")
Back to Journals » Drug Design, Development and Therapy » Volume 9
Profile of pridopidine and its potential in the treatment of Huntington disease: the evidence to date
Authors Squitieri F, de Yebenes JG
Received 19 July 2015
Accepted for publication 14 September 2015
Published 28 October 2015 Volume 2015:9 Pages 5827—5833
DOI https://doi.org/10.2147/DDDT.S65738
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Wei Duan
Ferdinando Squitieri,1 Justo Garcia de Yebenes2
1IRCCS Casa Sollievo della Sofferenza, San Giovanni Rotondo and Mendel Institute of Human Genetics, Rome, Italy; 2Fundación para Investigaciones Neurológicas, Madrid, Spain
Abstract: Huntington disease (HD) is a chronic, genetic, neurodegenerative disease for which there is no cure. The main symptoms of HD are abnormal involuntary movements (chorea and dystonia), impaired voluntary movements (ie, incoordination and gait balance), progressive cognitive decline, and psychiatric disturbances. HD is caused by a CAG-repeat expanded mutation in the HTT gene, which encodes the huntingtin protein. The inherited mutation results in the production of an elongated polyQ mutant huntingtin protein (mHtt). The cellular functions of the Htt protein are not yet fully understood, but the functions of its mutant variant are thought to include alteration of gene transcription and energy production, and dysregulation of neurotransmitter metabolism, receptors, and growth factors. The phenylpiperidines pridopidine (4-[3-methanesulfonyl-phenyl]-1-propyl-piperidine; formerly known as ACR16) and OSU6162 ([S]-[-]-3-[3-methane [sulfonyl-phenyl]-1-propyl-piperidine) are members of a new class of pharmacologic agents known as “dopamine stabilizers”. Recent clinical trials have highlighted the potential of pridopidine for symptomatic treatment of patients with HD. More recently, the analysis of HD models (ie, in vitro and in mice) highlighted previously unknown effects of pridopidine (increase in brain-derived neurotrophic factor, reduction in mHtt levels, and σ-1 receptor binding and modulation). These additional functions of pridopidine suggest it might be a neuroprotective and disease-modifying drug. Data from ongoing clinical trials of pridopidine will help define its place in the treatment of HD. This commentary examines the available preclinical and clinical evidence regarding the use of pridopidine in HD.
Keywords: Huntington disease, dopamine, neuroprotection, pridopidine
Introduction
Huntington disease (HD) is a chronic, neurodegenerative, dominantly transmitted genetic disease. Although the onset of HD generally occurs at around 40 years of age, it can, in rare cases, affect people younger than 20 years old (juvenile HD) and, even more rarely, children under 10 years of age. Patients with HD rapidly develop substantial mental and physical disability. No cure or treatment to prevent the progression of HD is available.1
From the time of its onset, HD is characterized by three main clinical patterns with subtle differences: psychiatric disturbances, often associated with a high rate of suicide; cognitive decline; and extrapyramidal signs and symptoms. All three progress to dementia and cachexia in five stages.1 Other sites are affected in addition to the brain, including muscles, bones, and testes. Inflammation contributes to the pathology in the central nervous and peripheral systems.2
From HTT-CAG mutation to complex biological mechanisms of HD
HD is associated with an expanded CAG-repeat mutation in the HTT gene that encodes the huntingtin protein (Htt).3,4 A CAG stretch expansion beyond 35 repeats is associated with the disease.4 Repeats in the range 36–39 are considered as low penetrance mutations (ie, the disease may start late or never during life), while full penetrance mutations (ie, all clinical manifestations are expected to present during life) are associated with ≥40 CAG repeats.4 The mutation is unstable during intergenerational transmission. In the offspring, the trinucleotide instability may cause further elongation of the translated polyQ stretch in the Htt protein, thus amplifying its toxic effects in neurons and other tissues in each new generation.3 The Htt protein has many cellular functions, but these have not yet been fully characterized. Mutant Htt (mHtt) with elongated polyQ may, instead, cause dysregulation of neurotransmitters, receptors, and growth factors.5 mHtt translocates from the cytoplasm to the nucleus and perinuclear regions where it interferes with gene transcription, energy production, and processing of abnormal proteins.6
The biology of HD is, therefore, very complex; mHtt has many, as yet unclear, functions and interacts with a large number of factors both within and outside the nervous system, especially in the striatum.5 One important, yet unsolved, issue is what causes the prominent early damage to the striatum in patients with HD. In addition to existing hypotheses for the potential toxic mechanisms of mHtt,5 recent studies have focused on the binding of the striatal-specific Rhes (Ras homologue enriched in striatum) protein to mHtt as a specific pathogenic mechanism leading to mHtt toxicity in the brain striatum,7,8 and regulation of the dopamine-D2 receptor activity by Rhes.9 Dopaminergic and glutamatergic signaling pathways act synergistically to enhance the sensitivity of striatal neurons to mHtt toxicity and may represent crucial therapeutic targets for HD treatment. Therefore, understanding dysfunction of dopamine and other (eg, cholinergic) receptors in HD is critical in interpreting the effects of pharmacotherapy.9
From Huntington “chorea” to Huntington “disease”
Historically, the description of HD typically referred to one of the most visible symptoms, ie, chorea,10 an involuntary, “dancelike”, uncontrolled movement disorder. Chorea is characterized by progressive jerking of the arms and trunk. It evolves to other abnormal movements, such as dystonia and Parkinsonism, generally in the advanced stages of the disease. Although chorea represents one of the main and most frequent symptoms of HD, it only marginally contributes to patients’ disability and loss of independence compared with other features, such as cognitive decline, psychiatric disturbances, and other neurological symptoms (incoordination, oculomotor abnormalities, ie, slow and saccade eye movement impairments, dysarthria, dysphagia, gait disturbance, and imbalance).11 Patients are frequently unaware of the symptoms of chorea, and approximately 8% of HD cases manifest atypical HD variants in which other signs and symptoms, such as parkinsonism and dystonia, predominate over choreic movements from the onset of the disease.12
In accordance with the original descriptions of HD, and taking into account both the prominence of chorea and the selective degeneration of the brain striatum as a key feature of the disease, therapeutic approaches to HD have included either the control of behavioral changes (eg, neuroleptics, benzodiazepines, and mood stabilizers)13,14 or attempts to reduce choreic movements with tetrabenazine, the only approved drug that inhibits dopamine release with a specific indication for chorea.13–15 However, knowledge of the natural history of HD has increased in recent years. The development of experimental HD models and new psychiatric drugs (eg, atypical neuroleptics and antidepressants), as well as novel findings regarding the neuropathology of the disease,16 have influenced HD management guidelines. The American Academy of Neurology’s most recent guidelines for the pharmacologic treatment of HD recommend symptomatic approaches.17 Curative treatment for HD would need to take the following into account: 1) HD is not only related to striatum degeneration, but also to more extensive brain damage, including the cortex and white matter;16 2) chorea is not the only symptomatic target;18 3) choreic movements make a marginal contribution to HD disability, and patients indicate that other abnormal voluntary movements, distinct from chorea (eg, gait imbalance, incoordination, dystonia) are the most disabling motor features (Squitieri and Mondher, personal communication); and 4) classical neuroleptics worsen HD progression.19
Dopidines: an example of a new class of drugs
Pridopidine (4-[3-methanesulfonyl-phenyl]-1-propyl-piperidine; formerly known as ACR16) and OSU6162 (S)-(−)-3-(3-methanesulfonyl-phenyl)-1-propyl-piperidine are phenylpiperidines.20 Phenylpiperidines are members of a new class of pharmacologic compounds known as “dopamine stabilizers” or “dopidines”,21 which are being widely investigated for neurodegenerative disorders such as HD.22,23 Pridopidine and OSU6162 are structurally related to 3-(3-hydroxyphenyl)-N-n-propylpiperidine (3-PPP), a compound with enantiomers that have different effects on dopamine receptors: the (+)3-PPP enantiomer is a weak dopamine receptor agonist, while the (−)3-PPP enantiomer is a weak dopamine receptor antagonist.24
Dopamine acts as a high-affinity neurotransmitter on dopamine receptors and exerts its action in the brain by means of both synaptic and extrasynaptic release. Dopamine stabilizers, such as pridopidine and OSU6162, are characterized by their actions at dopamine-D2 receptors, which result in state-dependent behavioral effects. These new dopamine–ligand compounds are thought to stabilize dysregulated psychomotor functions by modulating hyper- or hypoactive functioning in areas of the brain receiving dopamine input.25 Both pridopidine and OSU6162 bind in a dose-dependent manner to striatal D2 receptors and do not show partial agonism in functional assays.25 In this context, dopidines do not fit into the typical list of dopamine-D2 drugs classified as agonists, partial agonists, or antagonists.25 Both compounds inhibited amphetamine-induced locomotor activity and stimulated locomotion in habituated rats.26 They also resulted in preferential induction of Fos in the nucleus accumbens relative to the striatum, a characteristic of atypical antipsychotic efficacy.26 More recent studies have shown that pridopidine and OSU6162 reduce motor activity in animals during the period of exploration of the environment that takes place within the 1st minutes of depositing the animals in activity meters.27 However, the compounds increase activity in animals habituated to the equipment from within 15–60 minutes of analysis.27 In addition, these compounds increase activity in catecholamine-depleted animals treated with reserpine and α-methyl-para-tyrosine.27 These effects are mediated by the selective binding of these dopidines to high-affinity D2 dopamine receptors28 and their rapid dissociation constant.29 The original developer of pridopidine, Arvid Carlsson, has further explained the mechanisms of action of dopidines on dopamine receptors.30 According to the hypothesis proposed by Carlsson and Carlsson,30 pridopidine antagonizes the binding of other ligands to dopamine-D2 receptors in a similar manner to haloperidol, but with lower potency and with a more restricted receptor binding profile. While haloperidol blocks both synaptic and extrasynaptic receptors, pridopidine preferentially blocks dopamine neurotransmission at extrasynaptic receptors, including autoreceptors.30 Because normal dopamine function is limited to the synapses, dopidines – which do not block synaptic dopamine transmission – do not produce parkinsonism.30 The antagonism of extrasynaptic receptors by dopidines could improve psychosis and abnormal involuntary movements related to abnormal dopamine extrasynaptic transmission.30
In addition to its effects on dopamine receptors, pridopidine counteracts abnormal behaviors induced by the N-methyl-D-aspartate (NMDA) glutamate receptor antagonist MK-801 in mice.31 Both pridopidine and OSU6162 reversed withdrawal responses induced by MK-801 in rats.32 These results indicate an additional effect of dopidines on NMDA glutamate receptors and suggest that dopidines may have effects on cognitive processes. Furthermore, it has recently been shown that both pridopidine and OSU6162 bind with nanomolar affinities to the σ-1 receptor,33 a receptor that modulates interorganelle signaling.34 The σ-1 receptor has been implicated in the elimination of intranuclear inclusions in a cellular model of HD.35 This raises the possibility that dopidines could have disease-modifying effects in HD.
Pridopidine use in HD: lessons from patients and back to models
To date, three large multicenter clinical trials of pridopidine in HD have been published,36–38 and a meta-analysis of the two largest studies has been performed.39 A summary of the three trials and their outcomes is presented in Table 1. The first of these studies was a multicenter trial conducted in Scandinavia that included 28 patients with HD treated with pridopidine 50 mg/d for 4 weeks and 30 patients who received placebo.36 The primary endpoint of the study – change in cognitive scores – was not reached, but there was a statistically significant (P<0.05) improvement in some voluntary motor-symptom-related secondary endpoints in patients treated with pridopidine versus placebo (eg, the quality and speed of voluntary movements).
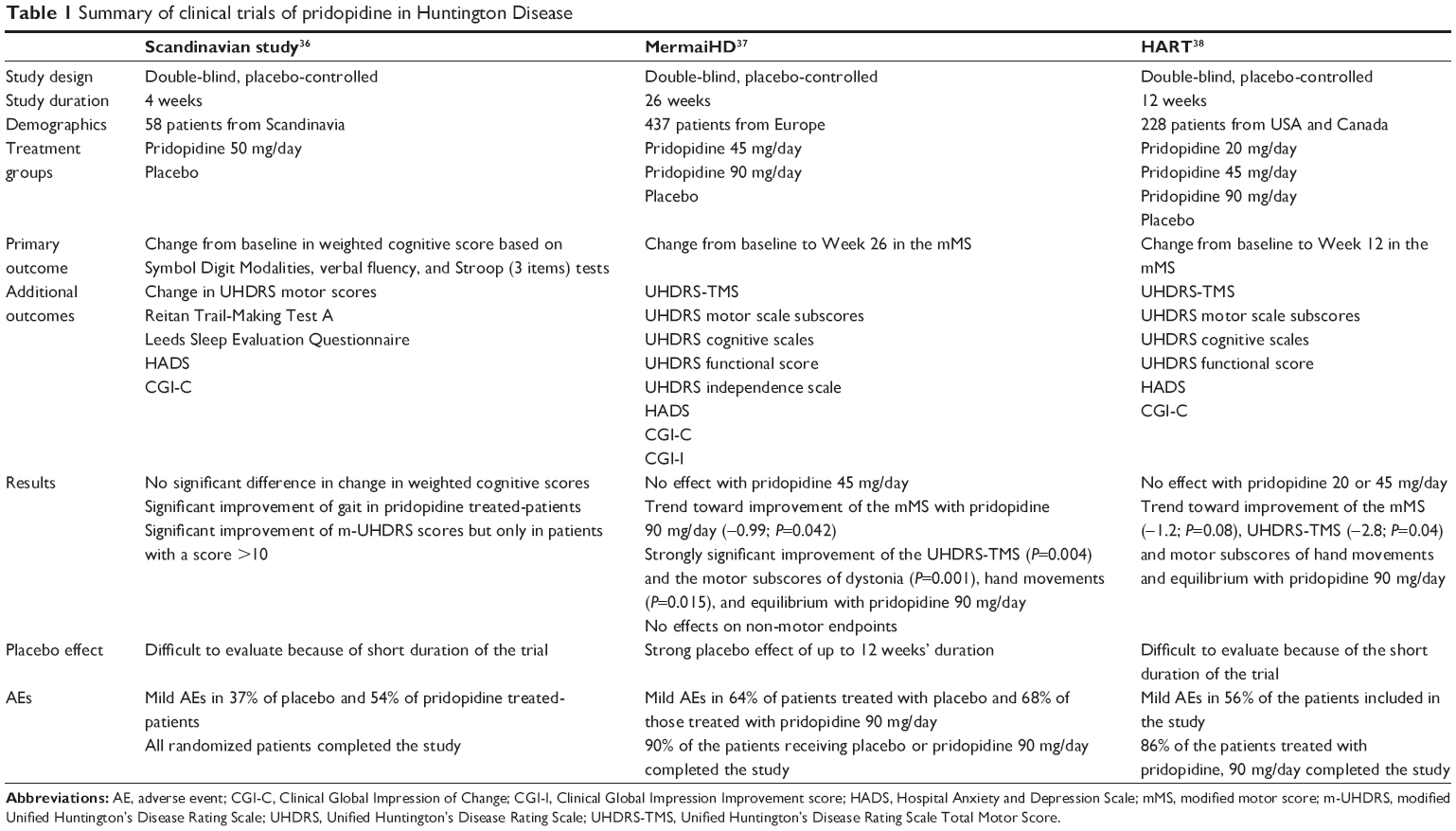 | Table 1 Summary of clinical trials of pridopidine in Huntington Disease |
The MermaiHD study – the largest clinical trial performed to date with pridopidine – was a multicenter European Phase III study that included 437 patients randomized (1:1:1) to three groups: pridopidine 90 mg/d, pridopidine 45 mg/d, or placebo for 26 weeks.37 The primary endpoint of the study was improvement from baseline in voluntary movements as determined by the modified motor score. The results of this study showed a strong trend toward improvement – close to statistical significance for the primary endpoint. Statistically significant improvements were reported for the global motor scores (P=0.004), as well as for specific items such as eye movements (P=0.007), dystonia (P=0.001), hand movements (P=0.015), and gait and balance (P=0.028) in the group of patients treated with pridopidine 90 mg/d, but not for the patients receiving pridopidine 45 mg/d. Pridopidine was well tolerated in this study37 and its safety has been confirmed in a 1-year, open-label extension study.40
The HART study was performed in the United States by the Huntington Study Group.38 It included 227 patients, randomized to four groups of similar size that received pridopidine 90, 45, or 20 mg/d, or placebo for 12 weeks. Although improvements in motor scores were reported in the group of patients treated with pridopidine 90 mg/d, the differences compared with placebo did not reach statistical significance. Pridopidine treatment was well tolerated, and despite the lack of statistically significant improvement in the primary motor endpoint, the authors considered that the overall results suggest that pridopidine may improve motor function in HD.38
A meta-analysis of the MermaiHD and HART studies has been published in part.39 The analysis showed that pridopidine treatment led to total motor score improvement, as well as improvement in specific items, such as hand movements, and gait and balance. Considering these effects, together with the benign safety and tolerability profile, pridopidine could be a potential new treatment for HD.
Careful consideration of the characteristics of the three multicenter studies is required in order to evaluate the potential utility of pridopidine in HD. The Scandinavian study36 was designed based on an expected rapid effect on cognitive deficits and dyskinesias without modification of voluntary movement. For this reason, the treatment duration was only 4 weeks, and voluntary movements and involuntary movements were measured independently, in spite of the fact that both types of deficits are part of the motor Unified Huntington’s Disease Rating Scale (UHDRS).41 The MermaiHD study37 was designed on the basis of the results reported in the Scandinavian study. The number of patients was increased to 437, and the duration of active treatment was 26 weeks. Unfortunately, responses to the modified and partial motor UHDRS scales were chosen as the primary endpoint because these were found to be the only motor endpoints improved by pridopidine in the Scandinavian study. However, the study design did not take into account the fact that the time taken to improve different motor deficits can vary in neurodegenerative disease. For instance, dystonia takes a long time to respond to treatment, regardless of the type of treatment (eg, pharmacotherapy or deep-brain stimulation). In the Scandinavian study, pridopidine did not improve dystonia, even after 4 weeks of treatment,36 but the dystonia score was reduced significantly after 26 weeks of treatment in the MermaiHD trial.37
The HART study38 was also designed based on the findings of the Scandinavian study; however, in this study, the treatment duration was only 12 weeks. Data obtained from the placebo arm of the MermaiHD study37 have shown that the placebo effect lasts for 12 weeks – the full duration of the HART trial. Therefore in the HART study, there is a strong placebo effect at the end of the evaluation period, which confounds the results of the trial. All of these observations suggest that any future trials of pridopidine should meet the following requirements: 1) pridopidine dose ≥90 mg/d; 2) duration of treatment ≥1 year; and 3) primary endpoint based on the total UHDRS scale for evaluation of motor results.
Interestingly, a pilot study using 18-fluoro-deoxy-glucose positron-emission tomography has documented improvement in glucose metabolism in some regions of the brains of patients with HD after 14 days of treatment with pridopidine.42 The changes in glucose metabolism correlated with clinical changes, suggesting that pridopidine may modify disease-related metabolic activity patterns in the brain.42
In all studies, pridopidine was well tolerated, with an adverse events profile similar of that of placebo.36–40 The most common adverse events reported in the MermaiHD Phase III study were falls, chorea, diarrhea, dizziness, and nausea.37 Similar results were described after 1 year of treatment in the open-label extension of MermaiHD with pridopidine 90 mg/d, with falls and worsening of chorea being the most commonly reported adverse events.40 No clinically meaningful changes in laboratory parameters nor relevant EKG-related safety concerns were identified after even 1 year of treatment.40
Recently, in addition to this promising clinical evidence, new preclinical data have emerged that show potential beneficial effects in HD. The administration of pridopidine induced an overall improvement of motor performance in HD R6/2 mice and showed antiapoptotic effects in vitro in striatal cells from 111/111 CAG-repeat knock-in mice. When tested in vivo, pridopidine treatment was associated with increases in striatal brain-derived neurotrophic factor, and dopamine- and cAMP-regulated neuronal phosphoprotein (DARPP32) levels, in addition to reduced mHtt aggregate size. Interestingly, this study highlights the involvement of σ-1 receptors in these beneficial effects.43 Indeed, the in vitro data showed no effect for pridopidine in striatal cells from 111/111 CAG-repeat mutant mice after using the striatal σ-1 receptor antagonist NE100.43 Collectively, these data suggest that pridopidine may have disease-modifying properties in HD, mediated at least in part through σ-1 receptors.
Conclusion
Advances in research and knowledge about the mechanisms underlying HD are opening exciting new fields of interest for pharmaceutical intervention in neurodegenerative diseases.44 The evidence that new molecular targets, such as the σ-1 receptor, may play a primary role in modulating the effects of the dopamine stabilizer pridopidine is an important finding, considering that this receptor also represents a target for drugs currently in use to counteract cognitive decline in Alzheimer disease.45 The σ-1 receptor modulation may, in theory, protect patients with HD from the progressive deterioration typical of the disease. Improvement in cognitive functions in HD is an important therapeutic goal because it may significantly modify the disease course and preserve patients’ independence and quality of life.22,23
A large, global, multicenter, double-blind Phase II trial of pridopidine – PRIDE-HD – is currently in progress (NCT02006472). The aim of this study is to assess the tolerability of higher drug doses (up to 112.5 mg twice daily) than previously tested and the potential effectiveness of pridopidine in managing motor manifestations (ie, total motor score). During the patient recruitment phase, Teva Pharmaceuticals (Frazer, PA, USA) amended the study protocol to prolong the double-blind experimental phase from 6 to 12 months in an attempt to capture potential disease-modifying effects. Patients who were recruited in the study and still receiving experimental treatment were asked to provide written informed consent to extend the double-blind treatment phase from 6 to 12 months. An open-label study due to be initiated soon – Open Pride HD (NCT02494778) – will investigate the effects of pridopidine 90 mg/d over 52 weeks. The results of these longer-term trials will be eagerly awaited to determine whether this new class of drug has the potential to improve the symptoms of HD and to prevent disease progression.
Acknowledgments
Editorial assistance (editing of final draft) was provided by Kirsteen Munn of Anthemis Consulting Ltd, funded by Teva Pharmaceutical Industries, Frazer, PA, USA. Teva provided a single medical accuracy review. The authors were not compensated and retained full editorial control over the content of the paper. We thank the Lega Italiana Ricerca Huntington (LIRH) Foundation (www.lirh.it) for providing constant support to research (FS) and assistance to HD patients and families.
Disclosure
The authors report no conflicts of interests in this work.
References
Dorsey ER, Beck CA, Darwin K, et al; for Huntington Study Group COHORT Investigators. Natural history of Huntington disease. JAMA Neurol. 2013;70(12):1520–1530. | ||
van der Burg JM, Björkqvist M, Brundin P. Beyond the brain: widespread pathology in Huntington’s disease. Lancet Neurol. 2009;8(8):765–774. | ||
Huntington’s Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell. 1993;72(6):971–983. | ||
Squitieri F. Neurodegenerative disease: fifty shades of grey in the Huntington’s disease gene. Nat Rev Neurol. 2013;9(8):421–422. | ||
Ross CA, Tabrizi SJ. Huntington’s disease: from molecular pathogenesis to clinical treatment. Lancet Neurol. 2011;10(1):83–98. | ||
Nasir J, Goldberg YP, Hayden MR. Huntington disease: new insights into the relationship between CAG expansion and disease. Hum Mol Genet. 1996;5(Spec. No.):1431–1435. | ||
Subramaniam S, Sixt KM, Barrow R, Snyder SH. Rhes, a striatal protein, mediates mutant huntingtin cytotoxicity. Science. 2009;324(5932):1327–1330. | ||
Swarnkar S, Chen Y, Pryor WM, et al. Ectopic expression of the striatal-enriched GTPase Rhes elicits cerebellar degeneration and an ataxia phenotype in Huntington’s disease. Neurobiol Dis. 2015;82:66–77. | ||
Sciamanna G, Napolitano F, Pelosi B, et al. Rhes regulates dopamine D2 receptor transmission in striatal cholinergic interneurons. Neurobiol Dis. 2015;78:146–162. | ||
Huntington G. On chorea. Med Surg Reporter Phila. 1872;26(15):317–321. | ||
Reilmann R, Leavitt BR, Ross CA. Huntington’s disease: a field on the move. Introduction. Mov Disord. 2014;29(11):1333–1334. | ||
Squitieri F, Berardelli A, Nargi E, et al. Atypical movement disorders in the early stages of Huntington’s disease: clinical and genetic analysis. Clin Genet. 2000;58(1):50–56. | ||
Mestre TA, Ferreira JJ. An evidence-based approach in the treatment of Huntington’s disease. Parkinsonism Relat Disord. 2012;18(4):316–320. | ||
Mestre T, Ferreira J, Coelho MM, Rosa M, Sampaio C. Therapeutic interventions for symptomatic treatment in Huntington’s disease. Cochrane Database Syst Rev. 2009;(3):CD006456. | ||
Burgunder JM. Orphan drugs in development for Huntington’s disease: challenges and progress. Orphan Drugs Res Rev. 2015;5:1–9. | ||
Hadzi TC, Hendricks AE, Latourelle JC, et al. Assessment of cortical and striatal involvement in 523 Huntington disease brains. Neurology. 2012;79(16):1708–1715. | ||
Armstrong MJ, Miyasaki JM; for the American Academy of Neurology. Evidence-based guideline: pharmacologic treatment of chorea in Huntington disease: report of the guideline development subcommittee of the American Academy of Neurology. Neurology. 2012;79(6):597–603. | ||
Reilmann R. Pharmacological treatment of chorea in Huntington’s disease – good clinical practice versus evidence-based guideline. Mov Disord. 2013;28(8):1030–1033. | ||
Tedroff J, Waters S, Barker AR, Roos R, Squitieri F. Antidopaminergic medication is associated with more rapidly progressive Huntington’s Disease. J Huntingtons Dis. 2015;4(2):131–140. | ||
Kara E, Lin H, Svensson K, Johansson AM, Strange PG. Analysis of the actions of the novel dopamine receptor-directed compounds (S)-OSU6162 and ACR16 at the D2 dopamine receptor. Br J Pharmacol. 2010;161(6):1343–1350. | ||
Pettersson F, Pontén H, Waters N, Waters S, Sonesson C. Synthesis and evaluation of a set of 4-phenylpiperidines and 4-phenylpiperazines as D2 receptor ligands and the discovery of the dopaminergic stabilizer 4-[3-(methylsulfonyl)phenyl]-1-propylpiperidine (huntexil, pridopidine, ACR16). J Med Chem. 2010;53(6):2510–2520. | ||
Feigin A. Pridopidine in treatment of Huntington’s disease: beyond chorea? Lancet Neurol. 2011;10(12):1036–1037. | ||
Reilmann R. The pridopidine paradox in Huntington’s disease. Mov Disord. 2013;28(10):1321–1324. | ||
Mulder AH, Draper R, Sminia P, Schoffelmeer AN, Stoof JC. Agonist and antagonist effects of 3-PPP enantiomers on functional dopamine autoreceptors and postsynaptic dopamine receptors in vitro. Eur J Pharmacol. 1985;107(3):291–297. | ||
Ponten H, Kullingsjö J, Lagerkvist S, et al. In vivo pharmacology of the dopaminergic stabilizer pridopidine. Eur J Pharmacol. 2010;644(1–3):88–95. | ||
Natesan S, Svensson KA, Reckless GE, et al. The dopamine stabilizers (S)-(−)-(3-methanesulfonyl-phenyl)-1-propyl-piperidine [(−)-OSU6162] and 4-(3-methanesulfonylphenyl)-1-propyl-piperidine (ACR16) show high in vivo D2 receptor occupancy, antipsychotic-like efficacy, and low potential for motor side effects in the rat. J Pharmacol Exp Ther. 2006;318(2):810–818. | ||
Rung JP, Rung E, Hegelson L, et al. Effects of (−)-OSU6162 and ACR16 on motor activity in rats, indicating a unique mechanism of dopaminergic stabilization. J Neural Transm. 2008;115(6):899–908. | ||
Seeman P, Tokita K, Matsumoto M, Matsuo A, Sasamata M, Miyata K. The dopaminergic stabilizer ASP2314/ACR16 selectively interacts with D2High receptors. Synapse. 2009;63(10):930–934. | ||
Dyhring T, Nielsen EØ, Sonesson C, et al. The dopaminergic stabilizers pridopidine (ACR16) and (−)-OSU6162 display dopamine D2 receptor antagonism and fast receptor dissociation properties. Eur J Pharmacol. 2010;628(1–3):19–26. | ||
Carlsson A, Carlsson ML. A dopaminergic deficit hypothesis of schizophrenia: the path to discovery. Dialogues Clin Neurosci. 2006;8(1):137–142. | ||
Nilsson M, Carlsson A, Markinhuhta KR, et al. The dopaminergic stabiliser ACR16 counteracts the behavioural primitivization induced by the NMDA receptor antagonist MK-801 in mice: implications for cognition. Prog Neuropsychopharmacol Biol Psychiatry. 2004;28(4):677–685. | ||
Rung JP, Carlsson A, Markinhuhta KR, Carlsson ML. The dopaminergic stabilizers (−)-OSU6162 and ACR16 reverse (+)-MK-801-induced social withdrawal in rats. Prog Neuropsychopharmacol Biol Psychiatry. 2005;29(5):833–839. | ||
Sahlholm K, Århem P, Fuxe K, Marcellino D. The dopamine stabilizers ACR16 and (−)-OSU6162 display nanomolar affinities at the σ-1 receptor. Mol Psychiatry. 2013;18(1):12–14. | ||
Su TP, Hayashi T, Maurice T, Buch S, Ruoho AE. The sigma-1 receptor chaperone as an inter-organelle signaling modulator. Trends Pharmacol Sci. 2010;31(12):557–566. | ||
Miki Y, Tanji K, Mori F, Wakabayashi K. Sigma-1 receptor is involved in degradation of intranuclear inclusions in a cellular model of Huntington’s disease. Neurobiol Dis. 2014;74:25–31. | ||
Lundin A, Dietrichs E, Haghighi S, et al. Efficacy and safety of the dopaminergic stabilizer pridopidine (ACR16) in patients with Huntington’s disease. Clin Neuropharmacol. 2010;33(5):260–264. | ||
de Yebenes JG, Landwehrmeyer B, Squitieri F, et al; for the MermaiHD study investigators. Pridopidine for the treatment of motor function in patients with Huntington’s disease (MermaiHD): a phase 3, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2011;10(12):1049–1057. | ||
Huntington Study Group HART Investigators. A randomized, double-blind, placebo-controlled trial of pridopidine in Huntington’s disease. Mov Disord. 2013;28(10):1407–1415. | ||
Landwehrmeyer B, Marder K, Billmann Ronn B, et al. Effects of the dopaminergic stabilizer pridopidine on motor symptoms in Huntington’s disease: a meta-analysis. Presented at: World Congress on Huntington Disease, September 11–14, 2011, Melbourne, Australia. | ||
Squitieri F, Landwehrmeyer B, Reilmann R, et al. One-year safety and tolerability profile of pridopidine in patients with Huntington disease. Neurology. 2013;80(12):1086–1094. | ||
Huntington Study Group. Unified Huntington’s Disease Rating Scale: reliability and consistency. Mov Disord. 1996;11(2):136–142. | ||
Esmaeilzadeh M, Kullingsjö J, Ullman H, Varrone A, Tedroff J. Regional cerebral glucose metabolism after pridopidine (ACR16) treatment in patients with Huntington disease. Clin Neuropharmacol. 2011;34(3):95–100. | ||
Squitieri F, Di Pardo A, Favellato M, Amico E, Maglione V, Frati L. Pridopidine, a dopamine stabilizer, improves motor performance and shows neuroprotective effects in Huntington disease R6/2 mouse model. J Cell Mol Med. Epub June 22, 2015 | ||
Wild EJ, Tabrizi SJ. Targets for future clinical trials in Huntington’s disease: what’s in the pipeline? Mov Disord. 2014;29(11):1434–1445. | ||
Ishikawa M, Hashimoto K. The role of sigma-1 receptors in the pathophysiology of neuropsychiatric diseases. J Receptor Ligand Channel Res. 2010;3:25–36. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.