")
Back to Journals » Drug Design, Development and Therapy » Volume 13
Preclinical pharmacodynamic and pharmacokinetic characterization of the major metabolites of cariprazine
Authors Kiss B, Némethy Z, Fazekas K, Kurkó D, Gyertyán I , Sághy K, Laszlovszky I, Farkas B, Kirschner N, Bolf-Terjéki E, Balázs O, Lendvai B
Received 26 September 2018
Accepted for publication 1 May 2019
Published 16 September 2019 Volume 2019:13 Pages 3229—3248
DOI https://doi.org/10.2147/DDDT.S188760
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Sukesh Voruganti
Video abstract of "Clinical efficacy of cariprazine" [ID 188760]
Views: 2943
Béla Kiss, Zsolt Némethy, Károly Fazekas, Dalma Kurkó, István Gyertyán, Katalin Sághy, István Laszlovszky, Bence Farkas, Norbert Kirschner, Etelka Bolf-Terjéki, Ottilia Balázs, Balázs Lendvai
Pharmacological and Drug Safety Research, Gedeon Richter Plc, Budapest, Hungary
Correspondence: Béla Kiss
Pharmacological and Drug Safety Research, Gedeon Richter Plc, P.O. Box 27, Budapest H-1475, Hungary
Tel +361 431 4226
Fax +361 889 8400
Email [email protected]
Introduction: Cariprazine, a dopamine D3-preferring D3/D2 receptor partial agonist and serotonin 5-HT1A receptor partial agonist, has two major human metabolites, desmethyl-cariprazine (DCAR) and didesmethyl-cariprazine (DDCAR). The metabolite pharmacology was profiled to understand the contribution to cariprazine efficacy.
Methods: In vitro receptor binding and functional assays, electrophysiology, animal models, microdialysis, and kinetic-metabolism approaches were used to characterize the pharmacology of DCAR and DDCAR.
Results: Similar to cariprazine, both metabolites showed high affinity for human D3, D2L, 5-HT1A, 5-HT2A, and 5-HT2B receptors, albeit with higher selectivity than cariprazine for D3 versus D2 receptors. In [35S]GTPγS binding assays, cariprazine and DDCAR were antagonists in membranes from rat striatum and from cells expressing human D2 and D3 receptors, and were partial agonists in membranes from rat hippocampus. In cAMP signaling assays, cariprazine, DCAR, and DDCAR acted as partial agonists at D2 and D3 receptors; cariprazine and DDCAR were full agonists, whereas DCAR was a partial agonist at 5-HT1A receptors. Cariprazine, DCAR, and DDCAR were pure antagonists at human 5-HT2B receptors. Cariprazine and DDCAR increased rat striatal dopamine and reduced cortical serotonin turnover. Cariprazine and DDCAR showed similar in vivo D3 receptor occupancy in rat brain; however, cariprazine was more potent for D2 receptor occupancy. Both cariprazine and DDCAR dose-dependently but partially suppressed the spontaneous activity of midbrain dopaminergic neurons in rats, with the parent compound being more potent but shorter acting than its metabolite. Consistent with the D2 receptor occupancy profile, DDCAR was 3- to 10-fold less potent than cariprazine in rodent models of antipsychotic-like activity. Following acute cariprazine administration, DDCAR was detected in the rodent brain but at much lower levels than cariprazine.
Conclusion: Overall, in vitro and in vivo pharmacological profiles of DCAR and DDCAR demonstrated high similarity with cariprazine, suggesting that the major metabolites of cariprazine contribute significantly to its clinical efficacy.
Keywords: dopamine, serotonin, schizophrenia, bipolar disorder, desmethyl-cariprazine, didesmethyl-cariprazine
Plain language summary
Cariprazine, an orally active and potent atypical antipsychotic, is a dopamine D3-preferring D3/D2 receptor partial agonist and serotonin 5-HT1A receptor partial agonist with two major active metabolites, desmethyl-cariprazine (DCAR) and didesmethyl-cariprazine (DDCAR). Due to their structural similarity to the parent compound, DCAR and DDCAR may play significant roles in the therapeutic profile of cariprazine. It was also previously determined in a pharmacokinetic clinical study that DCAR and DDCAR are substantially present in plasma under standard dosing conditions in humans. Using in vitro receptor binding and functional assays, electrophysiology, animal models, microdialysis, and kinetic-metabolism approaches, the in vitro and in vivo pharmacology of DCAR and DDCAR were characterized to gain a deeper understanding of the contribution of these metabolites to the effects of cariprazine. Based on their in vitro and in vivo pharmacological profiles, both DCAR and DDCAR demonstrated high degrees of similarity compared with cariprazine. Therefore, cariprazine’s clinical efficacy may include contributions from its two active metabolites.
Introduction
Cariprazine is an orally active, dopamine D3-preferring D3/D2 receptor partial agonist atypical antipsychotic. After demonstrating efficacy and safety in positive, randomized, placebo-controlled trials,1–6 cariprazine (USA: Vraylar®) was approved by the US Food and Drug Administration for the treatment of acute exacerbation of schizophrenia and manic or mixed episodes associated with bipolar I disorder in adults. Compared to placebo, cariprazine also significantly prolonged the time to relapse in patients with schizophrenia.7 In addition, it has demonstrated efficacy as monotherapy for predominant negative symptoms of schizophrenia in stable patients.8 Based on these data, cariprazine (Europe: Reagila®) was approved by the European Medicinal Agency for the treatment of schizophrenia in adults. Furthermore, cariprazine has demonstrated efficacy as monotherapy in bipolar depression9,10 and as an adjunctive therapy in major depressive disorder.11
Cariprazine exhibits a 6- to 8-fold greater in vitro affinity for D3 versus D2 receptors12 and has also demonstrated similar in vivo occupancy of both D3 and D2 receptors in rats, nonhuman primates, and the human brain13–17 at antipsychotic-effective doses. This is in contrast with some atypical antipsychotics (eg, risperidone, olanzapine, clozapine) which show significant in vitro affinity for D3 receptors but no (or minimal) in vivo occupancy for these receptors.18 In addition, cariprazine acts as a partial agonist at serotonin 5-HT1A receptors and as an antagonist at 5-HT2B receptors.12 Cariprazine shows moderate to low binding affinity for 5-HT2A, 5-HT2C, 5-HT7, and histamine H1 receptors, and low to negligible affinity for adrenergic and muscarinic receptors.12,19
Cariprazine undergoes demethylation to produce two major active metabolites, desmethyl-cariprazine (DCAR) and didesmethyl-cariprazine (DDCAR), in both humans and rodents.20,21 Cariprazine is metabolized by hydroxylation and dealkylation mainly via the enzyme CYP3A4 and, to a lesser extent, via CYP2D6. The metabolite DCAR is a primary metabolite formed by demethylation of cariprazine, and the metabolite DDCAR is formed from DCAR by a further demethylation reaction (Figure 1). Both metabolites maintain high structural similarity to the parent compound,22 and cariprazine and DDCAR display subnanomolar in vitro binding affinity for human D2 and D3 receptors with approximately 3- and 4-fold higher affinity for D3 receptors than for D2, with varying intrinsic activity depending on the level of receptor density, respectively.23 Substantial levels of the two metabolites were detected in the plasma samples of healthy volunteers and patients with schizophrenia after single or multiple dose(s) of cariprazine.21,24,25 DCAR and DDCAR are the major circulating metabolites in human plasma, representing approximately 30–40% of and 2- to 3-fold the parent drug exposure, respectively. Both metabolites are present in the human plasma at times comparable to (DCAR) or significantly longer than (DDCAR) cariprazine; the half-life of cariprazine, DCAR, and DDCAR in humans is 1–3 days, 1–2 days, and 2–3 weeks, respectively.24 Due to the high structural similarity, comparable plasma levels of cariprazine and its metabolites, and the relatively long half-life of DDCAR, pharmacological characterization of DCAR and DDCAR is essential to enhance the understanding of their roles in the efficacy of cariprazine for the treatment of schizophrenia and bipolar disorder. The purpose of this study was to characterize the in vitro and in vivo pharmacological profile of DCAR and DDCAR, the two major metabolites of cariprazine, in comparison with their parent compound, cariprazine.
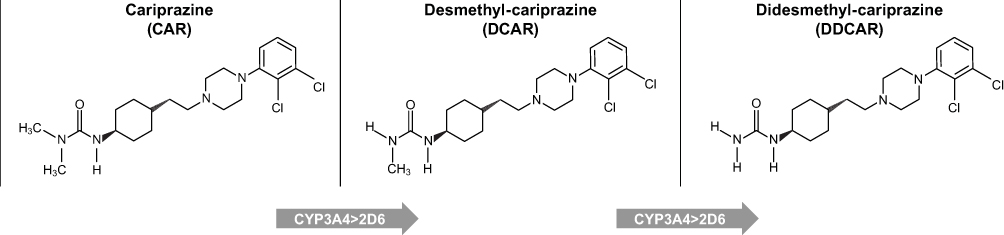 |
Figure 1 Formation of the main human metabolites of cariprazine.24 Abbreviations: CAR, cariprazine; DCAR, desmethyl-cariprazine; DDCAR, didesmethyl-cariprazine. |
Materials and methods
Brief descriptions of the materials and methods are presented below; additional details are provided in Supplementary materials.
Animals
Male Hannover Wistar rats weighing 180–220 g (for binding, occupancy, turnover, and 3,4-dihydroxyphenylalanine (DOPA) accumulation, rat striatal and hippocampal [35S]GTPγS binding), 160–220 g (for locomotor activity), 175–240 g (for scopolamine), 160–200 g (for catalepsy), 180–200 g (for conditioned avoidance response), 170–190 g (for bioavailability), 210–250 g (for cariprazine and DDCAR brain penetrability studies, DDCAR intravenous (i.v.) administration), 280–300 g (for microdialysis (MD)), and 280–350 g (for electrophysiology) were obtained from Toxicoop Hungary Ltd.
Male CD-1 mice weighing 20–35 g (for apomorphine climbing) were obtained from Wobe. Male NMRI mice weighing 24–30 g (for phencyclidine and MK-801 hypermotility) and male NMRI mice weighing 25–30 g (for pharmacokinetics) were obtained from Toxicoop Hungary Ltd.
Animals arrived at the laboratory 2–3 days prior to initiating experiments. They were kept under standard conditions (22±2 °C; 55–65% relative humidity; 12 h light–dark cycle) on commercial laboratory chow and tap water ad libitum. Animal maintenance and experiments were conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. All procedures using animals were approved by the local ethics committee (Institutional Animal Welfare Committee of Gedeon Richter Plc) and conformed to the rules and principles of Directive 86/609/EEC (the regulation in force at the time of the experiments).
Cell lines
CHO cells expressing recombinant human serotonin 5-HT1A receptors (cAMP Hunter™ CHO-K1 HTR1A Gi cell line) were purchased from DiscoverX (Fremont, CA, USA; Product Code: 95-0132C2). HEK293 cells expressing recombinant human D2 receptors (HEK/Gα15/D2) were purchased from HD Biosciences Corporation (Shanghai, China). The cloned gene for the full-length human dopamine D2 receptor (Genbank: NM_000795) was inserted into expression vectors (plasmid: pMT8-D2) as cDNA. CHO-K1 cells transfected with a gene for the human D3 receptor were from Euroscreen (Brussels, Belgium). The cloned genes for the human dopamine D3 receptor (Genbank: U32499) as cDNA inserted into eukaryotic expression vectors were also provided by Euroscreen. The ValiScreen™ human 5-HT2B cell line was obtained from PerkinElmer Inc. (Waltham, MA, USA; ES-314-C, lot 450-442-A). All cells expressing recombinant human receptors employed in this study were grown under sterile conditions.
Drugs
The following radioligands were purchased from Amersham Bioscience: [3H]prazosin [furanyl-5-3H] (TRK-843, specific activity 88 Ci/mmol), [3H]mesulergine [N6-methyl-3H] (TRK-845, specific activity 84 Ci/mmol), and [35S]GTPγS (SJ-1308, specific activity 1000–1200 Ci/mmol).
The following ligands were purchased from PerkinElmer Inc.: [3H]spiperone (NET-565, specific activity 15.7 Ci/mmol), [3H]8-OH-DPAT (NET-929, specific activity 129 Ci/mmol), and [3H]ketanserin (NET-791, specific activity 88.0 Ci/mmol).
[3H](+)PHNO (specific activity 63–64 Ci/mmol) was purchased from Moravek Biochemical Inc. (Brea, CA, USA).
Apomorphine HCl, butaclamol HCl, D-amphetamine sulphate, forskolin (FSK), phencyclidine, phentolamine, scopolamine HBr, (±)-2,5-dimethoxy-4-iodoamphetamine HCI, serotonin-creatinin sulphate, and isobutyl-methyl xanthine were purchased from Sigma-Aldrich Co. (St Louis, MO, USA).
Mianserin HCl was obtained from Organon, methysergide maleate from Sandoz, and MK-801 from RBI. Haloperidol, cariprazine, DDCAR (as base or HCl salt), and DCAR (as base or HCl salt) were synthesized at Gedeon Richter Plc (Budapest, Hungary).
Other chemicals used in the experiments were of analytical grade and obtained from commercial sources.
Treatments
For the in vivo experiments, cariprazine and DDCAR were administered either orally (per os (p.o.)) in a volume of 5 mL/kg for rats and 10 mL/kg for mice or by i.v. route in a maximal volume of 2 mL/kg. In the case of p.o. dosing, the test compounds were suspended in 2–6% Tween 80, depending on the dose and requirement for sufficient suspension homogeneity. Solutions required for doses in the low milligram per kilogram range were usually diluted from a stock solution or suspension. For the neurochemistry and occupancy experiments, stock solutions of cariprazine and DDCAR were made by dissolving their hydrochloride salt in a minimal amount (10–20 μL) of acetic acid and diluted with saline. Intravenous cariprazine and DDCAR doses, formulated in physiological saline, were injected into the femoral vein using a catheter fitted with a syringe. For further details, see Supplementary materials.
Assays
In vitro receptor binding assays
Radioligand binding assays for recombinant human and rat, and native rat receptors were performed by MDS Pharma Services and Gedeon Richter Plc. Binding affinities of cariprazine, DCAR, and DDCAR were determined for 76 targets representing receptors, channels, transporters, and enzymes (see Table 1).
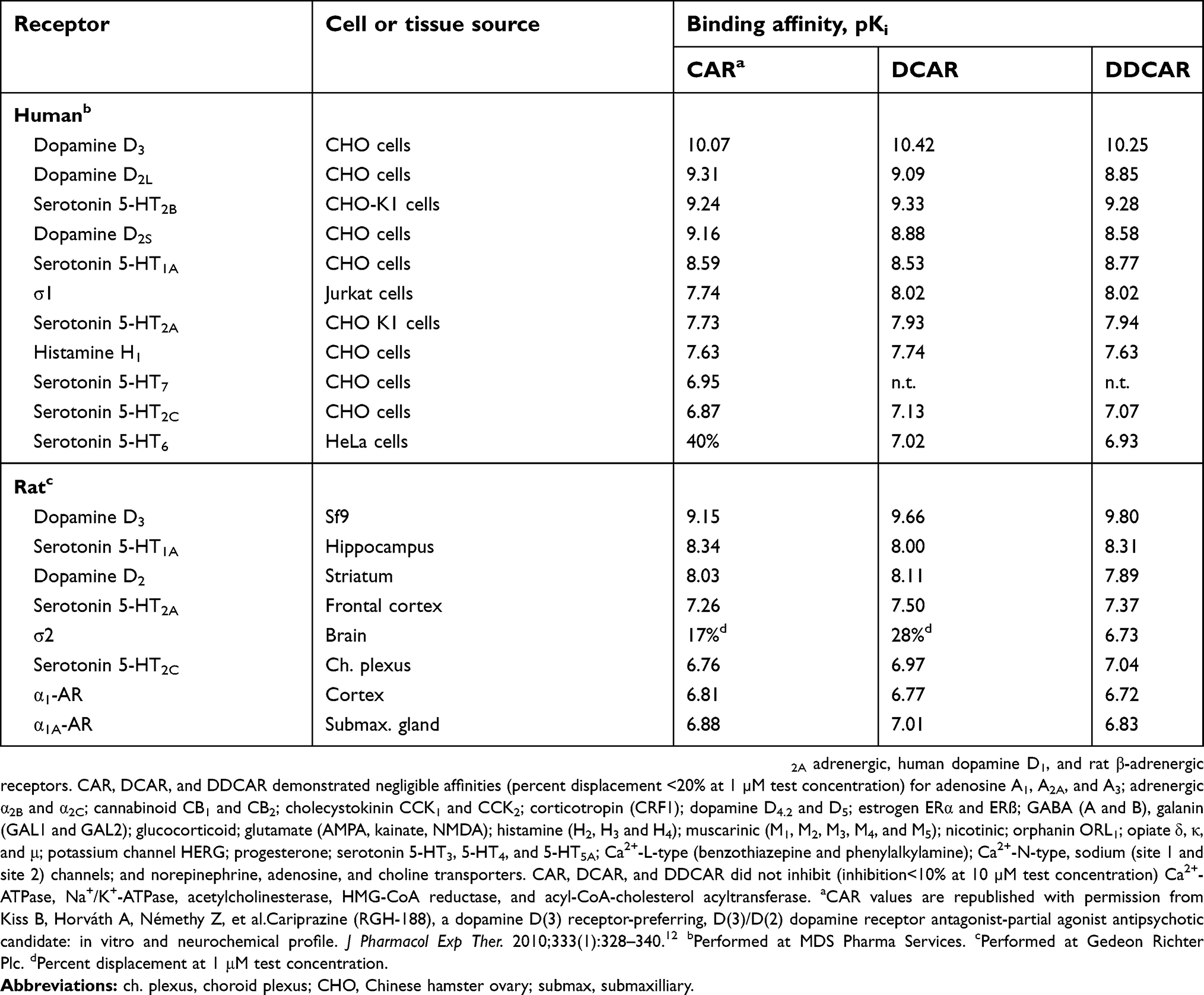 |
Table 1 In vitro receptor binding affinities of cariprazine (CAR), desmethyl-cariprazine (DCAR), and didesmethyl-cariprazine (DDCAR) |
Membrane preparation from cells expressing recombinant receptors and assays was performed at MDS Pharma Services. The experimental conditions of the assays reported in Table 1 are summarized in Table S1.
Rats were decapitated, the brain was removed, and the structures were dissected on a cold surface. Membranes from dissected brain structures were prepared and binding assays were performed as described in Table S2.
Experiments were performed in duplicate and repeated two or three times.
[35S]GTPγS binding, cAMP signaling, and Ca2+ release
The agonist/antagonist activities of cariprazine, DCAR, and DDCAR were determined in vitro using [35S]GTPγS binding, cAMP accumulation, and Ca2+-release assays. Determination of in vitro [35S]GTPγS binding and cAMP accumulation assays was carried out in membranes from rat striatum and at HEK293-hD2, CHO-hD2, CHO-hD3, and CHO-h5-HT1A receptors, as reported in the figure legends. Details are given in Supplementary materials.
Ca2+ release was assayed in CHO cells expressing recombinant human 5-HT2B receptors; compounds were tested with or without serotonin for their agonist or antagonist activities by the method previously described,26 with slight modifications. For further details, see Supplementary materials.
Brain dopamine receptor occupancy
Brain occupancy of D2 and D3 receptors by cariprazine and DDCAR was assessed by measuring inhibition of in vivo [3H](+)-PHNO binding in rat striatum and cerebellum lobes 9 and 10 (L9,10), respectively, according to the method published.27 For further details, see Supplementary materials.
Dopamine and serotonin turnover and DOPA accumulation
Cariprazine and DDCAR-induced changes in levels of dopamine, serotonin, their respective metabolites 3,4-dihydroxyphenyl acetic acid (DOPAC) and homovanillic acid (HVA), and 5-hydroxy-indolyl acetic acid (5-HIAA) in rat brain regions were determined using HPLC with electrochemical detection. Similar detection of the aromatic amino-acid decarboxylase inhibitor NSD-1015-induced accumulation of DOPA was used to assess the effects of the compounds on dopamine biosynthesis in the striatum of reserpine-treated rats.12 For further details, see Supplementary materials.
In vivo electrophysiology
Extracellular recordings of spontaneously active individual dopaminergic neurons were performed in the substantia nigra pars compacta (SNc) region of the midbrain after i.v. administration of cariprazine or DDCAR in anesthetized rats. Drug effects on neuronal firing rates were determined as previously described.28 For further details, see Supplementary materials.
Animal behavioral assessment
The effects of cariprazine and DDCAR on apomorphine (1.5 mg/kg, subcutaneous (s.c.))-induced climbing, conditioned avoidance response, scopolamine (3 mg/kg, intraperitoneal (i.p.))-induced learning deficits in a water labyrinth, spontaneous locomotor activity, and hypermotility induced by amphetamine (0.5 mg/kg, s.c.), phencyclidine (2.5 mg/kg, s.c.), or MK-801 (0.2 mg/kg, i.p.) and their cataleptogenic effects were assessed as described previously.14 The salt or base forms of cariprazine and DDCAR were used, but the ED50 data refer to the base form. For doses and number of animals per dose group, see Supplementary materials.
Pharmacokinetics
The pharmacokinetics of cariprazine was studied after single p.o. and i.v. administration of 1 mg/kg doses to male Wistar rats (n=3–5/group). The formulations were 0.1% acetic acid in deionized water (d.w.) for the bioavailability study (p.o.), 5% Tween 80/d.w. for the brain penetrability study (p.o.), and 0.1% acetic acid/5.5% glucose in d.w. (i.v.). The pharmacokinetics of cariprazine was also studied in male NMRI mice using a p.o. dose of 1 mg/kg in d.w. (n=5/group).
The pharmacokinetic characterization of DDCAR was performed after single p.o. administration at 0.9 mg/kg (equimolar to cariprazine) and i.v. dose of 1 mg/kg in male Wistar rats (n=3–4/group). The formulations were 5% Tween 80/d.w. and 1% lactic acid/0.9% NaCl, respectively.
Samples obtained after cariprazine or DDCAR administration were analyzed for cariprazine and its metabolite DDCAR or for DDCAR alone, respectively, by liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS).
Pharmacokinetic parameters were calculated from the concentration data with Kinetica software using the model independent approach. The following parameters were derived: maximum concentration (Cmax), with corresponding sampling time (Tmax), area under the concentration versus time curve from 0 (administration of the compound) to time t (AUC0–t) or extrapolated to infinity (AUC0–inf), terminal half-life (T1/2), systemic clearance (CL), and volume of distribution (Vd).
A dual-probe MD study was conducted to estimate the free extracellular levels of cariprazine and DDCAR in awake, freely moving, male Wistar rats (n=5). Cariprazine was administered intragastrically at 1 mg/kg dose in d.w. via surgically implanted catheter. The MD experiments were performed with the Culex automated sampling system (Bioanalytical Systems Inc.). Probes were implanted in the prefrontal cortex and striatum and perfused with artificial cerebrospinal fluid at a flow rate of 1 µL/min. Dialysate samples were collected in 20-min intervals from 60 min pre-dose to ≥300 min post-dose. Dialysate concentrations of cariprazine and DDCAR were determined by LC-MS/MS.
Brain extracellular levels were calculated by correction with the in vitro recovery of probes. AUC0–5 h, Cmax, and Tmax were calculated for extracellular concentrations with Kinetica software (Thermo Fischer Scientific, Waltham, MA, USA) using the model independent approach. Details of the pharmacokinetic and MD methodology are given in Supplementary materials.
Data analysis
Nonlinear least-squares regression analysis was used to calculate IC50 values (MDS Pharma Services, Data Analysis Toolbox; MDL Information Systems). Receptor binding isotherms and concentration or dose–response curves were analyzed by nonlinear regression using Origin 6.0 (OriginLab Corp.) or Prism 4.0 software (GraphPad Software Inc., La Jolla, CA, USA) or SoftMax Pro 5.2 (Molecular Devices LLC, Sunnyvale, CA, USA) software. Pharmacokinetic data were analyzed by Kinetica software (Thermo Fischer Scientific). Inhibition constants (Ki) were calculated from the IC50 values using the Cheng and Prusoff equation;29 antagonist affinity (KB) values were calculated according to the formula described by Craig.30 The Tukey–Kramer multiple comparison test was used to compare control and treatment groups in the rat turnover, biosynthesis, and occupancy experiments. In electrophysiological experiments, dose–response curves were constructed by plotting firing rates normalized to the mean firing rate of the “vehicle” period versus the tested doses. Repeated-measures ANOVA in a general linear model followed by Dunnett’s test was used for statistical analysis. ED50 values were calculated via sigmoidal curve regression. Time courses were compared using one-way ANOVA with Helmert contrast. ED50 values of the behavioral assays were calculated by linear regression; effective doses were determined by repeated-measures ANOVA followed by post-hoc Duncan test.
Details of data analysis for the relevant assays are given in Supplementary materials.
Results
In vitro receptor binding profile of cariprazine, DCAR, and DDCAR
The in vitro binding affinities of cariprazine, DCAR, and DDCAR were tested at 76 targets including human and rat receptors, transporters, and enzymes (Table 1). Both DCAR and DDCAR demonstrated higher affinity than cariprazine for human and rat D3 receptors. Similar to cariprazine, DCAR and DDCAR displayed subnanomolar/low nanomolar affinity for human 5-HT2B receptors and D2L receptors, low nanomolar affinity for human D2S and 5-HT1A receptors, and moderate (7.5<pKi<8.0) affinity for human 5-HT2A, H1, and σ1 receptors. It is of note that both metabolites showed higher selectivity for the human D3 receptor compared with cariprazine. All three compounds showed an overall lower affinity for native rat receptors than for recombinant human receptors.
[35S]GTPγS binding
Cariprazine and DDCAR acted as pure antagonists at D2 and D3 receptors by completely inhibiting dopamine-induced [35S]GTPγS binding in HEK293 cells expressing human D2 receptors (Figure 2) or CHO cells expressing human D3 receptors (Figure 3), without stimulating [35S]GTPγS binding in the absence of dopamine. Similar results were obtained in rat striatal membranes: cariprazine and DDCAR did not show stimulatory activity, but both antagonized dopamine-stimulated [35S]GTPγS binding (Table 2). Cariprazine and DDCAR were partial agonists at human 5-HT1A receptors (Figure 4) in the [35S]GTPγS binding assay, with DDCAR displaying greater intrinsic efficacy and lower potency than cariprazine. In rat hippocampal membranes, both cariprazine and DDCAR stimulated [35S]GTPγS binding with comparable potency, but similar to human 5-HT1A membranes, DDCAR demonstrated higher efficacy (Table 2).
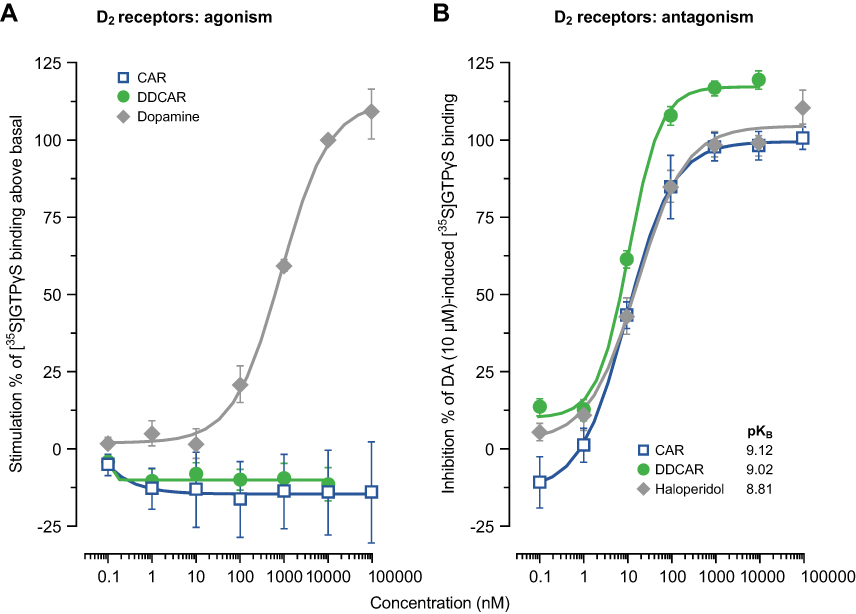 |
Figure 2 CAR and DDCAR do not stimulate [35S]GTPγS binding in membranes from HEK293 cells expressing human dopamine D2 receptors; however, both antagonize a dopamine (10 μM)-induced increase in [35S]GTPγS binding. (A) D2 receptor agonism; (B) D2 receptor antagonism. Abbreviations: CAR, cariprazine; DA, dopamine; DDCAR, didesmethyl-cariprazine; HEK, human embryonic kidney. |
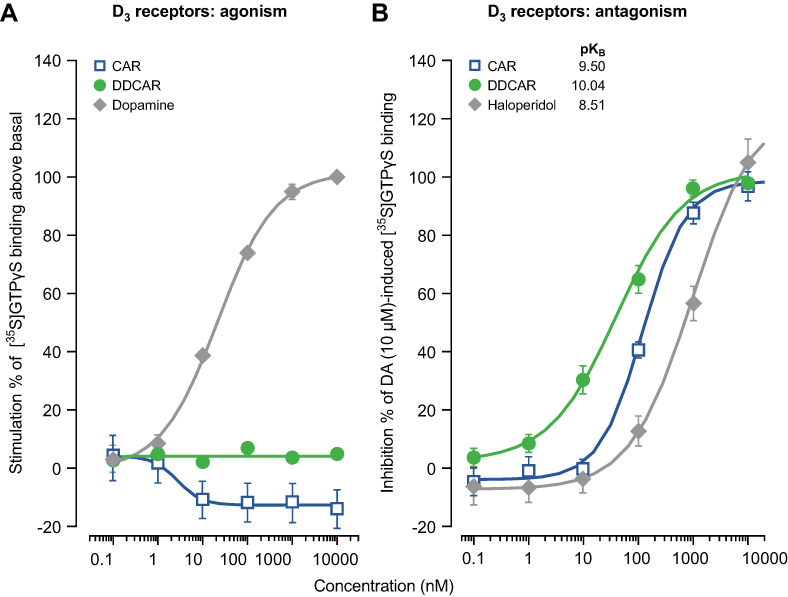 |
Figure 3 In the absence of dopamine, CAR and DDCAR do not stimulate [35S]GTPγS binding in membranes from CHO cells expressing human dopamine D3 receptors; however, both antagonize a dopamine (10 μM)-induced increase in [35S]GTPγS binding. (A) D3 receptor agonism; (B) D3 receptor antagonism. Abbreviations: CAR, cariprazine; CHO, Chinese hamster ovary; DA, dopamine; DDCAR, didesmethyl-cariprazine. |
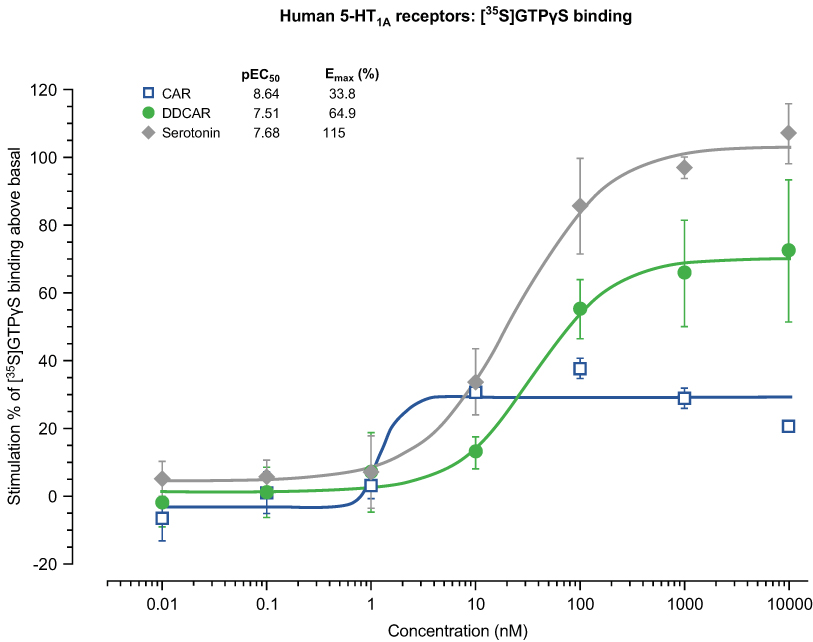 |
Figure 4 CAR and DDCAR partially stimulate [35S]GTPγS binding in membranes from CHO cells expressing human serotonin 5-HT1A receptors. Abbreviations: CAR, cariprazine; CHO, Chinese hamster ovary; DDCAR, didesmethyl-cariprazine; Emax (%), percent maximal stimulation; pEC50, negative logarithm of concentration of a drug that gives half-maximal response. |
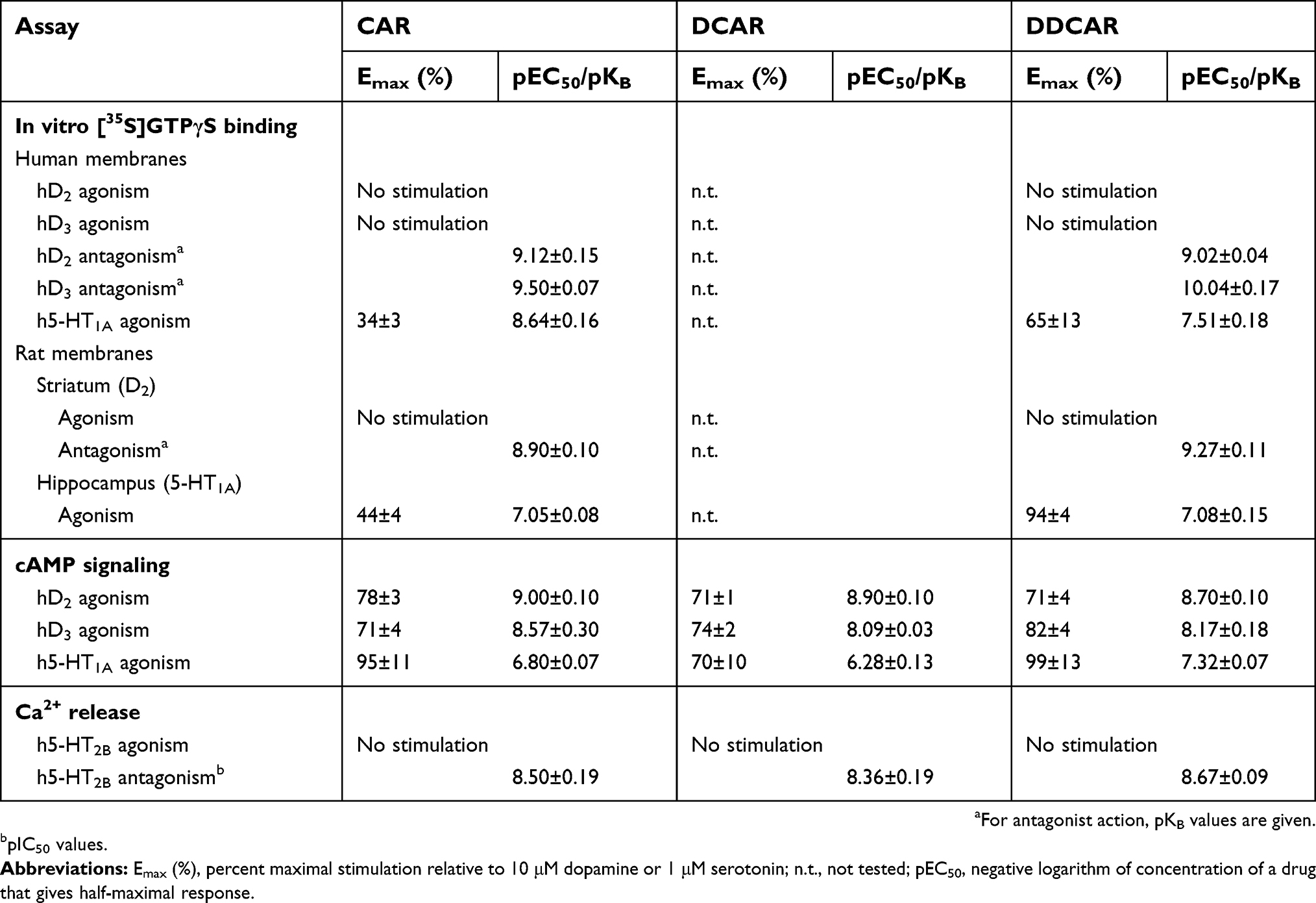 |
Table 2 Summary of functional activity data for cariprazine (CAR), desmethyl-cariprazine (DCAR), and didesmethyl-cariprazine (DDCAR) |
In vitro cAMP signaling at D2, D3, and 5-HT1A receptors
Cariprazine, DCAR, and DDCAR acted as partial agonists at the recombinant human D2 and D3 receptors by inhibiting FSK-induced cAMP production, with maximal levels that were 20–30% less than those seen with dopamine (Figure 5). Each showed comparable degrees of intrinsic efficacy at both D2 and D3 receptors. Cariprazine and its major metabolites showed comparable potencies at the D2 receptor (Figure 5A). At D3 receptors, cariprazine had the highest potency, followed by DDCAR and DCAR (Figure 5B). At the 5-HT1A receptor, cariprazine and DDCAR acted as full agonists, whereas DCAR displayed partial agonist activity in the cAMP assay at the human recombinant receptor expressed in CHO cells (Figure 6). DCAR displayed lower potency than cariprazine and DDCAR at the 5-HT1A receptor (Table 2).
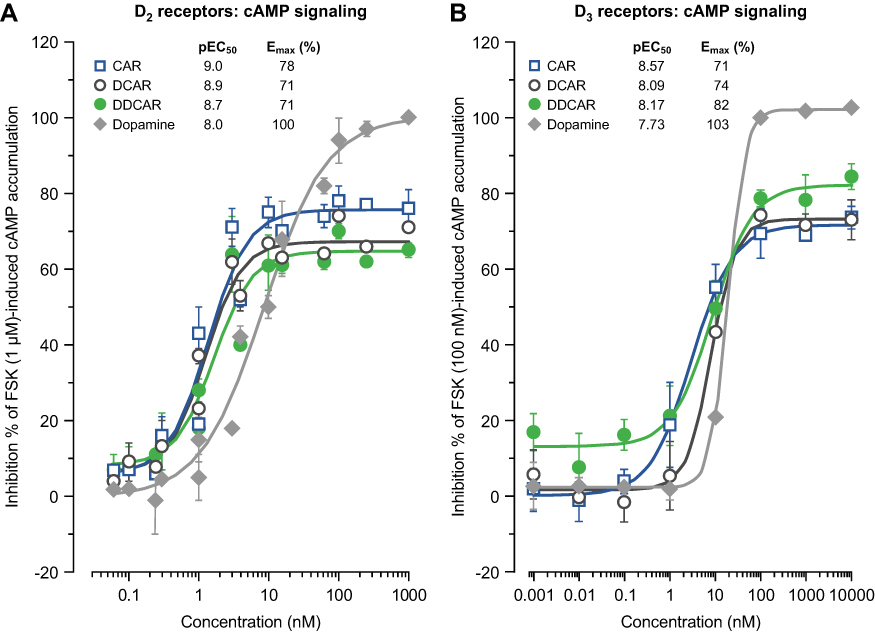 |
Figure 5 CAR, DCAR, and DDCAR are partial agonists in cAMP accumulation in CHO cells expressing human dopamine D2 or D3 receptors. cAMP signaling at (A) D2 receptors and (B) D3 receptors. Abbreviations: CAR, cariprazine; CHO, Chinese hamster ovary; DCAR, desmethyl-cariprazine; DDCAR, didesmethyl-cariprazine; Emax (%), percent maximal stimulation; FSK, forskolin; pEC50, negative logarithm of concentration of a drug that gives half-maximal response. |
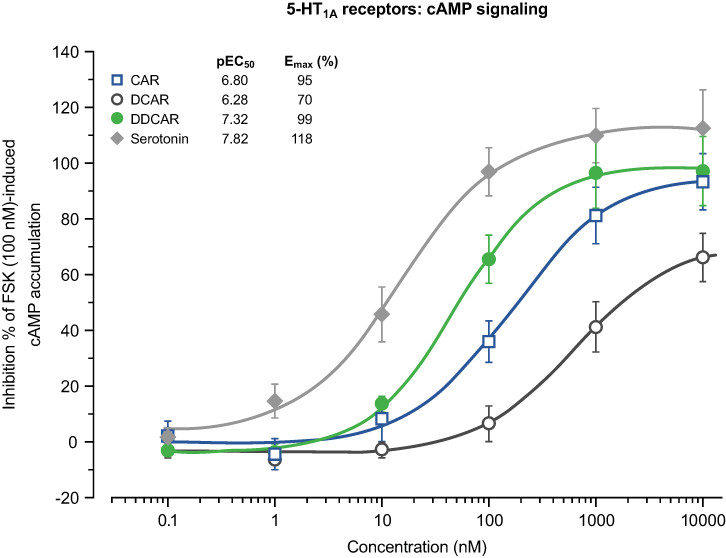 |
Figure 6 CAR and DDCAR are full agonists in cAMP accumulation assay in CHO cells expressing human serotonin 5-HT1A receptors, whereas DCAR demonstrates partial agonist activity. cAMP levels were determined by the HTRF method given by the vendor (Cisbio). Abbreviations: CAR, cariprazine; CHO, Chinese hamster ovary; DCAR, desmethyl-cariprazine; DDCAR, didesmethyl-cariprazine; Emax (%), percent maximal stimulation; FSK, forskolin; HTRF, homogeneous time-resolved fluorescence; pEC50, negative logarithm of concentration of a drug that gives half-maximal response. |
Ca2+ release in 5-HT2B receptor-expressing cells
Cariprazine, DCAR, and DDCAR did not stimulate serotonin-induced Ca2+ release in CHO-5-HT2B cells (data not shown) but inhibited serotonin-induced Ca2+release in these cells (Figure 7). The three compounds inhibited serotonin-induced Ca2+release with similarly high potency (Table 2).
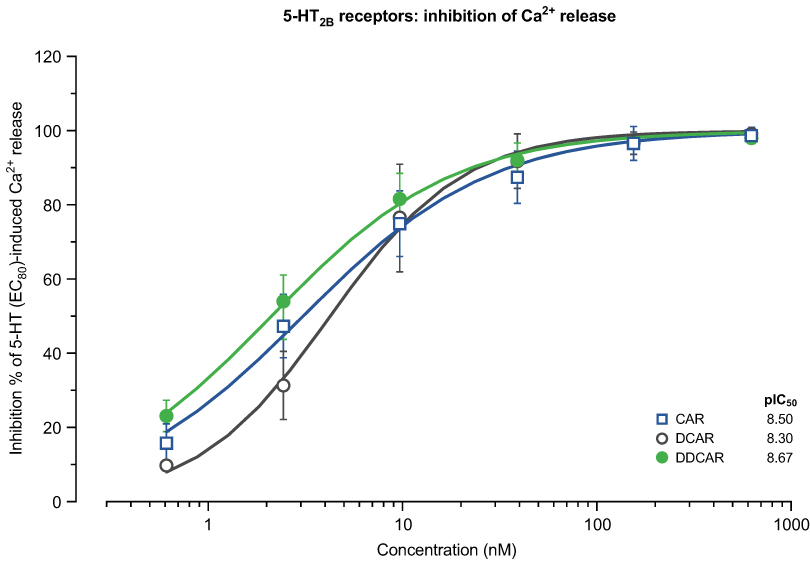 |
Figure 7 CAR, DCAR, and DDCAR are antagonists at human serotonin 5-HT2B receptors expressed in CHO cells in inhibiting 5-HT-evoked Ca2+ release. Abbreviations: CAR, cariprazine; CHO, Chinese hamster ovary; DCAR, desmethyl-cariprazine; DDCAR, didesmethyl-cariprazine; 5-HT, serotonin. |
Brain dopamine receptor occupancy
Cariprazine and DDCAR showed dose-dependent occupancy of D2 and D3 receptors in the rat brain, as determined by the displacement of [3H](+)-PHNO (Figure 8), a D3-preferring D2/D3 receptor agonist. Cariprazine showed 2.5-fold higher potency to occupy striatal D2 receptors than DDCAR (Figure 8A), whereas cariprazine and DDCAR showed similar potency for occupying D3 receptors in cerebellum L9,10, a D3 receptor-rich region (Figure 8B). In agreement with its higher D3 receptor selectivity in vitro, these in vivo brain occupancy results showed that DDCAR demonstrated higher D3 receptor occupancy compared to cariprazine (striatal D2 to cerebellar D3 ED50 ratio: cariprazine, 0.53; DDCAR, 1.41).
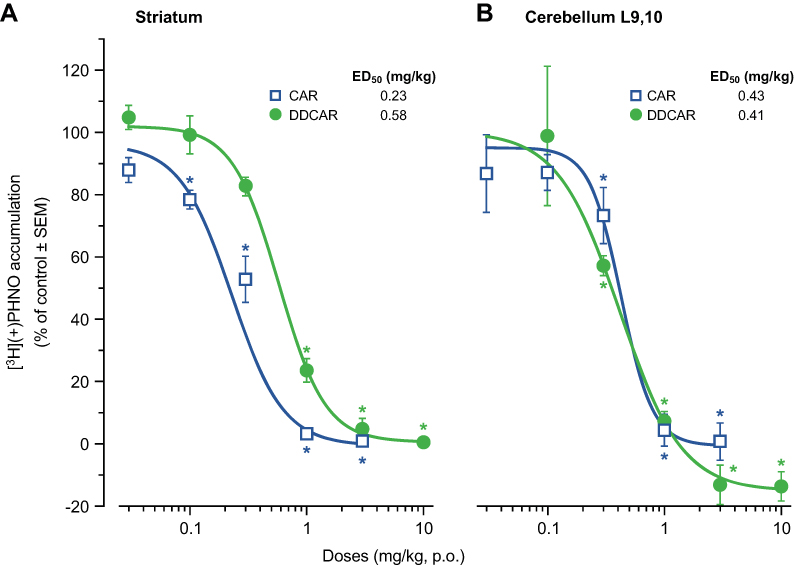 |
Figure 8 CAR and DDCAR inhibit [3H]-(+)PHNO binding in the rat brain. (A) Rat striatum; (B) cerebellum lobes 9 and 10 (L9,10). CAR data are also depicted in Gyertyán et al.14 *P<0.05 vs vehicle. Each dose group consisted of 5–6 animals. Abbreviations: CAR, cariprazine; DDCAR, didesmethyl-cariprazine; [3H](+)-PHNO, [3H](+)-trans-1a,2,3,4a,5,6-hexahydro-9-hydroxy-4-propyl-4H-naphtho[1,2-b]-1,4-oxazine; p.o., per os; SEM, standard error of the mean. |
There was a linear relationship between the cariprazine dose and the plasma or brain concentration (Figure S1A), which allowed data extrapolation to assess the relationship between plasma levels and brain receptor occupancy at the ED50 doses. The plasma concentration of cariprazine at its ED50 for striatal D2 and cerebellar D3 receptors was determined as 11.5 nM and 22.7 nM, respectively. Considering the (total) brain concentration is 13-fold higher than the plasma concentration and the free concentration is approximately 1.5% of the total brain concentration (as estimated from MD experiments), the free cariprazine levels were 2.2 nM and 4.4 nM at the 50% receptor occupancy for the rat striatum and cerebellum L9,10, respectively. These concentrations are in good agreement with the in vitro Ki values (ie, 2.3 nM for striatal and 3.9 nM for cerebellum L9,10 membranes, respectively) for cariprazine as determined with [3H]-(+)PHNO radioligand (Figure S1B).15
Neurochemistry of dopamine and serotonin
Cariprazine and DDCAR dose-dependently enhanced dopamine turnover (Figure 9A) and inhibited the NSD-1015-induced accumulation of DOPA (an index of dopamine biosynthesis; Figure 9B) in the striatum of reserpine-treated rats, with cariprazine having a greater partial agonist effect on dopamine biosynthesis than DDCAR. Both compounds slightly reduced serotonin turnover in the rat frontal cortex (Figure 9A).
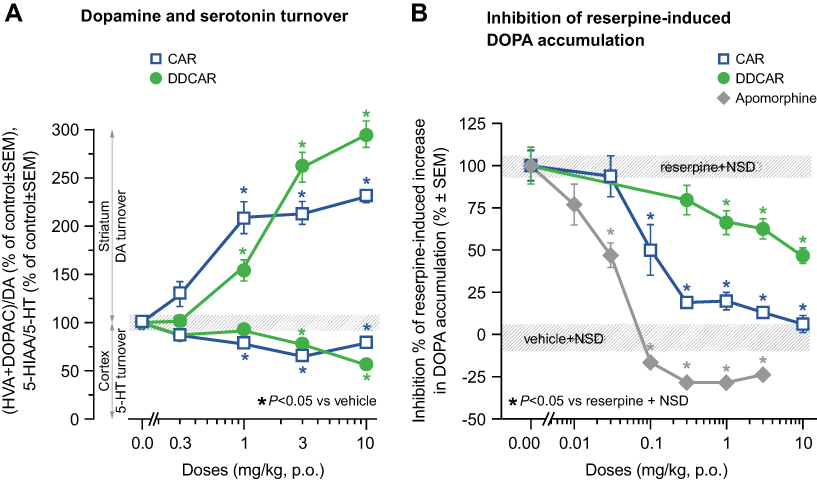 |
Figure 9 CAR and DDCAR enhance the dopamine turnover index ((HVA+DOPAC)/DA) in the rat striatum and slightly reduce the serotonin turnover index (5-HIAA/5-HT) in the rat frontal cortex; both partially inhibit reserpine-induced enhancement of dopamine biosynthesis (ie, NSD-1015-induced DOPA accumulation) in the rat striatum. (A) Dopamine and serotonin turnover index; (B) dopamine biosynthesis. Each group consisted of 5–6 animals.Abbreviations: CAR, cariprazine; DA, dopamine; DDCAR, didesmethyl-cariprazine; DOPA, 3,4-dihydroxyphenylalanine; DOPAC, 3,4-dihydroxyphenyl acetic acid; HVA, homovanillic acid; 5-HIAA, 5-hydroxy-indolyl acetic acid; 5-HT, serotonin; NSD, aromatic amino-acid decarboxylase inhibitor, NSD-1015; p.o., per os; SEM, standard error of the mean. |
Electrophysiology of substantia nigra dopamine neurons
Systemic cariprazine administration significantly (F6,48=23.11, P<0.0001) and dose-dependently suppressed the firing activity of SNc dopaminergic neurons, plateauing at less than 50% inhibition (Figure 10A). DDCAR also significantly diminished dopamine firing in the SNc (F7,42=16.35, P<0.0001; Figure 10A) in anesthetized rats. Development of the inhibitory effect was not significantly different between CAR and DDCAR when comparing roughly equiactive single doses (F1,9=0.92, P=0.36). The onset of neuronal suppression was followed by gradual spontaneous recovery of neuronal firing starting 10 min after dosing with CAR but not with DDCAR (Figure 10B).
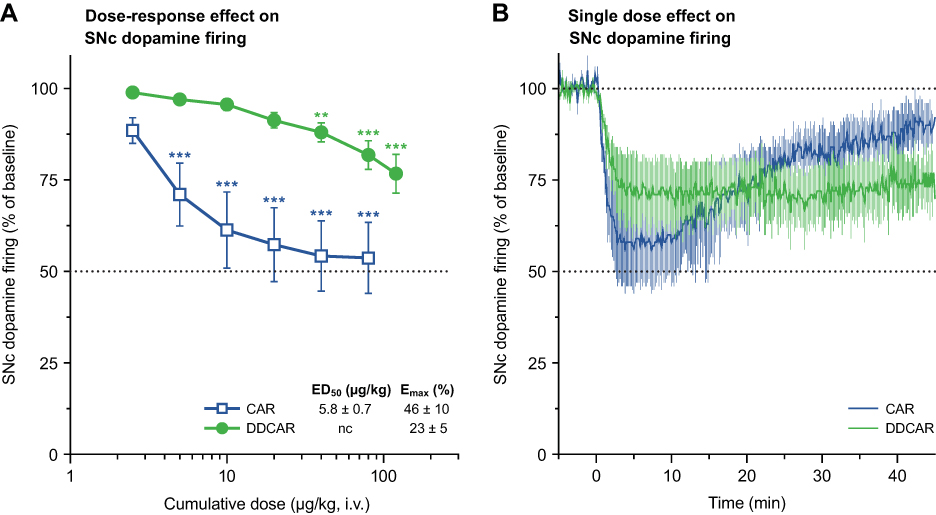 |
Figure 10 CAR and DDCAR inhibit spontaneous dopamine neuronal firing in the rat SNc. (A) Dose–response effect of systemic CAR (n=9) and DDCAR (n=7) administration on firing of SNc dopamine neurons. Inhibition of firing activity of dopamine neurons by CAR was more substantial than the effect exerted by DDCAR. **P<0.01; ***P<0.001 vs vehicle by Dunnett’s post hoc test. (B) Inhibition of SNc dopamine firing by a single dose (applied at time=0 min) of CAR (10 µg/kg, i.v.; n=6) or DDCAR (120 µg/kg, i.v.; n=5). Note the long-lasting inhibitory action of DDCAR on neuronal firing. CAR data are also depicted in Delcourte et al.28 The number of recorded neurons is indicated by n. Abbreviations: CAR, cariprazine; DDCAR, didesmethyl-cariprazine; Emax, maximal effect exerted by the drug; i.v., intravenous; nc, not calculable; SNc, substantia nigra pars compacta. |
Animal behavioral profile
In rodent models for antipsychotic-like and procognitive activity, DDCAR, the major human cariprazine metabolite, was studied in detail and compared with the results previously published for cariprazine.14 After oral administration, DDCAR showed similar antipsychotic-like and procognitive activity to cariprazine (Table 3). However, DDCAR was 3- to 10-fold less potent than cariprazine in the following assays: reduction of spontaneous locomotor activity; amphetamine, phencyclidine, and MK-801-induced hypermotility; apomorphine-induced climbing; conditioned avoidance response; and protection against scopolamine-induced learning deficit in the water labyrinth. Similar to cariprazine, DDCAR produced no catalepsy in rats at up to 50-fold its ED50 dose obtained in the conditioned avoidance response assay.
 |
Table 3 Effects of CAR and DDCAR in rodent models of psychosis, cognition, and EPS activity |
Pharmacokinetics
After p.o. administration of 1 mg/kg cariprazine to mice, the maximal concentration of the parent compound was attained at 2 h post dose in both the plasma and the brain. The formed DDCAR represented 15% of the cariprazine concentration in plasma and 5% in the brain. The brain-to-plasma ratio was high for cariprazine (B/P: 7.3) and much lower for the formed DDCAR (B/P: 2.7). Further, cariprazine showed rapid and effective absorption (Tmax: 0.5–1 h), with an absolute p.o. bioavailability of 52% in rats.31 After i.v. administration at a dose of 1 mg/kg to male rats, the systemic plasma clearance of cariprazine was 32 mL/min/kg, which is lower than the hepatic blood flow of this species. The high volume of distribution (Vd: 6.5 L/kg) indicates extensive tissue distribution of cariprazine in the rat. The elimination half-life of cariprazine was 2–3 h, regardless of the route of administration. Cariprazine (p.o.) penetrated rapidly and extensively into the brain, with a similar time to peak value as that of plasma (Tmax: 1 h) and a high brain-to-plasma ratio (B/P: 11). Formation of DDCAR was relatively slow (Tmax: 4 h), and the metabolite represented approximately 5–6% of cariprazine exposure both in the plasma and the brain (Table 4, Figure 11). After p.o. administration of DDCAR at a dose of 0.9 mg/kg (equimolar to 1 mg/kg cariprazine), high plasma and brain levels were attained shortly after administration (Tmax: 1–2 h). The absolute p.o. bioavailability of DDCAR was 34%, which was lower than that of cariprazine. When the metabolite DDCAR was i.v. administered to male rats at a dose of 1 mg/kg, the observed pharmokinetics parameters of DDCAR were similar to those of cariprazine (CL: 35 mL/min/kg; Vd: 9.2 L/kg). The elimination half-life of DDCAR was 3–4 h, regardless of the route of administration. The brain to plasma ratio showed high but somewhat lower brain partitioning than cariprazine (B/P: 5–6) (Table 4, Figure 11).
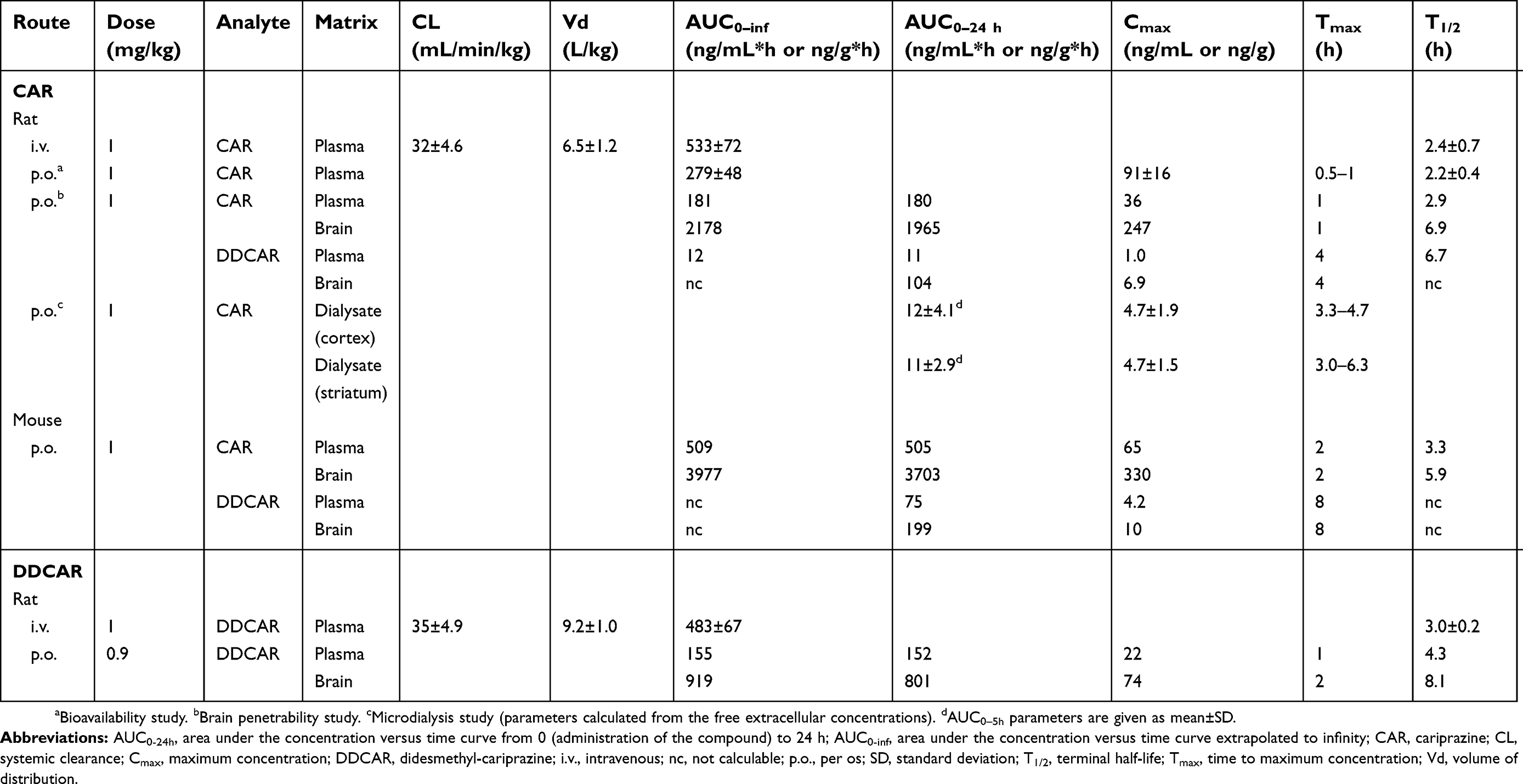 |
Table 4 Pharmacokinetic parameters in rats and mice after i.v. and p.o. administration |
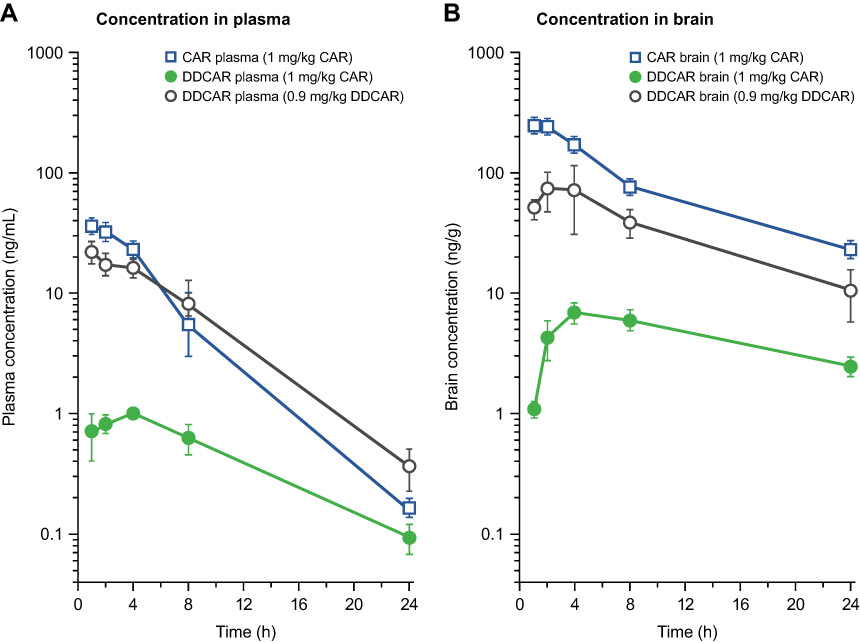 |
Figure 11 Concentration–time profiles of CAR and DDCAR in rats after equimolar p.o. administration of CAR and DDCAR. (A) Plasma; (B) brain. Abbreviations: CAR, cariprazine; DDCAR, didesmethyl-cariprazine; p.o., per os. |
In the rat MD study, extracellular levels of cariprazine in the brain ranged between 1.1 and 4.3 ng/mL (2.6–9.9 nM) after 1 mg/kg p.o. administration, which represented approximately 1–2% of the total brain exposure. No significant difference was observed between the prefrontal cortex and striatum. Cariprazine exhibited long-lasting brain exposure, up to 5 h; however, the concentration of DDCAR could not be detected throughout the collection period (<0.8 ng/mL in dialysate; Table 4, Figure 12). In general, the free extracellular concentration was found to be 1–5% of the total (bound+free) concentration in the rat brain. Moreover, there is an estimated technical loss of 80–85% during the procedure due to recovery. Assuming 5% free fraction and 20% recovery, we would expect the maximum amount of DDCAR in the dialysate to be 0.07 ng/mL (given that Cmax of DDCAR is ~7 ng/g; Table 4), which is lower than our limit of quantitation of 0.8 ng/mL. In summary, considering that only the free fraction of DDCAR is measured and other technical aspects, the concentrations could not be quantified even at the maximal concentration.
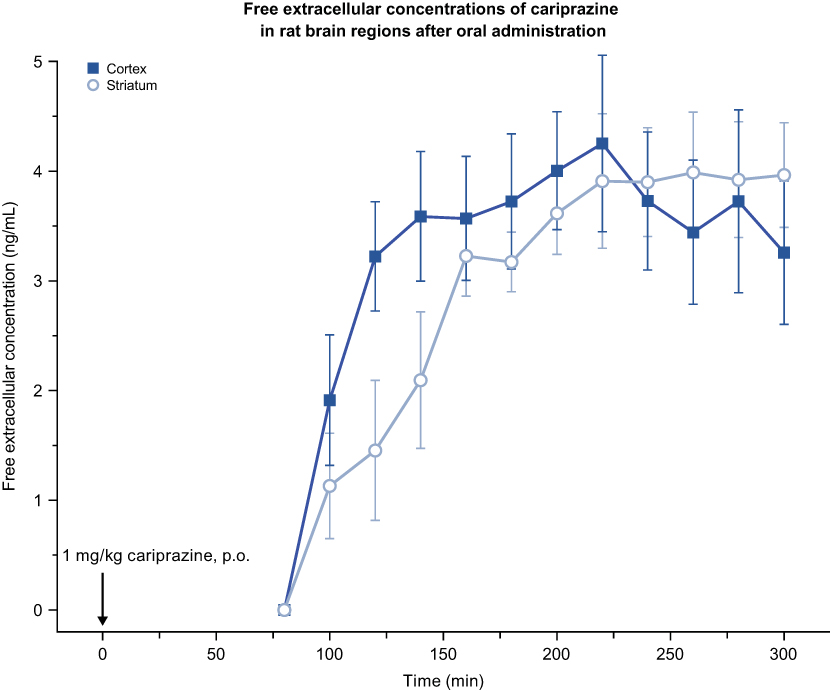 |
Figure 12 Free extracellular concentrations of cariprazine in rat brain regions after p.o. administration of CAR. CAR was dissolved in deionized water and administered at 1 mg/kg. Abbreviations: CAR, cariprazine; p.o., per os. |
Discussion
In this study, in vitro, in vivo, pharmacokinetic, and behavioral assays were used to compare the pharmacological profiles of cariprazine and its metabolites, DCAR and DDCAR. In general, these metabolites displayed similar receptor binding and occupancy, in vitro and in vivo functional effects, and behavioral effects as the parent compound.
Cariprazine and its metabolites exhibited comparable binding profiles at a variety of receptors (Figure 13). Similar to cariprazine, DCAR and DDCAR displayed high affinity for human and rat D3, D2L, and D2S receptors. Cariprazine and both metabolites showed greater affinity for human D3 receptors than for either of the D2 receptor isoforms, with DCAR and DDCAR having a considerably higher degree of in vitro selectivity for the human D3 receptor versus the D2L, D2S, 5-HT2B, and 5-HT1A receptors than was seen for cariprazine. Our results with cariprazine and DDCAR corroborate previous results, which found that cariprazine and DDCAR display subnanomolar in vitro binding affinity for human D2 and D3 receptors with approximately 3- and 4-fold higher affinity for D3 receptors than for D2 receptors, respectively.23 Cariprazine, DCAR, and DDCAR displayed similar subnanomolar or low nanomolar affinity for human 5-HT2B and 5-HT1A receptors, respectively. Both metabolites had a somewhat greater affinity for human 5-HT2A receptors than did cariprazine, and all three compounds displayed moderate to low affinities for human H1, 5-HT2C, and σ-1 receptors. All three compounds had subnanomolar affinity for (recombinant) rat D3 receptors and low nanomolar affinity for rat hippocampal 5-HT1A receptors, with DCAR having approximately half the affinity of cariprazine and DDCAR; they also showed low nanomolar affinity for rat dopamine D2, serotonin 5-HT2A, and adrenergic α receptors.
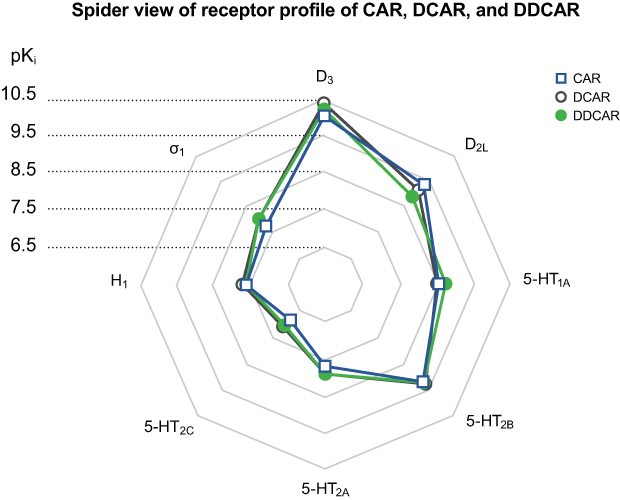 |
Figure 13 Spider plot of the receptor profile of CAR, DCAR, and DDCAR. Abbreviations: 5-HT, serotonin; CAR, cariprazine; D, dopamine; DCAR, desmethyl-cariprazine; DDCAR, didesmethyl-cariprazine; H, histamine. |
In agreement with the in vitro affinity, both cariprazine and DDCAR demonstrated in vivo occupancy at D2 and D3 receptors in rats, determined with the D3-preferring radioligand [3H]-(+)PHNO. Compared with DDCAR, cariprazine demonstrated higher occupancy at striatal D2 receptors and similar occupancy at D3 receptors in the cerebellum. The calculated free brain concentration of cariprazine at its ED50 doses corresponds with its in vitro Ki values, as determined by [3H]-(+)PHNO binding using rat striatum or cerebellum L9,10 membranes. The in vivo receptor occupancy data observed in cynomolgus monkeys confirmed the striatal occupancy data observed in the rat, indicating that cariprazine has approximately 2-fold higher affinity for dopamine receptors than DDCAR (ED50: 4.3 μg/kg13 vs 7.6 μg/kg; data on file).
In membranes from rat striatum or from CHO cells expressing human D2 or D3 receptors used for the G-protein recruitment assay (ie, [35S]GTPγS binding), neither cariprazine nor DDCAR showed agonist activity alone, but both acted as full antagonists at D2 and D3 receptors in the presence of dopamine. On the other hand, D2/D3 receptor partial agonism of all three compounds was demonstrated in assays in which cariprazine, DCAR, and DDCAR inhibited FSK-induced cAMP production to a lesser extent as compared to the full agonist, dopamine.
Under normal dopaminergic tone (eg, in healthy animals), the primary in vivo actions of cariprazine and DDCAR are antagonistic, resulting in dose-dependent increases in dopamine turnover in the rat striatum, with DDCAR increasing turnover to a slightly higher degree than cariprazine. However, under low dopaminergic tone (ie, when dopamine was depleted by reserpine pretreatment), both compounds showed partial agonist activity, with DDCAR showing a lesser degree of partial agonism than cariprazine. The results obtained in these two models indicate that while both compounds are D3/D2 receptor antagonists/partial agonists, DDCAR possesses lower in vivo intrinsic efficacy as compared to cariprazine.
At rat hippocampal and human 5-HT1A receptors, both cariprazine and DDCAR acted as partial agonists in the [35S]GTPγS binding assay. At this receptor, DDCAR showed higher intrinsic efficacy but slightly less potency than cariprazine. Although both cariprazine and DDCAR were partial agonists in the [35S]GTPγS assay, we showed they were full agonists at the human 5-HT1A receptor in inhibiting FSK-induced cAMP production. In contrast, DCAR acted as a partial agonist at 5-HT1A receptors, but with a lower potency than either cariprazine or DDCAR. Consistent with their 5-HT1A receptor activity in the [35S]GTPγS assay, both cariprazine and DDCAR slightly but dose-dependently reduced serotonin turnover in the rat frontal cortex and did so with equal potency. Cariprazine, DCAR, and DDCAR also acted as antagonists with similarly high potencies at the human 5-HT2B receptors.
In the rat SNc, partial inhibition of dopamine firing activity was seen with cariprazine after cumulative dosing, an effect reported to be mediated by partial agonism of midbrain autosomal D2 receptors.28 In addition, DDCAR partially diminished the firing activity of SNc dopaminergic neurons in a dose-dependent manner; however, it was less potent and steady state was not observed. Furthermore, while at 10 min after i.v. cariprazine administration the maximal inhibitory effect started to decline and full reversal of suppression on firing was observed within 60 min, on average,28 there was no sign of any setback in the inhibitory action of DDCAR within this time frame.
In previous studies, cariprazine demonstrated antipsychotic-like, procognitive, and antidepressant-like effects in rodent models.14,32–34 In this study, orally administered DDCAR demonstrated an antipsychotic-like and procognitive profile in multiple rodent behavioral assays in a manner similar to cariprazine. In all cases, the potency of DDCAR (p.o.) was 3- to 10-fold lower than that of cariprazine, perhaps reflecting the lower concentration of DDCAR found in the brain after acute administration and/or the lower affinity of DDCAR for occupying the D2 receptor in the rat brain. Monkey positron emission tomography data also showed higher affinity for brain D2/D3 receptor occupancy with cariprazine compared to DDCAR. Interestingly, the doses of cariprazine and DDCAR required to cause catalepsy in rats were 50- to 100-fold higher than those observed for their antipsychotic-like activity (attenuation of conditioned avoidance response), suggesting that efficacious antipsychotic activity may be achievable at doses considerably lower than those that induce extrapyramidal symptoms.
When administered p.o., cariprazine is rapidly absorbed and penetrates quickly into the brain in both rats and mice. Formation of the metabolite DDCAR resulted in plasma exposure of 6% of the parent compound’s exposure in rat and 15% in mouse. DDCAR represented 5% of the parent drug exposure in both mice and rat brains. These results show that the DDCAR metabolite, formed after cariprazine administration, probably does not contribute significantly to the pharmacological effect of cariprazine in rodents. Free extracellular concentrations of cariprazine were approximately 1–2% of the total concentration in rats in the MD study (a single dose of 1 mg/kg, p.o.), with a superabundance at receptors playing a role in its mechanism of action. However, free DDCAR concentrations were not detectable in rat brain. These data also support the minor (if any) role of DDCAR in cariprazine’s acute pharmacological effect in rats. The brain penetrability study of cariprazine and DDCAR at equimolar doses in rats revealed that cariprazine brain exposure was approximately 2.5-fold higher than that of DDCAR, which may explain the lower efficacy of DDCAR in pharmacology tests compared to cariprazine.
In a metabolite profiling study following p.o administration of 3 mg/kg radiolabelled cariprazine to rats, two major metabolites, DCAR and DDCAR, were identified, and their plasma exposure represented approximately 15% and 4% of the parent drug, respectively (data not shown). In another rat study, after 0.6 mg/kg p.o. administration of cariprazine, DCAR and DDCAR were also present in rat plasma and brain (DCAR, 13% in plasma and 9% in brain relative to the exposure of cariprazine; DDCAR, 3% in both matrices relative to the exposure of cariprazine; data not shown).
Despite the compelling evidence that DCAR and DDCAR are active metabolites of cariprazine and possess qualitatively similar in vitro and in vivo profiles compared with the parent compound, and may contribute to the established clinical efficacy of cariprazine, this study holds several limitations. Most notably, in the present experiments, the in vivo pharmacodynamic effects of cariprazine, DCAR, and DDCAR were investigated post acute dosing to animals; however, clinical efficacy of sustained cariprazine treatment has been established, and it has been determined that all of the active moieties are detectable under steady-state dosing in humans.25 Formation of DCAR and DDCAR after a single dose of cariprazine in rodents is limited; this implies that the direct contribution of DCAR and DDCAR to the acute pharmacological actions of the parent compound in rodents can only be minor.
While the plasma and brain levels of DCAR and DDCAR in rats suggested that their contribution to the rodent pharmacological action of the parent compound is likely minor, the situation may be different in humans, in which the major cariprazine moiety is DDCAR.25 As such, it has been hypothesized that DDCAR may be a major contributing factor to the clinical efficacy of cariprazine. Findings from a recent long-term cariprazine relapse-prevention study in patients with schizophrenia may provide some support for this hypothesis. In this clinical trial, DDCAR, with its extended half-life of 1–3 weeks, was found to be the primary compound in plasma following discontinuation of cariprazine.35 Additionally, there appeared to be a delayed incidence of relapse following discontinuation of cariprazine treatment in this study relative to that observed in similarly designed relapse prevention trials of other oral antipsychotics, further suggesting that DDCAR may contribute to the clinical efficacy of cariprazine.35
Conclusion
DCAR and DDCAR, the two major active metabolites of cariprazine, possess similar in vitro receptor binding profiles to the parent compound. Not surprisingly, given the structural similarity of the compounds, both the in vitro functional profiles of DCAR and DDCAR and the in vivo behavioral profileof DDCAR also demonstrate high degrees of similarity with those of cariprazine. Following acute administration of cariprazine, DCAR and DDCAR were detected in the rodent brain, but the levels were low relative to cariprazine at time points where most behavioral assays were performed. Thus, the metabolite contributions to the acute pharmacological actions of cariprazine appear to be minor in rodents. However, after repeated administration (eg, under steady-state conditions) in humans, both metabolites, especially DDCAR, are present in substantially higher levels in the plasma (and possibly in the brain). Based on the high degree of in vitro and in vivo similarity between cariprazine and its active metabolites, it can be concluded that these metabolites, especially DDCAR, could significantly contribute to the clinical efficacy of cariprazine.
Abbreviation list
Cariprazine (RGH-188), trans-N-[4-[2-[4-(2,3-dichlorophenyl)piperazin-1-yl]ethyl]cyclohexyl]-N’,N’-dimethylureahydrochloride; 5-HT, serotonin; hD2, human recombinant dopamine D2 receptor; hD3, human recombinant dopamine D3 receptor; h5-HT1A, human recombinant serotonin 5-HT1A receptor; h5-HT2B, human recombinant serotonin 5-HT2B receptor; DCAR, desmethyl-cariprazine; DDCAR, didesmethyl-cariprazine; DOPA, 3,4-dihydroxyphenylalanine; DOPAC, 3,4-dihydroxyphenyl acetic acid; HVA, homovanillic acid; 5-HIAA, 5-hydroxy-indolyl acetic acid; CHO, Chinese hamster ovary; HEK, human embryonic kidney; [3H](+)-PHNO, [3H](+)-trans-1a,2,3,4a,5,6-hexahydro-9-hydroxy-4-propyl-4H-naphtho[1,2-b]-1,4-oxazine.
Acknowledgments
The authors would like to thank Dr Éva Schmidt and Dr Anikó Gere of Gedeon Richter Plc (Budapest, Hungary) for conducting the receptor binding experiments, Dr Sándor Kolok and Dr Zoltán Kapui of Gedeon Richter Plc for conducting the functional experiments, Dr Judit Laszy of Gedeon Richter Plc for conducting the behavioral experiments, Dr Mónika Vastag of Gedeon Richter Plc for conducting the in vitro metabolism experiments, Dr Margit Kapás of Gedeon Richter Plc for conducting the pharmacokinetic experiments, Éva Ágai Csongor and Dr György Domány of Gedeon Richter Plc for synthesizing cariprazine and metabolites, Dr Christer Halldin and Dr Balázs Gulyás of Karolinska Institute (Stockholm, Sweden) for managing the positron emission tomography study, and Dr Nika Adham of Allergan (Madison, NJ, USA) and Dr Zsolt Szombathelyi of Gedeon Richter Plc (Ridgewood, NJ, USA) for their critical review and edits of the manuscript.
The authors express their sincere thanks for the expert technical assistance from all of our colleagues from Gedeon Richter Plc actively participating in the presented experiments.
Writing and editorial assistance was provided to the authors by Jennifer Fetting, PhD, and Katharine Fang, PhD, of Prescott Medical Communications Group (Chicago, IL, USA), a contractor of Allergan. These studies were sponsored by Gedeon Richter Plc. This manuscript was supported by funding from Allergan and Gedeon Richter Plc.
István Gyertyán's present affiliation is Semmelweis University, Institute of Pharmacology and Pharmacotherapy, NAP Cognitive Translational Behavioral Pharmacology Group, Budapest, Hungary.
Disclosure
Neither honoraria nor payments were made for authorship. All authors are or were employees of Gedeon Richter Plc. Mr. Béla Kiss, Dr. István Laszlovszky and Dr. Katalin Sághy are inventors of cariprazine and received a personal fee for it. Dr. István Gyertyán is an inventor of cariprazine and is entitled to receive a personal fee for it. All authors report nonfinancial support from Prescott Medical Communication Group for editorial assistance during preparation of the manuscript. The authors report no further conflicts of interest in this work.
References
1. Durgam S, Cutler AJ, Lu K, et al. Cariprazine in acute exacerbation of schizophrenia: a fixed-dose, phase 3, randomized, double-blind, placebo- and active-controlled trial. J Clin Psychiatry. 2015;76(12):e1574–e1582. doi:10.4088/JCP.15m09997
2. Durgam S, Starace A, Li D, et al. An evaluation of the safety and efficacy of cariprazine in patients with acute exacerbation of schizophrenia: a phase II, randomized clinical trial. Schizophr Res. 2014;152(2–3):450–457. doi:10.1016/j.schres.2013.11.041
3. Kane JM, Zukin S, Wang Y, et al. Efficacy and safety of cariprazine in acute exacerbation of schizophrenia: results from an international, phase III clinical trial. J Clin Psychopharmacol. 2015;35(4):367–373. doi:10.1097/JCP.0000000000000346
4. Durgam S, Starace A, Li D, et al. The efficacy and tolerability of cariprazine in acute mania associated with bipolar I disorder: a phase II trial. Bipolar Disord. 2015;17(1):63–75. doi:10.1111/bdi.12238
5. Calabrese JR, Keck PE
6. Sachs GS, Greenberg WM, Starace A, et al. Cariprazine in the treatment of acute mania in bipolar I disorder: a double-blind, placebo-controlled, phase III trial. J Affect Disord. 2015;174:296–302. doi:10.1016/j.jad.2014.11.018
7. Durgam S, Earley W, Li R, et al. Long-term cariprazine treatment for the prevention of relapse in patients with schizophrenia: a randomized, double-blind, placebo-controlled trial. Schizophr Res. 2016;176(2–3):264–271. doi:10.1016/j.schres.2016.06.030
8. Németh G, Laszlovszky I, Czobor P, et al. Cariprazine versus risperidone monotherapy for treatment of predominant negative symptoms in patients with schizophrenia: a randomised, double-blind, controlled trial. Lancet. 2017;389(10074):1103–1113. doi:10.1016/S0140-6736(17)30060-0
9. Durgam S, Earley W, Lipschitz A, et al. An 8-week randomized, double-blind, placebo-controlled evaluation of the safety and efficacy of cariprazine in patients with bipolar I depression. Am J Psychiatry. 2016;173(3):271–281. doi:10.1176/appi.ajp.2015.15020164
10. Earley W, Burgess M, Rekeda L, et al. Cariprazine treatment of bipolar I depression: a randomized, double blind, placebo-controlled phase 3 study.
11. Durgam S, Earley W, Guo H, et al. Efficacy and safety of adjunctive cariprazine in inadequate responders to antidepressants: a randomized, double-blind, placebo-controlled study in adult patients with major depressive disorder. J Clin Psychiatry. 2016;77(3):371–378. doi:10.4088/JCP.15m10070
12. Kiss B, Horváth A, Némethy Z, et al. Cariprazine (RGH-188), a dopamine D(3) receptor-preferring, D(3)/D(2) dopamine receptor antagonist-partial agonist antipsychotic candidate: in vitro and neurochemical profile. J Pharmacol Exp Ther. 2010;333(1):328–340. doi:10.1124/jpet.109.160432
13. Seneca N, Finnema SJ, Laszlovszky I, et al. Occupancy of dopamine D(2) and D(3) and serotonin 5-HT(1)A receptors by the novel antipsychotic drug candidate, cariprazine (RGH-188), in monkey brain measured using positron emission tomography. Psychopharmacology (Berl). 2011;218(3):579–587. doi:10.1007/s00213-011-2343-z
14. Gyertyán I, Kiss B, Sághy K, et al. Cariprazine (RGH-188), a potent D3/D2 dopamine receptor partial agonist, binds to dopamine D3 receptors in vivo and shows antipsychotic-like and procognitive effects in rodents. Neurochem Int. 2011;59(6):925–935. doi:10.1016/j.neuint.2011.07.002
15. Kiss B, Horti F, Bobok A. Cariprazine (RGH-188), a dopamine D(3) receptor-preferring, D(3)/D(2) dopamine receptor antagonist-partial agonist antipsychotic candidate: in vitro and neurochemical profile. Schizophr Res. 2012;136(Suppl1):
16. Girgis RR, Slifstein M, D’Souza D, et al. Preferential binding to dopamine D3 over D2 receptors by cariprazine in patients with schizophrenia using PET with the D3/D2 receptor ligand [(11)C]-(+)-PHNO. Psychopharmacology (Berl). 2016;233(19–20):3503–3512. doi:10.1007/s00213-016-4382-y
17. Girgis RR, Abi-Dargham A, Slifstein M, et al. In vivo dopamine D3 and D2 receptor occupancy profile of cariprazine versus aripiprazole: a PET study. Neuropsychopharmacology. 2017;42:S476–S652.
18. McCormick PN, Kapur S, Graff-Guerrero A, Raymond R, Nobrega JN, Wilson AA. The antipsychotics olanzapine, risperidone, clozapine, and haloperidol are D2-selective ex vivo but not in vitro. Neuropsychopharmacology. 2010;35(8):1826–1835. doi:10.1038/npp.2010.50
19. Citrome L. Cariprazine: chemistry, pharmacodynamics, pharmacokinetics, and metabolism, clinical efficacy, safety, and tolerability. Expert Opin Drug Metab Toxicol. 2013;9(2):193–206. doi:10.1517/17425255.2013.759211
20. Kirschner N, Kapás M, Colella G, et al. Metabolite profiles of cariprazine in plasma, urine, feces, and bile of rats.
21. Meszaros GP, Agai-Csongor E, Kapas M. Sensitive LC-MS/MS methods for the quantification of RGH-188 and its active metabolites, desmethyl- and didesmethyl-RGH-188 in human plasma and urine. J Pharm Biomed Anal. 2008;48(2):388–397. doi:10.1016/j.jpba.2007.12.016
22. Kirschner N, Gémesi L, Vastag M, et al. In vitro metabolism of RGH-188. Drug Metab Rev. 2008;40(Supplement1):
23. Tadori Y, Forbes RA, McQuade RD, Kikuchi T. In vitro pharmacology of aripiprazole, its metabolite and experimental dopamine partial agonists at human dopamine D2 and D3 receptors. Eur J Pharmacol. 2011;668(3):355–365. doi:10.1016/j.ejphar.2011.07.020
24. Nakamura T, Kubota T, Iwakaji A, Imada M, Kapas M, Morio Y. Clinical pharmacology study of cariprazine (MP-214) in patients with schizophrenia (12-week treatment). Drug Des Dev Ther. 2016;10:327–338. doi:10.2147/DDDT.S95100
25. Periclou A, Phillips L, Bihorel S, et al. Characterization of population pharmacokinetics of cariprazine and its major metabolites.
26. Kurko D, Bekes Z, Gere A, et al. Comparative pharmacology of adrenergic alpha(2C) receptors coupled to Ca(2+) signaling through different Galpha proteins. Neurochem Int. 2009;55(7):467–475. doi:10.1016/j.neuint.2009.04.015
27. Kiss B, Horti F, Bobok A. In vitro and in vivo comparison of [(3)H](+)-PHNO and [(3)H]raclopride binding to rat striatum and lobes 9 and 10 of the cerebellum: a method to distinguish dopamine D(3) from D(2) receptor sites. Synapse. 2011;65(6):467–478. doi:10.1002/syn.20867
28. Delcourte S, Ashby CR
29. Cheng Y, Prusoff WH. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol. 1973;22(23):3099–3108.
30. Craig DA. The Cheng-Prusoff relationship: something lost in the translation. Trends Pharmacol Sci. 1993;14(3):89–91.
31. Mészáros GP, Terjéki Bolf E, Gémesi L, Kapáas M, Tihanyi K, Szombathelyi Z. Pharmacokinetics of RGH-188, a novel dopamine D3/D2 antagonist/partial agonist atypical antipsychotic, in rats and dogs. Eur Neuropsychopharmacol. 2008;18:S456–S457. doi:10.1016/S0924-977X(08)70676-X
32. Duric V, Banasr M, Franklin T, et al. Cariprazine exhibits anxiolytic and dopamine D3 receptor-dependent antidepressant effects in the chronic stress model. Int J Neuropsychopharmacol. 2017;20(10):788–796. doi:10.1093/ijnp/pyx038
33. Zimnisky R, Chang G, Gyertyán I, Kiss B, Adham N, Schmauss C. Cariprazine, a dopamine D(3)-receptor-preferring partial agonist, blocks phencyclidine-induced impairments of working memory, attention set-shifting, and recognition memory in the mouse. Psychopharmacology (Berl). 2013;226(1):91–100. doi:10.1007/s00213-012-2896-5
34. Papp M, Gruca P, Lason-Tyburkiewicz M, Adham N, Kiss B, Gyertyán I. Attenuation of anhedonia by cariprazine in the chronic mild stress model of depression. Behav Pharmacol. 2014;25(5–6):567–574. doi:10.1097/FBP.0000000000000070
35. Correll CU, Jain R, Periclou A, et al. Potential relationship between relapse and plasma drug levels following discontinuation of cariprazine treatment in patients with schizophrenia.
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.