibrosis induced by BDL and CCl4 and declined after fucoidan treatment with statistically significant differences observed. We also focused on the phosphorylation of smads, such as smad2 and smad3, and found a significant increase in the BDL and CCl4 groups and a dose-dependent decrease following fucoidan treatment. Western blot and immunohistochemical staining showed consistent findings (Figure 4B and C). These results suggest that fucoidan reduced the expression of TGF-β1, which mediated negative activation of downstream smads signaling pathways, which may be an essential pathway in the induction of necrosis and autophagy during liver fibrosis.
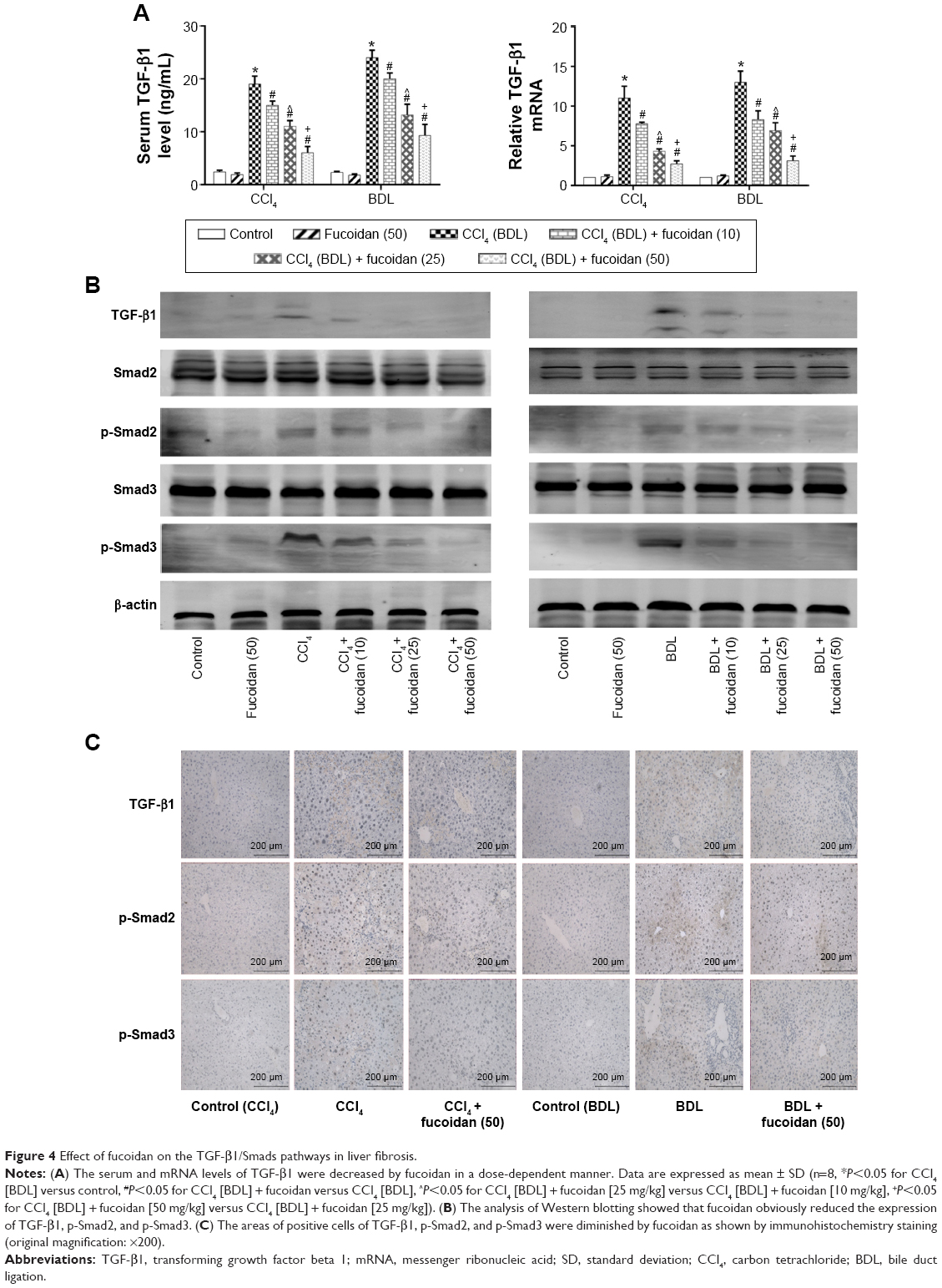 | Figure 4 Effect of fucoidan on the TGF-β1/Smads pathways in liver fibrosis. |
Discussion
Liver fibrosis is a scar formation induced by a compensatory response to a variety of chronic liver injuries. As the common pathological process in many liver diseases, liver fibrosis also leads to cirrhosis.34 It is important to identify new, safe, and effective drug therapies due to the lack of surgical procedures as a result of diffuse pathological changes. Fucoidan, a polysaccharide based on fucose, has attracted the attention of scientists.30,35,36 In the present study, two animal models of liver fibrosis (CCl4 and BDL) were established to determine the protective effect and mechanism of action of fucoidan by analyzing biochemical indicators (ALT, AST, and hydroxyproline), ECM components (collagenase I, MMPs, and TIMPs), and other key proteins located in signaling pathways (TGF-β1 and Smads).
The study by Senties-Gomez et al confirmed that increased synthesis and insufficient degradation of the ECM, which leads to excess deposition and interactions with other cytokines, occur in the initiation and development of liver fibrosis.37 Collagen type I, found in late pathological changes, is an important component of the ECM. As the characteristic amino acid of collagen type I is hydroxyproline, the serum level of this amino acid may be an important indicator of fibrosis. MMPs, a group of enzymes that contain Ca2+ and Zn2+, could play a role in ECM degradation, and TIMPs, secreted by activated HSCs, are specific inhibitors of MMPs.11 ECM metabolism is dependent on the balance between MMPs and TIMPs, mainly MMP-9/TIMP1.38,39 As the key cells in ECM production, HSCs can transform into MFBs, which express the unique molecular marker protein α-SMA.10 Hong et al and Moon et al verified that fucoidan increased MMP-9 secretion in the monocytic cell line U937 and downregulated type I procollagen and α-SMA synthesis in human skin fibroblasts.15,40 In our experiments, the expression of collagen type I and α-SMA detected by PCR, Western blot, and immunohistochemical staining in the CCl4 and BDL model groups showed an upward trend with TIMP1 regarded as an ECM metabolic inhibitory enzyme but decreased in a dose-dependent manner in the fucoidan-treated groups (10 mg/kg, 25 mg/kg, and 50 mg/kg). In contrast, MMP-9, the enzyme that promotes the metabolism of ECM, decreased in the model groups and increased after fucoidan treatment. These results showed that fucoidan can protect against liver injury induced by CCl4 and BDL through the effective inhibition of ECM production and preventing the change from HSCs to MFBs, resulting in reduced liver fibrosis. Based on detection of the above indicators, measurement of ALT, AST, and hydroxyproline suggested that liver function changed with ECM formation. Compared with the model groups, liver biopsy pathology in the fucoidan-treated groups showed less collagen fiber formation and focal necrosis that was dose dependent. In summary, fucoidan inhibited HSCs and reduced ECM formation and α-SMA release to protect against liver fibrosis.
Autophagy, referred to as type II programmed cell death widely exists in eukaryotic cell biology, occurs less frequently under normal circumstances but can be induced to maintain the balance between proteins and organelles under stress. However, excess activation of autophagy can also increase cell damage and death.41,42 In recent years, an increasing number of studies have verified the role of autophagy in chronic liver damage, such as nonalcoholic and alcoholic fatty liver disease, viral hepatitis, and cirrhosis.43–46 During liver fibrosis, the occurrence of autophagy is associated with the activation of HSCs acting on the TGF-β, platelet-derived growth factor, Wnt/β-catenin, and Toll-like receptor pathways.47–51 Therefore, we hypothesize that inhibition of autophagy may be a new drug target in the prevention of liver fibrosis. Beclin-1, LC3, and P62 were assessed to demonstrate the expression of autophagy in this study. The results showed that autophagy was upregulated in the CCl4 and BDL model groups and decreased with increased fucoidan concentration. Abundant autophagosomes and autophagolysosomes were seen in liver tissues of the model groups but were rarely seen following treatment with fucoidan. These findings show that fucoidan can effectively inhibit autophagy, which provides the energy for the activation of HSCs and thus slows down the process of liver fibrosis.
How does fucoidan regulate the formation of ECM and autophagy in liver fibrosis? Shen et al, He et al, and Ghavami et al found that autophagy is a regulator of the TGF-β1 pathway in atrial MFBs and HSCs, and these findings were verified by other researchers.12,23,28,29,45,47,48,50 The TGF-β family, which includes various functional cytokines, can regulate cell growth, proliferation, differentiation, migration, and apoptosis. Research evidence shows that TGF-β, mainly TGF-β1 with the highest biological activity located in intrahepatic cells, is highly expressed in HSCs, hepatic sinus, endothelial cells, and the inflammatory cells that affect liver fibrosis.52,53 HSCs were activated by CCl4 and cholestasis and transformed into MFBs, which secreted TGF-β1. TGF-β1 can expedite the processes of collagen, and TIMP1 in the ECM and TGFR1 on the surface of cell membranes can combine with TGF-β1 to stimulate Smad2/3 located in the cytoplasm.54–56 The examination of signal molecules in the TGF-β1/Smads pathway showed an increased trend in the model groups compared with the normal group, which declined following fucoidan treatment. The gene and protein expression results indicated that fucoidan effectively reduced secreted TGF-β1, thus inhibiting the downstream pathway. In summary, fucoidan inhibited the activation of HSCs, the formation of the ECM, and the release of TGF-β1. Insufficient TGF-β1 formed complexes with its receptor, which led to reduced phosphorylation of Smad2/3. Smad2/3 cannot be transferred from the pulp to the nucleus to combine with specific DNA sequences to promote Beclin-1 transcription due to its unphosphorylated state.51 Beclin-1 usually promotes the occurrence of autophagy through an interaction with PI3K, which induces the conversion of LC3-I to LC3-II.19,57 Following fucoidan treatment, the inhibition of TGF-β1 hindered the formation of autophagosomes, which degraded organelles to protect liver cells via the downregulated TGF-β1/Smad signaling pathways (Figure 5).
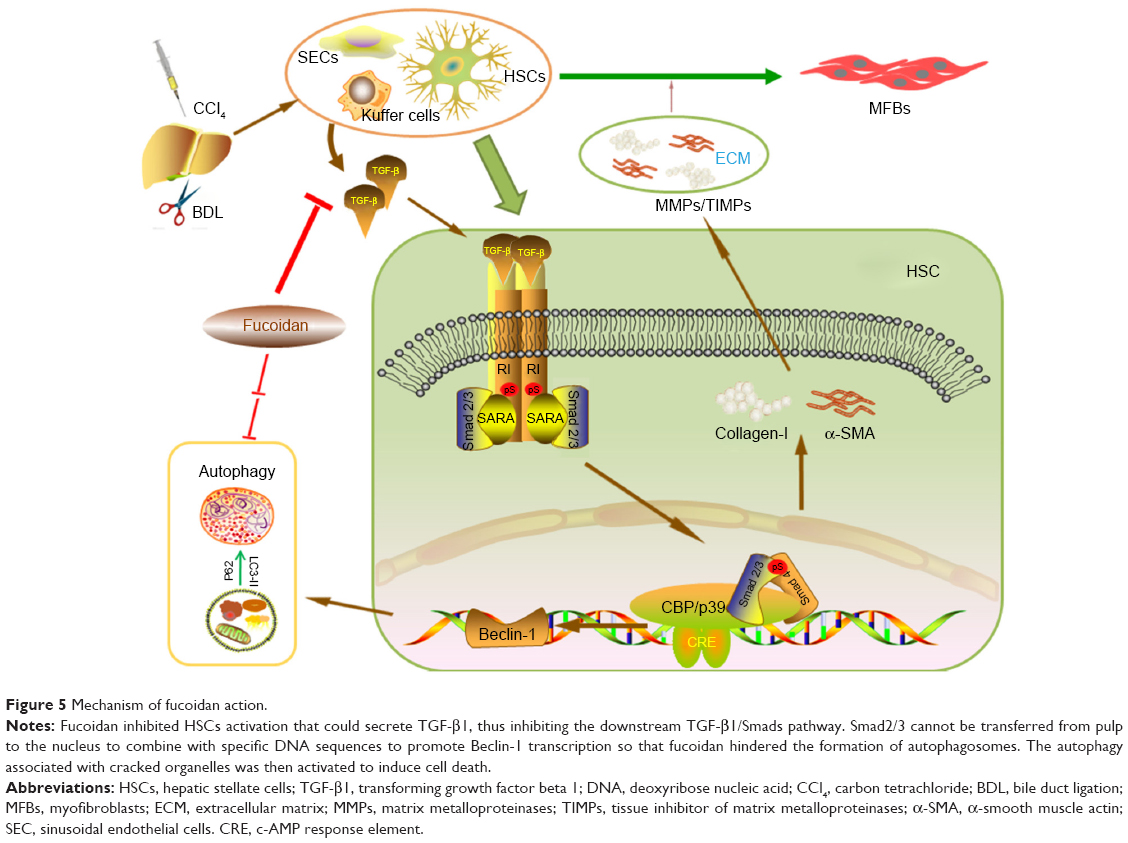 | Figure 5 Mechanism of fucoidan action. |
Conclusion
In conclusion, we preliminarily found that the activation of HSCs to secrete TGF-β1, which induced formation of the ECM and autophagy, was inhibited by fucoidan from F. vesiculosus during liver fibrosis in mice. As new drug targets, autophagy and TGF-β1 inhibitors could be promising potential therapeutic agents for liver fibrosis. Of course, the safety and other related mechanisms of fucoidan require further investigation prior to clinical application.
Author contributions
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in either drafting the article or revising it critically for important intellectual content; gave final approval of the version to be published; and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Friedman SL. Hepatic fibrosis – overview. Toxicology. 2008;254(3):120–129. | ||
Lim YS, Kim WR. The global impact of hepatic fibrosis and end-stage liver disease. Clin Liver Dis. 2008;12(4):733–746. | ||
Zhang Y, Li S, He L, et al. Combination therapy of fenofibrate and ursodeoxycholic acid in patients with primary biliary cirrhosis who respond incompletely to UDCA monotherapy: a meta-analysis. Drug Des Devel Ther. 2015;9:2757–2766. | ||
Zhang H, Yang J, Zhu R, et al. Combination therapy of ursodeoxycholic acid and budesonide for PBC-AIH overlap syndrome: a meta-analysis. Drug Des Devel Ther. 2015;9:567–574. | ||
Schwabe R, Bataller R. Liver fibrosis. Semin Liver Dis. 2015;35(2):95–96. | ||
Chen RJ, Wu HH, Wang YJ. Strategies to prevent and reverse liver fibrosis in humans and laboratory animals. Arch Toxicol. 2015;89(10):1727–1750. | ||
Lee UE, Friedman SL. Mechanisms of hepatic fibrogenesis. Best Pract Res Clin Gastroenterol. 2011;25(2):195–206. | ||
Schuppan D, Afdhal NH. Liver cirrhosis. Lancet. 2008;371(9615):838–851. | ||
Duval F, Moreno-Cuevas JE, Gonzalez-Garza MT, Rodriguez-Montalvo C, Cruz-Vega DE. Protective mechanisms of medicinal plants targeting hepatic stellate cell activation and extracellular matrix deposition in liver fibrosis. Chin Med. 2014;9(1):27. | ||
Reeves HL, Friedman SL. Activation of hepatic stellate cells – a key issue in liver fibrosis. Front Biosci. 2002;7:d808–d826. | ||
Puche JE, Saiman Y, Friedman SL. Hepatic stellate cells and liver fibrosis. Compr Physiol. 2013;3(4):1473–1492. | ||
Shen M, Chen K, Lu J, et al. Protective effect of astaxanthin on liver fibrosis through modulation of TGF-beta1 expression and autophagy. Mediators Inflamm. 2014;2014:954502. | ||
Hayashi S, Itoh A, Isoda K, Kondoh M, Kawase M, Yagi K. Fucoidan partly prevents CCl4-induced liver fibrosis. Eur J Pharmacol. 2008;580(3):380–384. | ||
Tung H, Lee F, Wang S, et al. The beneficial effects of P2X7 antagonism in rats with bile duct ligation-induced cirrhosis. PLoS One. 2015;10(5):e124654. | ||
Hong SW, Jung KH, Lee HS, et al. Suppression by fucoidan of liver fibrogenesis via the TGF-beta/Smad pathway in protecting against oxidative stress. Biosci Biotechnol Biochem. 2011;75:833–840. | ||
Montfort I, Perez-Tamayo R. Collagenase in experimental carbon tetrachloride cirrhosis of the liver. Am J Pathol. 1978;92(2):411–420. | ||
Tag CG, Sauer-Lehnen S, Weiskirchen S, et al. Bile duct ligation in mice: induction of inflammatory liver injury and fibrosis by obstructive cholestasis. J Vis Exp. 2015;10(96):e52438. | ||
Ashford TP, Porter KR. Cytoplasmic components in hepatic cell lysosomes. J Cell Biol. 1962;12:198–202. | ||
Gordy C, He YW. The crosstalk between autophagy and apoptosis: where does this lead? Protein Cell. 2012;3(1):17–27. | ||
Cheng P, Chen K, Xia Y, et al. Hydrogen sulfide, a potential novel drug, attenuates concanavalin A-induced hepatitis. Drug Des Devel Ther. 2014;8:1277–1286. | ||
Mao Y, Wang J, Yu F, et al. Ghrelin reduces liver impairment in a model of concanavalin A-induced acute hepatitis in mice. Drug Des Devel Ther. 2015;9:5385–5396. | ||
Jia G, Sowers JR. Autophagy: a housekeeper in cardiorenal metabolic health and disease. Biochim Biophys Acta. 2015;1852(2):219–224. | ||
Doria A, Gatto M, Punzi L. Autophagy in human health and disease. N Engl J Med. 2013;368(19):1845. | ||
Li J, Xia Y, Liu T, et al. Protective effects of astaxanthin on ConA-induced autoimmune hepatitis by the JNK/p-JNK pathway-mediated inhibition of autophagy and apoptosis. PLoS One. 2015;10(3):e120440. | ||
Shen M, Lu J, Dai W, et al. Ethyl pyruvate ameliorates hepatic ischemia-reperfusion injury by inhibiting intrinsic pathway of apoptosis and autophagy. Mediators Inflamm. 2013;2013:461536. | ||
Cheng P, Wang F, Chen K, et al. Hydrogen sulfide ameliorates ischemia/reperfusion-induced hepatitis by inhibiting apoptosis and autophagy pathways. Mediators Inflamm. 2014;2014:935251. | ||
Wang C, Chen K, Xia Y, et al. N-acetylcysteine attenuates ischemia-reperfusion-induced apoptosis and autophagy in mouse liver via regulation of the ROS/JNK/Bcl-2 pathway. PLoS One. 2014;9(9):e108855. | ||
Hernandez-Gea V, Ghiassi-Nejad Z, Rozenfeld R, et al. Autophagy releases lipid that promotes fibrogenesis by activated hepatic stellate cells in mice and in human tissues. Gastroenterology. 2012;142(4):938–946. | ||
Liu M, He Y, Zhang J. [Effect of autophagy inhibitor 3-methyladenine on proliferation and activation of hepatic stellate cells]. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi. 2013;29(8):809–812. Chinese. | ||
Fitton JH. Therapies from fucoidan; multifunctional marine polymers. Mar Drugs. 2011;9(12):1731–1760. | ||
Kwak J. Fucoidan as a marine anticancer agent in preclinical development. Mar Drugs. 2014;12(2):851–870. | ||
Senthilkumar K, Manivasagan P, Venkatesan J, Kim S. Brown seaweed fucoidan: biological activity and apoptosis, growth signaling mechanism in cancer. Int J Biol Macromol. 2013;60:366–374. | ||
Pan RL, Xiang LX, Wang P, et al. Low-molecular-weight fibroblast growth factor 2 attenuates hepatic fibrosis by epigenetic down-regulation of delta-like1. Hepatology. 2015;61(5):1708–1720. | ||
Tsukada S, Parsons CJ, Rippe RA. Mechanisms of liver fibrosis. Clin Chim Acta. 2006;364(1–2):33–60. | ||
Lim J, Lee S, Kim T, et al. Fucoidan from Fucus vesiculosus protects against alcohol-induced liver damage by modulating inflammatory mediators in mice and HepG2 cells. Mar Drugs. 2015;13(2):1051–1067. | ||
Li C, Gao Y, Xing Y, Zhu H, Shen J, Tian J. Fucoidan, a sulfated polysaccharide from brown algae, against myocardial ischemia-reperfusion injury in rats via regulating the inflammation response. Food Chem Toxicol. 2011;49(9):2090–2095. | ||
Senties-Gomez MD, Galvez-Gastelum FJ, Meza-Garcia E, Armendariz-Borunda J. [Hepatic fibrosis: role of matrix metalloproteases and TGFbeta]. Gac Med Mex. 2005;141(4):315–322. Spanish. | ||
Jackson PL, Xu X, Wilson L, et al. Human neutrophil elastase-mediated cleavage sites of MMP-9 and TIMP-1: implications to cystic fibrosis proteolytic dysfunction. Mol Med. 2010;16(5–6):159–166. | ||
Fu W, Wang Y, Jin Z, et al. Losartan alleviates renal fibrosis by down-regulating HIF-1alpha and up-regulating MMP-9/TIMP-1 in rats with 5/6 nephrectomy. Ren Fail. 2012;34(10):1297–1304. | ||
Moon HJ, Lee SH, Ku MJ, et al. Fucoidan inhibits UVB-induced MMP-1 promoter expression and down regulation of type I procollagen synthesis in human skin fibroblasts. Eur J Dermatol. 2009;19(2):129–134. | ||
Murrow L, Debnath J. Autophagy as a stress-response and quality-control mechanism: implications for cell injury and human disease. Annu Rev Pathol. 2013;8:105–137. | ||
Li J, Wang F, Xia Y, et al. Astaxanthin pretreatment attenuates hepatic ischemia reperfusion-induced apoptosis and autophagy via the ROS/MAPK pathway in mice. Mar Drugs. 2015;13(6):3368–3387. | ||
Kwanten WJ, Martinet W, Michielsen PP, Francque SM. Role of autophagy in the pathophysiology of nonalcoholic fatty liver disease: a controversial issue. World J Gastroenterol. 2014;20(23):7325–7338. | ||
Osna NA, Thomes PG, Donohue Jr TM. Involvement of autophagy in alcoholic liver injury and hepatitis C pathogenesis. World J Gastroenterol. 2011;17(20):2507–2514. | ||
Wang J, Kang R, Huang H, et al. Hepatitis C virus core protein activates autophagy through EIF2AK3 and ATF6 UPR pathway-mediated MAP1LC3B and ATG12 expression. Autophagy. 2014;10(5):766–784. | ||
Sasaki M, Yoshimura-Miyakoshi M, Sato Y, Nakanuma Y. A possible involvement of endoplasmic reticulum stress in biliary epithelial autophagy and senescence in primary biliary cirrhosis. J Gastroenterol. 2015;50(9):984–995. | ||
He W, Wang B, Yang J, et al. Chloroquine improved carbon tetrachloride-induced liver fibrosis through its inhibition of the activation of hepatic stellate cells: role of autophagy. Biol Pharm Bull. 2014;37(9):1505–1509. | ||
He Y, Zhu J, Huang Y, Gao H, Zhao Y. Advanced glycation end product (AGE)-induced hepatic stellate cell activation via autophagy contributes to hepatitis C-related fibrosis. Acta Diabetol. 2015;52(5):959–969. | ||
Thoen LF, Guimaraes EL, Grunsven LA. Autophagy: a new player in hepatic stellate cell activation. Autophagy. 2012;8(1):126–128. | ||
Ghavami S, Cunnington RH, Gupta S, et al. Autophagy is a regulator of TGF-β1-induced fibrogenesis in primary human atrial myofibroblasts. Cell Death Dis. 2015;6(3):e1696. | ||
Pan CC, Kumar S, Shah N, et al. Endoglin regulation of Smad2 function mediates beclin1 expression and endothelial autophagy. J Biol Chem. 2015;290(24):14884–14892. | ||
Gressner AM, Weiskirchen R. Modern pathogenetic concepts of liver fibrosis suggest stellate cells and TGF-beta as major players and therapeutic targets. J Cell Mol Med. 2006;10(1):76–99. | ||
Massague J, Blain SW, Lo RS. TGFbeta signaling in growth control, cancer, and heritable disorders. Cell. 2000;103(2):295–309. | ||
Schnabl B, Kweon YO, Frederick JP, Wang XF, Rippe RA, Brenner DA. The role of Smad3 in mediating mouse hepatic stellate cell activation. Hepatology. 2001;34(1):89–100. | ||
Rao C, Lin SL, Ruan WJ, Wen H, Wu DJ, Deng H. High expression of IGFBP7 in fibroblasts induced by colorectal cancer cells is co-regulated by TGF-beta and Wnt signaling in a Smad2/3-Dvl2/3-dependent manner. PLoS One. 2014;9(1):e85340. | ||
Samarakoon R, Chitnis SS, Higgins SP, Higgins CE, Krepinsky JC, Higgins PJ. Redox-induced Src kinase and caveolin-1 signaling in TGF-beta1-initiated SMAD2/3 activation and PAI-1 expression. PLoS One. 2011;6(7):e22896. | ||
Nikoletopoulou V, Markaki M, Palikaras K, Tavernarakis N. Crosstalk between apoptosis, necrosis and autophagy. Biochim Biophys Acta. 2013;1833(12):3448–3459. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.