")
Back to Journals » Drug Design, Development and Therapy » Volume 12
Profile of asfotase alfa in the treatment of hypophosphatasia: design, development, and place in therapy
Received 23 June 2018
Accepted for publication 21 August 2018
Published 24 September 2018 Volume 2018:12 Pages 3147—3161
DOI https://doi.org/10.2147/DDDT.S154922
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Qiongyu Guo
Sasigarn A Bowden,1 Brian L Foster2
1Division of Endocrinology, Department of Pediatrics, Nationwide Children’s Hospital/The Ohio State University College of Medicine, Columbus, OH 43205, USA; 2Division of Biosciences, College of Dentistry, The Ohio State University, Columbus, OH 43205, USA
Abstract: Hypophosphatasia (HPP) is a multi-systemic metabolic disorder caused by loss-of-function mutations in the ALPL gene that encodes the mineralization-associated enzyme, tissue-nonspecific alkaline phosphatase (TNSALP). HPP is characterized by defective bone and dental mineralization, leading to skeletal abnormalities with complications resulting in significant morbidity and mortality. Management of HPP has been limited to supportive care until the introduction of a recently approved enzyme replacement therapy employing bone-targeted recombinant human TNSALP, asfotase alfa (AA). This new therapy has been transformative as it improves survival in severely affected infants, and overall quality of life in children and adults with HPP. This review provides an overview of HPP, focusing on important steps in the development of AA enzyme replacement therapy, including the drug design, preclinical studies in the HPP mouse model, and outcomes from clinical trials and case report publications to date, with special attention given to response to therapy of skeletal manifestations, biochemical features, and other clinical manifestations. The limitations, adverse effects, and outcomes of AA are outlined and the place in therapy for individuals with HPP is discussed.
Keywords: alkaline phosphatase, bone mineralization, rickets, osteomalacia, teeth, enzyme replacement therapy, inorganic pyrophosphate
Tissue-non-specific alkaline phosphatase (TNSALP) and hypophosphatasia (HPP)
The enzyme, tissue-nonspecific alkaline phosphatase (TNSALP, TNALP, or TNAP), was first reported by Dr Robert Robison in 1923.1,2 TNSALP is encoded by the ALPL gene on chromosome 1 in humans and is highly expressed in bones, teeth, liver, and kidney (and at lower levels in fibroblasts, endothelial cells, and nervous system), thus its nomenclature as a “nonspecific” enzyme.3,4 TNSALP is bound to cell surfaces by a glycosylphosphatidylinositol (GPI) anchor that can be cleaved to release the enzyme into circulation, where alkaline phosphatase activity (ALP) can be detected in plasma and is a useful metric for diagnosis of some conditions. The enzyme active site is located in the extracellular domain making TNSALP an ectoenzyme. While a broad substrate specificity has been demonstrated in vitro, physiological substrates indicated by TNSALP loss-of-function include inorganic pyrophosphate (PPi), phosphoethanolamine (PEA), and pyridoxal 5′-phosphate (PLP).
The ability of TNSALP to hydrolyze and inactivate PPi was found to be an important function of TNSALP in mineralization. PPi is a potent inhibitor of calcium phosphate (hydroxyapatite [HAP]) crystal growth that represents the inorganic mineral component of bones and teeth.5–11 PPi is a byproduct of cellular metabolism, and local levels of PPi are increased by proteins including the progressive ankylosis protein (ANK in mice/ANKH in humans) that regulates the transport of PPi into the extracellular space and ectonucleotide pyrophosphatase phosphodiesterase 1 (ENPP1), an ectoenzyme that cleaves nucleotide triphosphates to generate PPi (Figure 1).12–23 Altogether, TNSALP, ANK/ANKH, ENPP1, and other regulators comprise a complex feedback system that directs the location and extent of mineralization in the body.
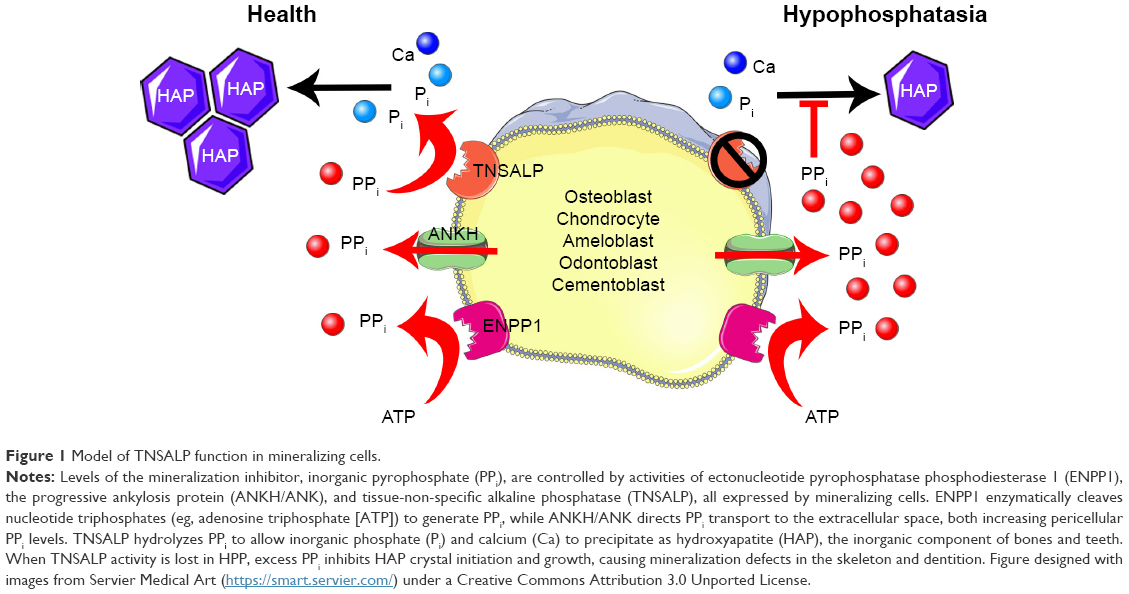 | Figure 1 Model of TNSALP function in mineralizing cells. |
Loss-of-function mutations in ALPL cause HPP (OMIM# 241500, 241510, and 146300), an inherited disorder characterized by defective mineralization of bones and teeth.3,24,25 To date, 361 HPP-causing ALPL mutations have been reported (http://www.sesep.uvsq.fr/03_hypo_mutations.php), and these are predominantly (~72%) missense mutations. The mode of inheritance of HPP can be either autosomal recessive or autosomal dominant.26,27 Deficiency of functional TNSALP is reflected as low circulating ALP levels.25 Deficiency of TNSALP in HPP leads to increased PPi concentrations that impair skeletal and dental mineralization. The deficiency of TNSALP secondarily leads to a disturbance of mineral homeostasis. Elevation of serum calcium or phosphorus levels is sometimes noted and thought to result from the combination of unaffected gut absorption of the ions, but lack of phosphate and calcium deposition in the skeleton, resulting in elevated circulating levels. In addition to increased PPi, deficiency of TNSALP also leads to increased extracellular accumulation of the two other known physiological substrates, PEA and PLP.
Clinical manifestations of HPP are highly variable, with increased severity typically correlating with earlier onset. In its most severe presentation, HPP manifests in utero and causes stillbirth due to catastrophically hypomineralized skeleton. In its mildest presentation, individuals suffer from premature loss of deciduous teeth with little or no other systemic or skeletal manifestations. HPP is currently classified into six clinical forms, based on the age at the onset of first signs or symptoms. This classification delineates disease severity and may correlate with prognosis, as well as the levels of ALP and biochemical substrates, although there is considerable overlap in clinical phenotypes and biochemical hallmarks in HPP, and strong genotype–phenotype correlations have not been established for the majority of mutations.28 The six different forms of HPP are outlined as follows.
Perinatal HPP
This is the most severe form of HPP and is generally lethal due to an almost complete absence of bone mineralization. Short and deformed extremities and polyhydramnios (excess of amniotic fluid) can be detected in utero. Affected infants often die shortly after birth due to respiratory compromise arising from hypoplastic lungs and skeletal deformities of the thorax. Perinatal HPP is diagnosed on prenatal imaging by severe generalized osteopenia and non-ossified cranial vault.
Benign prenatal HPP
This form manifests in utero, with skeletal deformities including poorly mineralized bone or short and severely bowed legs detected prenatally by ultrasound, which would typically signify the perinatal lethal form of HPP. However, spontaneous improvement in the skeletal disease is observed starting from the third trimester of pregnancy and after birth, with a clinical outcome ranging in severity from infantile to odonto-HPP phenotypes.29 Therefore, abnormal prenatal ultrasound findings before the third trimester are not always predictive of perinatal lethal HPP.
Infantile HPP
The infantile form has an onset prior to six months, and infants might initially appear healthy immediately after birth. Affected infants soon develop hypotonia, poor feeding, failure to thrive, and generalized hypomineralization with severe skeletal deformities, including rachitic defects of the chest that lead to respiratory insufficiency. Unlike other forms of genetic and nutritional rickets, serum calcium levels are generally high at diagnosis of HPP due to blocked mineral entry into the skeleton caused by elevated extracellular PPi levels, and hypercalciuria and nephrocalcinosis can occur as consequences of hypercalcemia. Infants may also have muscle pain and weakness from a static myopathy, possibly as a result of an accumulation of PPi.30 Craniosynostosis and skull abnormalities (Figure 2A, B) occur in approximately 40% of infants, and may require neurosurgical intervention due to intracranial hypertension.31 Irritability and vomiting are common, which can be attributed to hypercalcemia or increased intracranial pressure with papilledema secondary to craniosynostosis. The infantile form is associated with approximately 50% mortality due to respiratory failure.
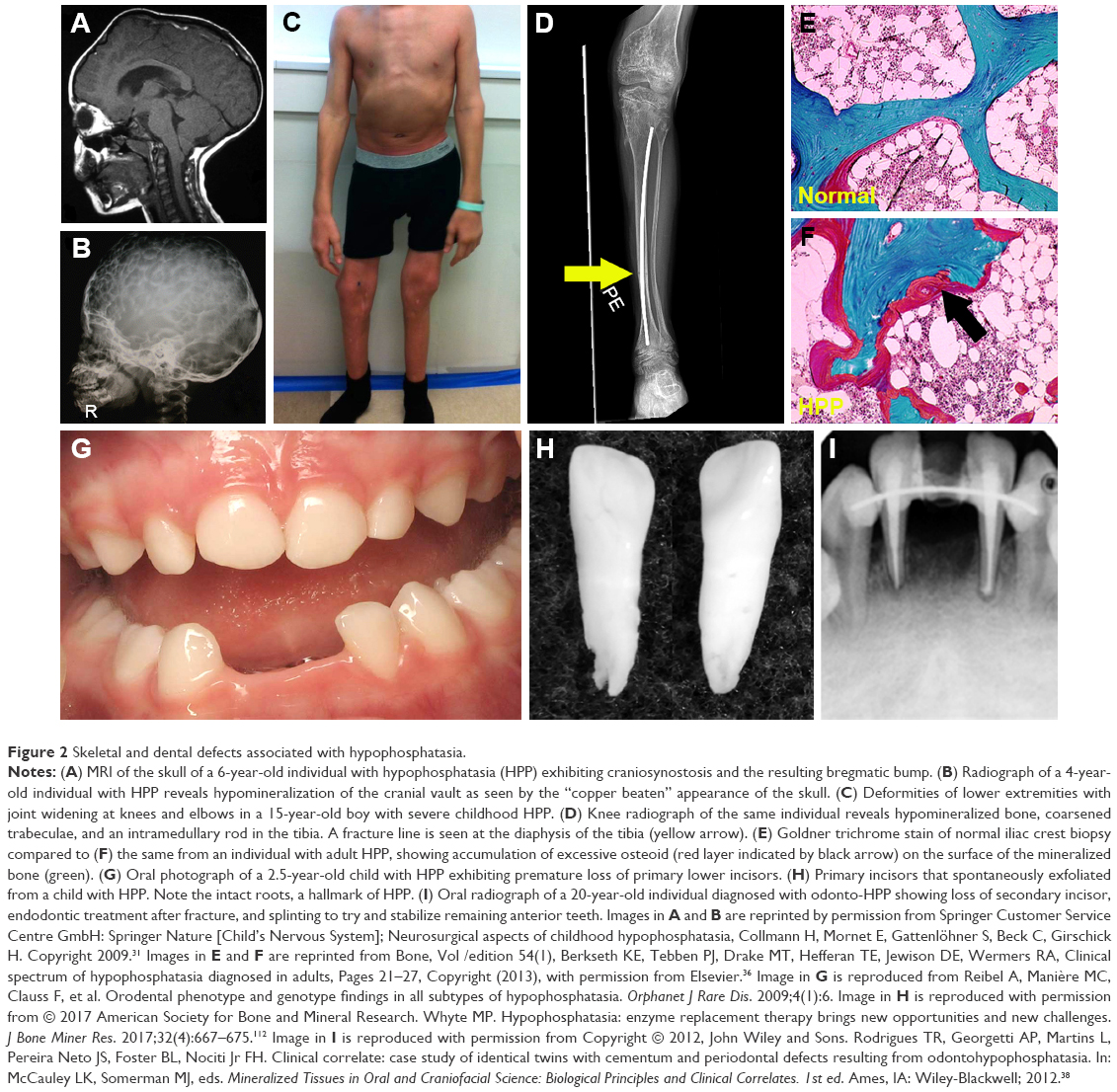 | Figure 2 Skeletal and dental defects associated with hypophosphatasia. |
Childhood HPP
This form features skeletal abnormalities that appear after 6 months, especially rickets manifesting as bowed legs and bony enlargement near joints due to widened metaphyses (Figure 2C and D).32 Chronic skeletal pain, recurrent fractures, short stature, muscle weakness, abnormal ambulation or gait, and premature loss of deciduous teeth are common presenting signs or symptoms. Rarely, childhood HPP can present with chronic multifocal non-bacterial osteomyelitis mimicking malignancy,33 due to marrow edema secondary to PPi crystal deposition.34 Craniosynostosis can also be a presenting sign accompanied by chronic elevated intracranial pressure, papilledema, and impaired visual acuity.35 In 2015, it was proposed to separate childhood HPP into “mild” and “severe” forms to further delineate the wide range of severity of this form.32 Severe childhood HPP presents with significant skeletal abnormalities like those described above, whereas mild childhood HPP allows good physical function with minimal symptoms or skeletal changes.
Adult HPP
This form can manifest at any point of time in the adult life (≥18 years) with a wide range of symptoms, ranging from fractures or osteomalacia (Figure 2E and F) to dental symptoms, to being completely asymptomatic.36 Individuals with adult HPP are usually diagnosed in middle age, and commonly present with skeletal symptoms including fractures, osteomalacia, chondrocalcinosis, osteoarthropathy, and stress fractures. Fractures can be at the hip or femoral neck, spine, or small bones in the wrists or feet. Femoral pseudofractures and subtrochanteric fractures have been reported.36,37 Those patients with lower ALP levels and higher PLP and PEA levels tend to experience greater numbers of fractures. Other symptoms include musculoskeletal pain, joint swelling, pseudogout, low bone density, and hearing loss. Musculoskeletal pain can be debilitating with restricted range of motion as a result of chondrocalcinosis, sometimes leading to severe disability.
Odontohypophosphatasia (odonto-HPP)
While individuals diagnosed with odonto-HPP exhibit the biochemical characteristics of HPP, this form is characterized by dental manifestations with minimal or no observable skeletal manifestations. Though this mildest form of HPP is classified according to dental abnormalities, all the more severe clinical forms described above feature similar dental manifestations; tooth loss and other dental problems are nearly universal among individuals with any form of HPP. Premature loss of primary (deciduous) teeth (prior to 5 years of age) is the most commonly reported dental manifestation of HPP, with teeth presenting full roots (rather than resorbed roots typical of exfoliated primary teeth) (Figure 2G–I). This is the direct result of high levels of PPi inhibiting formation or mineralization of tooth root acellular cementum. Other dental abnormalities reported include loss of secondary (permanent) teeth, tooth mobility, abnormal dentin, thin roots, wide pulp chambers, alveolar bone loss, malocclusion, delayed eruption, and enamel defects.38–42 It should be noted that a subset of patients with the diagnosis of odonto-HPP may develop bone manifestations of HPP later in life, evolving to a mild form of childhood- or adult-onset HPP with bone pain and fractures. Therefore, long-term follow-up care for individuals with odonto-HPP is recommended.43
Attempted treatments of HPP prior to asfotase alfa (AA)
Historically and prior to implementation of AA recombinant TNSALP enzyme replacement therapy (ERT), management of HPP was largely limited to supportive care. The scope of supportive care varies with the wide ranging severity of disease and may include ventilator support for infants with respiratory insufficiency (ranging from oxygen supplementation with nasal cannula to mechanical ventilation), treatment of hypercalcemia with fluid hydration and low-calcium formula or diet, pain relief, and orthopedic surgeries for fractures. This standard-of-care remains for patients who cannot access AA therapy.
Recognition that individuals with HPP exhibited very low ALP levels prompted the first interventions to attempt to ameliorate the effects of severe HPP. Paget’s disease of bone (PDB; OMIM# 167250, 239000, 602080, 616833), a metabolic disease of increased and disorganized bone remodeling that typically manifests in adulthood, dramatically increases ALP in affected subjects.44 In a clever approach, Whyte et al collected ALP-rich plasma from individuals with PDB and administered this by intravenous (IV) infusion to four subjects with severe, life-threatening infantile HPP.45,46 Treatment over ≥5 weeks elevated ALP into the normal range and improved hypercalcemia for all subjects and two subjects showed radiographic or histological improvement of skeletal manifestations. However, urinary PPi and PEA remained elevated, and hypomineralization and skeletal deformities persisted and worsened in most subjects over time. Additional attempted interventions included administration of parathyroid hormone (PTH), prednisone, and normal plasma over several months; however, all of these approaches failed to make long-term improvements.47 A different approach employing bone marrow cell transplantation (BMT) yielded similar disappointing results. BMT of T-cell depleted marrow from a healthy sister to an 18-month-old female subject with severe infantile HPP initially spurred improved skeletal mineralization; however, the return of host hematopoiesis was accompanied by worsening rickets, scoliosis, and fractures by 6 months post-BMT.48 A modified BMT approach employing marrow, implantation of bone fragments, and IV delivery of primary osteoblasts to a 9.5-month-old female with severe infantile HPP promoted slight improvements in severity of rickets, scoliosis, and skeletal mineralization, though minimal engraftment was evident.49 A similar approach was taken in Japan to treat an 8-month-old child with perinatal HPP using father-derived BMT, osteoblasts cultured from her father’s mesenchymal stem cells (MSCs), and osteogenic constructs.50 Four months after treatment, skeletal and respiratory improvements were apparent though biochemical features were unchanged, and the patient’s condition declined precipitously shortly thereafter. An MSC booster at 15 months again improved skeletal and respiratory parameters, and the individual survived, though featured serious HPP-associated pathologies including craniosynostosis. A follow-up clinical trial tested the ability of BMT combined with multiple courses of allogeneic MSC transplants to treat perinatal HPP in two male infants.51 Similar to earlier approaches, biochemical markers were not normalized, though improvements to bone mineralization and respiratory function were realized, and both patients survived past the age of 3 years.
Based on observations of the short-term, limited improvements resulting from these experimental infusion, BMT, and MSC transfer approaches, the hypothesis arose that perhaps ALP in circulation was not sufficiently elevated over the long term, was not physiologically active, or was not accessing the tissues where its activity is most required.
Other than the attempts to directly or indirectly supply functional ALP described above, few treatment attempts for HPP have been reported. Bisphosphonates, a group of drugs that prevent bone loss by inhibiting osteoclast function, have been administered to some HPP patients, usually due to misunderstanding of pathological mechanisms of HPP or in the absence of proper diagnosis.52–56 Because bisphosphonates do not promote bone formation and may disrupt mineralization, these drugs are strongly contraindicated for use in HPP patients. Teriparatide (TPTD), a recombinant peptide based on human PTH, is an anabolic agent used to treat osteoporosis. Administration of TPTD to an adult with HPP and multiple stress fractures increased ALP levels, reduced bone pain, and was associated with fracture healing,57 and some additional case reports on TPTD use in subjects with HPP have indicated potentially positive effects.53–55 Relative success of TPTD treatment may depend on the phenotype and biochemistry of each individual HPP patient, as increased numbers and activity of osteoblasts will still produce defective TNSALP enzyme. TPTD is generally not used in the pediatric age group due to concerns about its association with osteosarcoma development in animal studies.
Drug design and preclinical testing of recombinant TNSALP for ERT
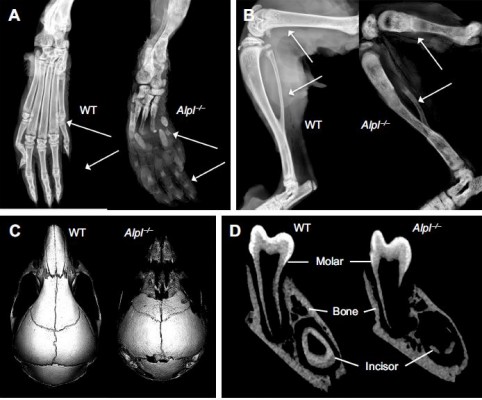
In order to study the underlying mechanisms of the disease, a mouse model of HPP was created in the 1990s in the laboratory of Dr Jose Luis Millán, targeting deletion of exons 5–8 from the Alpl gene.58 These Alpl knockout (Alpl−/−) mice recapitulated manifestations of severe infantile HPP, displaying bone hypomineralization and fractures, osteoid accumulation, rachitic changes and growth plate abnormalities in long bones, and severe seizures, with mice typically dying by 2–3 weeks of age (Figure 3A and B).59 The Millán laboratory employed this HPP mouse model extensively to explore the role of TNSALP in bone mineralization, vitamin B metabolism, and other organ systems, as well as to elucidate the interactions of TNSALP with other mineralization regulators.14,19,60–67 While an earlier publication confirmed that Alpl−/− mice featured cementum hypoplasia consistent with observations from the HPP case literature,68 a series of additional reports focused in greater detail on other dental and craniofacial defects in Alpl−/− mice, documenting disturbed enamel mineralization, failure of crystal growth at initial sites of dentin mineralization, defective cranial base mineralization, abnormal cranial shape, and craniosynostosis (Figure 3C–F).21,23,69–73
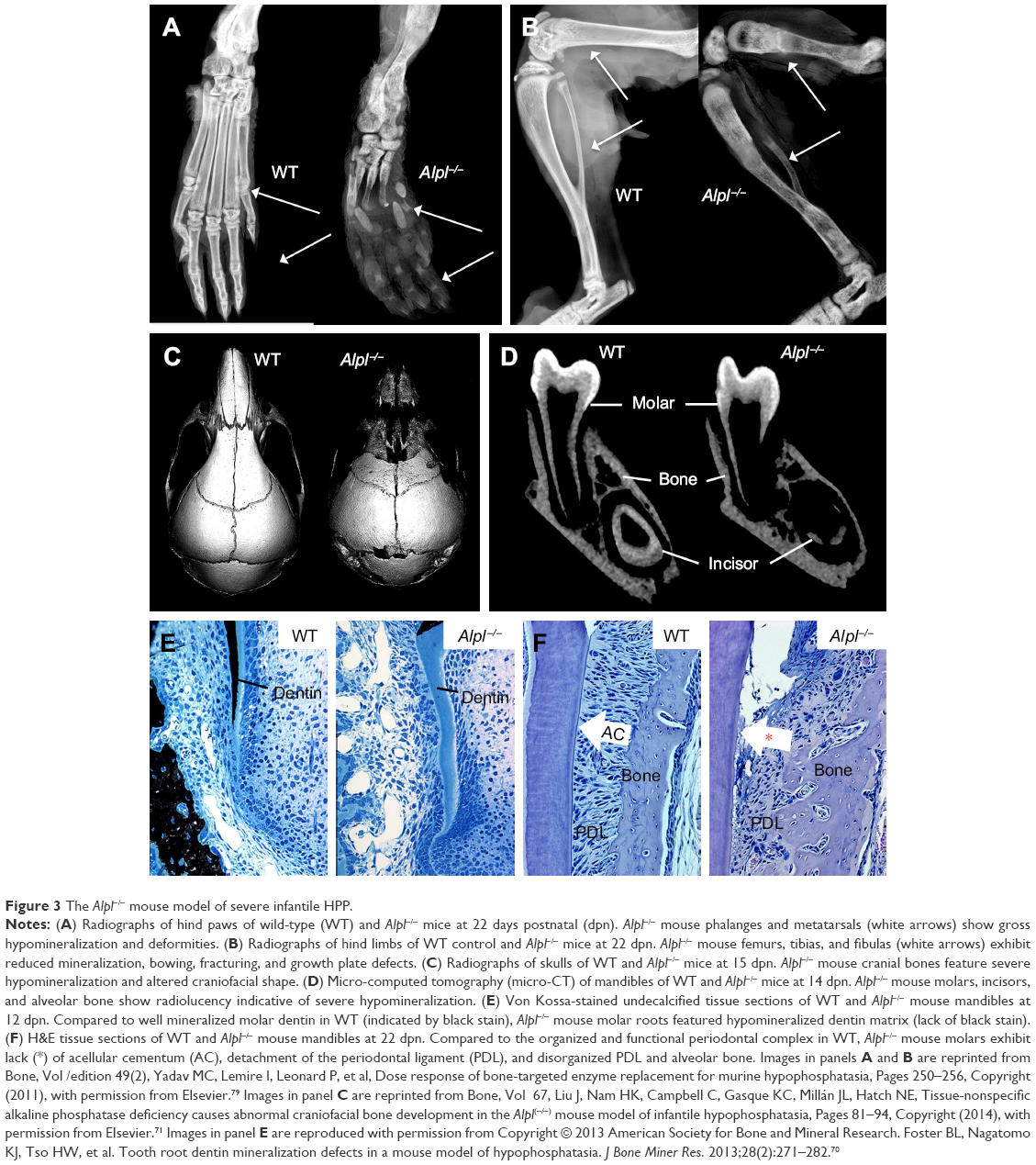 | Figure 3 The Alpl−/− mouse model of severe infantile HPP. |
Based on the above-described unsuccessful attempts to treat HPP in severely affected patients, a new goal was set to develop a recombinant TNSALP enzyme that could be directed to sites of mineralization in the skeleton and dentition, where it was hypothesized that PPiase activity was required. Dr Philippe Crine had begun exploring this concept with scientists at Enobia Pharma Inc. (Montreal, Canada), focusing on another enzyme, PHEX, and its associated mineralization disorder, X-linked hypophosphatemia (XLH; OMIM# 307800).74,75 In order to engineer a soluble, secreted, and stable TNSALP that could be expressed in vitro and easily purified, the GPI anchor was removed from the protein and the Fc region of human IgG antibody was added.76 In order to enhance delivery of recombinant TNSALP to mineralizing tissues, a deca-aspartate (D10) extension with high negative charge density was added to the C-terminus of the recombinant TNSALP (Figure 4A). The concept of an acidic amino acid “tail” had been previously shown to significantly improve in vivo delivery to and retention of TNSALP in bones in mice.77 The high negative charge density of the D10 tail mimicked naturally occurring bone- and tooth-associated proteins with acidic amino acid motifs, such as members of the Small Integrin-Binding LIgand N-Linked Glycoprotein (SIBLING) protein family, including osteopontin, dentin phosphoprotein, and bone sialoprotein.78
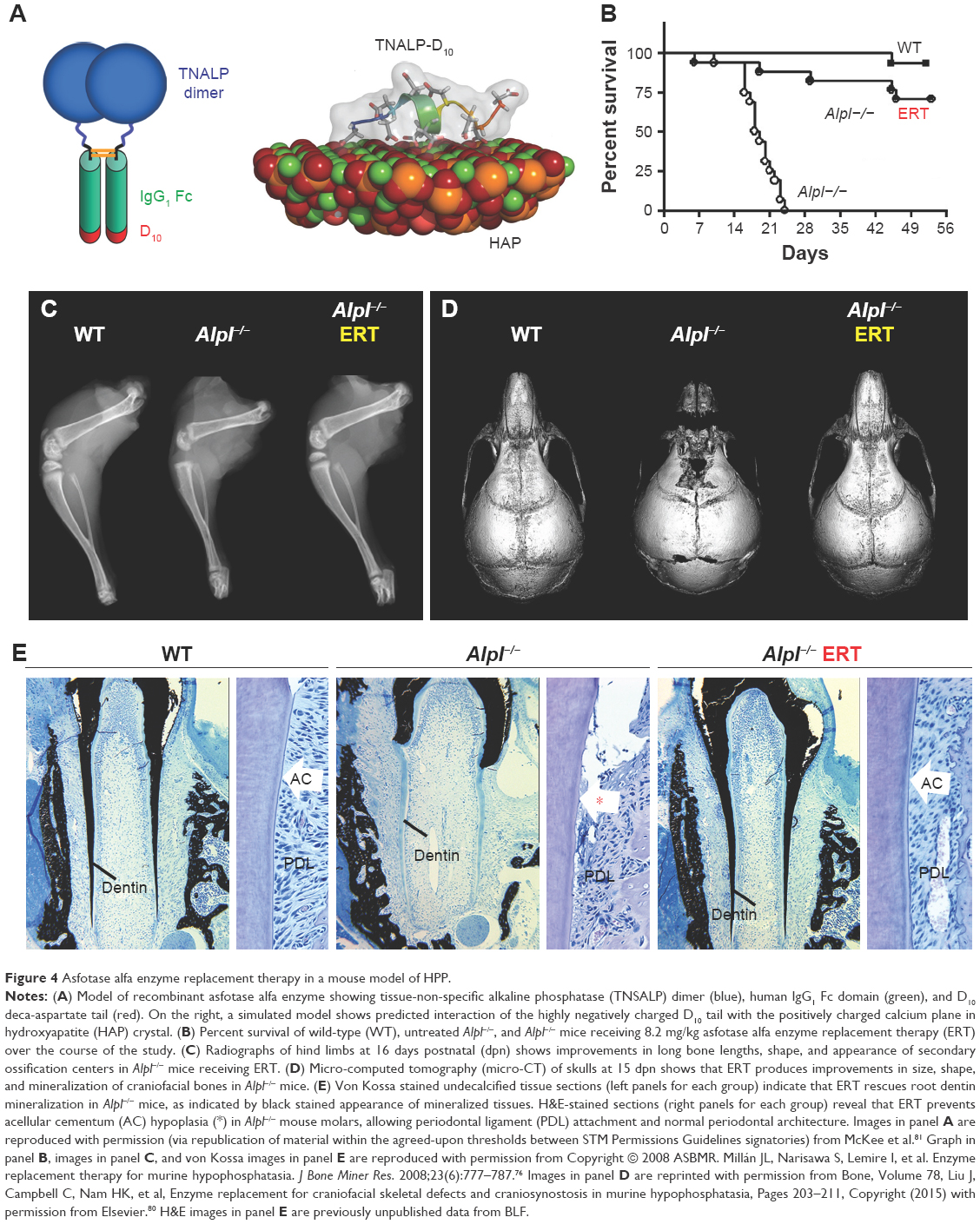 | Figure 4 Asfotase alfa enzyme replacement therapy in a mouse model of HPP. |
Recombinant TNSALP-D10 expressed in vitro in Chinese hamster ovary cells was purified and its molecular mass was confirmed by Western blotting to be consistent with homodimer formation.76 TNSALP-D10 bound to HAP 32-fold more efficiently than unmodified bovine kidney TNSALP control and apparently retained enzymatic catalytic activity. Pharmacokinetics and tissue distribution of TNSALP-D10 were investigated in adult and newborn mice. A single IV bolus of 5 mg/kg given to adult mice confirmed proof-of-concept by demonstrating retention of radiolabeled enzyme in bone, whereas significant accumulation was not observed in other tissues.76 Repeated daily subcutaneous (SC) injection of 10 mg/kg enzyme into newborns reproducibly elevated circulating ALP levels to about 50-fold higher than normal.
Following these encouraging results from in vitro assays and administration of TNSALP-D10 to normal mice, the necessary next step was to test the ability of ERT to ameliorate manifestations of HPP. The Alpl−/− mouse model of severe infantile HPP was well characterized and an ideal model to test the efficacy of TNSALP-D10 ERT. Based on daily SC injections to newborn pups over the course of 15 days, 2 mg/kg was determined to be the minimal efficacious dose, as supported by normalized growth rate, increased vertebral bone mineral density (BMD), and positive changes in cortical and trabecular bone.76 The higher dose of 8.2 mg/kg injected SC to newborns demonstrated great potency in increasing lifespans and preventing skeletal defects in Alpl−/− mice (Figure 4B and C). Untreated Alpl−/− mice died by median 18.5 days, while 75% of treated Alpl−/− mice lived to the conclusion of the experiment at 52 days, showing normal physical activity and dramatic improvement in all measured bone parameters (eg, long bone length, appearance of secondary ossification centers, and prevention of fractures). Further dose–response studies established that the dose preventing 80% of bone defects in mice (ED80) was ~3 mg/kg for sites including feet, lower limbs, ribs, and jaws.79 High-dose TNSALP-D10 prevented craniosynostosis and improved cranial shape and mineralization of craniofacial bones (Figure 4D).72,73,80 Additionally, TNSALP-D10 treatment significantly improved dental mineralization and function in Alpl−/− mice, preventing enamel and dentin defects and allowing establishment of acellular cementum to normalize periodontal structure and function (Figure 4E).69,70,76,81
While these preclinical mouse studies indicated tremendous potential for ERT to prevent skeletal and dental manifestations of HPP, an important limitation in the study designs and their interpretation was that ERT was initiated prior to onset of the majority of the defects. Because the skeleton is in a constant state of remodeling, ERT would be expected to promote mineralization of osteoid, resulting in replacement of poor quality bone with improved bone tissue with superior mechanical properties. However, the enamel, dentin, and cementum tissues of teeth do not remodel, and dentin and cementum have limited ability for repair, therefore timing of therapeutic intervention is likely a critical factor to improve the development and function of teeth. This lesson on early intervention of treatment has been learned through other endocrine disorders and metabolic disorders affecting teeth, such as nutritional vitamin D deficiency and XLH.82–84
Clinical trials and case reports on TNSALP ERT
Based on successful preclinical studies in Alpl−/− mice, clinical trials for TNSALP-D10 were planned. In 2008, Alexion Pharmaceuticals, Inc. (New Haven, CT, USA) initiated the first clinical trials (NCT00739505 Phase 1; NCT00744042 Phases 1 and 2) for TNSALP-D10, renamed as asfotase alfa (AA; also known as ALXN1215 or previously ENB-0040 under Enobia Pharma Inc.). Additional clinical trials that followed were: in the US, Canada, Europe, and Australia: NCT01205152 Phase 2 in 2009, NCT00952484 Phase 2 in 2009, NCT01203826 and NCT01163149 Phase 2 in 2010, NCT01176266 Phases 2 and 3 in 2010, NCT02797821 Phase 2 in 2016; In Japan: NCT02456038 Phase 2 in 2014 and NCT02531867 Phase 4 in 2015. In 2015, regulatory bodies in the US, Canada, the European Union, and Japan approved AA for the treatment of pediatric onset HPP. This new ERT with recombinant human TNSALP has been transformative in the short time since its approval. Below we outline the treatment outcomes of AA reported from clinical trials85–88 and case reports89–95 reported to date.
Perinatal and infantile HPP
The first clinical trial study reported by Whyte et al in 2012 described 1-year efficacy of AA therapy in 11 infants and young children aged ≤3 years with life-threatening perinatal and infantile forms of HPP.85 Treatment with one IV infusion at 2 mg/kg, followed by a dose of 1–3 mg/kg delivered by SC injections three times per week, resulted in dramatic healing of the skeletal manifestations of HPP and improvement in respiratory and motor function. All treated patients with gross motor delay were bearing weight or walking by 48 weeks of treatment. Improvement in skeletal mineralization was apparent by radiograph as early as 3 weeks and was striking by 24 weeks of treatment. Specifically, radiographs revealed improved mineralization, healing of rickets, resolution of radiolucency and sclerosis, fracture healing, and reduced deformity.85 The survival rate in 37 infants and young children (≤5 years) with perinatal or infantile HPP (including eleven patients in the first cohort and additional patients) treated with AA was compared with that of a historical cohort.86 The treated group had significantly improved the survival rate compared to the historical control group (95% vs 42% at 1-year-old and 84% vs 27% at 5-year-old, respectively). Another study in Japan reported 100% survival rate in perinatal and infantile HPP treated with AA.87 Early treatment with AA starting on the first day of life resulted in survival in an infant with lethal perinatal HPP.89 In the infantile HPP cohort, AA therapy results in improved mineralization of the skeleton, including ribs, contributing to improved respiratory function and survival rate.
Children (6–12 years)
In 2016, Whyte et al reported on a separate cohort of eleven older children, aged 6–12 years, treated with AA for 5 years.96 Similar to the younger children, radiographic improvement was noted by 6 months and persisted through 5 years. A significant increase in weight Z scores was noted at 6 weeks, while significantly increased height Z scores were noted after 1.5 years of treatment. After 5 years of treatment, most children exhibited increased muscle strength and motor skills that were on par with healthy children, as well as normal ambulation, and some resolution of pain and disability.
Adolescents (≥12 years) and adults
In randomized, open-label studies in six adolescents (aged 13–18 years) and 13 adults (aged 13–65 years) with HPP,88,97,98 SC administration of AA at 0.3 or 0.5 mg/kg/day was compared with control (no treatment) subjects over the course of 6 months. After 6 months, all patients received active treatment at 0.5 mg/kg/day, and the dose was again increased to 1 mg/kg administered six times per week after 6–12 months, and completed at 5 years of treatment. AA treatment resulted in significantly decreased PLP levels at 6 months and decreases in serum PPi and PLP were sustained through 5 years of treatment. ERT allowed significant improvement in physical function with increases in the 6-minute walking test, and improvement in health-related quality of life measured by the Childhood Health Assessment Questionnaire Disability Index and the Pediatric Outcomes Data Collection Instrument global function. Improved mobility without a walking assistive device (that was previously used) was reported in an adolescent after 6 months of AA treatment,93 and striking improvement in bone radiograph appearance was observed over the same period (Figure 5). In a 59-year-old adult with childhood-onset HPP, treatment with AA significantly increased the quality of life (increased mobility, reduced pain medication) and improved bone mineralization with healing of non-union fractures and no occurrence of new fractures.90 A recent report showed that treatment with AA could promote more rapid and successful fracture healing in adults with HPP.95 One subject had sustained non-healing fragility fracture for 3 years and another subject for 8 years, and both showed resolution of fractures after 11–16 months of therapy. Even more dramatically, 11 months of AA therapy resulted in near resolution of a non-healing femoral pseudofracture sustained 17 years prior in a patient who was previously wheelchair-bound. Fracture healing and other benefits of AA allowed this individual to stand and provided dramatic improvement in walking tolerance up to distances of 4 miles.
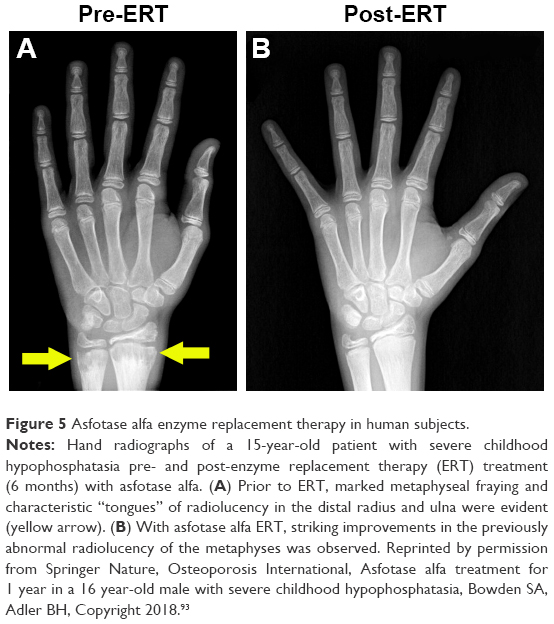 | Figure 5 Asfotase alfa enzyme replacement therapy in human subjects. |
Specific clinical features of HPP and therapeutic response to AA
While AA treatment causes dramatic reversal and improvement of skeletal defects from HPP, effects on other manifestations are not uniform. Below we highlight the response to treatment of neurological manifestations, nephrocalcinosis, chronic pain, and craniosynostosis.
Neurological manifestations
In severe perinatal HPP, seizures resulting from defective PLP metabolism are considered to be an ominous sign, associated with 100% mortality rate.86 PLP is an active form of vitamin B6 that cannot cross the plasma membrane or blood–brain barrier. TNSALP dephosphorylates PLP into the pyridoxal form of vitamin B6, which is able to cross the blood–brain barrier, subsequently becomes re-phosphorylated, and serves as a co-factor for many enzymes. PLP in the brain is required for biosynthesis of many neurotransmitters (eg, dopamine, norepinephrine, gamma-aminobutyric acid). Absence of TNSALP therefore results in elevated serum PLP and vitamin B6-dependent seizures, and administration of a high dose of vitamin B6 can temporarily prevent seizures. To date, it has not been clearly defined whether AA treatment has immediate effects on pyridoxine-responsive seizures, in part because severely affected neonates that manifest with seizures have been treated with both AA and vitamin B6 in the clinical trial.85 Nevertheless, treatment with AA in infants with seizures (the group at the greatest risk of death) resulted in markedly improved survival rate of 77%.86 Other neurological manifestations observed in infants or young children with infantile HPP include delayed motor milestones and cognitive development, likely to be secondary to their overall poor health associated with severe HPP. Treatment with AA improved gross motor, fine motor, and cognitive development as assessed by the Bayley Scales of Infant Development.85
Nephrocalcinosis
Nephrocalcinosis can be observed in individuals with HPP at the time of diagnosis and is thought to be secondary to hypercalcemia with hypercalciuria. This can cause impaired renal function. During clinical trials, no progression of nephrocalcinosis was noted in patients with infantile HPP,85,87 and the status even improved in some patients.85 Monitoring of nephrocalcinosis by renal ultrasound is recommended at baseline and every 3 months in perinatal/infantile HPP and at baseline, 6 months, and then annually in childhood and adult HPP.99
Chronic pain
Musculoskeletal pain in HPP is thought to be, in part, due to chronic inflammation with elevated prostaglandin levels100 caused by calcium pyrophosphate dihydrate crystal depositions.101 This condition can result in incapacitating bone and joint pain that severely diminishes quality of life. Resolution of chronic musculoskeletal pain with discontinuation of daily analgesic use was reported in an adolescent male with severe childhood HPP after 3 months of AA treatment started at the age of 15 years.93
Craniosynostosis
Premature fusion of cranial sutures may occur in cases of perinatal, infantile, and childhood HPP, the mechanism of which is poorly understood. Late clinical sequelae of craniosynostosis include papilledema and increased intracranial pressure with a copper-beaten skull on a skull X-ray (Figure 2B).31,102 To date, treatment with AA did not appear to have an effect on craniosynostosis, as progression of the condition was noted over the course of treatment, requiring neurosurgical intervention.85 However, when treatment starts at birth, AA treatment can prevent craniosynostosis in mice.80 The benefit of early treatment for prevention of craniosynostosis may hold true for human subjects, as has been reported in a neonate with lethal perinatal HPP receiving AA treatment from the first day after birth.89
Dental manifestations
Unfortunately, clinical trials to date have not incorporated dental examinations into data collection plans, representing a missed opportunity of sorts in determining the efficacy for AA to prevent or reverse HPP-associated dental-periodontal defects. These outcomes would have been especially instructive in determining how the timing of intervention affects the development and retention of primary and secondary teeth, and whether AA is equally effective at improving cementum, dentin, enamel, and alveolar bone/periodontal manifestations of HPP. The opportunity to collect quantitative and qualitative dental findings in treated HPP subjects remains feasible, is a high priority for clinicians and researchers concerned with dental effects of HPP, and could prove important in justifying early administration of AA to prevent lifelong oral health problems and improve quality of life for individuals with severe dental manifestations of HPP. A registry has been initiated to collect qualitative and quantitative data on dental effects of HPP, allowing pre- and post-ERT comparisons to begin to determine efficacy of AA on ameliorating dental defects (http://u.osu.edu/hppstudy/).
Biochemical changes resulting from AA treatment
Circulating ALP represents the most widely used metric to diagnose HPP, and ALP levels inversely correlate with severity of disease. Serum ALP increased rapidly and markedly after AA treatment from <100 to several thousand units per liter, to as high as ~24,000 IU/L within 4 weeks of starting ERT therapy,87 remaining at dramatically elevated levels of 3,000–6,000 IU/L after 5 years of continuous therapy.96 To date, these markedly elevated ALP levels do not appear to have harmful effects. Elevated TNSALP substrate levels (serum PPi and PLP) decrease significantly with AA treatment.85,87,93,96
While calcium abnormalities are rare in adults with HPP, hypercalcemia is common in infants and children and often presents at diagnosis. Some infants continue to experience hypercalcemia and hyperphosphatemia after AA therapy, requiring continued use of low-calcium formula and/or a low-phosphorus formula.87 Hypocalcemic (<8 mg/dL) seizures with serum calcium of 4.7 mg/dL were reported in one neonate with perinatal HPP after 3 weeks of AA therapy initiated on the first day of life.87 Seizures resolved after hypocalcemia was corrected by increasing calcium supplementation. Hypocalcemia during AA treatment suggests a “hungry bone” syndrome-like condition as AA stimulates rapid mineralization of bone, requiring availability of large quantities of calcium for HAP deposition. Dietary calcium restriction should be liberalized once hypercalcemia is no longer present, or when serum PTH levels increase, to prevent hungry bones.85 Serum PTH increases during AA treatment, coinciding with the skeletal remineralization as evidenced by the radiographic improvements.
Adverse effects from AA treatment
Results from clinical trials to date show very good safety profiles for AA treatment.85,87,96 The most common adverse effect associated with AA treatment is injection site reaction, occurring in about 75% of patients,99 consisting of mild, localized, transient erythema, pain, induration, and lipohypertrophy or lipohypotrophy at injection sites. Other rare adverse effects include anaphylactoid reaction.97 All patients in clinical trials developed detectable levels of anti-AA antibodies, but no tachyphylaxis has been reported to date.85,99
Adverse outcomes associated with AA treatment
Substantial benefits of AA therapy are evident in infants, children, and adults with HPP reported in the clinical trials and case reports described above. Infants with life-threatening HPP survived, while those individuals who died, succumbed to sepsis not attributed to AA therapy.85,92 Excellent clinical outcomes in all areas, including physical function and quality of life, continue after ≥5 years of follow-up in those children and adults.88 There has been one case of a term infant with perinatal HPP experiencing unsuccessful treatment outcome due to irreversible pulmonary hypoplasia, despite improvement in rib mineralization after the AA dose was increased to 9 mg/kg/day.91 The poor outcome in this case may not be wholly attributed to treatment failure, but possibly due to an underlying genetic factor associated with failure of postnatal alveolar development.
Adverse effects from prolonged interrupted therapy have not been widely reported to date. Interruption of AA therapy for up to 1 year due to non-adherence by a teenager with severe childhood HPP resulted in the reappearance of hypomineralization of metaphyses, similar to the pre-treatment appearance of bone radiographs.94 This finding suggests that long-term uninterrupted therapy with AA is critical to maintain optimal TNSALP levels and mineralization activity, perhaps especially during rapid skeletal growth in adolescents.
Future directions of therapy for HPP
The frequency of administration (three or six times weekly), injection site reactions in the majority of subjects, production of costly recombinant enzyme, potential for adverse effects such as exacerbation of pathological calcification (eg, vascular calcification, kidney stones, eye calcifications; though these have not been documented to date), possibility of developing immunity against treatment, and potential lifetime requirement for AA all support investigation into alternative delivery systems and/or therapies. Preclinical animal studies to date have included gene therapies employing lentivirus (LV) and adeno-associated virus delivery of TNSALP-D10 that have achieved dramatic correction of HPP manifestations with a single injection.103–106 An alternative to the gene therapy approach is transduction of bone marrow cells ex vivo with LV-TNSALP-D10, and preclinical studies have indicated that engraftment of such cells could significantly improve (though not completely correct) HPP manifestations and lifespan in Alpl−/− mice.107 Another alternative approach to the concept of bone-targeted treatment is to achieve sustained high levels of soluble, circulating TNSALP. An engineered chimeric alkaline phosphatase (RecAP or ChimAP) engineered from human intestinal phosphatase and the placental alkaline phosphatase108 was able to improve growth and skeletal parameters, however, was unable to completely normalize trabecular bone, osteoid accumulation, or dental manifestations in HPP mice.109 As of this writing, RecAP is being explored as a treatment for acute kidney injury; however, HPP studies remain in the preclinical stage. A recent exploratory, uncontrolled proof-of-concept, Phase IIA escalating-dose trial of bone anabolic therapy with a monoclonal anti-sclerostin antibody (BPS804) demonstrated increased bone formation markers, as well as a transient decrease in the bone resorption markers in eight adults with HPP.110 ALP levels slightly increased during the treatment period, while lumbar spine BMD showed a mean increase of 3.9% by the end of the study.
Conclusion: place of AA in HPP therapy
AA is an approved treatment for pediatric-onset HPP. Treatment in clinical trials and case reports thus far has been targeted to individuals with high disease burden from perinatal, infantile, childhood, and adult HPP, specifically in those with significant skeletal manifestations of HPP. Improved skeletal mineralization in response to AA treatment leads to improvement in respiratory status and survival in infants, thus revolutionizing the outcome of a once fatal form of the disease. The improvement of skeletal manifestations after AA treatment also alleviates other complications related to bone abnormalities, such as improved physical function, mobility, and growth. Currently, no data have been reported on AA treatment of individuals with less symptomatic HPP (mild childhood HPP or odonto-HPP). Risk–benefit ratio, feasibility, and safety of treatment need to be considered in these patients, and long-term follow-up care is needed to monitor for late musculoskeletal complications of HPP.43 In those patients with symptomatic and disabling HPP, with or without fractures, for whom AA is considered, clinicians should establish treatment goals in order to monitor treatment response and determine what clinical outcomes are to be achieved.99 Given the wide clinical variability in disease manifestations of adult HPP, it has recently been suggested that AA treatment should be considered for adult HPP if one or more of the following is present (and determined to be attributable to HPP): musculoskeletal pain requiring prescription pain medications, disabling polyarthropathy, disabling functional impairment assessed by validated measures, low-trauma fracture, delayed or incomplete fracture healing, repeated orthopedic surgeries for HPP bone disease, low BMD T-score ≤−2.5 in postmenopausal women and men aged ≥50 years, or Z-score ≤−2.0 in younger adult female and male patients with fractures or nephrocalcinosis.111 Ultimately, how AA will be employed in the long-term management of severely and mildly affected HPP patients, whether any additional adverse effects or outcomes are discovered, and whether alternative approaches for delivery or achieving ERT are successful and transform HPP therapy yet again remain to be studied.
Acknowledgments
We thank the subjects and their families for their cooperation and assistance in this research. This research was supported by two seed grants from Soft Bones, Inc. to BLF and grant AR 066110 to BLF from the National Institute of Arthritis and Musculoskeletal and Skin Diseases. For more information on hypophosphatasia for affected individuals, researchers, or health care professionals, see the Soft Bones, Inc. website at https://www.softbones.org/.
Disclosure
BLF has served as a consultant and speaker for Alexion Pharmaceuticals, Inc., and received two research grants from Soft Bones, Inc., a nonprofit patient advocacy, support, and education group for families with hypophosphatsia. The authors report no other conflicts of interest in this work.
References
Robison R. The Possible Significance of Hexosephosphoric Esters in Ossification. Biochem J. 1923;17(2):286–293. | ||
Siller AF, Whyte MP. Alkaline Phosphatase: Discovery and Naming of Our Favorite Enzyme. J Bone Miner Res. 2018;33(2):362–364. | ||
Millán JL, Whyte MP. Alkaline Phosphatase and Hypophosphatasia. Calcif Tissue Int. 2016;98(4):398–416. | ||
Millán JL. Mammalian Alkaline Phosphatases: From Biology to Applications in Medicine and Biotechnology. Weinheim, Germany: Wiley-VCH; 2006. | ||
Meyer JL, Fleisch H. Calcification inhibitors in rat and human serum and plasma. Biochim Biophys Acta. 1984;799(2):115–121. | ||
Meyer JL. Can biological calcification occur in the presence of pyrophosphate? Arch Biochem Biophys. 1984;231(1):1–8. | ||
Bisaz S, Russell RG, Fleisch H. Isolation of inorganic pyrophosphate from bovine and human teeth. Arch Oral Biol. 1968;13(6):683–696. | ||
Fleisch H, Russell RG, Straumann F. Effect of pyrophosphate on hydroxyapatite and its implications in calcium homeostasis. Nature. 1966;212(5065):901–903. | ||
Fleisch H, Schibler D, Maerki J, Frossard I. Inhibition of aortic calcification by means of pyrophosphate and polyphosphates. Nature. 1965;207(5003):1300–1301. | ||
Fleisch H, Bisaz S. Isolation from urine of pyrophosphate, a calcification inhibitor. Am J Physiol. 1962;203(4):671–675. | ||
Fleisch H, Bisaz S. Mechanism of calcification: inhibitory role of pyrophosphate. Nature. 1962;195(4844):911. | ||
Gurley KA, Chen H, Guenther C, et al. Mineral formation in joints caused by complete or joint-specific loss of ANK function. J Bone Miner Res. 2006;21(8):1238–1247. | ||
Murshed M, Harmey D, Millan JL, Mckee MD, Karsenty G. Unique coexpression in osteoblasts of broadly expressed genes accounts for the spatial restriction of ECM mineralization to bone. Genes Dev. 2005;19(9):1093–1104. | ||
Harmey D, Hessle L, Narisawa S, Johnson KA, Terkeltaub R, Millan JL. Concerted regulation of inorganic pyrophosphate and osteopontin by akp2, enpp1, and ank: an integrated model of the pathogenesis of mineralization disorders. Am J Pathol. 2004;164(4):1199–1209. | ||
Rutsch F, Ruf N, Vaingankar S, et al. Mutations in ENPP1 are associated with ‘idiopathic’ infantile arterial calcification. Nat Genet. 2003;34(4):379–381. | ||
Nociti FH, Berry JE, Foster BL, et al. Cementum: a phosphate-sensitive tissue. J Dent Res. 2002;81(12):817–821. | ||
Terkeltaub RA. Inorganic pyrophosphate generation and disposition in pathophysiology. Am J Physiol Cell Physiol. 2001;281(1):C1–C11. | ||
Rutsch F, Vaingankar S, Johnson K, et al. PC-1 nucleoside triphosphate pyrophosphohydrolase deficiency in idiopathic infantile arterial calcification. Am J Pathol. 2001;158(2):543–554. | ||
Johnson KA, Hessle L, Vaingankar S, et al. Osteoblast tissue-nonspecific alkaline phosphatase antagonizes and regulates PC-1. Am J Physiol Regul Integr Comp Physiol. 2000;279(4):R1365–R1377. | ||
Ho AM, Johnson MD, Kingsley DM. Role of the mouse ank gene in control of tissue calcification and arthritis. Science. 2000;289(5477):265–270. | ||
Foster BL, Nagatomo KJ, Nociti FH, et al. Central role of pyrophosphate in acellular cementum formation. PLoS One. 2012;7(6):e38393. | ||
Millán JL. The role of phosphatases in the initiation of skeletal mineralization. Calcif Tissue Int. 2013;93(4):299–306. | ||
Zweifler LE, Patel MK, Nociti FH, et al. Counter-regulatory phosphatases TNAP and NPP1 temporally regulate tooth root cementogenesis. Int J Oral Sci. 2015;7(1):27–41. | ||
Mornet E. Hypophosphatasia. Metabolism. 2018;82:142–155. | ||
Whyte MP. Hypophosphatasia – aetiology, nosology, pathogenesis, diagnosis and treatment. Nat Rev Endocrinol. 2016;12(4):233–246. | ||
Thakker RV, Whyte MP, Eisman J, Igarashi T. Genetics of Bone Biology and Skeletal Disease. London: Academic Press; 2017. | ||
Mornet E. Genetics of hypophosphatasia. Archives de Pédiatrie. 2017;24(5):S551–S555. | ||
Whyte MP, Coburn SP, Ryan LM, Ericson KL, Zhang F. Hypophosphatasia: Biochemical hallmarks validate the expanded pediatric clinical nosology. Bone. 2018;110:96–106. | ||
Wenkert D, Mcalister WH, Coburn SP, et al. Hypophosphatasia: nonlethal disease despite skeletal presentation in utero (17 new cases and literature review). J Bone Miner Res. 2011;26(10):2389–2398. | ||
Seshia SS, Derbyshire G, Haworth JC, Hoogstraten J. Myopathy with hypophosphatasia. Arch Dis Child. 1990;65(1):130–131. | ||
Collmann H, Mornet E, Gattenlöhner S, Beck C, Girschick H. Neurosurgical aspects of childhood hypophosphatasia. Childs Nerv Syst. 2009;25(2):217–223. | ||
Whyte MP, Zhang F, Wenkert D, et al. Hypophosphatasia: validation and expansion of the clinical nosology for children from 25 years experience with 173 pediatric patients. Bone. 2015;75:229–239. | ||
Girschick HJ, Mornet E, Beer M, Warmuth-Metz M, Schneider P. Chronic multifocal non-bacterial osteomyelitis in hypophosphatasia mimicking malignancy. BMC Pediatr. 2007;7(1):3. | ||
Whyte MP, Wenkert D, Mcalister WH, et al. Chronic recurrent multifocal osteomyelitis mimicked in childhood hypophosphatasia. J Bone Miner Res. 2009;24(8):1493–1505. | ||
Kosnik-Infinger L, Gendron C, Gordon CB, Pan BS, van Aalst JA, Vogel TW. Enzyme replacement therapy for congenital hypophosphatasia allows for surgical treatment of related complex craniosynostosis: a case series. Neurosurg Focus. 2015;38(5):E10. | ||
Berkseth KE, Tebben PJ, Drake MT, Hefferan TE, Jewison DE, Wermers RA. Clinical spectrum of hypophosphatasia diagnosed in adults. Bone. 2013;54(1):21–27. | ||
Genest F, Seefried L. Subtrochanteric and diaphyseal femoral fractures in hypophosphatasia-not atypical at all. Osteoporos Int. 2018;29(8):1815–1825. | ||
Rodrigues TR, Georgetti AP, Martins L, Pereira Neto JS, Foster BL, Nociti Jr FH. Clinical correlate: case study of identical twins with cementum and periodontal defects resulting from odontohypophosphatasia. In: McCauley LK, Somerman MJ, eds. Mineralized Tissues in Oral and Craniofacial Science: Biological Principles and Clinical Correlates. 1st ed. Ames, IA: Wiley-Blackwell; 2012. | ||
Reibel A, Manière MC, Clauss F, et al. Orodental phenotype and genotype findings in all subtypes of hypophosphatasia. Orphanet J Rare Dis. 2009;4(1):6. | ||
Foster BL, Nociti FH, Somerman MJ. The rachitic tooth. Endocr Rev. 2014;35(1):1–34. | ||
Foster BL, Ramnitz MS, Gafni RI, et al. Rare bone diseases and their dental, oral, and craniofacial manifestations. J Dent Res. 2014;93(7 Suppl):7S–19S. | ||
Bloch-Zupan A. Hypophosphatasia: diagnosis and clinical signs – a dental surgeon perspective. Int J Paediatr Dent. 2016;26(6):426–438. | ||
Mori M, Dearmey SL, Weber TJ, Kishnani PS. Case series: Odontohypophosphatasia or missed diagnosis of childhood/adult-onset hypophosphatasia? – Call for a long-term follow-up of premature loss of primary teeth. Bone Rep. 2016;5:228–232. | ||
Vallet M, Ralston SH. Biology and Treatment of Paget’s Disease of Bone. J Cell Biochem. 2016;117(2):289–299. | ||
Whyte MP, Mcalister WH, Patton LS, et al. Enzyme replacement therapy for infantile hypophosphatasia attempted by intravenous infusions of alkaline phosphatase-rich Paget plasma: Results in three additional patients. J Pediatr. 1984;105(6):926–933. | ||
Whyte MP, Valdes R, Ryan LM, Mcalister WH. Infantile hypophosphatasia: Enzyme replacement therapy by intravenous infusion of alkaline phosphatase-rich plasma from patients with Paget bone disease. J Pediatr. 1982;101(3):379–386. | ||
Whyte MP, Magill HL, Fallon MD, Herrod HG. Infantile hypophosphatasia: normalization of circulating bone alkaline phosphatase activity followed by skeletal remineralization. Evidence for an intact structural gene for tissue nonspecific alkaline phosphatase. J Pediatr. 1986;108(1):82–88. | ||
Whyte MP, Kurtzberg J, Mcalister WH, et al. Marrow cell transplantation for infantile hypophosphatasia. J Bone Miner Res. 2003;18(4):624–636. | ||
Cahill RA, Wenkert D, Perlman SA, et al. Infantile hypophosphatasia: transplantation therapy trial using bone fragments and cultured osteoblasts. The Journal of Clinical Endocrinology & Metabolism. 2007;92(8):2923–2930. | ||
Tadokoro M, Kanai R, Taketani T, Uchio Y, Yamaguchi S, Ohgushi H. New bone formation by allogeneic mesenchymal stem cell transplantation in a patient with perinatal hypophosphatasia. J Pediatr. 2009;154(6):924–930. | ||
Taketani T, Oyama C, Mihara A, et al. Ex Vivo Expanded Allogeneic Mesenchymal Stem Cells with Bone Marrow Transplantation Improved Osteogenesis in Infants with Severe Hypophosphatasia. Cell Transplant. 2015;24(10):1931–1943. | ||
Deeb AA, Bruce SN, Morris AA, Cheetham TD. Infantile hypophosphatasia: disappointing results of treatment. Acta Paediatr. 2000;89(6):730–743. | ||
Cundy T, Michigami T, Tachikawa K, et al. Reversible Deterioration in Hypophosphatasia Caused by Renal Failure With Bisphosphonate Treatment. J Bone Miner Res. 2015;30(9):1726–1737. | ||
Righetti M, Wach J, Desmarchelier R, Coury F. Teriparatide treatment in an adult patient with hypophosphatasia exposed to bisphosphonate and revealed by bilateral atypical fractures. Joint Bone Spine. 2018;85(3):365–367. | ||
Doshi KB, Hamrahian AH, Licata AA. Teriparatide treatment in adult hypophosphatasia in a patient exposed to bisphosphonate: a case report. Clin Cases Miner Bone Metab. 2009;6(3):266–269. | ||
Sutton RA, Mumm S, Coburn SP, Ericson KL, Whyte MP. “Atypical femoral fractures” during bisphosphonate exposure in adult hypophosphatasia. J Bone Miner Res. 2012;27(5):987–994. | ||
Whyte MP, Mumm S, Deal C. Adult hypophosphatasia treated with teriparatide. J Clin Endocrinol Metab. 2007;92(4):1203–1208. | ||
Narisawa S, Fröhlander N, Millán JL. Inactivation of two mouse alkaline phosphatase genes and establishment of a model of infantile hypophosphatasia. Dev Dyn. 1997;208(3):432–446. | ||
Fedde KN, Blair L, Silverstein J, et al. Alkaline phosphatase knock-out mice recapitulate the metabolic and skeletal defects of infantile hypophosphatasia. J Bone Miner Res. 1999;14(12):2015–2026. | ||
Shao J-S, Engle M, Xie Q, et al. Effect of tissue non-specific alkaline phosphatase in maintenance of structure of murine colon and stomach. Microsc Res Tech. 2000;51(2):121–128. | ||
Wennberg C, Hessle L, Lundberg P, et al. Functional characterization of osteoblasts and osteoclasts from alkaline phosphatase knockout mice. J Bone Miner Res. 2000;15(10):1879–1888. | ||
Narisawa S, Wennberg C, Luis Millán J, Millan JL. Abnormal vitamin B6 metabolism in alkaline phosphatase knock-out mice causes multiple abnormalities, but not the impaired bone mineralization. J Pathol. 2001;193(1):125–133. | ||
Hessle L, Johnson KA, Anderson HC, et al. Tissue-nonspecific alkaline phosphatase and plasma cell membrane glycoprotein-1 are central antagonistic regulators of bone mineralization. Proc Natl Acad Sci U S A. 2002;99(14):9445–9449. | ||
Narisawa S, Huang L, Iwasaki A, Hasegawa H, Alpers DH, Millán JL. Accelerated fat absorption in intestinal alkaline phosphatase knockout mice. Mol Cell Biol. 2003;23(21):7525–7530. | ||
Anderson HC, Sipe JB, Hessle L, et al. Impaired calcification around matrix vesicles of growth plate and bone in alkaline phosphatase-deficient mice. Am J Pathol. 2004;164(3):841–847. | ||
Anderson HC, Harmey D, Camacho NP, et al. Sustained osteomalacia of long bones despite major improvement in other hypophosphatasia-related mineral deficits in tissue nonspecific alkaline phosphatase/nucleotide pyrophosphatase phosphodiesterase 1 double-deficient mice. Am J Pathol. 2005;166(6):1711–1720. | ||
Harmey D, Johnson KA, Zelken J, et al. Elevated skeletal osteopontin levels contribute to the hypophosphatasia phenotype in Akp2(−/−) mice. J Bone Miner Res. 2006;21(9):1377–1386. | ||
Beertsen W, Vandenbos T, Everts V. Root development in mice lacking functional tissue non-specific alkaline phosphatase gene: inhibition of acellular cementum formation. J Dent Res. 1999;78(6):1221–1229. | ||
Yadav MC, de Oliveira RC, Foster BL, et al. Enzyme replacement prevents enamel defects in hypophosphatasia mice. J Bone Miner Res. 2012;27(8):1722–1734. | ||
Foster BL, Nagatomo KJ, Tso HW, et al. Tooth root dentin mineralization defects in a mouse model of hypophosphatasia. J Bone Miner Res. 2013;28(2):271–282. | ||
Liu J, Nam HK, Campbell C, Gasque KC, Millán JL, Hatch NE. Tissue-nonspecific alkaline phosphatase deficiency causes abnormal craniofacial bone development in the Alpl(−/−) mouse model of infantile hypophosphatasia. Bone. 2014;67:81–94. | ||
Nam HK, Sharma M, Liu J, Hatch NE. Tissue Nonspecific Alkaline Phosphatase (TNAP) Regulates Cranial Base Growth and Synchondrosis Maturation. Front Physiol. 2017;8:161. | ||
Durussel J, Liu J, Campbell C, Nam HK, Hatch NE. Bone mineralization-dependent craniosynostosis and craniofacial shape abnormalities in the mouse model of infantile hypophosphatasia. Dev Dyn. 2016;245(2):175–182. | ||
Campos M, Couture C, Hirata IY, et al. Human recombinant endopeptidase PHEX has a strict S1’ specificity for acidic residues and cleaves peptides derived from fibroblast growth factor-23 and matrix extracellular phosphoglycoprotein. Biochem J. 2003;373(1):271–279. | ||
Boileau G, Tenenhouse HS, Desgroseillers L, Crine P. Characterization of PHEX endopeptidase catalytic activity: identification of parathyroid-hormone-related peptide 107–139 as a substrate and osteocalcin, PPi and phosphate as inhibitors. Biochem J. 2001;355(3):707–713. | ||
Millán JL, Narisawa S, Lemire I, et al. Enzyme replacement therapy for murine hypophosphatasia. J Bone Miner Res. 2008;23(6):777–787. | ||
Nishioka T, Tomatsu S, Gutierrez MA, et al. Enhancement of drug delivery to bone: characterization of human tissue-nonspecific alkaline phosphatase tagged with an acidic oligopeptide. Mol Genet Metab. 2006;88(3):244–255. | ||
Fisher LW, Fedarko NS. Six genes expressed in bones and teeth encode the current members of the SIBLING family of proteins. Connect Tissue Res. 2003;44(1):33–40. | ||
Yadav MC, Lemire I, Leonard P, et al. Dose response of bone-targeted enzyme replacement for murine hypophosphatasia. Bone. 2011;49(2):250–256. | ||
Liu J, Campbell C, Nam HK, et al. Enzyme replacement for craniofacial skeletal defects and craniosynostosis in murine hypophosphatasia. Bone. 2015;78:203–211. | ||
Mckee MD, Nakano Y, Masica DL, et al. Enzyme replacement therapy prevents dental defects in a model of hypophosphatasia. J Dent Res. 2011;90(4):470–476. | ||
Davit-Béal T, Gabay J, Antoniolli P, Masle-Farquhar J, Wolikow M. Dental complications of rickets in early childhood: case report on 2 young girls. Pediatrics. 2014;133(4):e1077–e1081. | ||
Biosse Duplan M, Coyac BR, Bardet C, et al. Phosphate and Vitamin D Prevent Periodontitis in X-Linked Hypophosphatemia. J Dent Res. 2017;96(4):388–395. | ||
Foster BL, Hujoel PP, Vitamin D in dentoalveolar and oral health. In: Feldman D, Pike JW, Bouillon R, eds. Vitamin D. Vol 1. Fourth edition. London, UK: Academic Press; 2018:497–520. | ||
Whyte MP, Greenberg CR, Salman NJ, et al. Enzyme-replacement therapy in life-threatening hypophosphatasia. N Engl J Med. 2012;366(10):904–913. | ||
Whyte MP, Rockman-Greenberg C, Ozono K, et al. Asfotase alfa treatment improves survival for perinatal and infantile hypophosphatasia. The Journal of Clinical Endocrinology & Metabolism. 2016;101(1):334–342. | ||
Kitaoka T, Tajima T, Nagasaki K, et al. Safety and efficacy of treatment with asfotase alfa in patients with hypophosphatasia: Results from a Japanese clinical trial. Clin Endocrinol. 2017;87(1):10–19. | ||
Kishnani PS, Rockman-Greenberg C, Denker AE, Moseley S, Whyte MP. Biochemical and physical function outcomes after 5 years of treatment with asfotase Alfa in adolescents and adults with hypophosphatasia: Phase 2 study results. International Conference on Children’s Bone Health; 2017. Available from: http://www.bone-abstracts.org/ba/0006/ba0006OC25.htm. Last accessed September 6, 2018. | ||
Okazaki Y, Kitajima H, Mochizuki N, Kitaoka T, Michigami T, Ozono K. Lethal hypophosphatasia successfully treated with enzyme replacement from day 1 after birth. Eur J Pediatr. 2016;175(3):433–437. | ||
Remde H, Cooper MS, Quinkler M. Successful Asfotase Alfa Treatment in an Adult Dialysis Patient with Childhood-Onset Hypophosphatasia. J Endocr Soc. 2017;1(9):1188–1193. | ||
Costain G, Moore AM, Munroe L, et al. Enzyme replacement therapy in perinatal hypophosphatasia: Case report of a negative outcome and lessons for clinical practice. Mol Genet Metab Rep. 2018;14:22–26. | ||
Vidmar AP, Ng C, Ganster A, Pitukcheewanont P. Asfotase alfa treatment of an African-American infant with perinatal hypophosphatasia and homozygous hemoglobin SC disease. IBMS BoneKEy. 2017;14:849. | ||
Bowden SA, Adler BH. Asfotase alfa treatment for 1year in a 16year-old male with severe childhood hypophosphatasia. Osteoporos Int. 2018;29(2):511–515. | ||
Bowden SA, Adler BH. Reappearance of hypomineralized bone after discontinuation of asfotase alfa treatment for severe childhood hypophosphatasia. Osteporos Int. 2018;29(9):2155–2156. | ||
Klidaras P, Severt J, Aggers D, Payne J, Miller PD, Ing SW. Fracture healing in two adult patients with hypophosphatasia after asfotase alfa therapy. JBMR Plus. 2018;98(4). | ||
Whyte MP, Madson KL, Phillips D, et al. Asfotase alfa therapy for children with hypophosphatasia. JCI Insight. 2016;1(9): e85971. | ||
Kishnani PS, Madson KL, Whyte MP, Gayron M, Fujita K, Rockman-Greenberg C. Biochemical and physical function outcomes in adolescents and adults with hypophosphatasia treated with asfotase alfa for up to 4 years: interim results from a phase II study. In: Metabolic and Genetic Bone Disorders. Endocrine Society. 2016:OR26-23. | ||
Tomazos IC, Moseley S, L’Italien G, da Silva HG, Phillips D. Improvements in the 6-minute walk test and correlation with quality-of-life measures in children and adults with hypophosphatasia treated with asfotase alfa. ENDO 2017. 2017, Orlando, FL, USA. Available from: https://www.endocrine.org/meetings/endo-annual-meetings/abstract-details?ID=29927. Last accessed September 6, 2018. | ||
Kishnani PS, Rush ET, Arundel P, et al. Monitoring guidance for patients with hypophosphatasia treated with asfotase alfa. Mol Genet Metab. 2017;122(1–2):4–17. | ||
Girschick HJ, Schneider P, Haubitz I, et al. Effective NSAID treatment indicates that hyperprostaglandinism is affecting the clinical severity of childhood hypophosphatasia. Orphanet J Rare Dis. 2006;1(1):24. | ||
Beck C, Morbach H, Richl P, Stenzel M, Girschick HJ. How can calcium pyrophosphate crystals induce inflammation in hypophosphatasia or chronic inflammatory joint diseases? Rheumatol Int. 2009;29(3):229–238. | ||
Poryo M, Meyer S, Eymann R, Yilmaz U, Nemat S, Rohrer T. Clinical Images: A Cloudy Skull-Hypophosphatasia as Reason for Copper-Beaten Skull. Neuropediatrics. 2016;47(6):410. | ||
Yamamoto S, Orimo H, Matsumoto T, et al. Prolonged survival and phenotypic correction of Akp2(−/−) hypophosphatasia mice by lentiviral gene therapy. J Bone Miner Res. 2011;26(1):135–142. | ||
Matsumoto T, Miyake K, Yamamoto S, et al. Rescue of severe infantile hypophosphatasia mice by AAV-mediated sustained expression of soluble alkaline phosphatase. Hum Gene Ther. 2011;22(11):1355–1364. | ||
Sugano H, Matsumoto T, Miyake K, et al. Successful gene therapy in utero for lethal murine hypophosphatasia. Hum Gene Ther. 2012;23(4):399–406. | ||
Nakamura-Takahashi A, Miyake K, Watanabe A, et al. Treatment of hypophosphatasia by muscle-directed expression of bone-targeted alkaline phosphatase via self-complementary AAV8 vector. Mol Ther Methods Clin Dev. 2016;3:15059. | ||
Iijima O, Miyake K, Watanabe A, et al. Prevention of Lethal Murine Hypophosphatasia by Neonatal Ex Vivo Gene Therapy Using Lentivirally Transduced Bone Marrow Cells. Hum Gene Ther. 2015;26(12):801–812. | ||
Kiffer-Moreira T, Sheen CR, Gasque KC, et al. Catalytic signature of a heat-stable, chimeric human alkaline phosphatase with therapeutic potential. PLoS One. 2014;9(2):e89374. | ||
Gasque KC, Foster BL, Kuss P, et al. Improvement of the skeletal and dental hypophosphatasia phenotype in Alpl−/− mice by administration of soluble (non-targeted) chimeric alkaline phosphatase. Bone. 2015;72:137–147. | ||
Seefried L, Baumann J, Hemsley S, et al. Efficacy of anti-sclerostin monoclonal antibody BPS804 in adult patients with hypophosphatasia. J Clin Invest. 2017;127(6):2148–2158. | ||
Shapiro JR, Lewiecki EM. Hypophosphatasia in adults: clinical assessment and treatment considerations. J Bone Miner Res. 2017;32(10):1977–1980. | ||
Whyte MP. Hypophosphatasia: Enzyme Replacement Therapy Brings New Opportunities and New Challenges. J Bone Miner Res. 2017;32(4):667-675. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.