")
Back to Journals » Drug Design, Development and Therapy » Volume 9
Profile of paritaprevir/ritonavir/ombitasvir plus dasabuvir in the treatment of chronic hepatitis C virus genotype 1 infection
Received 5 August 2015
Accepted for publication 16 September 2015
Published 13 November 2015 Volume 2015:9 Pages 6083—6094
DOI https://doi.org/10.2147/DDDT.S80226
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Wei Duan
Video abstract presented by Alice Lim.
Views: 1047
Michael A Smith, Alice Lim
Department of Pharmacy Practice and Pharmacy Administration, Philadelphia College of Pharmacy, University of the Sciences, Philadelphia, PA, USA
Abstract: Over the last several years, many advances have been made in the treatment of chronic hepatitis C virus (HCV) infection with the development of direct-acting antivirals. Paritaprevir/ritonavir/ombitasvir with dasabuvir (PrOD) is a novel combination of a nonstructural (NS) 3/4A protein inhibitor boosted by ritonavir, an NS5A protein inhibitor, and an NS5B nonnucleoside polymerase inhibitor. This review aims to discuss the pharmacology, efficacy, safety, drug interactions, and viral drug resistance of PrOD in the treatment of HCV genotype 1 infections. Phase I, II, and III human and animal studies that describe the pharmacology, pharmacokinetics, efficacy, and safety of PrOD for HCV were identified and included. Studies that evaluated patients without cirrhosis (n=2,249) and with cirrhosis (n=422) demonstrated that PrOD for 12 or 24 weeks was effective at achieving sustained virologic response rates (>90%) in patients with genotype 1a or 1b HCV infection. Although indicated for the treatment of HCV genotype 1 infection, PrOD is also recommended for the treatment of HCV in patients coinfected with HIV. Additionally, promising data exist for the use of PrOD in liver-transplant recipients. The most common adverse drug events associated with PrOD included nausea, pruritus, insomnia, diarrhea, asthenia, dry skin, vomiting, and anemia. The high efficacy rates seen coupled with a favorable side effect profile seen with PrOD with or without ribavirin have led to its addition as a recommended treatment regimen for HCV genotype 1 infection.
Keywords: direct-acting antiviral, interferon-free, ribavirin-free
Introduction
The World Health Organization and the Centers for Disease Control and Prevention have estimated global and national rates of chronic hepatitis C virus (HCV) infection to be approximately 180 million and 4 million people, respectively.1,2 Given the large number of patients infected with HCV, there has been great interest in drug development to improve on the sustained virologic response (SVR) rates of peginterferon (PegIFN)/ribavirin (RBV). In 2011, the first direct-acting antiviral (DAA) was approved and, since then, the field has grown so rapidly that guideline recommendations have been moved to a website (www.hcvguidelines.org).3 The landscape of DAA approval and use has evolved at such a rate that the first-generation DAAs (eg, telaprevir and boceprevir) are practically obsolete, giving way to new combinations of treatments.
The combination of paritaprevir (a nonstructural [NS] 3/4a protein inhibitor), ritonavir, ombitasvir (an NS5A protein inhibitor), and dasabuvir (an NS5B nonnucleoside polymerase inhibitor) with or without RBV has been approved to treat HCV genotype 1 infections.4 This combination, PrOD, is currently recommended as a first-line regimen for patients who are treatment-naïve with genotype 1a (with or without cirrhosis + RBV), 1b (with or without cirrhosis + RBV in cirrhosis), and 4 (without dasabuvir + RBV). It is also recommended for patients who have previously failed PegIFN/RBV with genotype 1a (with or without cirrhosis + RBV), 1b (with or without cirrhosis + RBV), and 4 (without dasabuvir + RBV).3 This review will focus on the use of PrOD to treat HCV genotype 1 infections.
Clinical pharmacology
Paritaprevir, previously known as ABT-450, inhibits the function of NS3/4A protease, which is an essential component of HCV viral replication. The half-maximal effective concentrations (EC50s) and intracellular concentrations of paritaprevir needed for potent antiviral activity against HCV genotype 1a and 1b were 1.0 and 0.21 nmol/L and 0.18 and 0.43 nM, respectively.5,6 When combined with the cytochrome P-450 (CYP-450) 3A4 inhibitor ritonavir, which has no HCV inhibitory properties, the area under the curve (AUC) of paritaprevir was increased approximately 48-fold while peak concentrations increased approximately 28-fold. The addition of ritonavir also prolonged the elimination half-life of paritaprevir, allowing for once-daily dosing.5 After oral administration, paritaprevir/ritonavir reached maximal exposure, above a dose-proportional response, in approximately 4 hours. Paritaprevir has an absolute bioavailability of approximately 50%, is highly protein bound, and has a moderate volume of distribution (16.7 L). Paritaprevir is metabolized by CYP3A4 and 3A5 and is mostly excreted in the feces (nearly 90%).4,6,7
Ombitasvir, previously known as ABT-267, is an inhibitor of NS5A, which is a phosphoprotein without enzymatic function but remains vital to HCV replicase.8,9 Its role in the HCV life cycle has been previously detailed in the journal Drug Design, Development and Therapy.10 The EC50 of ombitasvir in genotype 1a and 1b replicons is 14 and 5 pmol/L. The in vitro activity of ombitasvir has not been reported, given its lack of enzymatic function.9 As with paritaprevir, the time to maximal concentration is approximately 4 hours in a dose-proportional manner with an absolute bioavailability of 50%. The drug is highly protein bound with a large volume of distribution (50 L) and undergoes amide hydrolysis followed by oxidative metabolism producing antiviral inactive metabolites. Ombitasvir is primarily excreted in the feces (90%) with an elimination half-life of over 20 hours.4,11,12
Dasabuvir, previously known as ABT-333, is a nonnucleoside NS5B polymerase inhibitor that showed potent antiviral activity with EC50s and intracellular concentrations of 7.7 and 1.8 nmol/L and 2.8 and 3.7 nM to HCV genotype 1a and 1b, respectively.4,13,14 Dasabuvir and ombitasvir have similar absorption pharmacokinetics, with an absolute bioavailability of approximately 50% after oral administration. Dasabuvir is also highly protein bound with extensive distribution (396 L). It is metabolized to an active metabolite, M1, by CYP2C8. This metabolite has similar efficacy to that of dasabuvir. Nearly the entire dose (94%) is eliminated in the feces with an elimination half-life of 6 hours.4,13,14
Given the large role of hepatic metabolism, the pharmacokinetics of PrOD in patients with hepatic impairment is important. In a Phase I, single-dose trial of healthy participants with varying degrees of hepatic impairment, the pharmacokinetics of PrOD were evaluated.15 The study showed that mild hepatic impairment (Child-Pugh class A) did not significantly affect the pharmacokinetic parameters assessed (Cmax, Tmax, AUC, half-life, clearance, and unbound fraction of drug). The authors defined comparable as a ±30% difference from healthy controls. Drug exposure in mild hepatic impairment was within this range with the exception of ritonavir, which was decreased (34%). In moderate hepatic impairment (Child-Pugh class B), drug exposure was within this range with the exception of paritaprevir, which was increased (62%). Compared to the healthy controls, patients with severe hepatic impairment (Child-Pugh class C), drug exposure of paritaprevir was increased (920%), ritonavir was within range, ombitasvir was decreased (55%), and dasabuvir was increased (320%). Elimination half-lives were not particularly affected by hepatic impairment in mild and moderate disease; however, in severe disease, the half-lives of ritonavir and dasabuvir were significantly prolonged (approximately threefold).15 Based on this data, PrOD is contraindicated in patients with severe hepatic impairment and not recommended in moderate hepatic impairment.
No dosage adjustments are recommended in patients with any stage of renal dysfunction based on another Phase I pharmacokinetic study. Although the study showed changes in AUC in patients with creatinine clearances above 15 mL/min, elimination half-lives were not significantly different.4
Clinical trials
To date, there have been seven Phase III clinical trials evaluating the safety and efficacy of PrOD, with or without RBV, for HCV genotype 1 infection.16–21 For the primary outcome, each trial evaluated SVR at 12 weeks after the end of treatment (SVR12) as compared to the historical SVR12 rates of telaprevir/PegIFN/RBV therapy. Virologic failure and virologic relapse were also assessed. Virologic failure during treatment was defined as an HCV RNA level of 25 IU/mL or more during treatment, increase in HCV RNA level of more than 1 log10 IU/mL above nadir during treatment, or HCV RNA level of 25 IU/mL or more at any time during treatment in patients who received at least 6 weeks of treatment. Virologic relapse was defined as an HCV RNA level of 25 IU/mL or more after the end of completed treatment. All trials included patients aged 18–70 years (GIFT-I included patients aged 18–75 years) with HCV RNA levels >10,000 IU/mL and who did not have coinfection with hepatitis B or HIV-1. In studies of noncirrhotic patients, absence of cirrhosis was confirmed either by liver biopsy (Metavir score of 3 or less or Ishak score of 4 or less), FibroTest score of ≤0.72 and aspartate aminotransferase-to-platelet ratio index (APRI) of ≤2, or FibroScan® result of <9.6 kPa (<12.6 kPa in the GIFT-I study). Most of the studies were conducted in North America and Europe,16–19,21 and some studies included patients from Australia.16,17 One study was conducted in Japan.20 In each trial, treatment doses were paritaprevir 150 mg, ritonavir 100 mg, and ombitasvir 25 mg given once daily as a co-formulated tablet, along with dasabuvir 250 mg twice daily. If RBV was given, the dose was weight-based (1,000 mg daily if body weight <75 kg, 1,200 mg daily if body weight was ≥75 kg).
Historically, interleukin 28B (IL28B) non-CC genotype, black race, higher fibrosis scores, and higher baseline HCV RNA levels were associated with decreased response to HCV therapy, specifically with PegIFN/RBV therapy. Thus, each trial performed analyses of these characteristics to determine their influence on the efficacy of PrOD. The study characteristics and results are summarized in Table 1.
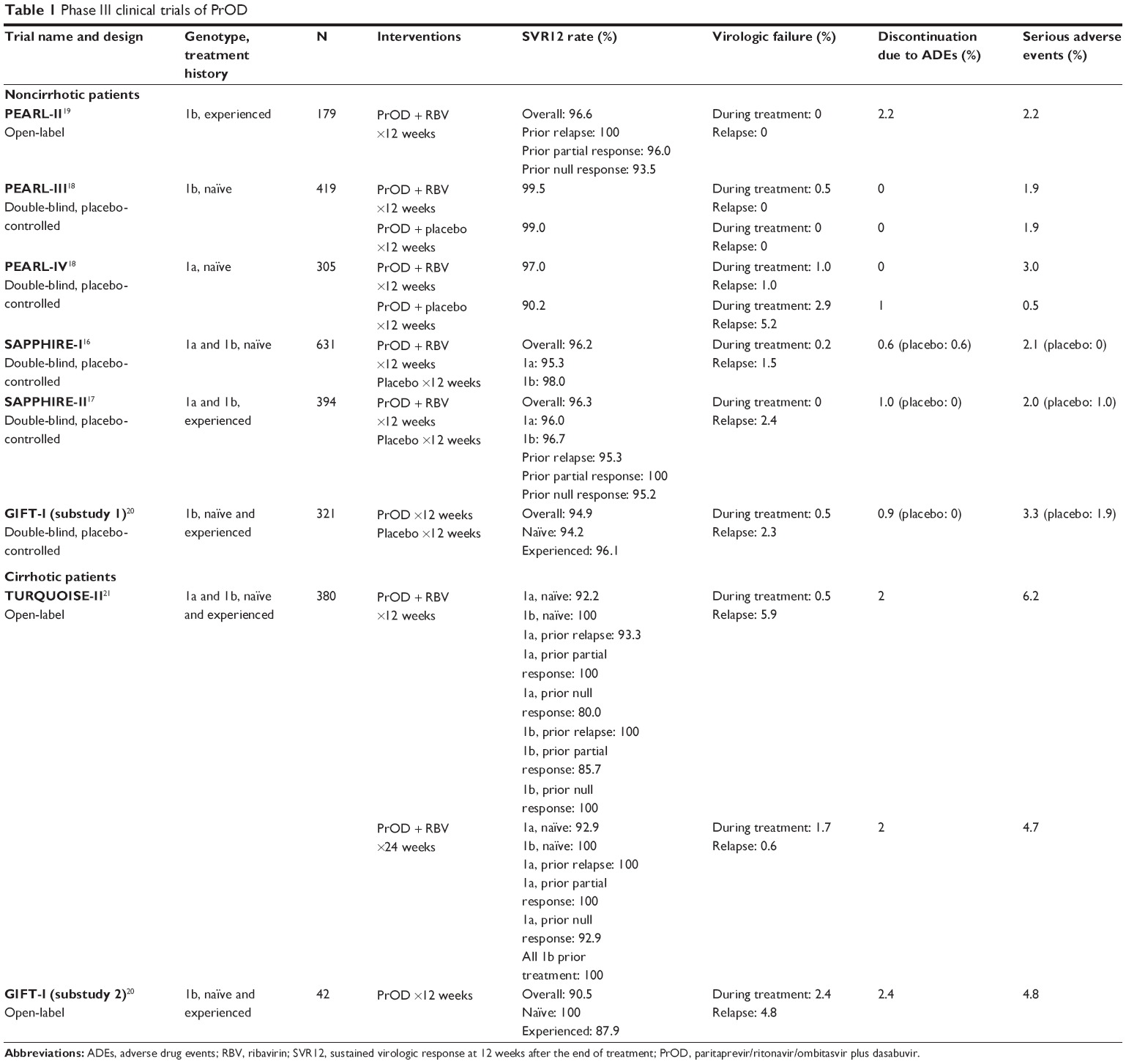 | Table 1 Phase III clinical trials of PrOD |
Noncirrhotic patients
SAPPHIRE-I was a randomized, double-blind, placebo-controlled trial that evaluated the use of PrOD plus RBV in treatment-naïve, noncirrhotic patients with either genotype 1a or 1b infection. Six hundred and thirty-one patients were randomized in a 3:1 ratio to receive active treatment or placebo; 96.2% of patients in the active treatment arm achieved SVR12. Response rates by genotype were 95.3% in patients with genotype 1a infection and 98.0% in patients with genotype 1b infection. Compared to the historical response rate with telaprevir/PegIFN/RBV therapy (SVR12 78%), this regimen was found to be both noninferior and superior. The high SVR rates were unaffected by IL28B genotype, race, fibrosis score, and baseline HCV RNA level. However, elevated body mass index (BMI) was significantly associated with reduced SVR rates (odds ratio 0.89, P=0.02). Still, those with BMI >30 kg/m2 experienced a high response rate of 91.5%. Overall, the study demonstrated high SVR12 rates with a regimen of PrOD plus RBV.16
SAPPHIRE-II compared PrOD plus RBV versus placebo in genotype 1a and 1b infection for those who previously failed PegIFN and RBV dual therapy. Three hundred and ninety-four patients were stratified by type of treatment failure and were randomized 3:1 to receive active treatment versus placebo for 12 weeks. Overall, the response rate was high at 96.3%, which was superior compared to the rates seen historically with telaprevir/PegIFN/RBV (SVR12 65%). Patients with genotype 1a and 1b experienced similarly high response rates, at 96% and 96.7%, respectively. SVR rates were 95.3% for those with prior relapse, 100% for those with prior partial response, and 95.2% for those with prior null response. Patient characteristics such as age, race, fibrosis score, HCV RNA level, and IL28B genotype did not impact SVR12 rates. No virologic failures occurred during treatment; however, seven patients (2.4%) experienced treatment relapse. Six of these patients had a prior null response to dual therapy. Similarly to treatment-naïve patients, SAPPHIRE-II demonstrated that treatment-experienced patients also experienced excellent efficacy with PrOD plus RBV.17
While all patients in the SAPPHIRE trials received RBV along with PrOD, PEARL-II, -III, and -IV evaluated the necessity of RBV therapy in HCV genotype 1 infection without cirrhosis.18,19 In PEARL-III, 419 treatment-naïve patients with genotype 1b infection were randomized to receive PrOD with or without RBV. Both groups did well, with 99.5% of patients with RBV achieving SVR12 and 99.0% of patients without RBV achieving SVR12. The two patients who did not achieve SVR in the RBV-free group were lost to follow-up. One patient taking the regimen with RBV experienced virologic failure during treatment. There were no treatment relapses. Given the very high response rates in each group, IL28B genotype status did not appear to affect treatment efficacy.18
PEARL-IV utilized the same study design as PEARL-III but in genotype 1a patients. Three hundred and five patients were randomized in a 1:2 ratio to receive PrOD with or without RBV and were stratified by IL28B genotype. Response rates were 97.0% in the RBV group compared to 90.2% without RBV. While both rates were noninferior and superior to historical rates found with telaprevir/PegIFN/RBV (SVR12 72%), the RBV-free regimen failed to meet noninferiority when compared to treatment with RBV (difference −6.8%, 95% confidence interval: −12.0 to −1.5). Virologic failures occurred more frequently in the RBV-free group compared to the RBV group (7.8% versus 2.0%). These patients were all found to have resistant variants. Unlike the genotype 1b patients in PEARL-III, IL28B genotype did have a statistically significant impact on response, with IL28B CC type experiencing higher rates of SVR (P=0.03). PEARL-III and -IV suggest that, while the omission of RBV appears to spare patients from added adverse drug events (ADEs) without sacrificing efficacy in patients with genotype 1b infection, RBV may be a necessary addition to decrease the risk of virologic failure in patients with genotype 1a infection.18
PEARL-II evaluated the efficacy of PrOD with or without RBV in treatment-experienced, noncirrhotic patients with genotype 1b infection. This was an open-label design with 179 patients stratified by type of prior failure to PegIFN and RBV dual therapy. Response rates were 96.6% in those with RBV and 100% in those without RBV. These responses were both superior to the historical response with telaprevir/PegIFN/RBV (SVR12 64%). The three patients who did not achieve SVR12 did not experience treatment failure, but rather discontinued treatment due to ADEs (n=2) or were lost to follow-up (n=1). High response rates were seen regardless of previous failure type, IL28B genotype, or sex. PEARL-II confirmed that PrOD treatment without RBV was noninferior to treatment with RBV in treatment-experienced patients with genotype 1b infection.19
Treatment-experienced patients with genotype 1a infection were not included in PEARL-II. However, given the results of PEARL-IV that suggested the necessity of RBV therapy in treatment-naïve patients with genotype 1a hepatitis C infection, it is likely that RBV is likewise important in treatment-experienced patients with genotype 1a infection.
GIFT-I was a clinical trial conducted in treatment-naïve and -experienced Japanese patients with genotype 1b infection. The study was divided by noncirrhotic patients (substudy 1) and cirrhotic patients (substudy 2). In substudy 1, 321 patients were randomized in a 2:1 ratio and given either PrOD or placebo for 12 weeks. The overall SVR12 rate was 94.9%, with response rates by treatment history at 94.2% in treatment-naïve patients and 96.1% in treatment-experienced patients. The placebo group received open-label therapy after the study treatment period and saw a similarly high SVR rate, at 98.1%. GIFT-I offers insight into the utility of PrOD in the previously understudied Japanese population.20
Cirrhotic patients
TURQUOISE-II attempted to evaluate the safety and efficacy of PrOD plus RBV in both treatment-naïve and treatment-experienced (prior treatment with PegIFN and RBV dual therapy) patients with compensated cirrhosis, defined as documentation of cirrhosis by liver biopsy (Metavir score >3 or Ishak score >4) or a FibroScan® result of ≥14.6 kPa within the last 6 months and a Child-Pugh class A score of <7 at screening. Patients with genotype 1a and 1b infection were both included. The study excluded patients with hepatocellular carcinoma, current or past evidence of Child-Pugh score B or C, platelet count <60,000/m3, serum albumin <2.8 g/dL, total bilirubin >3 mg/dL, international normalized ratio >2.3, and serum alpha-fetoprotein level >100 ng/mL. Three hundred and eighty patients were randomized to receive open-label PrOD plus RBV for 12 or 24 weeks. Compared to the historical SVR rate of telaprevir/PegIFN/RBV of 47%, both 12 weeks and 24 weeks were superior in efficacy, at 91.8% and 95.9%, respectively. The difference in SVR rates between the two duration groups was not significant (P=0.09); however, the difference was more pronounced in patients with genotype 1a infection who had a prior null response (12 weeks: 80%, 24 weeks: 92.9%). In treatment-naïve patients, the response rates for genotype 1a and 1b infection were 92.2%–92.9% and 100%, respectively. Response rates were unaffected by race, BMI, diabetes diagnosis, baseline HCV RNA level, platelet count, and albumin level. On the other hand, genotype 1a infection, former injection drug use, and prior null response were associated with lower SVR12 rates. Significantly more patients in the 12-week duration group experienced a relapse compared to the 24-week group. A majority of these patients had HCV genotype 1a infection and a prior null response, suggesting that longer durations of therapy are warranted in this population.21
In substudy 2 of GIFT-I, which included treatment-naïve and -experienced cirrhotic patients, 42 Japanese patients received open-label PrOD for 12 weeks. Cirrhosis was confirmed by liver biopsy (Metavir Score or New Inuyama score >3 or Ishak score >4), a FibroTest result of ≥0.73 and APRI >2, FibroScan® score ≥14.6 kPa, or screening discriminant score >0. Patients must also have had a Child-Pugh score of ≤6 at screening. A majority of patients were treatment-experienced (78.6%). Overall, 90.5% of patients reached SVR12, with treatment-naïve patients and treatment-experienced patients achieving response rates of 100% and 87.9%, respectively.20
TURQUOISE-II studied the utility of PrOD with RBV in cirrhotic patients, while GIFT-I demonstrated high efficacy of PrOD without RBV in genotype 1b patients with cirrhosis. Data from the TURQUOISE-III study confirmed the safety and efficacy of PrOD without RBV for 12 weeks in patients with genotype 1b infection and compensated cirrhosis. All 60 patients achieved SVR12.22
All seven trials demonstrated superior efficacy compared to the previous standard therapy of a protease inhibitor with PegIFN/RBV in the treatment HCV genotype 1. These trials were instrumental in determining the most recent guidelines for HCV genotype 1 infection, and that treatment should be dictated by genotype, treatment history, and presence of cirrhosis. RBV may be omitted in treatment-naïve and -experienced patients with genotype 1b infection with or without cirrhosis. On the other hand, RBV therapy is necessary in treatment-naïve and -experienced genotype 1a infection. In compensated cirrhosis, patients with genotype 1a infection require a 24-week duration of PrOD plus RBV, while patients with genotype 1b infection require a 12-week duration of PrOD without RBV regardless of treatment history.3
The strengths of these studies include their overall consistency in trial design, primary outcomes, and stratifications of characteristics historically shown to influence treatment efficacy. The placebo-controlled designs of SAPPHIRE-I, SAPPHIRE-II, and substudy 1 of GIFT-I allowed for an unobstructed view of the ADE profile of PrOD with or without RBV.16,17,20 Given the clear benefit of therapy over placebo, patients randomized to the placebo arms received open-label active treatment after the study period. However, several limitations must be considered. While the trials were conducted in multiple centers across different countries, they were widely Western populations with little representation of non-Caucasian races.16–19,21 GIFT-I and results from a Phase II study show promising results in the Japanese population, but results for other races and ethnicities remain to be clarified.20,23 These trials also excluded patients with hepatitis B or HIV-1 coinfection, decompensated cirrhosis, uncontrolled diabetes with hemoglobin A1c >8.5%, and creatinine clearance <60 mL/min.
Notably, BMI and IL28B genotype status significantly impacted efficacy rates in SAPPHIRE-I and PEARL-IV, respectively.16,18 The PEARL studies excluded patients with BMI >38 kg/m2, and thus may not have had enough patients with elevated BMI levels to detect a difference in efficacy rates.18,19 PEARL-IV only included genotype 1a patients, therefore the influence of IL28B genotype on response rates may be limited to this population, since IL28B status did not appear to impact other studies that included both genotype 1a and 1b infected patients.16–18,21 However, without further investigation, this has yet to be confirmed. Currently available trials also do not elucidate the utility of PrOD in the retreatment of patients who failed therapies other than PegIFN/RBV dual therapy and how this regimen compares to the other first-line recommended therapy of ledipasvir/sofosbuvir.
Special populations
Phase II clinical trial data are available for patients with HCV and HIV coinfection and HCV patients who have undergone liver transplantation.24–26 HCV/HIV coinfection is of particular clinical interest, as these patients not only experience greater risk for liver disease progression but have also experienced relatively low SVR rates with treatment.27 Complicating treatment further is the inherent risk of drug interactions with many HIV medications. TURQUOISE-I enrolled patients from the USA and Puerto Rico with HCV genotype 1 and HIV infection. It included patients who were treatment-naïve or treatment-experienced to HCV medications with or without cirrhosis, with HIV-1 RNA <40 copies/mL, and CD4+ T-cell count ≥200/mm3 or CD4+ T-cell percentage ≥14%. Patients were stable on atazanavir or raltegravir plus two nucleos(t)ide analog reverse transcriptase inhibitors for at least 8 weeks. Sixty-three patients received open-label PrOD with RBV for 12 or 24 weeks. SVR12 rates were 94% (29 of 31 patients) and 91% (29 of 32 patients) for 12 and 24 weeks of active treatment, respectively. The difference between treatment groups was not significant. Of the five patients who did not achieve SVR, two patients had genotype 1a infection, compensated cirrhosis, and prior null response to PegIFN and RBV. Two patients were treatment-naïve with noncirrhotic genotype 1a infection. One patient withdrew consent. Three patients in the 12-week group and five patients in the 24-week group had an HIV-1 RNA level higher than 40 copies/mL during treatment. Still, this was lower than the rate typically seen in HIV-1 monoinfection, and all achieved resuppression without a change in either antiviral regimen. No patients experienced an HIV-1 RNA level >200 copies/mL, suggesting there are minimal clinically relevant effects on each regimen caused by drug interactions. The most common ADEs were fatigue, insomnia, nausea, and headache. The only severe ADE deemed related to study treatment was insomnia. One patient taking atazanavir had a grade 4 elevation in total bilirubin which improved to grade 3 without the need for interruption in therapy. There were no discontinuations due to ADEs. These results demonstrate promising efficacy and safety of PrOD with RBV in HCV/HIV coinfected patients, although there are a significant number of drug interactions with PrOD and antiretroviral therapies.24
HCV infection is the leading cause of liver transplantation. Unfortunately, patients commonly have recurrence of infection, and consequently experience accelerated fibrosis and cirrhosis. In the open-label CORAL-1 trial, PrOD with RBV for 24 weeks was evaluated in liver-transplant recipients with no or mild fibrosis, defined by a Metavir score ≤2 on liver biopsy at least 9 months posttransplant and within 6 months of screening. Patients were included if they received a liver transplant at least 12 months prior to screening due to chronic HCV infection, had no evidence of advanced fibrosis within 6 months of screening, and were on a stable regimen of tacrolimus or cyclosporine with or without glucocorticosteroids. Concomitant cyclosporine or tacrolimus doses were decreased due to the boosting effects of ritonavir therapy. A majority of the patients had genotype 1a infection and non-CC IL28B genotype and had been previously treated with PegIFN and RBV. Out of 34 patients enrolled, 33 achieved SVR12 and SVR24 (97%). One patient with genotype 1a infection experienced a relapse and was found to have several resistance-associated variants. The most common ADEs were fatigue, headache, and cough. One patient achieved SVR12 despite discontinuing treatment after week 18 due to rash, memory impairment, and anxiety. Of note, RBV dosing was individualized and determined by the investigator due to the risk of hematologic toxicity in transplant recipients. A majority of patients received 600 or 800 mg daily at initiation and by completion. Nineteen patients decreased RBV during treatment, mostly due to hemoglobin declines. Five patients who had initial RBV doses of 1,000 or 1,200 mg daily required erythropoietin.25
Preliminary data from a Phase III study of genotype 1 patients with chronic kidney disease (stage 4 or 5) without cirrhosis or coinfection was reported in the Ruby-I trial.28 Thirteen genotype 1a patients were given PrOD plus RBV for 12 weeks while seven genotype 1b patients were given PrOD without RBV for 12 weeks. The majority of patients were male (85%), black (70%), without fibrosis (F0–F1 50%), and on dialysis (65%). Virologic response was assessed at the end of treatment (100% for n=14), SVR4 (100% for n=10), and SVR12 (100% for n=2). The Ruby-I trial showed that the use of PrOD was safe and effective in patients with end-stage renal disease, including those on dialysis.28
Drug interactions
Paritaprevir and ritonavir are mostly metabolized through the CYP3A system, while dasabuvir is mostly metabolized by CYP2C8 and, to a lesser extent, by CYP3A4. Ombitasvir undergoes amide hydrolysis followed by oxidative metabolism. Paritaprevir is a substrate and inhibitor of organic anion transporting polypeptide (OATP) IB1/B3, P-glycoprotein (P-gp), and breast cancer resistance protein (BCRP). Ritonavir’s primary role in the regimen is to inhibit CYP3A, but it also inhibits P-gp and BCRP. Dasabuvir is a P-gp and BCRP inhibitor. Ombitasvir, paritaprevir, ritonavir, and dasabuvir are all substrates of P-gp and BCRP.4,29
Mechanism-based drug–drug interactions and interactions with commonly prescribed medications in the HCV-infected population have been studied in a total of 228 subjects.29,30 Findings and recommendations are detailed in Table 2.29–32 Importantly, the presented information does not include every drug–drug interaction, and more interactions are likely to be revealed with future studies and Phase IV data.
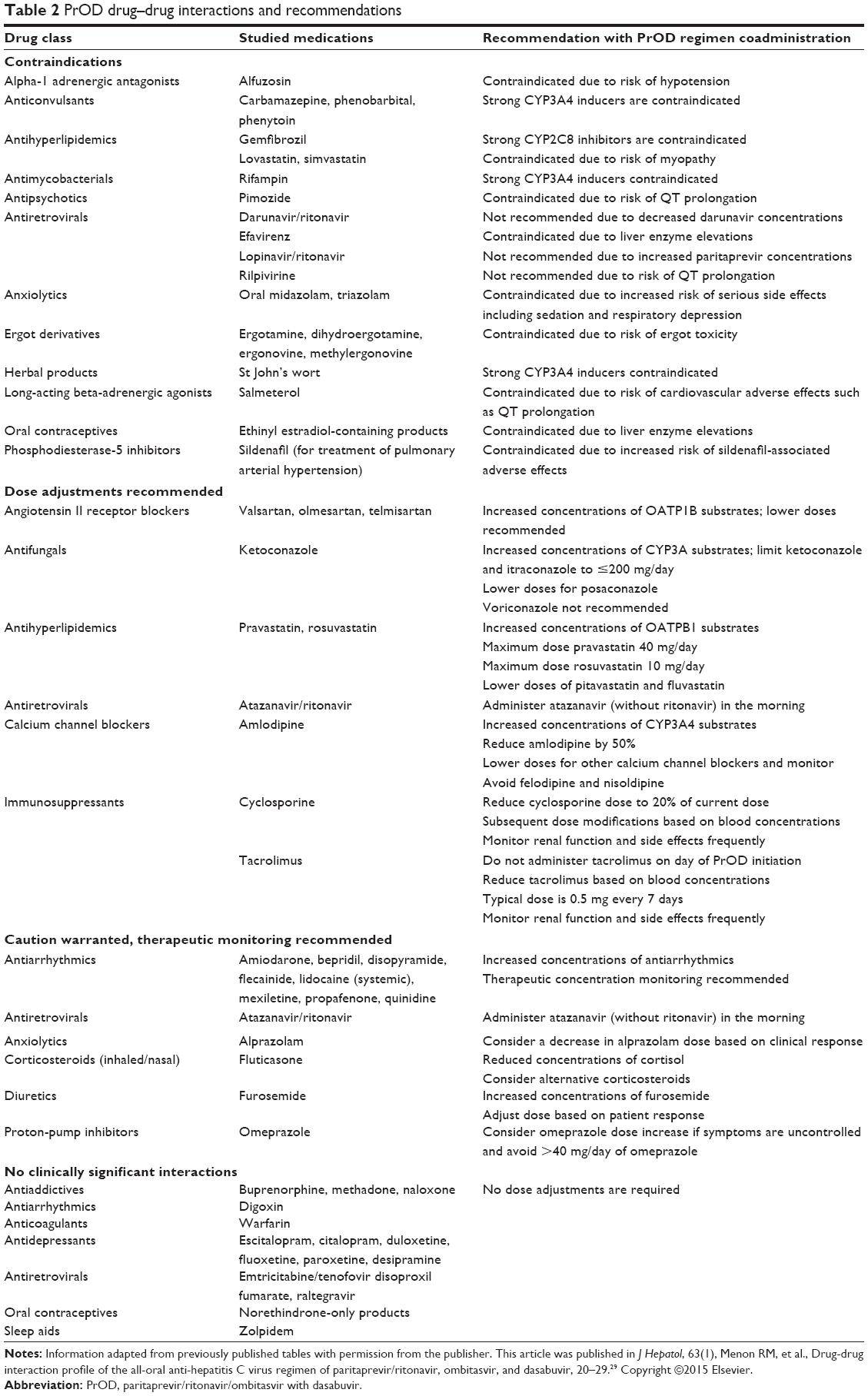 | Table 2 PrOD drug–drug interactions and recommendations |
Interactions specifically with HIV antiretroviral medications and immunosuppressants have been studied. In healthy volunteers, the AUC and Cmax of paritaprevir increased by 46% and 94% with atazanavir, and 119% and 216% with lopinavir, respectively. Meanwhile, trough concentrations increased for atazanavir and lopinavir and decreased for darunavir. As a result, lopinavir and darunavir are not recommended for coadministration with PrOD. Efavirenz toxicity increased significantly with PrOD, and coadministration is contraindicated. Rilpivirine AUC, Cmax, and Cmin increased by 225%, 155%, and 262%, respectively, and should not be given with PrOD due to increased risk of QT prolongation. In contrast, no interactions were found with emtricitabine, raltegravir, or tenofovir. Studies with dolutegravir and elvitegravir have not yet been conducted.26,31
The narrow therapeutic window of immunosuppressants makes posttransplant patients especially susceptible to toxicity or decreased efficacy from these drugs. Cyclosporine, tacrolimus, and sirolimus are CYP3A4 and P-gp substrates. Cyclosporine moderately inhibits CYP3A4 and P-gp. While cyclosporine was shown to increase the AUC and Cmax of paritaprevir and decrease the Cmax of dasabuvir in 12 healthy volunteers, these changes were considered clinically insignificant. In 72 healthy individuals participating in a Phase I study, PrOD increased the AUC and half-life of cyclosporine and tacrolimus.31 Based on these findings, the CORAL-1 study adjusted the doses of cyclosporine and tacrolimus in liver-transplant recipients. Participants taking cyclosporine had their dose reduced to 20% of their current dose.25 Dose adjustments were made throughout the study based on trough concentration levels. Tacrolimus doses were reduced to 0.5 mg every 7 days and adjusted throughout the study based on trough concentrations. Mycophenolate mofetil was permitted in the study and required no dose adjustments. Though sirolimus was not permitted in the study, it is known that ritonavir may increase concentrations of sirolimus, thus warranting therapeutic drug monitoring. Ritonavir also has the potential to increase prednisolone, therefore doses of prednisolone (and prednisone) did not exceed 5 mg daily in CORAL-1.25,26,31
Safety
The most common ADEs associated with PrOD treatment are nausea, pruritus, insomnia, diarrhea, asthenia, dry skin, vomiting, and anemia. The majority of patients treated in clinical trials experienced at least one ADE without the use of RBV (67%–82%); however, the use of RBV increases the risk of experiencing an ADE (79%–97%). Patients treated with RBV are more likely to suffer from fatigue, nausea, pruritus/rash, insomnia (P≤0.08), increased bilirubin, exertional dyspnea, anemia, dry skin, and cough (P<0.02, except where noted). Duration of treatment also affected rates of ADEs, with those being treated for longer more likely to suffer from fatigue, dyspnea, back pain, memory impairment, and upper respiratory tract infections. Presence or absence of cirrhosis had no bearing on the rate of ADEs. Serious ADEs and therapy discontinuations were reported in 0%–3% and 2.2% of patients treated with PrOD plus RBV and 0%–6.2% and 2.3% of patients treated with PrOD, respectively. Similar rates were seen in treatment-experienced patients and patients coinfected with HIV.16–25
Viral drug resistance
During HCV replication, RNA-dependent RNA polymerase lacks the editing function to remove errors in base pairs, resulting in amino acid substitutions in HCV viral proteins.32,33 Coupled with high virion production, this potentiates a vastly heterogeneous viral population. As anti-HCV treatments work to eliminate wild-type HCV, resistant variants survive and reproduce to eventually overtake the viral population, thus inciting virologic breakthrough and resistance to treatment.34–36 Mutations can develop in all three viral protein targets of PrOD, including NS3/4A, NS5A, and NS5B. The rate and type of resistance differs between genotype 1a and 1b.37 Given the lack of data on long-term effects of resistant variants, it remains to be determined whether resistance persists long enough to impact retreatment, and whether baseline resistance testing is valuable in informing treatment decisions.
Paritaprevir
In an in vitro study, HCV genotype 1a and 1b viral colonies were exposed to paritaprevir at concentrations ten-, 100-, and 500-fold greater than the EC50. In genotype 1a, no colonies survived at concentrations 100- or 500-fold greater. The surviving colonies contained NS3 substitutions at R155K and D168E/N. R155K almost universally results in resistance to HCV NS3/4A protease inhibitors, therefore the risk of cross-resistance is high. In genotype 1b, no colonies survived at 500-fold greater than EC50. The prevailing resistant variants observed were A156T and D168H/V/Y. In both genotypes, D168Y confers the highest level of resistance at >200-fold resistance.5
Ombitasvir
After in vitro exposure to ombitasvir at ten-, 100-, or 1,000-fold greater than the EC50, the predominant variants selected in genotype 1a were M28T/V, Q30R, and Y93C/H. When these resistant colonies were expanded, M28V resulted in 58-fold resistance, while M28T, Q30R, and Y93C/H resulted in >800-fold resistance. In vivo analysis of ombitasvir monotherapy in treatment-naïve genotype 1a infection led to resistant substitutions at amino acid positions M28, Q30, and Y93. None of the 12 patients had resistant variants detected at baseline. In genotype 1b, Y93H was the most prevalent surviving variant, resulting in 77-fold resistance. Similar to other NS5A inhibitors, ombitasvir demonstrates a low genetic barrier to resistance and requires the coadministration of other agents to minimize the development of resistance.12
Dasabuvir
In one in vitro study, HCV colonies were exposed to dasabuvir at levels ten- or 100- fold greater than the EC50. The predominant surviving colony of HCV genotype 1a at concentrations tenfold greater was S556G (43%). At concentrations 100-fold greater, C316Y, Y448C, C451R, and S556G were selected. Variants A395G, M414T, N444K, S556G/N, and S565F resulted in ten- to 32-fold resistance to dasabuvir, while C316Y and Y448C/H resulted in >940-fold resistance. In genotype 1b colonies, the predominant variants selected after exposure to dasabuvir at tenfold higher than EC50 were C316Y (33%) and M414T (25%). At concentrations 100-fold greater, all 12 surviving colonies selected for C316Y. Again, C316Y substitution conferred the highest level of resistance, at 1,569-fold. S368T, N411S, M414T, and A553V produced 47- to 139-fold resistance. Additionally, C316Y, M414T, Y448H, and S556G variants were found to have the highest replication efficiency rate, further compounding the resistance rate against dasabuvir.13
Clinical trial data report the emergence of virologic resistance in patients receiving PrOD treatment who either failed therapy during treatment or experienced relapse. In patients with genotype 1a infection who failed treatment, the most frequently observed amino acid variants in genotype 1a infection were D168A/V/Y in NS3, M28T and Q30R in NS5A, and M414T and S556G in NS5B.16–18,21,38 The most frequently observed amino acid variants in patients with genotype 1b infection who failed therapy were Y56H and D168V in NS3, Y93H in NS5A, and S556G in NS5B.16–18,21,38
Conclusion
PrOD has been shown to be effective for the treatment of genotype 1a and 1b chronic HCV infection in treatment-naïve and treatment-experienced patients with and without cirrhosis. The addition of RBV to this regimen is still required in any patient with genotype 1a and in cirrhotic patients with genotype 1b infections, which, along with its higher pill burden, may limit its use in comparison to other available agents. There is also evidence to support the use of PrOD in the treatment of special populations, including HCV/HIV coinfection, post-liver-transplant HCV genotype 1 infections, and those with renal impairment. PrOD has an advantage over other antivirals on the market for patients with end-stage renal disease given the results of the Ruby-I study; however, its use in coinfection may be limited by the drug–drug interaction potential with antiretrovirals, and it should not be used in decompensated liver disease or in patients who previously failed a protease inhibitor. Clinical trial data have also shown PrOD to have a favorable ADE profile compared to historical regimens, with a higher risk of adverse events if used along with RBV. More clinical data are still needed to elucidate its use in those who failed prior therapies other than pegIFN/RBV, additional drug interactions, the impact of resistance on retreatment, and the utility of baseline resistance testing. Given the regimen’s high efficacy and relatively mild side effect profile, PrOD with or without RBV is currently considered one of several first-line DAA regimens for the treatment of HCV genotype 1 infection.
Disclosure
The authors report no conflicts of interest in this work.
References
World Health Organization. Guidelines for the Screening, Care and Treatment of Persons with Hepatitis C Infection. Geneva: World Health Organization; 2014. | ||
Smith BD, Morgan RL, Beckett GA, et al; Centers for Disease Control and Prevention. Recommendations for the identification of chronic hepatitis C virus infection among persons born during 1945–1965. MMWR Recomm Rep. 2012;61(RR-4):1–32. | ||
Hepatitis C Guidance: Recommendations for Testing, Managing, and Treating Hepatitis C [webpage on the Internet]. American Association for the Study of Liver Diseases (AASLD), Infectious Diseases Society of America (IDSA). Available from: http://www.hcvguidelines.org/. Accessed September 10, 2015. | ||
Viekira Pak (ombitasvir, paritaprevir, and ritonavir tablets; dasabuvir tablets) copackaged for oral use [prescribing information]. North Chicago: AbbVie Inc; 2015. | ||
Pilot-Matias T, Tripathi R, Cohen D, et al. In vitro and in vivo antiviral activity and resistance profile of the hepatitis C virus NS3/4A protease inhibitor ABT-450. Antimicrob Agents Chemother. 2015;59(2):988–997. | ||
Gentile I, Borgia F, Buonomo AR, Zappulo E, Castaldo G, Borgia G. ABT-450: a novel protease inhibitor for the treatment of hepatitis C virus infection. Curr Med Chem. 2014;21(28):3261–3270. | ||
Badri P, Dutta S, Coakley E, et al. Pharmacokinetics and dose recommendations for cyclosporine and tacrolimus when coadministered with ABT-450, ombitasvir, and dasabuvir. Am J Transplant. 2015;15(5):1313–1322. | ||
Fridell RA, Qiu D, Valera L, Wang C, Rose RE, Gao M. Distinct functions of NS5A in hepatitis C virus RNA replication uncovered by studies with the NS5A inhibitor BMS-790052. J Virol. 2011;85(14):7312–7320. | ||
DeGoey DA, Randolph JT, Liu D, et al. Discovery of ABT-267, a pan-genotypic inhibitor of HCV NS5A. J Med Chem. 2014;57(5):2047–2057. | ||
Lee C. Daclatasvir: potential role in hepatitis C. Drug Des Devel Ther. 2013;7:1223–1233. | ||
Gentile I, Buonomo AR, Borgia G. Ombitasvir: a potent pan-genotypic inhibitor of NS5A for the treatment of hepatitis C virus infection. Expert Rev Anti Infect Ther. 2014;12(9):1033–1043. | ||
Krishnan P, Beyer J, Mistry N, et al. In vitro and in vivo antiviral activity and resistance profile of ombitasvir, an inhibitor of hepatitis C virus NS5A. Antimicrob Agents Chemother. 2015;59(2):979–987. | ||
Kati W, Koev G, Irvin M, et al. In vitro activity and resistance profile of dasabuvir, a nonnucleoside hepatitis C virus polymerase inhibitor. Antimicrob Agents Chemother. 2015;59(3):1505–1511. | ||
Gentile I, Buonomo AR, Borgia G. Dasabuvir: a non-nucleoside inhibitor of NS5B for the treatment of hepatitis C virus infection. Rev Recent Clin Trials. 2014;9(2):115–123. | ||
Khatri A, Menon R, Marbury TC, et al. Pharmacokinetics and safety of coadministered paritaprevir plus ritonavir (paritaprevir/r), ombitasvir, and dasabuvir in hepatic impairment. J Hepatol. Epub 2015 Jun 10. | ||
Feld JJ, Kowdley KV, Coakley E, et al. Treatment of HCV with ABT-450/r-ombitasvir and dasabuvir with ribavirin. N Engl J Med. 2014;370(17):1594–1603. | ||
Zeuzem S, Jacobson IM, Baykal T, et al. Retreatment of HCV with ABT-450/r-ombitasvir and dasabuvir with ribavirin. N Engl J Med. 2014;370(17):1604–1614. | ||
Ferenci P, Bernstein D, Lalezari J, et al; PEARL-III Study; PEARL-IV Study. ABT-450/r-ombitasvir and dasabuvir with or without ribavirin for HCV. N Engl J Med. 2014;370(21):1983–1992. | ||
Andreone P, Colombo MG, Enejosa JV, et al. ABT-450, ritonavir, ombitasvir, and dasabuvir achieves 97% and 100% sustained virologic response with or without ribavirin in treatment-experienced patients with HCV genotype 1b infection. Gastroenterology. 2014; 147(2):359–365.e1. | ||
Kumada H, Chayama K, Rodrigues L Jr, et al. Randomized phase 3 trial of ombitasvir/paritaprevir/ritonavir for hepatitis C virus genotype 1b-infected Japanese patients with or without cirrhosis. Hepatology. Epub 2015 Jul 3. | ||
Poordad F, Hezode C, Trinh R, et al. ABT-450/r-ombitasvir and dasabuvir with ribavirin for hepatitis C with cirrhosis. N Engl J Med. 2014;370(21):1973–1982. | ||
Feld JJ, Moreno C, Trinh R, et al. Turquoise-III: safety and efficacy of 12-week ribavirin-free treatment for patients with HCV genotype 1b and cirrhosis. [Abstract P226.] 15th Annual International Symposium on Viral Hepatitis and Liver Disease. June 26–28, 2015; Berlin, Germany. | ||
Chayama K, Notsumata K, Kurosaki M, et al. Randomized trial of interferon- and ribavirin-free ombitasvir/paritaprevir/ritonavir in treatment-experienced hepatitis C virus-infected patients. Hepatology. 2015;61(5):1523–1532. | ||
Sulkowski MS, Eron JJ, Wyles D, et al. Ombitasvir, paritaprevir co-dosed with ritonavir, dasabuvir, and ribavirin for hepatitis C in patients co-infected with HIV-1: a randomized trial. JAMA. 2015;313(12):1223–1231. | ||
Kwo PY, Mantry PS, Coakley E, et al. An interferon-free antiviral regimen for HCV after liver transplantation. N Engl J Med. 2014;371(25):2375–2382. | ||
Toussaint-Miller KA, Andres J. Treatment considerations for unique patient populations with HCV genotype 1 infection. Ann Pharmacother. 2015;49(9):1015–1030. | ||
Graham CS, Baden LR, Yu E, et al. Influence of human immunodeficiency virus infection on the course of hepatitis C virus infection: a meta-analysis. Clin Infect Dis. 2001;33(4):562–569. | ||
Pockros PJ, Reddy KR, Mantry PS, et al. L01 : safety of ombitasvir/paritaprevir/ritonavir plus dasabuvir for treating HCV GT1 infection in patients with severe renal impairment or end-stage renal disease: the Ruby-I study. J Hepatol. 2015;62:S257–S261. | ||
Menon RM, Badri PS, Wang T, et al. Drug-drug interaction profile of the all-oral anti-hepatitis C virus regimen of paritaprevir/ritonavir, ombitasvir, and dasabuvir. J Hepatol. 2015;63(1):20–29. | ||
Lalezari J, Sullivan JG, Varunok P, et al. Ombitasvir/paritaprevir/r and dasabuvir plus ribavirin in HCV genotype 1-infected patients on methadone or buprenorphine. J Hepatol. 2015;63(2):364–369. | ||
Burgess S, Partovi N, Yoshida EM, Erb SR, Azalgara VM, Hussaini T. Drug interactions with direct-acting antivirals for hepatitis C: implications for HIV and transplant patients. Ann Pharmacother. 2015;49(6):674–687. | ||
Soriano V, Labarga P, Barreiro P, et al. Drug interactions with new hepatitis C oral drugs. Expert Opin Drug Metab Toxicol. 2015;11(3):333–341. | ||
Simmonds P. Genetic diversity and evolution of hepatitis C virus – 15 years on. J Gen Virol. 2004;85(Pt 11):3173–3188. | ||
Rong L, Dahari H, Ribeiro RM, Perelson AS. Rapid emergence of protease inhibitor resistance in hepatitis C virus. Sci Transl Med. 2010;2(30):30ra32. | ||
Halfon P, Locarnini S. Hepatitis C virus resistance to protease inhibitors. J Hepatol. 2011;55(1):192–206. | ||
Kuntzen T, Timm J, Berical A, et al. Naturally occurring dominant resistance mutations to hepatitis C virus protease and polymerase inhibitors in treatment-naïve patients. Hepatology. 2008;48(6):1769–1778. | ||
Wyles DL, Gutierrez JA. Importance of HCV genotype 1 subtypes for drug resistance and response to therapy. J Viral Hepat. 2014;21(4):229–240. | ||
Lontok E, Harrington P, Howe A, et al. Hepatitis C virus drug resistance-associated substitutions: state of the art summary. Hepatology. Epub 2015 Jun 10. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.