")
Back to Archived Journals » Gastrointestinal Cancer: Targets and Therapy » Volume 5
Profile of lanreotide autogel and its potential in the treatment of gastroenteropancreatic neuroendocrine tumors
Received 11 May 2015
Accepted for publication 20 July 2015
Published 2 September 2015 Volume 2015:5 Pages 123—130
DOI https://doi.org/10.2147/GICTT.S70701
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Eileen O'Reilly
Anja Rinke, Daniela Müller
Department of Internal Medicine, Division of Gastroenterology and Endocrinology, Philipps University, Marburg, Germany
Abstract: Gastroenteropancreatic neuroendocrine tumors (GEPNETs) comprise a heterogenous group of neoplasm, and their incidence is increasing in the population. Approximately one-third of these tumors are associated with characteristic hormonal syndromes like flushing and diarrhea in carcinoid syndrome. Most GEPNETs express receptors for somatostatin. The somatostatin analogs octreotide and lanreotide constituted a major therapeutic advance in palliating hypersecretion syndromes. Lanreotide autogel is a viscous aqueous solution of lanreotide that is usually administered deep subcutaneously every 4 weeks, but extended dosing intervals have also been proposed. In recent years, increasing evidence has supported the use of somatostatin analogs (SSA) as antitumor agents. In the double-blind, placebo-controlled, Phase III CLARINET trial, lanreotide autogel 120 mg every 4 weeks was associated with a significant prolongation of progression-free survival in patients with nonfunctioning GEPNETs, with a Ki-67 of <10%. This antiproliferative effect and the favorable toxicity profile make lanreotide autogel attractive for long-term treatment in patients with well-differentiated GEPNETs. Further studies are needed to evaluate the role of lanreotide autogel as a combination partner and the efficacy of high-dose lanreotide.
Keywords: neuroendocrine tumor, somatostatin analogs, lanreotide, symptomatic treatment, antiproliferative treatment
Introduction
Neuroendocrine tumors of gastrointestinal (GI) or pancreatic origin – also called gastroenteropancreatic neuroendocrine tumors (GEPNETs) – are clinically and prognostically heterogeneous neoplasms derived from the diffuse neuroendocrine cell system. Historically regarded as rare disease, several registries demonstrated an increase in incidence in recent years.1,2 In terms of estimated prevalence according to the American SEER data, the GEPNETs are the second most common GI cancers.1
The heterogeneous nature of these tumors is reflected in a broad range of clinical presentations as well as disease courses. Approximately 20%–30% of GEPNETs are functioning, which means that they are associated with a hormonal syndrome like carcinoid syndrome or Zollinger–Ellison syndrome. Stage at diagnosis and proliferative activity (Ki-67 index) have been shown to be prognostically significant.3–7 A new classification system, the WHO 2010 classification, which is mainly based on the proliferative activity, has been established.8
In terms of treatment, operative removal of tumors when there is no evidence of (nonresectable) metastases is the only curative therapeutic approach, but still 25%–50% of disease cases are not diagnosed until metastases are already present, thus requiring additional treatment options.1,9
The nonmedical approaches include surgical tumor debulking, (chemo-) embolization of liver metastases, radiofrequency ablation, and selective internal radiotherapy of liver metastases and treatment with 90yttrium- or 177lutetium-labeled somatostatin analogs (SSAs; peptide receptor radionuclide therapy [PRRT]).
In the palliative situation, various substances have been tested in recent years with the aim of inhibiting or delaying tumor growth. For instance, certain chemotherapeutic drugs have exhibited an antiproliferative effect, especially against neuroendocrine pancreatic tumors (pNETs).10,11 These include, in particular, streptozocin combined with 5-fluorouracil or doxorubicin. Temozolomide has also been tried in combination with capecitabine with encouraging results.12
In extrapancreatic tumors, especially midgut and hindgut tumors, however, the results of chemotherapy are disappointing.
Recently, the therapeutic options in patients with metastatic GEPNETs have increased with the availability of the mTOR inhibitor everolimus and the multikinase inhibitor sunitinib. Both drugs demonstrated a significant prolongation of progression-free survival (PFS) in placebo-controlled randomized trials that included patients with progressive, metastatic, well-differentiated pancreatic NETs.13,14 Both drugs were approved for pNETs.
Everolimus in combination with octreotide long-acting repeatable (LAR) has also been investigated in patients with carcinoid syndrome15 and also as monotherapy in patients with nonfunctioning NETs of the lung and intestine (RADIANT-4 trial, results pending), but has not been approved for extrapancreatic NETs so far.
Simultaneously with the development of new drugs, the role of the SSAs has expanded significantly. Originally developed in the 1980s to control hypersecretion syndromes such as the carcinoid syndrome,16 SSAs have recently been shown to prolong PFS in patients with metastasized NETs.17,18 These antiproliferative effects and the favorable toxicity profile make SSAs attractive for long-term treatment in patients with well-differentiated NETs.
The depot formulation of the SSA lanreotide – lanreotide autogel – has been available in Europe for approximately 10 years. While there is also comprehensive literature on its role in acromegaly, this review focuses on the role of lanreotide in GEPNETs.
Somatostatin and somatostatin receptors
In 1972, somatostatin (SST) was incidentally discovered in sheep hypothalami during the search for a growth hormone (GH)-releasing hormone.19 It was described as an inhibitor of GH and analysis of the structure revealed a 14 amino-acid peptide. In addition, a second bioactive form of SST – the less predominant but more potent SST-28 – was identified.20 SST was subsequently discovered to be a pan inhibitor not only of GH but also of GI hormone secretion. It additionally reduces exocrine secretion and motility, portal blood flow, and intestinal absorption.20 Immunomodulatory effects of SST by regulation of cytokine secretion in B- and T-cells have been described.21
The physiological effects of SSTs are a result of specific interaction with SST receptors, a family of G-coupled proteins with seven transmembrane domains and five receptor subtypes (SSTR1–5).
GEPNETs often express SSTRs at supraphysiological levels, with SSTR2 being the most prevalent subtype in these tumors.22,23
SSAs – developmental history
As the natural SSTs have a very short circulation half-life of approximately 2 minutes, their clinical utility is very limited. In the early 1980s, several synthetic peptides, including the octapeptides octreotide and lanreotide, were developed.24 These SSAs retain the binding affinity for SSTRs – at least with high affinity for subtype 2 receptor and moderate affinity for subtype 5 receptor – and are more resistant to peptidases resulting in longer half-lives. Octreotide with subcutaneous (sc) injections three times daily was the first clinically available SSA approved in 1988 for the treatment of hormone syndromes like carcinoid syndrome. To provide more sustained drug levels and further improve quality of life, a LAR formulation of octreotide was developed and introduced in 1995 by slow release of octreotide through a biodegradable microsphere suspension. Octreotide LAR is typically administered intramuscularly (im) every 4 weeks. The same year, a slow release formulation of lanreotide – lanreotide MP – was licensed in France. This formulation was injected im every 14 days. Lanreotide autogel is a viscous aqueous solution composed of only lanreotide and water that is usually administered deep sc every 4 weeks. This formulation was licensed in France in 2001, in Germany in 2007, and was very recently also approved by the US Food and Drug Administration (FDA).
In 2004, an SSA with broader SSTR binding affinity, the cyclohexapeptide SOM 230 or pasireotide, was developed. At the moment, this drug is approved for Cushing’s syndrome and acromegaly and is under investigation in several NET trials.
Table 1 summarizes the pharmacokinetics of SST isoforms and SSAs.
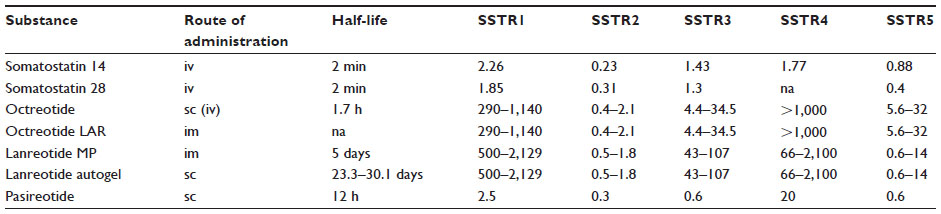 | Table 1 Summary of pharmacokinetics in somatostatin and somatostatin analogs and binding affinity profiles |
Pharmacokinetic features of lanreotide autogel
After a single sc injection, a dose-dependent peak serum concentration (Cmax) was reached the first day. After repeated administrations, lanreotide autogel exhibited linear pharmacokinetics over the dose range. After reaching Cmax, there was a slow decline in serum concentrations, with an elimination half-life of 23, 27, and 30 days. A steady state was reached after four injections.25 The absolute bioavailability after deep sc administration of lanreotide autogel in healthy adults was 63%. The rate of absorption and bioavailability of lanreotide autogel were independent of the administered dose in the range from 60 to 120 mg, and no significant effect of sex, age, or bodyweight was found.26
At the last American Society of Clinical Oncology GI cancer symposium, the pharmacokinetic analysis of 1,541 lanreotide serum concentrations obtained from 290 GEPNET patients treated with lanreotide autogel within four study protocols was presented: None of the patients characteristics tested (including demographics, age, renal function markers, sex, and body weight) were found to be clinically relevant in terms of requiring dose adjustment in clinical practice.27
As there are still effective serum concentrations 56 days after injection, extended dosing intervals have been proposed.26 In GEPNET patients, the vast majority of trials used injection intervals of 4 weeks. In a randomized trial in 60 patients with well-differentiated neuroendocrine tumors, 120 mg lanreotide autogel every 6 weeks was as effective for tumor control as 60 mg lanreotide MP every 3 weeks.28
Mechanism of biological activity
Binding of SSA mainly to the receptor subtypes SSTR1, SSTR2, and SSTR5 results in inhibition of calcium channels and stimulation of various potassium channels. This leads to depletion of intracellular calcium concentration and inhibition of adenylate cyclase, with consequent decrease in cyclic adenosine monophosphate levels and inhibition of protein phosphatases that inhibit exocytosis. This mechanism and also direct effects on regulation of vesicle exocytosis explain the antisecretory effects of SSA.20,29
Regarding the antiproliferative effects of SSA, a variety of different mechanisms depending on SSTR subtype and cell type have been described. Indirect mechanisms include inhibition of circulating growth factors such as vascular endothelial growth factor, insulin-like growth factor (IGF), gastrin, and epidermal growth factor. Inhibition of tumor angiogenesis through interaction with SSTRs on resident cells of the surrounding microenvironment may also contribute to the antiproliferative effect.20
Upon activation, all SSTRs induce cell cycle inhibitors such as p27 and p21, thus leading to cell cycle arrest.30 Suppression of mitogenic pathways like the ERK1/2/PI3K/AKT and the nitric oxide (NO)/cyclic guanosine monophosphate (cGMP) pathway may result from activation of several phosphotyrosine phosphatases after binding of SSA to SSTRs. SSA can also directly induce apoptosis in tumor cells through different mechanisms.31,32 Restoration of functional gap junctions has been suggested to be another mechanism of SSA-induced cell growth inhibition.31 Changes in natural killer cell activity have been reported during SSA treatment of patients with metastatic GEPNET that might contribute to the antiproliferative effects.31
Lanreotide for symptom control
Lanreotide was initially licensed for the treatment of hormonal symptoms associated with advanced NETs, and most data are related to the carcinoid syndrome. A crossover study on 33 patients with carcinoid syndrome compared the efficacy of 30 mg lanreotide MP every 10 days im and 200 μg octreotide three times a day sc in controlling flushing and diarrhea. Both drugs were equally efficacious in terms of symptom control and reduction in biomarkers.33 Seventy-one patients with carcinoid syndrome were included in an open multicentric trial of lanreotide autogel for 6 months.34 The response to the first injection was rapid and sustained. The clinical effect further increased after the second and third injection and reached a plateau after four injections. At the end of the study, 80% of patients with flushing and 75% of patients with diarrhea had an improvement of symptoms compared to baseline. A release of at least 50% of flushing episodes was reported in 65%, a 50% reduction of diarrhea in 18% of patients. Less effect on diarrhea may be explained by other causes for diarrhea than hormone hypersecretion, like bile salt release after ileal resection, short bowel syndrome, and side effects of SSA. The greater benefit for flushing than for diarrhea was also reported with other SSA formulations.33,35
More recently Khan et al36 provided data on long-term treatment with lanreotide autogel in 69 patients with carcinoid syndrome treated in their center over a period of 9 years with a median follow-up of 33 months.36 In this retrospective trial, 94% of patients achieved a symptomatic response at the first follow-up visit. Twenty patients required an increase in dose during long-term follow-up, from the starting dose to 120 mg, to achieve adequate symptomatic control. In 74% of patients, the carcinoid syndrome was well controlled with lanreotide autogel alone throughout the study period.
In the ELECT study (NCT00774930), 115 patients with carcinoid syndrome and morphological stable disease for at least 6 months were randomized to either lanreotide autogel 120 mg every 4 weeks sc or placebo. The blinded treatment phase of 16 weeks was followed by an open label phase for 32 weeks. Primary endpoint of the trial was the use of rescue octreotide sc during the blinded phase as a surrogate parameter for symptom control. The percentage of days in which octreotide was used was significantly lower in the lanreotide autogel group than in the placebo group, but the predefined absolute treatment difference of 30% was not met. A complete treatment success (no use of octreotide) was seen more often in patients treated with lanreotide compared to placebo (40.7% versus 23.2%).37
The international noninterventional SymNET trial (NCT01234168) assessed patient-reported satisfaction with diarrhea control in 273 patients with carcinoid syndrome. The majority of included patients (75.7%) was satisfied with the control of diarrhea after lanreotide autogel treatment.38
Biochemical response with lanreotide autogel – in most trials defined as a ≥50% reduction of baseline tumor markers (chromogranin A and urinary 5-hydroxyindoleacetic acid [5-HIAA]) – was reported in 24%–55%.28,34,36 High levels of 5-HIAA are associated with increased risk for carcinoid heart syndrome characterized by plaque-like endocardial deposits of fibrous tissue on the tricuspid valve leaflets, the pulmonic valve, and also right atrium and right ventricle. Therefore, decrease in hormone levels under SSA treatment could contribute to reduced risk or delay in development of carcinoid heart syndrome. A clear proof of an effect with regard to carcinoid heart disease is lacking so far.
From a clinical point of view, both available long-acting formulations of SSA (lanreotide autogel and octreotide LAR) seem to be equally effective and tolerated. There is no randomized clinical study available in patients with functioning GEPNETs; in patients with acromegaly, a small randomized cross-over study reported similar efficacy on growth hormone and IGF-1 levels.39
Lanreotide for control of tumor growth
Besides their recognized suppressive effects on secretory symptoms, SSA also demonstrated inhibition of growth in cancer cell lines. Direct antitumor action after binding to SST receptors may include inhibition of cell cycle, inhibition of growth factor effects, and proapoptotic effects.32 Additionally, indirect effects including inhibition of the release of growth factors and hormones that drive tumor growth, antiangiogenic effects, and immunomodulatory effects have been described.32
Initial evidence of antiproliferative efficacy of SSA in vivo was provided by several Phase II trials. In general, these studies were limited by including small numbers and heterogeneous cohorts, and a lack of a randomized study design. Most trials reported a low rate of morphologic response (<10%) but a high rate of stabilization (30%–80%). The rate of stabilization was higher in intestinal than in pancreatic NETs, and extrahepatic distant metastases were associated with nonresponse to SSA.40 Nonresponders to SSA treatment had a worse survival.40,41
The results for lanreotide are summarized in Table 2. The study of Martín-Richard et al42 reported a PFS of 12.9 months in GEP-NET patients with documented tumor progression within 6 months prior to study entry.
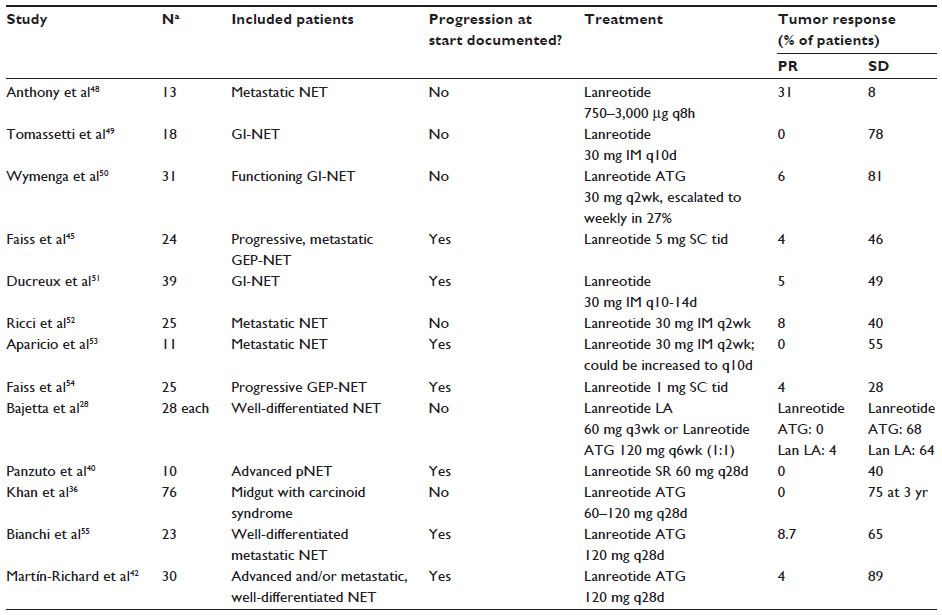 | Table 2 Clinical trials with data on morphological response of lanreotide in NET |
The first placebo-controlled trial that proved the antiproliferative efficacy of SSA was the so-called PROMID study in a cohort of patients with treatment naïve metastatic midgut tumors or tumors of unknown origin believed to derive from the midgut: Treatment with octreotide LAR significantly lengthened time to tumor progression as compared with placebo.17
This result was recently confirmed and expanded with lanreotide autogel 120 mg every 28 days in the double-blind, placebo-controlled, Phase III CLARINET trial.43 Within this protocol, 204 patients with well or moderately differentiated, nonfunctioning, SST receptor-positive GEPNETs with a Ki-67 of <10% were randomized to receive either lanreotide autogel 120 mg every 4 weeks or placebo for 96 weeks. Prior to randomization, the protocol included an observation period of 3–6 months to document progression status.
The vast majority of patients (96%) had radiographically stable disease at randomization and initiation of study treatment. Lanreotide was associated with a significant prolongation of PFS, with a median not reached versus a median of 18 months in the placebo arm (hazard ratio [HR] 0.47; P<0.001). The estimated rates of PFS at 24 months were 65.1% in the lanreotide group and 33% in the placebo group. The therapeutic effect in preplanned subgroups (hepatic tumor load less versus greater than 25%; G1 versus G2 and division by primary site) was generally consistent with that in the overall study population. The benefit in the patients with midgut NET (HR 0.35; P=0.009) was greater than in the pancreatic subset (HR 0.58; P=0.06). This borderline significance in patients with pancreatic neuroendocrine tumor still reflects a clinically meaningful prolongation of PFS (not reached versus 12.1 months in the placebo arm), and it is important to note that the study was not powered for statistical significance of the subgroups. No benefit could be demonstrated for the small subgroup of patients with hindgut NET (HR 1.47). Patients stable after 96 weeks on both treatment arms of the CLARINET trial and patients progressing under placebo were offered open treatment with 120 mg lanreotide autogel in an extension trial (open label extension study – OLE NCT00842348). Eighty-eight patients from the CLARINET core study (41 of the lanreotide arm and 47 of the placebo arm) participated in the OLE study. For the subset of patients who had progressive disease while on placebo treatment in the core study, median time to further progression with lanreotide was 14 months.44
According to the data of the CLARINET trial, lanreotide autogel was recently approved by FDA, and the European Medicines Agency agreed to a label extension for use of lanreotide 120 mg in antiproliferative purpose in patients with metastatic GEPNET (hindgut excluded).
Probably due to the small number of deaths, the crossover to open treatment, and the impact of other poststudy treatments, neither the PROMID trial nor the CLARINET trial was able to prove an overall survival benefit. Therefore, it is a matter of debate whether there is still a place for a “watch-and-wait” approach or whether the treatment with SSA should start immediately to prevent tumor progression.
Role of dose escalation (above label doses)
As SSA are generally well tolerated (see ”Side effects of lanreotide” section), dose escalation to above the standard dose could be an option for insufficient controlled carcinoid syndrome or better tumor control. Only two small trials investigated high dose lanreotide (1.2 mg sc daily and 5 mg three times a day sc, respectively) in GEPNET patients.45,46 Tumor control was achieved in 75% and 43%, respectively. A clear conclusion cannot be drawn because of the heterogeneous and small-sized study cohorts. There are no data available for high-dose lanreotide in the autogel formulation. Dose escalation can also be achieved by shortening of injection intervals. While this strategy is used in the clinical setting of insufficient symptom control, the association of shorter injection intervals of the SSA depot formulations and tumor control has not been systematically analyzed so far.
Planned and ongoing clinical trials
In the so-called CLARINET Forte study – an open multicentric Phase II trial – the effect on tumor control of a high dose of lanreotide autogel (120 mg every 2 weeks) in GEPNET patients progressing under the standard dose of 120 mg lanreotid autogel every 4 weeks will be investigated.
Another European multicenter trial open for recruitment evaluates the role of lanreotide autogel as maintenance therapy in patients with duodenopancreatic NETs after first-line treatment with chemotherapy, everolimus, or sunitinib (REMINET study, NCT02288377).
The role of lanreotide autogel as a combination partner is further investigated in the French so-called Sunland-Trial (NCT02231762 lanreotide autogel in combination with sunitinib or placebo in midgut NETs) and the German multicenter trial SONNET (NCT02231762 in combination with temozolomide).
Side effects of lanreotide
Lanreotide is generally well tolerated. Most common side effects are diarrhea, abdominal discomfort, flatulence, nausea, and local reactions at the site of injection including subcutaneous nodules.
Cholelithiasis is reported in 3%–10%36,42,43 of patients, asymptomatic in most cases but can rarely cause complications like acute cholezystitis, bilious attacks, or acute pancreatitis. Steatorrhea is reported in very different frequencies, and most cases can be sufficiently treated with pancreas enzymes. GI side effects often decrease in intensity with the duration of treatment. Lanreotide can cause hyperglycemia in approximately 5%–10% of treated patients. Rare side effects include hypoglycemia, hair loss, headache, myalgia, acute hepatitis, hyperbilirubinemia, obstipation, and paralytic ileus.
In the studies with the new formulation lanreotide autogel, the frequencies of side effects were comparable and no unexpected side effects were reported.28,34,36,42,43 In the CLARINET trial, the total rate of adverse events was similar in the lanreotide and the placebo group (88% versus 90%). The most common related adverse event was diarrhea in 26% of lanreotide-treated patients.43 In the ELECT study also, the rate of treatment-emergent adverse events was not different between lanreotide autogel and placebo (53.4% versus 59.6%).37
Conclusion
Lanreotide autogel is a well-tolerated drug initially approved for the control of hormonal symptoms in acromegaly and GEPNETs. In recent years, several trials, especially the placebo-controlled CLARINET study, supported its role for growth control in well-differentiated GEPNETs with proliferation index below 10%. Further studies are needed to evaluate its efficacy as combination partner and in patients with lung or thymic NETs.
Disclosure
Anja Rinke has performed consultation for Novartis, Ipsen, and Pfizer and received honoraria for presentations from Novartis and Ipsen. Daniela Müller has received honoraria for presentations from Novartis. Both authors report no other conflicts of interest in this work.
References
Yao JC, Hassan M, Phan A, et al. One hundred years after “carcinoid”: epidemiology of and prognostic factors for neuroendocrine tumors in 35,825 cases in the United States. J Clin Oncol. 2008;26:3063–3072. | |
Hauso O, Gustafsson BI, Kidd M, et al. Neuroendocrine tumor epidemiology: contrasting Norway and North America. Cancer. 2008;113:2655–2664. | |
Pape UF, Berndt U, Müller-Nordhorn J, et al. Prognostic factors of long-term outcome in gastroenteropancreatic neuroendocrine tumours. Endocr Relat Cancer. 2008;15:1083–1097. | |
Jann H, Roll S, Couvelard A, et al. Neuroendocrine tumors of midgut and hindgut origin: tumor-node-metastasis classification determines clinical outcome. Cancer. 2011;117:3332–3341. | |
Rindi G, D’Adda T, Froio E, et al. Prognostic factors in gastrointestinal endocrine tumors. Endocr Pathol. 2007;18:145–149. | |
Rindi G, Falconi M, Klersy C, et al. TNM staging of neoplasms of the endocrine pancreas: results from a large international cohort study. J Natl Cancer Inst. 2012;104:764–777. | |
Strosberg JR, Weber JM, Feldman M, et al. Prognostic validity of the American Joint Committee on Cancer staging classification for midgut neuroendocrine tumors. J Clin Oncol. 2013;31:420–425. | |
Rindi G, Arnold R, Bosman FT, et al. Nomenclature and classification of neuroendocrine neoplasms of the digestive system. In: WHO Classification of Tumors of the Digestive System. Geneva, Switzerland: WHO; 2010:13–14. | |
Modlin IM, Oberg K, Chung DC, et al. Gastroenteropancreatic neuroendocrine tumours. Lancet Oncol. 2008;9:61–72. | |
Moertel CG, Lefkopoulo M, Lipsitz S, et al. Streptozocin-doxorubicin, streptozocin-fluorouracil or chlorozotocin in the treatment of advanced islet-cell carcinoma. N Engl J Med. 1992;326:519–523. | |
Kouvaraki MA, Ajani JA, Hoff P, et al. Fluorouracil, doxorubicin, and streptozocin in the treatment of patients with locally advanced and metastatic pancreatic endocrine carcinomas. J Clin Oncol. 2004;22:4762–4771. | |
Strosberg JR, Fine RL, Choi J, et al. First-line chemotherapy with capecitabine and temozolomide in patients with metastatic pancreatic endocrine carcinomas. Cancer. 2011;117:268–275. | |
Yao JC, Shah MH, Ito T, et al. Everolimus for advanced pancreatic neuroendocrine tumors. N Engl J Med. 2011;364:514–523. | |
Raymond E, Dahan L, Raoul JL, et al. Sunitinib malate for the treatment of pancreatic neuroendocrine tumors. N Engl J Med. 2011;364:501–513. | |
Pavel ME, Hainsworth JD, Baudin E, et al. Everolimus plus octreotide long-acting repeatable for the treatment of advanced neuroendocrine tumours associated with carcinoid syndrome (RADIANT-2): a randomised, placebo-controlled, phase 3 study. Lancet. 2011;378:2005–2012. | |
Kvols LK, Moertel CG, O’Connell MJ, et al. Treatment of the malignant carcinoid syndrome. Evaluation of a long-acting somatostatin analogue. N Engl J Med. 1986;315:663–666. | |
Rinke A, Müller HH, Schade-Brittinger C, et al. Placebo-controlled, double-blind, prospective, randomized study on the effect of octreotide LAR in the control of tumor growth in patients with metastatic neuroendocrine midgut tumors: a report from the PROMID Study Group. J Clin Oncol. 2009;27:4656–4663. | |
Caplin ME, Pavel M, Ćwikła JB, et al. Lanreotide in metastatic enteropancreatic neuroendocrine tumors. N Engl J Med. 2014;371:224–233. | |
Brazeau P, Vale W, Burgus R, et al. Hypothalamic polypeptide that inhibits the secretion of immunoreactive pituitary growth hormone. Science. 1973;179:77–79. | |
Modlin IM, Pavel M, Kidd M, et al. Review article: somatostatin analogues in the treatment of gastroenteropancreatic neuroendocrine (carcinoid) tumours. Aliment Pharmacol Ther. 2010;31:169–188. | |
Lichtenauer-Kaligis EG, Dalm VA, Oomen SP, et al. Differential expression of somatostatin receptor subtypes in human peripheral blood mononuclear cell subsets. Eur J Endocrinol. 2004;150:565–577. | |
Caplin M, Sundin A, Nillson O, et al. ENETS Consensus Guidelines for the management of patients with digestive neuroendocrine neoplasms: colorectal neuroendocrine neoplasms. Neuroendocrinology. 2012;95:88–97. | |
Kulaksiz H, Eissele R, Rössler D, et al. Identification of somatostatin receptor subtypes 1, 2A, 3, and 5 in neuroendocrine tumours with subtype specific antibodies. Gut. 2002;50:52–60. | |
Bauer W, Briner U, Doepfner W, et al. SMS 201-995: a very potent and selective octapeptide analogue of somatostatin with prolonged action. Life Sci. 1982;31:1133–1140. | |
Bronstein M, Musolino N, Jallad R, et al. Pharmacokinetic profile of lanreotide Autogel in patients with acromegaly after four deep subcutaneous injections of 60, 90 or 120 mg every 28 days. Clin Endocrinol (Oxf). 2005;63:514–519. | |
Trocóniz IF, Cendrós JM, Peraire C, et al. Population pharmacokinetic analysis of lanreotide autogel in healthy subjects: evidence for injection interval of up to 2 months. Clin Pharmacokinet. 2009;48:51–62. | |
Buil-Bruna N, Garrido MJ, Dehez M, et al. Population pharmacokinetic (PK) analysis of lanreotide autogel/depot in the treatment of gastroenteropancreatic (GEP) neuroendocrine tumors (NETs): pooled analysis of four clinical trials. J Clin Oncol. 2015;33(Suppl 3):abstr 397. ASCO Meeting Abstracts. | |
Bajetta E, Procopio G, Catena L, et al. Lanreotide autogel every 6 weeks compared with Lanreotide microparticles every 3 weeks in patients with well differentiated neuroendocrine tumors: a Phase III Study. Cancer. 2006;107:2474–2481. | |
Betke KM, Wells CA, Hamm HE. GPCR mediated regulation of synaptic transmission. Prog Neurobiol. 2012;96:304–321. | |
Weckbecker G, Lewis I, Albert R, et al. Opportunities in somatostatin research: biological, chemical and therapeutic aspects. Nat Rev Drug Discov. 2003;2:999–1017. | |
Guillermet J, Saint-Laurent N, Rochaix P, et al. Somatostatin receptor subtype 2 sensitizes human pancreatic cancer cells to death ligand-induced apoptosis. Proc Natl Acad Sci U S A. 2003;100:155–160. | |
Susini C, Buscail L. Rationale for the use of somatostatin analogs as antitumor agents. Ann Oncol. 2006;17:1733–1742. | |
O’Toole D, Ducreux M, Bommelaer G, et al. Treatment of carcinoid syndrome: a prospective crossover evaluation of lanreotide versus octreotide in terms of efficacy, patient acceptability, and tolerance. Cancer. 2000;88:770–776. | |
Ruszniewski P, Ish-Shalom S, Wymenga M, et al. Rapid and sustained relief from the symptoms of carcinoid syndrome: results from an open 6-month study of the 28-day prolonged-release formulation of lanreotide. Neuroendocrinology. 2004;80:244–251. | |
Rubin J, Ajani J, Schirmer W, et al. Octreotide acetate long-acting formulation versus open-label subcutaneous octreotide acetate in malignant carcinoid syndrome. J Clin Oncol. 1999;17:600–606. | |
Khan MS, El-Khouly F, Davies P, et al. Long-term results of treatment of malignant carcinoid syndrome with prolonged release Lanreotide (Somatuline Autogel). Aliment Pharmacol Ther. 2011;34:235–242. | |
Vinik A, Wolin EM, Audry H, et al. ELECT: a phase 3 study of efficacy and safety of lanreotide autogel/depot (LAN) treatment for carcinoid syndrome in patients with neuroendocrine tumors (NETs). J Clin Oncol. 2014;32(Suppl 3):abstr 268. ASCO Meeting Abstracts. | |
Ruszniewski PB, Caplin ME, Valle JW, et al. Patient-reported satisfaction with symptom control during lanreotide autogel/depot (LAN) treatment for carcinoid syndrome (CS) in gastroenteropancreatic neuroendocrine tumor (GEP-NET) patients: symnet, a large multinational, cross-sectional, observational study. J Clin Oncol. 2014;32(Suppl 3): abstr 273. ASCO Meeting Abstracts. | |
Andries M, Glintborg D, Kvistborg A, et al. A 12-month randomized crossover study on the effects of lanreotide autogel and octreotide long-acting repeatable on GH and IGF-l in patients with acromegaly. Clin Endocrinol (Oxf). 2008;68:473–480. | |
Panzuto F, Di Fonzo M, Iannicelli E, et al. Long-term clinical outcome of somatostatin analogues for treatment of progressive, metastatic, well-differentiated entero-pancreatic endocrine carcinoma. Ann Oncol. 2006;17:461–466. | |
Arnold R, Rinke A, Klose K, et al. Octreotide versus octreotide plus interferon-alpha in endocrine gastroenteropancreatic tumors: a randomized trial. Clin Gastroenterol Hepatol. 2005;3:761–771. | |
Martín-Richard M, Massutí B, Pineda E, et al. Antiproliferative effects of lanreotide autogel in patients with progressive, well-differentiated neuroendocrine tumours: a Spanish, multicentre, open-label, single arm phase II study. BMC Cancer. 2013;13:427. | |
Caplin ME, Pavel M, Ruszniewski P. Lanreotide in metastatic enteropancreatic neuroendocrine tumors. N Engl J Med. 2014;371:1556–1557. | |
Caplin M, Ruszniewski P, Pavel M, et al. Progression-free survival (PFS) with lanreotide autogel/depot (LAN) in enteropancreatic NETs patients: the CLARINET extension study. J Clin Oncol. 2014;32(18 Suppl): abstr 4107. | |
Faiss S, Räth U, Mansmann U, et al. Ultra-high-dose lanreotide treatment in patients with metastatic neuroendocrine gastroenteropancreatic tumors. Digestion. 1999;60:469–476. | |
Eriksson B, Renstrup J, Imam H, et al. High-dose treatment with lanreotide of patients with advanced neuroendocrine gastrointestinal tumors: clinical and biological effects. Ann Oncol. 1997;8:1041–1044. | |
Schmid HA. Pasireotide (SOM230): development, mechanism of action and potential applications. Mol Cell Endocrinol. 2008;286:69–74. | |
Anthony L, Johnson D, Hande K, et al. Somatostatin analogue phase I trials in neuroendocrine neoplasms. Acta Oncol. 1993;32:217–223. | |
Tomassetti P, Migliori M, Gullo L. Slow-release lanreotide treatment in endocrine gastrointestinal tumors. Am J Gastroenterol. 1998;93:1468–1471. | |
Wymenga AN, Eriksson B, Salmela PI, et al. Efficacy and safety of prolonged-release lanreotide in patients with gastrointestinal neuroendocrine tumors and hormone-related symptoms. J Clin Oncol. 1999;17:1111. | |
Ducreux M, Ruszniewski P, Chayvialle JA, et al. The antitumoral effect of the long-acting somatostatin analog lanreotide in neuroendocrine tumors. Am J Gastroenterol. 2000;95:3276–3281. | |
Ricci S, Antonuzzo A, Galli L, et al. Long-acting depot lanreotide in the treatment of patients with advanced neuroendocrine tumors. Am J Clin Oncol. 2000;23:412–415. | |
Aparicio T, Ducreux M, Baudin E, et al. Antitumour activity of somatostatin analogues in progressive metastatic neuroendocrine tumours. Eur J Cancer. 2001;37:1014–1019. | |
Faiss S, Pape UF, Böhmig M, et al. Prospective, randomized, multicenter trial on the antiproliferative effect of lanreotide, interferon alfa, and their combination for therapy of metastatic neuroendocrine gastroenteropancreatic tumors – the International Lanreotide and Interferon Alfa Study Group. J Clin Oncol. 2003;21:2689–2696. | |
Bianchi A, De Marinis L, Fusco A, et al. The treatment of neuroendocrine tumors with long-acting somatostatin analogs: a single center experience with lanreotide autogel. J Endocrinol Invest. 2011;34:692–697. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.