")
Back to Journals » Journal of Blood Medicine » Volume 6
Profile of efraloctocog alfa and its potential in the treatment of hemophilia A
Received 24 February 2015
Accepted for publication 30 March 2015
Published 24 April 2015 Volume 2015:6 Pages 131—141
DOI https://doi.org/10.2147/JBM.S54632
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Martin H Bluth
Lindsey A George,1,2 Rodney M Camire1–3
1Division of Hematology, The Children's Hospital of Philadelphia, Philadelphia, PA, USA; 2Center for Cellular and Molecular Therapeutics, The Children's Hospital of Philadelphia, Philadelphia, PA, USA; 3Department of Pediatrics, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA, USA
Abstract: Hemophilia care has improved dramatically over the past 50 years, evolving from plasma concentrates, to purified plasma proteins, to recombinant clotting factors. These collective developments allowed for home delivery of on-demand and prophylactic treatment, resulting in the reduction of hemophilia morbidity and mortality and improved quality of life. Although efficacious in treating bleeding, conventional factor products' half-lives require frequent venipuncture, which remains a significant burden to patients. Despite the remarkable advances in hemophilia care, no improvements have, until now, been made to the pharmacokinetic properties of factor products. Multiple strategies have more recently been employed to generate novel bioengineered products that, with great hope, represent the next wave of progress in hemophilia care. The use of these products will undoubtedly raise important discussion about choosing conventional factor over new long-acting factor products. Incorporation of these therapies into clinical care is accompanied by unanswered safety questions that will likely be evaluated only in postmarketing surveillance analysis. Further, these products may change current treatment paradigms with unclear cost repercussions and feasibility. This paper will review efraloctocog alfa (FVIII-Fc) and its role in the treatment of hemophilia A.
Keywords: hemophilia A, factor VIII, Fc fusion, bioengineered products, efraloctocog alfa
Introduction
Hemophilia A is an X-linked congenital bleeding disorder resulting from deficiency of plasma coagulation factor VIII (FVIII), with an incidence of 1 in 5,000 male births.1,2 Bleeding manifestations of hemophilia are approximated based on circulating plasma factor activity. Patients with plasma FVIII levels <1% of normal are classified as severe and constitute approximately 60% of the hemophilia A population. The severe hemophilia phenotype is characterized by spontaneous hemarthrosis, soft tissue hematomas, postsurgical bleeding, and retroperitoneal and intracerebral hemorrhage. Moderate hemophilia patients, with FVIII levels of 1%–5% of normal, typically only bleed in response to minor or major trauma, while patients with mild hemophilia A (FVIII levels >5%–40% of normal) typically only bleed in response to surgery, tooth extractions, or major injuries. In the absence of appropriate factor replacement treatment, these disease manifestations can have disabling or even fatal consequences. Over time, sequelae from recurrent joint bleeding and soft tissue hematomas can result in hemophilic arthropathy, muscle contractures, and pseudotumors, leading to chronic pain and disability.3 Thankfully, in countries that have routine access to clotting factors, great strides in hemophilia care over the preceding half century have made such complications the exception rather than the rule.
Transfusion technologies developed for World War II were incorporated for the treatment of hemophilia; however, fresh whole blood or fresh frozen plasma transfusions were typically reserved for acute manifestations and not readily available to all patients. Dr Judith Graham Pool’s 1964 publication4 detailing methods of cryoprecipitation and subsequent fractionation procedures allowed for the storage of a therapeutic form of clotting FVIII. Thereafter, freeze-dried plasma-derived (pd) factor concentrates were developed, allowing patients to treat themselves at home and thereby revolutionizing hemophilia care. By 1980, the life expectancy of a person with hemophilia was 60 years, which was in contrast to routine death during childhood or adolescence just a few decades before. Unfortunately, these advances were not without complications. Cryoprecipitate and pd factor, pooled from multiple donors, were contaminated with blood-borne pathogens (HIV, hepatitis B and C) before the risk of viral transmission was recognized. Reports of hemophilia patients falling ill to Pneumocystis carinii, in 1982, raised initial concerns for HIV transmission through factor products.5 By the late 1980s, contaminated blood products resulted in HIV transmission in nearly half of all the hemophilia patients and an estimated 90% of severe hemophilia patients. With the selective pressure of blood-borne pathogens affecting a large portion of the hemophilia population, improved viral inactivation techniques emerged in the late 1980s.
With increasing recognition of the risks associated with pd factor concentrates, coupled with the beginning of the molecular biology revolution, the FVIII gene was cloned in 1984 and provided a foundation for the development of recombinant factor products. In 1992, the first recombinant FVIII product obtained US Food and Drug Administration (FDA) approval. Between the combined efforts of viral inactivation techniques and emergence of recombinant products, no person with hemophilia in the United States has contracted HIV by factor contamination since 1987.6 Soon after the development of the first recombinant FVIII product, second- and third-generation recombinant FVIII products followed, each with increasingly minimal risks of viral transmission.
The development of factor products allowed for patients with hemophilia to be infused on a regular basis to prevent bleeding (prophylaxis), such that the phenotype of severe hemophilia could be augmented to that of moderate hemophilia. The idea of prophylaxis was pioneered by a group of physicians in Malmö, Sweden, who demonstrated that, when prophylaxis was initiated early and regularly (primary prophylaxis), patients had significantly reduced bleeding frequency and improved joint health.7 Thereafter, the superiority of prophylaxis over on-demand treatment (factor given in the setting of bleeding) was supported in several cohort studies.8–10 This was confirmed in Manco-Johnson et al’s11 landmark randomized controlled clinical trial demonstrating the superiority of prophylaxis over on-demand treatment for joint health in hemophilia A. While the timing of prophylaxis initiation is still debated, available data demonstrate that earlier prophylaxis initiation is associated with better long-term joint outcome results.12
In addition to effects on joint health, prophylaxis, when compared to on-demand therapy, decreases time lost from school or work and improves the quality of life for patients and their families.13 As a direct result of these developments, primary factor prophylaxis is considered optimal care for severe hemophilia patients in developed countries. This is, in part, responsible for improved hemophilia life expectancy that now approaches that of the general population in developed countries.14,15 Presently, however, 70%–80% of the world’s hemophilia population (approximately 300,000 people) do not have regular or any access to factor, which is attributed to cost rather than to availability.16
Among those countries that do have access to factor for prophylaxis, general recommendations for prophylaxis are to maintain FVIII trough levels at 1% of normal.17 For hemophilia A, this is generally achieved by prophylactic factor replacement approximately three to four times a week. FVIII trough maintained at ≥1% of normal is typically effective at preventing spontaneous bleeding; however, some patients experience bleeding despite maintaining levels of ≥1% of normal, which speaks to the heterogeneity of the disease and the importance of individualized prophylaxis regimens when appropriate.16,18,19 Full discussion of prophylaxis is outside the scope of this review. Further information and definitions may be found in publications from the National Hemophilia Foundation and the World Federation of Hemophilia.17,20
The rationale for FVIII replacement therapy in hemophilia is based on the therapeutic principles of 1) volume of clotting factor distribution, of which the amount of extravascular FVIII is thought to be negligible; 2) factor survival and half-life in plasma; and 3) the minimal hemostatic factor level required to control the type and extent of hemorrhage. Traditional factor half-life is biphasic and averages approximately 12 hours. The biggest disadvantage of currently available factor concentrates relates to their relatively short half-lives, which result in the need for frequent factor infusions. Current FVIII prophylaxis regimens generally coalesce around factor administration three to four times/week, with thrice-weekly FVIII prophylaxis to prevent joint bleeds and arthropathy recommended by the Medical and Scientific Advisory Committee of the National Hemophilia Foundation.17,20,21 Half-lives of current FVIII products typically range from 8 to 12 hours, with interpatient variability (6–24 hours). This variability is yet to be completely explained or easily predictable. Some available information suggests that individual patient FVIII product pharmacokinetic variability may in part be related to endogenous clearance of von Willebrand factor (vWF), blood group (type O blood group may have a shorter half-life related to vWF), and age-associated alterations in volume of distribution.22,23 Irrespective of individual patient pharmacokinetics, requirement for frequent painful and inconvenient factor administration is associated with incomplete adherence and delay or avoidance of prophylaxis.24,25 This issue is particularly pronounced in the pediatric population, wherein factor delivery may necessitate central venous access devices with intendant risks of infection and, not infrequently, thrombosis.26
With the adoption of pd or recombinant factor use and the eradication of known blood-borne pathogen transmission, the major cause of morbidity and mortality in hemophilia care is now attributable to the development of alloimmune inhibitory antibodies (inhibitors) to exogenous factor. Inhibitors develop in approximately 30% of severe hemophilia A patients and roughly 10% of mild hemophilia A patients.1,27 Thus far, the only demonstrated means to eradicate inhibitors is through repeated antigenic exposure to FVIII. Termed immune tolerance induction, the regular infusion of specific clotting factor over the course of months to years with or without concomitant immunomodulation is successful in approximately 80% of severe FVIII patients with inhibitors.1,27
The development of inhibitors with FVIII replacement therapy led to concern and subsequent debate about the relative immunogenicity of pd versus rFVIII and individual rFVIII products. Numerous studies and systematic reviews have not demonstrated any difference in the risk of inhibitor development between pd and recombinant FVIII products.28–31 Additionally, prospective, but not randomized, studies evaluated the risk of inhibitor development in previously untreated patients (PUPs), with conflicting results. Repeated separate investigations demonstrated that second-generation full-length recombinant products were associated with an increased risk of inhibitor development compared with third-generation products.32–34 Skeptics of this evidence suspect that initial marketing studies of second-generation products included subjects with a lesser likelihood of inhibitor development, thereby resulting in initial lower incidence of inhibitor development. Subsequently, those initial findings may have influenced prescriber behavior such that patients with preexisting risk factors for inhibitor development were treated with second-generation products. This may provide an alternative explanation for increased inhibitor incidence observed among second-generation factor use in PUP studies.32–34 This has not been confirmed and is unlikely to be investigated in future studies. Importantly, these studies demonstrate the potential vulnerabilities of marketing studies to predict inhibitor risk and highlight the importance of postmarketing safety surveillance. This point is particularly relevant now, with multiple novel factor products in development or recently approved.
Despite the prolific advances in hemophilia care over the preceding 50 years (Table 1), all conventional FVIII products have indistinguishable pharmacokinetics with half-lives that necessitate prophylactic dosing approximately three to four times a week or more. Recently approved, efraloctocog alfa (rBDD FVIII-Fc) and several other treatment strategies in preclinical or early-phase clinical development will undoubtedly challenge current treatment paradigms. With increasingly diverse biochemical characteristics, challenges of incorporating these strategies will include determining potencies, monitoring in the face of currently available clinical assays, careful surveillance for the potentially increased incidence of alloantibody formation, and, in the case of rBDD FVIII-Fc, safety of long-term, frequent Fc administration. Despite these dilemmas, new products come with great promise to improve treatment and quality of life for patients with hemophilia.
 | Table 1 Major therapeutic events in hemophilia A history |
Efraloctocog alfa
Efraloctocog alfa is the first of a cadre of novel therapeutics with extended half-life in development for hemophilia A to achieve FDA approval. Marketed under the trade name Eloctate by Biogen Idec (Cambridge, MA, USA), rBDD FVIII-Fc obtained FDA approval for on-demand and prophylactic treatment of children and adults with hemophilia A in the summer of 2014. Efraloctocog alfa is a fusion of a recombinant B-domain-deleted FVIII and the dimeric constant region (Fc) of IgG1 resulting in an approximate 1.5-fold increase in half-life relative to traditional recombinant products without alteration of FVIII specific activity.
Modulating the interaction of IgG with FcRn
Fusion of the Fc domain of IgG to a therapeutic protein prolongs half-life through a mechanism involving binding to the neonatal Fc receptor (FcRn). Among other roles, FcRn is the homeostatic receptor that plays a critical role in IgG catabolism and salvage, thereby protecting IgG from degradation.8 Contrary to what its name would imply, the FcRn receptor is present throughout life in various tissues. Work examining the interaction of FcRn with the Fc portion of IgG has paved the way for mechanism-based modifications of existing therapeutics, resulting in an emerging class of Fc-fusion therapeutics (eg, etanercept and romiplostim) that alter pharmacokinetics of Fc-coupled biological compounds, thereby improving the half-life of existing therapeutic proteins.
Specific to the field of coagulation, Fc–FcRn interaction has been exploited to develop extended half-life factor products, including Alprolix (Biogen Idec; recombinant FIX-IgG1 Fc-fusion protein) and rBDD FVIII-Fc. In the case of rBDD FVIII-Fc, recombinant B-domain-deleted FVIII has been covalently linked to the N-terminus of the IgG1 Fc-dimer to form a recombinant Fc-fusion protein (rBDD FVIII-Fc) (Figure 1). Importantly, covalent linkage of the rFVIII moiety with IgG1 Fc-dimer confers an approximate 1.5-fold increase in half-life.35–39
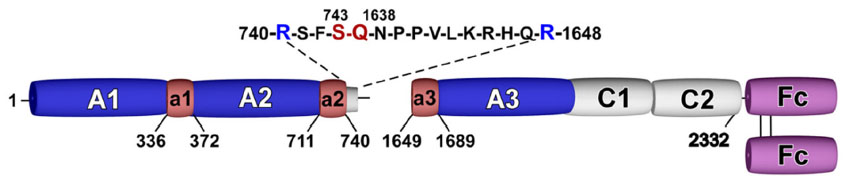 | Figure 1 Schematic of efraloctocog alfa. |
Many cell types, including vascular endothelial cells, express FcRn, which is thought to be the site at which most FcRn–IgG interaction occurs and therefore most important for IgG protection. The Fc portion of IgG binds FcRn with high affinity at an acidic pH (<6.5) within endosomes, but not at physiological pH (7.4). The pH dependence of the FcRn–Fc interaction is mediated by a hydrophobic interaction between FcRn and Fc, which is stabilized when salt bridges form between the two molecules at an acidic pH. When endosomes fuse back to the plasma cell membrane, the neutral pH causes dissociation of the Fc domain from FcRn, thereby releasing IgG or Fc-fusion proteins back into the bloodstream, thus avoiding lysosomal degradation (Figure 2).40 Importantly for safety considerations, the Fc fragment degrades and does not accumulate in the body.41 However, the long-term effects of repeated frequent Fc infusions, such as would be employed with rBDD FVIII-Fc, are not known. Efraloctocog alfa is a second-generation Fc-fusion monomer, which describes the second generation of Fc-fusion therapies that are dimeric with respect to Fc but monomeric with respect to their therapeutic protein, ie, rBDD-FVIII. Monomeric Fc-fusion molecules were developed with greater efficiency and half-life extension than first-generation dimeric fusion proteins due to decreased molecular weight resulting in increased endosomal transport efficiency.42
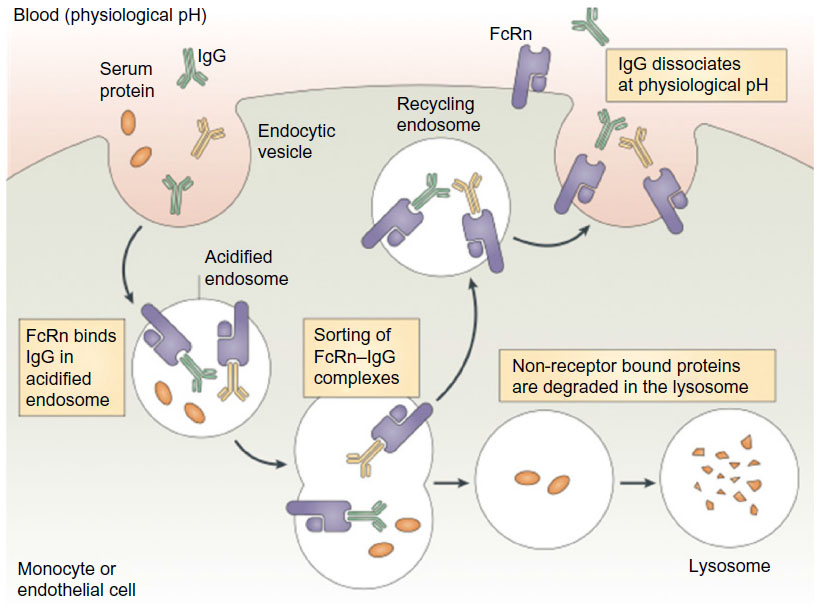 | Figure 2 Proposed role of the the neonatal Fc receptor for IgG (FcRn) in the vascular endothelial cells. |
Biochemical and functional studies
Like rFIX-Fc, rBDD FVIII-Fc is produced in human embryonic kidney 293 cells, a human cell line for the purposes of preserving posttranslational modifications, including glycosylation. Appropriate glycosylation is thought to be essential in ensuring optimal stability and specific activity and minimizing risk of neoepitope formation.43 rBDD FVIII-Fc posttranslational modification was compared to current rFVIII products and found to have the same N-linked glycosylation. Further, analysis of rBDD FVIII-Fc via mass spectrometry following lysyl endopeptidase and thrombin digestion demonstrated comparable results to both B-domain-deleted FVIII (ReFacto and Xyntha, Wyeth Pharmaceuticals, Philadelphia, PA, USA) and full-length FVIII products (Advate, Baxter Healthcare, Westlake Village, CA, USA).35 Thus, extensive efforts have been made to characterize rBDD FVIII-Fc biochemically without evidence in alteration of the biochemical characteristics relative to recombinant B-domain-deleted FVIII.
Similar functional characteristics of rBDD FVIII-Fc were obtained in preclinical studies. Using a chromogenic assay, rBDD FVIII-Fc and rFVIII were found to have similar activity, supporting the idea that the FVIII protein moiety of rBDD FVIII-Fc is not altered by Fc fusion. However, vWF affinity for rBDD FVIII-Fc was approximately 30% less than rBDD hFVIII. The relevance of this finding to rBDD FVIII-Fc pharmacokinetics is unclear, but thought to be insignificant.35
In vivo rBDD FVIII-Fc hemostatic efficacy was evaluated in murine and canine models of hemophilia A. rBDD FVIII-Fc was equally effective as rFVIII in murine tail clip injury model, supporting similar hemostatic efficacy. Further, a twofold extension in rBDD FVIII-Fc half-life was supported by prolonged protection from bleeding in hemophilia A mice and extended correction of whole blood clotting times in canine models of hemophilia A.36 Lastly, to ensure Fc–FcRn was indeed responsible for the extended half-life of rBDD FVIII-Fc, the protein was evaluated in FcRn-knockout mice. As expected, half-life extension of rBDD FVIII-Fc was abrogated in these knockout mice and was similar to rFVIII, thereby confirming the role of FcRn in rBDD FVIII-Fc half-life improvement.36 Current evidence supports that the net result of rBDD FVIII-Fc has improved pharmacokinetics due to Fc–FcRn interaction without compromised biochemical properties, including specific activity, of the bound moiety.
Clinical data
Pharmacokinetics of rBDD FVIII-Fc
Phase I/IIa study (ClinicalTrials.gov; NCT01027377) investigating the safety and pharmacokinetic properties of rBDD FVIII-Fc was completed in previously treated severe hemophilia A males. Half-life data for two-dose cohorts of rBDD FVIII-Fc and rFVIII demonstrated a 1.5- to 1.7-fold longer elimination half-life relative to rFVIII (18.8 hours in both doses of rBDD FVIII-Fc versus 12.2 and 11 hours for rFVIII). Time to reach 1% FVIII activity was 1.5- to 1.8-fold longer in rBDD FVIII-Fc than in rFVIII. Dose-dependent plasma recoveries of rBDD FVIII-Fc and rFVIII roughly approximated each other, corroborating animal data. Further, the work demonstrated no change in specific activity in the Fc-bound rFVIII protein, and the half-life extension results were confirmed.37
These findings were recapitulated in Phase III trial (A-Long Study; ClinicalTrials.gov; NCT01458106). The terminal half-life of rBDD FVIII-Fc (mean: 19.0 hours) was 1.5-fold longer, and significantly different, from that observed in rFVIII (mean: 12.4 hours). Further, the chromogenic activity assay again demonstrated similar specific activity of rBDD FVIII-Fc relative to rFVIII. Consistent with preclinical and Phase I data, rBDD FVIII-Fc half-life demonstrated a significant positive correlation with baseline vWF antigen levels. vWF protects FVIII from degradation, thereby playing a critical role in the half-life of FVIII. However, although normally a positive outcome, it turns out that the interaction between vWF and FVIII-Fc likely limits the ability of the Fc region to extend the half-life of the new protein beyond 1.5-fold. Consistent with this theory, rBDD FVIII-Fc failed to extend FVIII half-life beyond that of vWF (10–25 hours) and has been observed in other FVIII-fusion strategies, such as PEGylation.38 The interaction and half-life of vWF and FVIII appear to limit the ability to extend FVIII half-life.
Efficacy
In the Phase III trial, rBDD FVIII-Fc was evaluated in three cohorts: individualized twice-weekly prophylaxis with 25–65 IU/kg every 3–5 days (arm 1), weekly prophylaxis of 65 IU/kg (arm 2), and on-demand treatment for bleeding with 10–50 IU/kg (arm 3). Subject annualized bleeding rate (ABR) was a primary efficacy end point. As expected, subjects on prophylaxis (arms 1 and 2) had significantly reduced ABR relative to subjects treated on demand (arm 3). Based on a negative binomial regression model, ABR for arms 1, 2, and 3 (2.91, 8.92, and 37.2, respectively; P<0.001) significantly increased across groups. With respect to arm 2, the investigators noted that ABR data were substantially influenced by four subjects with underlying preenrollment hemophilic arthropathy with baseline ABR>20 that may have influenced findings.38 Among subjects in arm 1, the median dosing interval was 3.5 days at study completion for a median total weekly factor dose of 77.7 IU/kg. Across all arms of the study, approximately 90% of bleeds resolved with a single factor dose and nearly all resolved after two factor doses. The median dose per injection to treat bleeding was 27.35 IU/kg and was consistent with recommended dosing for treating hemarthrosis.17,38 Among the nine subjects who underwent surgery, the hemostatic response to rBDD FVIII-Fc was rated by investigators/surgeons as excellent or good. Results supported rBDD FVIII-Fc hemostatic efficacy and an approximately 1.5-fold increase in half-life.38 Formal recommendations for incorporation of rBDD FVIII-Fc into prophylaxis have not yet been made; however, clinical trial findings support likely reduced frequency of prophylaxis, but the data support a minimum twice-weekly rBDD FVIII-Fc dosing to maintain FVIII >1% of normal.
Pediatric data
A Phase III open-label multicenter trial (Kids A-Long, ClinicalTrials.gov; NCT01458106) is evaluating the safety, efficacy, and pharmacokinetics of rBDD FVIII-Fc in previously treated (>50 factor exposures) children <12 years with severe hemophilia A. The preliminary information was presented at the American Society of Hematology 2014 annual meeting. Among the 61 subjects with ≥50 rBDD FVIII-Fc exposure days, there were no serious adverse events and no subject developed an inhibitor. Mean terminal half-life in subjects <6 years was 11.23 and 14.88 hours for subjects 6 to <12 years. Subject half-life ratios (rFVIII-Fc: prestudy FVIII) were observed to be 0.79–2.98, which is roughly approximate to the 1.5-fold increase in half-life observed in adults. Similar to adult findings, median on-study dosing interval was every 3.5 days, wherein nearly 75% of subjects reduced prophylactic dosing interval from their prestudy FVIII product. Compared to prestudy measures, on-study subjects had similar breakthrough bleeding tendency and overall factor consumption. The preliminary results of Kids A-Long appeared consistent with findings in the adult A-Long Phase III study; however, formal analysis has not yet been made available.39
Safety
Among all subjects studied in the clinical trials, no hypersensitivity, anaphylactic, or thrombotic events occurred. Adverse events related to the drug were most commonly arthralgia and malaise and affected a small percentage of study participants.37,38 No subjects developed inhibitors, consistent with data supporting no observed risk of increased inhibitor development when switching between rFVIII products.34,38 Thus, the theoretical concern that Fc fusion with FVIII may generate FVIII neoepitopes and result in inhibitory alloantibody formation in tolerant patients was not supported by Phase III available findings. Notably, available rBDD FVIII-Fc clinical trial data represent subjects with >50 factor exposure days, after which the risk of inhibitor is exceedingly rare. Thus, it is not yet clear whether rBDD FVIII-Fc may result in increased (or decreased) inhibitor incidence in PUPs and will be important to further understand the safety profile.
The proposed Hemophilia Inhibitor Prevention Trial – INHIBIT will evaluate whether rBDD FVIII-Fc use in PUPs may result in decreased incidence of inhibitor development and is not yet enrolling (ClinicalTrials.gov; NCT02196207). Rationale for the trial includes the following: 1) Fc may have immunoregulatory properties; 2) sustained higher FVIII levels can be achieved with long-acting products and may result in increased tolerance; and 3) the protein-bound FVIII moiety of rBDD FVIII-Fc is the same as other B-domain-deleted FVIII products.44 Further, prior work supports that B-domain-deleted FVIII has no increased risk of inhibitor development.45,46 Thus, while the Fc-bound FVIII of rBDD FVIII-Fc is indistinguishable from standard recombinant FVIII, addition of Fc may have immunoregulatory properties. Previous evidence demonstrated that IgG-bound haptens induce antigen-specific tolerance and that infusions of IgG have increased total regulatory T cells.47,48 Importantly, when FVIII peptide epitopes or FVIII immunogenic regions are coupled to IgG Fc, they induce regulatory T cells.47 Although it is possible that, in addition to prolonging FVIII half-life, rBDD FVIII-Fc may promote tolerance, prior immunoregulatory properties of Fc-fusion and IgG should be extrapolated with caution as they have not yet been demonstrated in humans.44,47,48
rBDD FVIII-Fc impact on care
Prophylaxis
Consistent with pharmacokinetic data obtained in Phase I–II trial, published and preliminary Phase III data support rBDD FVIII-Fc results in moderate half-life prolongation that is approximately 1.5-fold that of traditional FVIII products, resulting in an ability to space prophylaxis to an approximately twice-weekly schedule. Traditional prophylaxis in severe hemophilia generally coalesces around a frequency of three to four times/week. Although rBDD FVIII-Fc will likely allow for some decrease in prophylaxis and venipuncture frequency, results are, overall, less dramatic than findings observed in the FIX-Fc fusion product, Alprolix (Biogen Idec). Optimistically, these seemingly small changes may reduce the need for central venous access devices and their intendant complications or translate to earlier initiation of prophylaxis and fewer missed doses. Conversely, a prolonged interval between dosing may result in greater time spent with lower factor levels relative to traditional factor prophylaxis; however, given the only minimal decrease in prophylaxis administration frequency, it is likely that this will be of lesser concern for rBDD FVIII-Fc than for FIX-Fc.
Until the development of longer-acting factor products, setting the trough level of ≥1% of normal was chosen due to both cost considerations as well as inconvenience of more frequent factor delivery. Although conventional prophylaxis has demonstrated the ability to reduce spontaneous bleeding frequency, it does not protect against moderate phenotype bleeding, ie, trauma-induced hemorrhage. Given the longer half-life of rBDD FVIII-Fc, patients and caregivers may opt to maintain higher troughs, which may facilitate a more active lifestyle not previously available to hemophilia patients. At this time, it is not clear what trough level to target and what threshold of higher cost will be permitted or what this new target may be.49,50 In situations of a more sedentary lifestyle or in the case of difficult venipuncture, eg, infants, patients, and caregivers may choose fewer infusions over higher troughs. Although the emergence of rBDD FVIII-Fc and other long-acting therapies will undoubtedly result in increased opportunity for individualized prophylaxis and improved quality of life, the practical implementation of these therapies into clinical care is not yet clear.
Although clinical trial data in adult and preliminary pediatric previously treated patients data support the concept that rBDD FVIII-Fc does not break tolerance, such results should be interpreted with caution and regarded as preliminary with enthusiasm for continued postmarketing surveillance. Further, data regarding risk of inhibitor development in PUPs treated with rBDD FVIII-Fc are not yet available. Given the lack of PUP or postmarketing inhibitor incidence data, strong consideration should be given to avoid rBDD FVIII-Fc in PUPs.
Monitoring
Most clinical-based laboratories employ one-stage activated partial thromboplastin time (aPTT) based clotting assays for FVIII activity analysis. The optimal testing for FVIII activity with rBDD FVIII-Fc therapy is not yet clear. Clinical trial analysis used a chromogenic-based assay that is not widely available in clinical coagulation laboratories. While unlikely given the available data, it is possible that the Fc-fusion protein may interfere with phospholipids used in traditional aPTT-based assays, rendering it a suboptimal assay for monitoring rBDD FVIII-Fc therapy. If this is the case, available testing may complicate both routine and emergent rBDD FVIII-Fc monitoring. Formal recommendations for monitoring rBDD FVIII-Fc are not yet available.
Cost
The impact of rBDD FVIII-Fc on cost is not yet clear. Preliminary studies suggest that the overall factor use is approximately the same for rBDD FVIII-Fc and traditional factor; however, this is targeting FVIII troughs of traditional prophylaxis.38,39 If goals of prophylaxis were to evolve to target high troughs, use of rBDD FVIII-Fc would undoubtedly be far more expensive than traditional factor products. Although factor use varies per individual patient, protein replacement therapy for hemophilia is currently grossly approximated at US$200,000/year/patient. Furthermore, it is unclear how funders/payers will react to these new costs.51 Thus, although targeting higher FVIII troughs may be ideal, they are not likely to be attainable in the short term due to prohibitive therapeutic cost.52 An important consideration, however, is that although higher troughs will necessitate increased factor use and expense, they may reduce bleeding complications and sequelae, thereby reducing the overall cost of care. Further, it is possible that long-acting factor products may result in decreased pricing of traditional factors making them more affordable to patients who currently do not have access to factor.
Other novel products for hemophilia A
There are several other novel methodologies being pursued for half-life extension of FVIII products as well as other means to treat hemophilia. Although beneficial to endogenous FVIII by protecting it from proteolytic cleavage, the vWF–FVIII interaction clearly limits further FVIII half-life extension beyond that of vWF.37,53 To date, all strategies utilizing the FVIII protein are subject to the limitation imposed by the vWF–FVIII interaction. Thus, alternative strategies that do not employ FVIII or result in permanent restoration of the FVIII protein, ie, gene therapy, may ultimately be more efficacious than FVIII half-life extension strategies. Select novel therapeutics for the treatment of hemophilia A are highlighted in what follows and outlined in Table 2.
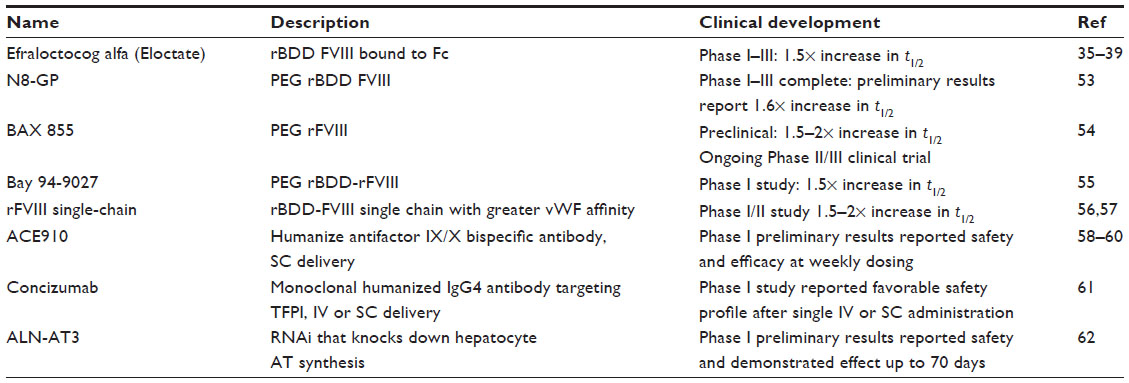 | Table 2 Recently approved or in clinical trial novel hemophilia A therapeutics |
FVIII fusion products and FVIII modifications
FVIII glycopegylation
PEGylation is the covalent coupling of hydrophilic polyethylene glycol (PEG) to active pharmaceutical ingredients, ie, FVIII. By binding a target protein, PEGylation reduces glomerular filtration, proteolytic degradation, and clearance of the PEGylated protein specific receptors resulting in increased half-life. Renal and hepatic pathways eliminate PEGs; however, renal tubular vacuolization has occurred due to renal PEG accumulation in animal models. Thus, a major safety concern of these molecules is the unknown consequence of repeated and frequent administration of PEGylated products and risk of accumulation. Several PEGylated FVIII products are in development. Each of the described products has demonstrated in either preclinical or clinical investigation to have similar half-lives that are 1.5- to 1.6-fold greater than rFVIII. Like rBDD FVIII-Fc, PEGylated FVIII product half-life extensions appear to be limited by the modulating effects of vWF.53–55
Single-chain rFVIII
Endogenous FVIII circulates as a heterodimer comprised of a heavy and light chain held together by a metal-ion bridge, which makes the FVIII molecule relatively unstable. A rBDD product modified for greater vWF affinity incorporates a covalent linkage of the heavy and light chains, resulting in a single-chain protein, and is designed to improve stability. Similar to other FVIII fusion therapies, preliminary results from the AFFINITY Phase I/II clinical trial supported twice-weekly prophylactic therapy, again demonstrating a 1.5- to 2.0-fold increase in half-life (ClinicalTrials.gov; NCT01486927).56,57
Monoclonal antibodies
ACE910
A humanized antifactor IX/X bispecific antibody, ACE910, is delivered subcutaneously and places the two factors in spatially appropriate positions to mimic FVIII cofactor function and exert in vivo hemostatic efficacy. Importantly, ACE910 can improve the intrinsic pathway of coagulation in the presence of an inhibitor. Thus, this molecule is a viable therapeutic for patients with or without an inhibitor.58,59 Initial clinical data were presented at the American Society of Hematology 2014 meeting reporting on once-weekly ACE910 prophylaxis in hemophilia A subjects with or without inhibitors. Results demonstrated a promising efficacy profile without major safety concerns.60 The combined subcutaneous availability, possibility for weekly prophylaxis, and efficacy in patients with or without inhibitors make ACE910 a potentially important therapeutic advance in the treatment of hemophilia.
Monoclonal antibody 2021: blocking FXa and TFPI
Tissue factor mediates thrombin generation by binding factor VIIa (FVIIa), thereby promoting the activation of FX. Factor Xa (FXa) generation is limited by the feedback of tissue factor pathway inhibitor (TFPI). Concizumab is a high-affinity, monoclonal, humanized IgG4 antibody targeting the kunitz-2 domain of human TFPI. Inhibition of TFPI may allow enough thrombin to be generated through the TF:FVIIa:FXa complex to overcome deficiencies of FVIII or FIX. Concizumab showed a favorable safety profile after intravenous or subcutaneous administration with a concentration-dependent procoagulant effect. Nonlinear pharmacokinetics was observed due to target-mediated clearance.61
Molecular biology approaches
RNA interference (RNAi)
The antithrombin (AT) pathway serves as an important anticoagulant effect by inhibiting FXa and thrombin. ALN-AT3 is a short-acting, subcutaneously administered RNAi that knocks down hepatocyte AT synthesis. In animal models of hemophilia, including nonhuman primate, ALN-AT3 has demonstrated efficacy at weekly dosing intervals. This molecule may be used for the treatment of hemophilia A and B with or without inhibitors as well as rare bleeding disorders. A Phase I study in healthy volunteers or patients with hemophilia is currently recruiting (ClinicalTrials.gov; NCT02035605). Preliminary results presented at the American Society of Hematology 2014 meeting supported AT reduction was stable for up to 70 days after a single dose.62
Gene therapy
Obtaining stable endogenous FVIII levels by transgene expression following a single-dose vector administration presents the ultimate solution to overcoming FVIII pharmacokinetic barriers. Proof of concept for FIX adeno-associated viral (AAV)-mediated gene transfer in humans was initially reported in 2011 and long-term follow-up data were recently published.63,64 Several programs in FVIII gene transfer are in preclinical development employing AAV vectors (Pfizer/Spark Therapeutics, Biomarin, Bayer/Dimension Therapeutics, UniQure). Thus far, obtaining therapeutic FVIII levels has been limited by inefficient FVIII expression profiles and immune responses to the AAV vector. Inefficient FVIII expression may necessitate the use of FVIII variants with increased specific activity.65 Caution is being executed as use of a variant FVIII transgene may itself result in unclear risks of an immune or inhibitory response to the expressed protein. Importantly, prior work has demonstrated that liver-directed gene therapy has the capacity to induce transgene tolerance.66,67 Notably, this has specifically been demonstrated for AAV-mediated FVIII gene transfer in canine models hemophilia A.68 Other strategies include ex vivo megakaryocyte transduction using a lentiviral cassette for ectopic platelet expression of FVIII; however, this approach requires a syngeneic transplant with reduced intensity conditioning that may not be widely appealing to patients with hemophilia.69
Conclusion
Much is likely to change in hemophilia care in both the short term, with the incorporation of now-licensed Fc-fusion products into clinical care, and the long term, with potentially multiple long-acting treatment strategies coming to market. The transition of these novel therapeutics to clinical practice represents the next wave of hemophilia therapeutic advances. Where rBDD FVIII-Fc will emerge among the many upcoming therapies is not clear. The consistent inability to dramatically improve FVIII half-life through various approaches represents an inherent limitation to FVIII. How this will impact current therapy and the future development of new FVIII therapeutics in a competitive market remains to be determined. Given the many emerging agents, hemophilia care will be marked by both significant changes and need for careful evaluation of optimal treatment among these new therapeutics and better understanding of their inherent risks and benefits. Consistent with the precedent set by the previous advances in hemophilia, these novel therapeutics represent a continued trend of expeditious incorporation of rationally designed therapeutics to improve hemophilia care.
Acknowledgments
We are grateful to Leslie Raffini and Valder Arruda for their thoughtful input.
Disclosure
RM Camire receives research support from Pfizer Pharmaceuticals. LA George is a National Hemophilia Foundation fellow funded, in part, by Baxter. The authors report no other conflicts of interest in this work.
References
Goodnight SH, Hathaway W. Disorders of Hemostasis and Thrombosis. 2nd ed. Lancaster, PA: McGraw-Hill, Inc.; 2001. | |
Mannucci PM, Tuddenham EG. The hemophilias – from royal genes to gene therapy. N Engl J Med. 2001;344(23):1773–1779. | |
White GC 2nd, Rosendaal F, Aledort LM, et al. Definitions in hemophilia. Recommendation of the scientific subcommittee on factor VIII and factor IX of the scientific and standardization committee of the International Society on Thrombosis and Haemostasis. Thromb Haemost. 2001;85(3):560. | |
Pool JG, Gershgold EJ, Pappenhagen AR. High-potency antihaemophilic factor concentrate prepared from cryoglobulin precipitate. Nature. 1964;203:312. | |
Evatt BL. The tragic history of AIDS in the hemophilia population, 1982–1984. J Thromb Haemost. 2006;4(11):2295–2301. | |
Pipe SW. Recombinant clotting factors. Thromb Haemost. 2008; 99(5):840–850. | |
Nilsson IM, Berntorp E, Lofqvist T, Pettersson H. Twenty-five years’ experience of prophylactic treatment in severe haemophilia A and B. J Intern Med. 1992;232(1):25–32. | |
Yoshida M, Kobayashi K, Kuo TT, et al. Neonatal Fc receptor for IgG regulates mucosal immune responses to luminal bacteria. J Clin Invest. 2006;116(8):2142–2151. | |
Liesner RJ, Khair K, Hann IM. The impact of prophylactic treatment on children with severe haemophilia. Br J Haematol. 1996;92(4):973–978. | |
van den Berg HM, Fischer K, Mauser-Bunschoten EP, et al. Long-term outcome of individualized prophylactic treatment of children with severe haemophilia. Br J Haematol. 2001;112(3):561–565. | |
Manco-Johnson MJ, Abshire TC, Shapiro AD, et al. Prophylaxis versus episodic treatment to prevent joint disease in boys with severe hemophilia. N Engl J Med. 2007;357(6):535–544. | |
Fischer K, van der Bom JG, Mauser-Bunschoten EP, et al. The effects of postponing prophylactic treatment on long-term outcome in patients with severe hemophilia. Blood. 2002;99(7):2337–2341. | |
Molho P, Rolland N, Lebrun T, et al. Epidemiological survey of the orthopaedic status of severe haemophilia A and B patients in France. The French Study Group. [email protected]. Haemophilia. 2000;6(1):23–32. | |
Mannucci PM, Franchini M. Present and future challenges in the treatment of haemophilia: a clinician’s perspective. Blood Transfus. 2013;11(Suppl 4):S77–S81. | |
Plug I, Van Der Bom JG, Peters M, et al. Mortality and causes of death in patients with hemophilia, 1992–2001: a prospective cohort study. J Thromb Haemost. 2006;4(3):510–516. | |
Ljung R, Auerswald G, Benson G, et al. Novel coagulation factor concentrates: issues relating to their clinical implementation and pharmacokinetic assessment for optimal prophylaxis in haemophilia patients. Haemophilia. 2013;19(4):481–486. | |
World Federation of Hemophilia. Guidelines for the Management of Hemophilia. Quebec, Canada: World Federation of Hemophilia. Available from: http://www1.wfh.org/publication/files/pdf-1472.pdf. Accessed October 2, 2015. | |
Collins PW. Personalized prophylaxis. Haemophilia. 2012;18(Suppl 4):131–135. | |
Carcao M. Changing paradigm of prophylaxis with longer acting factor concentrates. Haemophilia. 2014;20(Suppl 4):99–105. | |
Srivastava A, Brewer AK, Mauser-Bunschoten EP, et al. Guidelines for the management of hemophilia. Haemophilia. 2013;19(1):e1–e47. | |
Vehar G, Keyt B, Eaton D, et al. Structure of human factor VIII. Nature. 1984;312:337–342. | |
Fischer K, Pendu R, van Schooten CJ, et al. Models for prediction of factor VIII half-life in severe haemophiliacs: distinct approaches for blood group O and non-O patients. PLoS One. 2009;4(8):e6745. | |
Fijnvandraat K, Peters M, ten Cate JW. Inter-individual variation in half-life of infused recombinant factor VIII is related to pre-infusion von Willebrand factor antigen levels. Br J Haematol. 1995;91(2):474–476. | |
Hacker MR, Geraghty S, Manco-Johnson M. Barriers to compliance with prophylaxis therapy in haemophilia. Haemophilia. 2001;7(4):392–396. | |
De Moerloose P, Urbancik W, Van Den Berg HM, Richards M. A survey of adherence to haemophilia therapy in six European countries: results and recommendations. Haemophilia. 2008;14(5):931–938. | |
Journeycake JM, Buchanan GR. Catheter-related deep venous thrombosis and other catheter complications in children with cancer. J Clin Oncol. 2006;24(28):4575–4580. | |
Hay CR, DiMichele DM; International Immune Tolerance Study. The principal results of the International Immune Tolerance Study: a randomized dose comparison. Blood. 2012;119(6):1335–1344. | |
Goudemand J, Rothschild C, Demiguel V, et al. Influence of the type of factor VIII concentrate on the incidence of factor VIII inhibitors in previously untreated patients with severe hemophilia A. Blood. 2006; 107(1):46–51. | |
Gouw SC, van der Bom JG, Auerswald G, Ettinghausen CE, Tedgard U, van den Berg HM. Recombinant versus plasma-derived factor VIII products and the development of inhibitors in previously untreated patients with severe hemophilia A: the CANAL cohort study. Blood. 2007;109(11):4693–4697. | |
Iorio A, Halimeh S, Holzhauer S, et al. Rate of inhibitor development in previously untreated hemophilia A patients treated with plasma-derived or recombinant factor VIII concentrates: a systematic review. J Thromb Haemost. 2010;8(6):1256–1265. | |
Franchini M, Tagliaferri A, Mengoli C, Cruciani M. Cumulative inhibitor incidence in previously untreated patients with severe hemophilia A treated with plasma-derived versus recombinant factor VIII concentrates: a critical systematic review. Crit Rev Oncol Hematol. 2012;81(1):82–93. | |
Collins PW, Palmer BP, Chalmers EA, et al. Factor VIII brand and the incidence of factor VIII inhibitors in previously untreated UK children with severe hemophilia A, 2000–2011. Blood. 2014;124(23):3389–3397. | |
Calvez T, Chambost H, Claeyssens-Donadel S, et al. Recombinant factor VIII products and inhibitor development in previously untreated boys with severe hemophilia A. Blood. 2014;124(23):3398–3408. | |
Gouw SC, van der Bom JG, Ljung R, et al. Factor VIII products and inhibitor development in severe hemophilia A. N Engl J Med. 2013; 368(3):231–239. | |
Peters RT, Toby G, Lu Q, et al. Biochemical and functional characterization of a recombinant monomeric factor VIII-Fc fusion protein. J Thromb Haemost. 2013;11(1):132–141. | |
Dumont JA, Liu T, Low SC, et al. Prolonged activity of a recombinant factor VIII-Fc fusion protein in hemophilia A mice and dogs. Blood. 2012;119(13):3024–3030. | |
Powell JS, Josephson NC, Quon D, et al. Safety and prolonged activity of recombinant factor VIII Fc fusion protein in hemophilia A patients. Blood. 2012;119(13):3031–3037. | |
Mahlangu J, Powell JS, Ragni MV, et al. Phase 3 study of recombinant factor VIII Fc fusion protein in severe hemophilia A. Blood. 2014;123(3):317–325. | |
Young G, Mahlangu JN, Kulkarni R, et al. Safety, efficacy, and pharmacokinetics of recombinant factor VIII Fc fusion protein (rFVIIIFc) in previously-treated children with severe hemophilia a (Kids-ALONG). Presented at: 56th Annual Meeting and Exposition, December 17, 2014, San Francisco, CA. Available from: https://ash.confex.com/ash/2014/webprogram/Paper70146.html. Accessed February 10, 2015. | |
Roopenian DC, Akilesh S. FcRn: the neonatal Fc receptor comes of age. Nat Rev Immunol. 2007;7(9):715–725. | |
McGarry T, Hough R, Rogers S, Rechsteiner M. Intracellular distribution and degradation of immunoglobulin G and immunoglobulin G fragments injected into HeLa cells. J Cell Biol. 1983;96(2):338–346. | |
Peters RT, Low SC, Kamphaus GD, et al. Prolonged activity of factor IX as a monomeric Fc fusion protein. Blood. 2010;115(10):2057–2064. | |
Ghaderi D, Zhang M, Hurtado-Ziola N, Varki A. Production platforms for biotherapeutic glycoproteins. Occurrence, impact, and challenges of non-human sialylation. Biotechnol Genet Eng Rev. 2012;28:147–175. | |
Ragni MV, Malec LM. Design of the INHIBIT trial: preventing inhibitors by avoiding ‘danger’, prolonging half-life and promoting tolerance. Expert Rev Hematol. 2014;7(6):747–755. | |
Hay CR, Palmer BP, Chalmers EA, et al. The incidence of factor VIII inhibitors in severe haemophilia A following a major switch from full-length to B-domain-deleted factor VIII: a prospective cohort comparison. Haemophilia. 2015;21(2):219–226. | |
Gringeri A, Tagliaferri A, Tagariello G, et al. Efficacy and inhibitor development in previously treated patients with haemophilia A switched to a B domain-deleted recombinant factor VIII. Br J Haematol. 2004;126(3):398–404. | |
De Groot AS, Moise L, McMurry JA, et al. Activation of natural regulatory T cells by IgG Fc-derived peptide “Tregitopes”. Blood. 2008; 112(8):3303–3311. | |
Lei TC, Scott DW. Induction of tolerance to factor VIII inhibitors by gene therapy with immunodominant A2 and C2 domains presented by B cells as Ig fusion proteins. Blood. 2005;105(12):4865–4870. | |
Broderick CR, Herbert RD, Latimer J, et al. Association between physical activity and risk of bleeding in children with hemophilia. JAMA. 2012;308(14):1452–1459. | |
Petrini P, Valentino LA, Gringeri A, Re WM, Ewenstein B. Individualizing prophylaxis in hemophilia: a review. Expert Rev Hematol. 2015;8(2):237–246. | |
Colombo GL, Di Matteo S, Mancuso ME, Santagostino E. Cost-utility analysis of prophylaxis versus treatment on demand in severe hemophilia A. Clinicoecon Outcomes Res. 2011;3:55–61. | |
Skinner MW. WFH: closing the global gap – achieving optimal care. Haemophilia. 2012;18(Suppl 4):1–12. | |
Tiede A, Brand B, Fischer R, et al. Enhancing the pharmacokinetic properties of recombinant factor VIII: first-in-human trial of glycoPEGylated recombinant factor VIII in patients with hemophilia A. J Thromb Haemost. 2013;11(4):670–678. | |
Turecek PL, Bossard MJ, Graninger M, et al. BAX 855, a PEGylated rFVIII product with prolonged half-life. Development, functional and structural characterisation. Hamostaseologie. 2012;32(Suppl 1):S29–S38. | |
Coyle TE, Reding MT, Lin JC, Michaels LA, Shah A, Powell J. Phase I study of BAY 94-9027, a PEGylated B-domain-deleted recombinant factor VIII with an extended half-life, in subjects with hemophilia A. J Thromb Haemost. 2014;12(4):488–496. | |
Zollner SB, Raquet E, Muller-Cohrs J, et al. Preclinical efficacy and safety of rVIII-SingleChain (CSL627), a novel recombinant single-chain factor VIII. Thromb Res. 2013;132(2):280–287. | |
Zollner S, Raquet E, Claar P, et al. Non-clinical pharmacokinetics and pharmacodynamics of rVIII-SingleChain, a novel recombinant single-chain factor VIII. Thromb Res. 2014;134(1):125–131. | |
Kitazawa T, Igawa T, Sampei Z, et al. A bispecific antibody to factors IXa and X restores factor VIII hemostatic activity in a hemophilia A model. Nat Med. 2012;18(10):1570–1574. | |
Muto A, Yoshihashi K, Takeda M, et al. Anti-factor IXa/X bispecific antibody (ACE910): hemostatic potency against ongoing bleeds in a hemophilia A model and the possibility of routine supplementation. J Thromb Haemost. 2014;12(2):206–213. | |
Shima M, Hanabusa H, et al. Safety and prophylactic efficacy profiles of ACE910, a humanized bispecific antibody mimicking the FVIII cofactor function, in Japanese hemophilia A patients both without and with FVIII inhibitors: first-in-patient Phase 1 Study. Presented at: 56th Annual Meeting and Exposition, December 17, 2014, San Francisco, CA. Available from: https://ash.confex.com/ash/2014/webprogram/Paper67797.html. Accessed February 19, 2015. | |
Chowdary P, Lethagen S, Friedrich U, et al. Safety and pharmacokinetics of anti-TFPI antibody (concizumab) in healthy volunteers and patients with hemophilia: a randomized first human dose trial. J Thromb Haemost. Epub January 31, 2015. | |
Sorensen B, Mant T, Akinc A, et al; Aln-AT3 Investigators. A subcutaneously administered RNAi therapeutic (ALN-AT3) targeting antithrombin for treatment of hemophilia: interim Phase 1 study results in healthy volunteers and patients with hemophilia A or B. Presented at: 56th Annual Meeting and Exposition, December 17, 2014, San Francisco, CA. Available from: https://ash.confex.com/ash/2014/webprogram/Paper75077.html. Accessed February 19, 2015. | |
Nathwani AC, Tuddenham EG, Rangarajan S, et al. Adenovirus-associated virus vector-mediated gene transfer in hemophilia B. N Engl J Med. 2011;365(25):2357–2365. | |
Nathwani AC, Reiss UM, Tuddenham EG, et al. Long-term safety and efficacy of factor IX gene therapy in hemophilia B. N Engl J Med. 2014;371(21):1994–2004. | |
Siner JI, Iacobelli NP, Sabatino DE, et al. Minimal modification in the factor VIII B-domain sequence ameliorates the murine hemophilia A phenotype. Blood. 2013;121(21):4396–4403. | |
Mingozzi F, Liu YL, Dobrzynski E, et al. Induction of immune tolerance to coagulation factor IX antigen by in vivo hepatic gene transfer. J Clin Invest. 2003;111(9):1347–1356. | |
Crudele JM, Finn JD, Siner JI, et al. AAV liver expression of FIX-Padua prevents and eradicates FIX inhibitor without increasing thrombogenicity in hemophilia B dogs and mice. Blood. 2015;125(10):1553–1561. | |
Finn JD, Ozelo MC, Sabatino DE, et al. Eradication of neutralizing antibodies to factor VIII in canine hemophilia A after liver gene therapy. Blood. 2010;116(26):5842–5848. | |
Shi Q, Fahs SA, Wilcox DA, et al. Syngeneic transplantation of hematopoietic stem cells that are genetically modified to express factor VIII in platelets restores hemostasis to hemophilia A mice with preexisting FVIII immunity. Blood. 2008;112(7):2713–2721. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.