")
Back to Journals » Drug Design, Development and Therapy » Volume 9
Pharmacokinetics, pharmacodynamics, and tolerability of LC350189, a novel xanthine oxidase inhibitor, in healthy subjects
Authors Yoon S , Shin D, Lee H , Jang I , Yu K
Received 18 April 2015
Accepted for publication 2 July 2015
Published 31 August 2015 Volume 2015:9 Pages 5033—5049
DOI https://doi.org/10.2147/DDDT.S86884
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Professor Shu-Feng Zhou
Seonghae Yoon,1 Donghoon Shin,1 Howard Lee,1,2 In-Jin Jang,1 Kyung-Sang Yu1
1Department of Clinical Pharmacology and Therapeutics, Seoul National University College of Medicine, Seoul National University Hospital, 2Department of Transdisciplinary Studies, Graduate School of Convergence Science and Technology, Seoul National University, Seoul, Republic of Korea
Introduction: LC350189 is a novel selective xanthine oxidase inhibitor under clinical development for the management of hyperuricemia in gout patients. The aim of this study was to evaluate the pharmacokinetics, pharmacodynamics, and tolerability of the drug in healthy subjects.
Methods: A dose-block randomized, double-blind, active and placebo-controlled, single- and multiple-dosing study was conducted. A single ascending dose (SAD) study (10–600 mg) and a multiple ascending dose (MAD) study with once-daily doses (100–800 mg) for 7 days were conducted. Serial samples of blood and urine for pharmacokinetics/pharmacodynamics analysis were collected, and tolerability and adverse events were assessed throughout the study.
Results: Sixty-seven and 58 subjects were enrolled in the SAD and MAD studies, respectively. The mean Cmax and AUClast values increased with increasing doses, and exposure to LC350189 was dose proportional. The 24-hour mean serum uric acid (Cmean,24) decreased by 8.7%–31.7% (day 1) and 53.5%–91.2% (day 7) from baseline in the SAD and MAD studies, respectively, and the percentage decrease in Cmean,24 increased with higher doses.
Conclusion: LC350189 was well tolerated in the dose range of 10–800 mg. It lowered the serum and urine uric acid levels substantially in this dose range; the extent of the decrease in the serum uric acid level in the 200 mg dose group was similar or higher compared to that of febuxostat 80 mg group in the MAD study. It is expected that LC350189 could be safely administered once daily to patients with hyperuricemia or gout, leading to a sufficient decrease in uric acid levels.
Keywords: gout, xanthine oxidase inhibitor, pharmacokinetics, pharmacodynamics, LC350189
Background
Gout is a common inflammatory joint disease characterized by recurrent attacks of joint destruction, pain, reduced mobility, and decreased quality of life.1–3 Gout is caused by elevated levels of serum uric acid (SUA) (hyperuricemia), usually due to reduced excretion.4
Epidemiological studies suggest that gout is becoming more prevalent across the world;5 in Korea, the prevalence has grown 2.3-fold since 2001, up to 0.4% in 2008.6 The disease burden in terms of cost and health-related quality of life due to gout also increased.7 The humanistic burden of gout is largely due to physical disability and pain resulting from chronic clinical manifestations. In addition, patients with refractory gout exert a significant burden economically to society compared to those who are gout free,8 and gout inflicts a substantial burden of illness on the employed population due to not only medical costs but also sick leave, short- and long-term disability, and workers’ compensation.9,10 Considering this increase in prevalence and humanistic and economic burden, clinicians will need more therapeutic options than the limited ones they have now. Uric acid is the final product of purine metabolism in humans, which involves xanthine oxidase (XO), an enzyme that converts hypoxanthine to xanthine and xanthine to uric acid (Figure 1). Therefore, XO inhibitors can decrease the formation of uric acid in patients with gout; allopurinol and febuxostat are the only XO inhibitors that have been used for this purpose in the clinical setting. Even though allopurinol has sufficient efficacy in most gout patients, there are some rare but severe toxicities, including allopurinol hypersensitivity syndrome.11–13 Furthermore, its dosage needs to be adjusted in patients with chronic kidney disease.14–17 According to recent studies comparing allopurinol and febuxostat, the overall efficacy and safety profile was comparable. In addition, febuxostat was the superior drug in patients with mild and moderate renal insufficiency.18–21
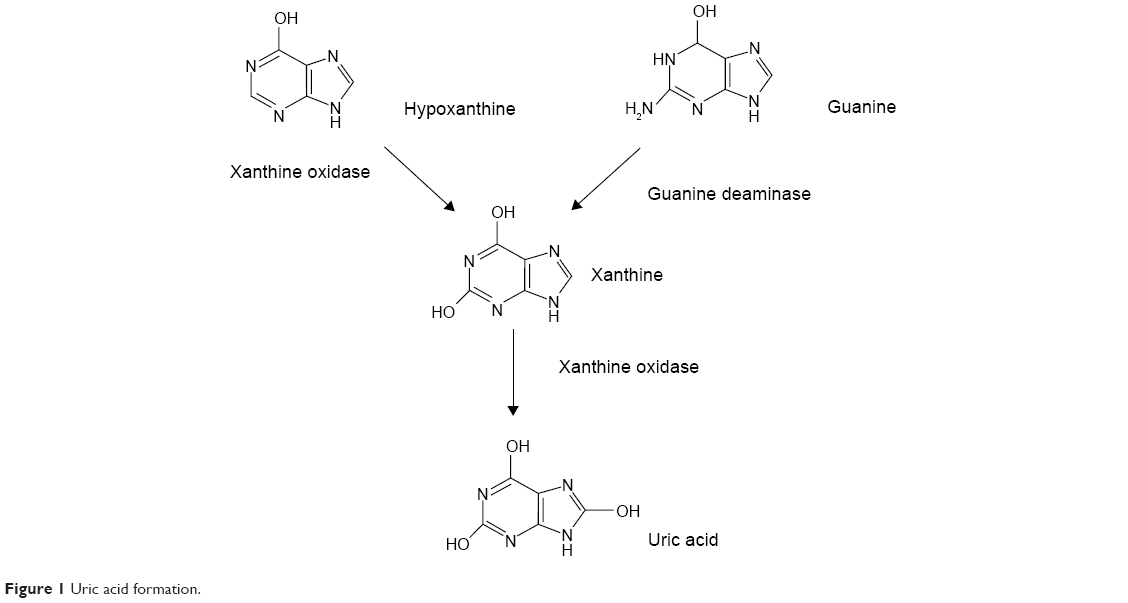 | Figure 1 Uric acid formation. |
LC350189 is a novel non-purine selective inhibitor of XO under development for the treatment of gout and hyperuricemia. In an in vitro assay, LC350189 inhibited XO enzyme activity at a level that was comparable to febuxostat (data on file at LG Life Sciences). Additionally, in an in vivo study, LC350189 sufficiently reduced SUA (data on file at LG Life Sciences). Furthermore, no significant toxicity was found in the preclinical studies up to 12.5 mg/kg and 200 mg/kg in the rat and dog, respectively, rendering the clinical development of LC350189 worthwhile. Based on this understanding, the present study aimed to investigate the pharmacodynamics (PDs), pharmacokinetics (PKs), and safety and tolerability of LC350189 after a single oral dose and multiple oral doses in healthy subjects.
Methods
Study participants
This study enrolled male subjects of ages 20–50 if their body weight was 55–90 kg (both non-inclusive) with a body mass index (BMI) of 18.0–27.0 kg/m2. Study participants had to be healthy based on physical examination, vital signs, 12-lead electrocardiography (ECG), serology (hepatitis B surface antigen, anti-hepatitis C virus antibody, anti-HIV antibody), urinary drug screening test, and routine clinical laboratory tests (hematology, blood coagulation, clinical chemistry, and urinalysis), which were performed within 4 weeks prior to the first administration of the study drug. Study participants whose SUA level was <10.5 mg/dL, which is 1.5 times the upper limit of normal for uric acid or 7.0 mg/dL, but without a history of gout were eligible for this study. Study participants were excluded if they had taken any prescription medication or herbal medicines within 2 weeks or any over-the-counter medication or vitamin supplement within 1 week, prior to the study.
This study was conducted at the Clinical Trials Center, Seoul National University Hospital, Seoul, Korea, in full compliance with the principles stipulated in the ICH Good Clinical Practices guideline (clinicaltrials.gov registry number: NCT01361646). The protocol was approved by the Institutional Review Board at the Seoul National University Hospital. Participants provided written informed consent after the investigator explained the study and before any study procedure was performed, including screening for eligibility.
Study design
This study was conducted using a randomized (in each dose group), double-blind, active and placebo-controlled, dose-escalation design after a single dose (part I) and multiple doses (part II). In part I, study participants randomly received a single oral dose of LC350189 or placebo in the fasted state after an overnight fast at a ratio of 6:2 (10 mg and 25 mg) or 8:2 (50 mg, 100 mg, 200 mg, 400 mg, and 600 mg). Furthermore, those assigned to 200 mg repeated the study in the fed state after a 7-day washout to assess the effect of a high-fat diet on the PK profile of LC350189. In part II, study participants randomly received multiple oral doses of LC350189, febuxostat at 80 mg, or placebo once daily for 7 days in the fasted state after an overnight fast at a ratio of 8:2:2 (100 mg, 200 mg, 400 mg, and 600 mg) or 6:2 (LC350189: placebo, 800 mg). Subjects were admitted to the Clinical Trials Center on 3 days before drug administration and discharged after finishing scheduled procedures. Each subject was administered placebo to evaluate baseline PD characteristics (day -1). During the confinement period, the subjects were provided with standardized meals (except for the high-fat diet on day 8 for the 200 mg dose group) to minimize the effect of food on PK/PD evaluations.
In the single ascending dose (SAD) study, PK blood collections were scheduled right before drug administration (0 hour) and at 16 time points afterward. During the study, PK results of the 10 mg single-dose group revealed that the median Tmax was 2.8 hours (minimum: 2.5 hours, maximum: 4.0 hours). Therefore, the 0.25-hour and 0.75-hour time points were excluded, and the 3.5-hour and 5-hour time points were added to 50 mg or higher dose groups. In the multiple ascending dose (MAD) study, blood samples were collected before and after the first dose (day 1), just prior to dosing on days 3, 4, 5, and 6 and after the last dose on day 7. Additional blood samples were collected on days -1 and 1 in part I to determine of the serum concentrations of uric acid, xanthine, and hypoxanthine and plasma concentrations, which were repeated on day 8 for the 200 mg food effect study. On the other hand, blood samples were collected daily from days -1 to 10 in part II. Urine samples were collected over the prespecified time intervals to evaluate PK and PD. Details of the sample collection time are shown in Table S1.
Analytical methods
Details of the analytical methods used to determine the concentrations of LC350189 and PD markers are presented in the Supplementary materials.
Analysis of PKs and PDs and PKs/PDs relationship
The individual PK and PD parameters were derived using the noncompartmental methods. The actual collection times were used in the PK analysis. The area under the curve (AUC0–24) for uric acid, xanthine, and hypoxanthine were calculated by noncompartmental methods and the 24-hour mean serum concentrations (Cmean,24) were calculated as AUC0–24 divided by 24. The % change from baseline (day -1) in Cmean,24 for uric acid was calculated by (Cmean,1d(or 7d) − Cmean,base)/Cmean,base ×100. The other details of the PK and PD analysis are presented in the Supplementary materials.
To evaluate PK/PD relationship, the AUCtau,ss and the % change in Cmean,24 were fitted using an Emax model as follows: E = Emax × C/(C + EC50), where Emax is the maximum effect, and EAUC50 (EC50) is the AUCtau,ss of LC350189 at which 50% of the maximum effect is achieved.
The PK and PD evaluations were performed using Phoenix™ WinNonlin® (Version 6.2; Certara, Princeton, NJ, USA).
Safety evaluation
Safety and tolerability were evaluated based on physical examination, vital signs (systolic and diastolic blood pressure, pulse rate), 12-lead ECG (including continuous ECG monitoring), impedance cardiography, and laboratory tests (hematology, clinical chemistry, thyroid function test, coagulation, urinalysis) throughout the study. The Triage® NGAL test was used to evaluate renal toxicity. Adverse events (AEs) were collected using a questionnaire or based on the subjects’ spontaneous reports.
Statistical analysis
Given the exploratory nature of the present study, the sample size was not calculated based on formal hypotheses testing. Instead, eight subjects for active drug per dose group were deemed appropriate because six to 15 is the number of subjects commonly enrolled per dose group in the conventional first in man studies that evaluate the PK characteristics and safety profiles.22 The SPSS® software, Version 21.0 (SPSS Inc., Chicago, IL), was used for the statistical analysis.
Demographic characteristics and PK and PD parameters were summarized using descriptive statistics. Using linear regression analysis (power regression model) for log-transformed Cmax and AUClast (or AUCtau,ss in part II),23,24 dose-linearity was declared when the slope was not different from 1, and its confidence limit fell entirely within the range of 0.8 and 1.25.24 To affirm steady state has been attained for LC350189, its trough concentrations on days 2–6 were compared to that on day 7 using the paired t-test. P-values <0.05 were considered to be statistically significant. The effect of food on the PK of LC350189 was examined using the geometric mean ratio of AUClast and Cmax with and without food and the corresponding 90% confidence interval (CI). To this end, Cmax and AUClast were log-transformed and entered into a mixed-effects analysis of variance model, where period and treatment were fixed effects and subject was random effect. The PD parameters were compared statistically by dose and treatment groups using the Kruskal–Wallis test and Jonckheere–Terpstra test.
Results
Study participants
In part I, 71 subjects were randomized with 66 subjects completing the study as defined in the study protocol (the PK/PD data set), and their mean (standard deviation) age, body weight, and BMI were 26.6 (3.9) years, 69.7 (6.7) kg, and 22.8 (1.9) kg/m2, respectively. In part II, 58 subjects were randomized with 56 subjects completing the study as defined in the study protocol (the PK/PD data set), and their mean (standard deviation) age, body weight, and BMI were 26.6 (3.6) years, 68.0 (7.3) kg, and 22.5 (2.0) kg/m2, respectively. On the other hand, the safety analysis data set included 67 and 58 subjects in parts I and II, respectively, including a subject who took only placebo on day -1 (part I) and two subjects who dropped out (part II).
Pharmacodynamics
After a single administration of LC350189, SUA decreased in a dose-dependent manner, which lasted at least 24 hours post dose (Figure 2A). Furthermore, the 24-hour mean concentration (Cmean,24) of SUA decreased in a dose-dependent manner (Figure 3A) such that the percentage decrease from baseline in Cmean,24 in the 600 mg dose group was almost 10% points greater than that of 200 mg group (-31.7% vs -22.9%, Table 1). Similarly, after multiple oral administrations of LC350189, SUA concentrations decreased steadily in a dose-dependent manner while on medication, which appeared to return to the baseline value over several days after discontinuing the medication (Figure 2C). Additionally, percentage decrease from baseline in Cmean,24 in the LC350189 800 mg group was almost double of that in the febuxostat group (-91.2% vs -58.8%, Figure 4A). As the SUA concentration decreased after the drug administration, the amount of uric acid excreted in the urine also decreased in a dose-dependent manner (Table 1, Figures 3B and 4B), although the renal clearance of uric acid was comparable before and after the drug administration (Table 1). After food intake, uric acid level changed similarly between under fasting and fed status when we calculated changes from 0 hour of day 1 and day 8 in the 200 mg dose group (Figure 2B). The percentage decrease in Cmean,24 of uric acid was a little higher after food intake (Table 1).
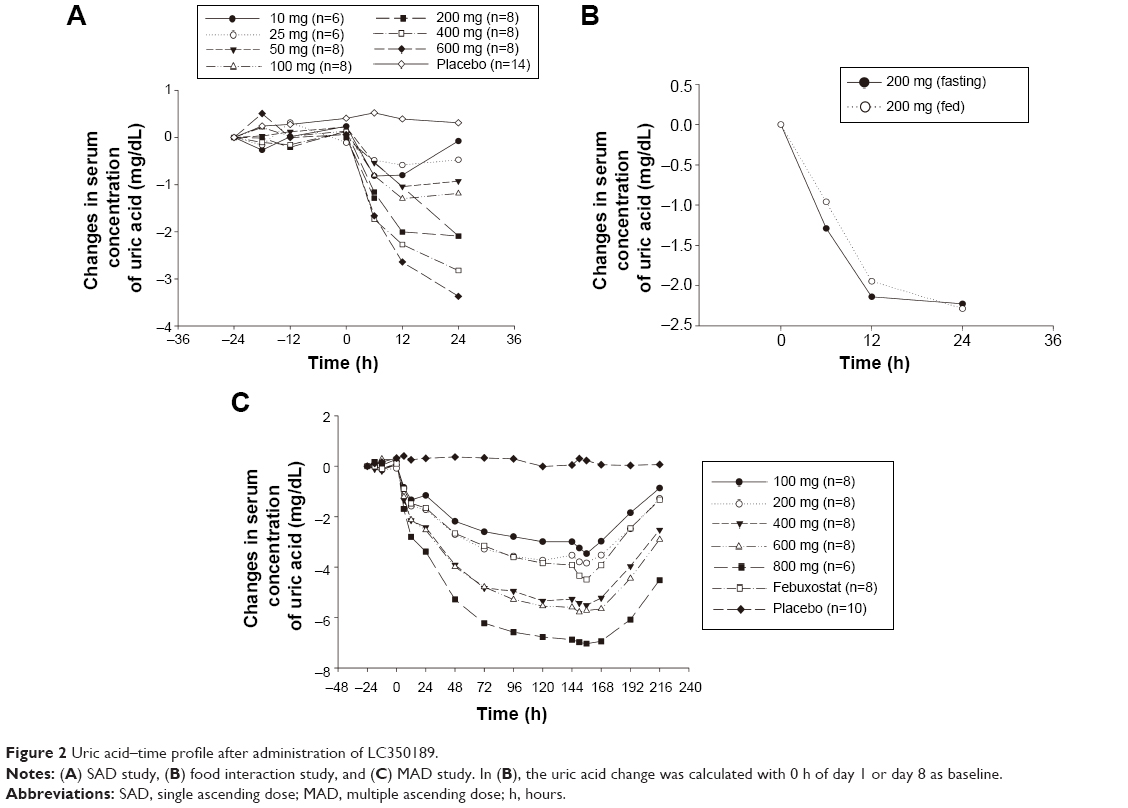 | Figure 2 Uric acid–time profile after administration of LC350189. |
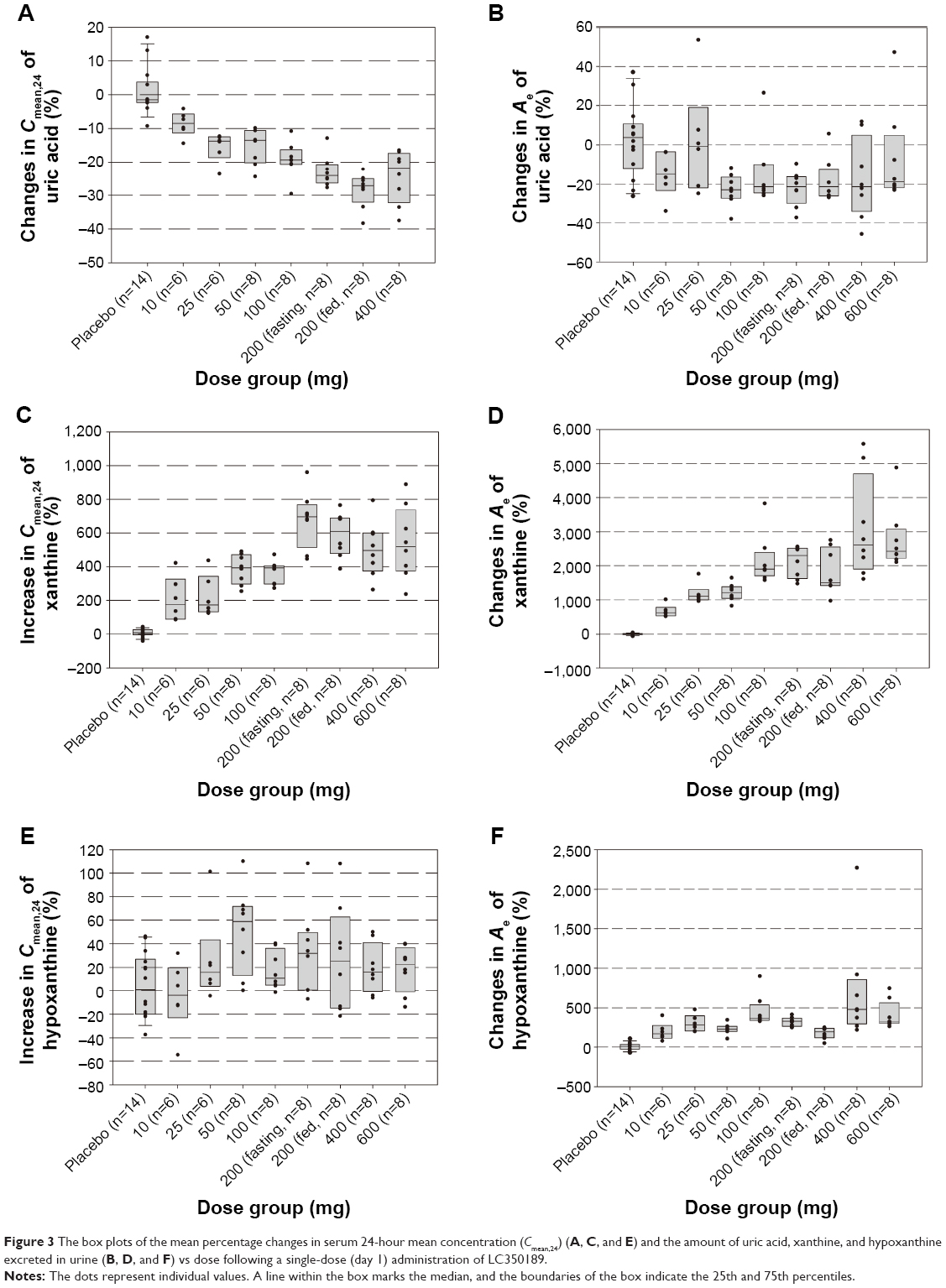 | Figure 3 The box plots of the mean percentage changes in serum 24-hour mean concentration (Cmean,24) (A, C, and E) and the amount of uric acid, xanthine, and hypoxanthine excreted in urine (B, D, and F) vs dose following a single-dose (day 1) administration of LC350189. |
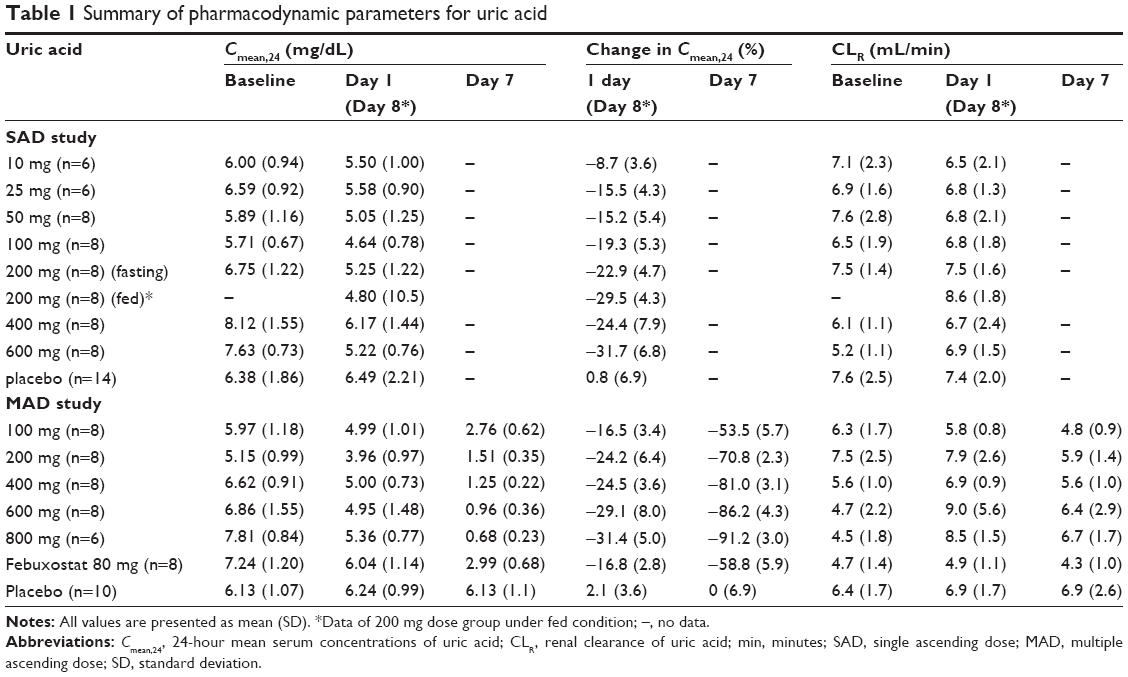 | Table 1 Summary of pharmacodynamic parameters for uric acid |
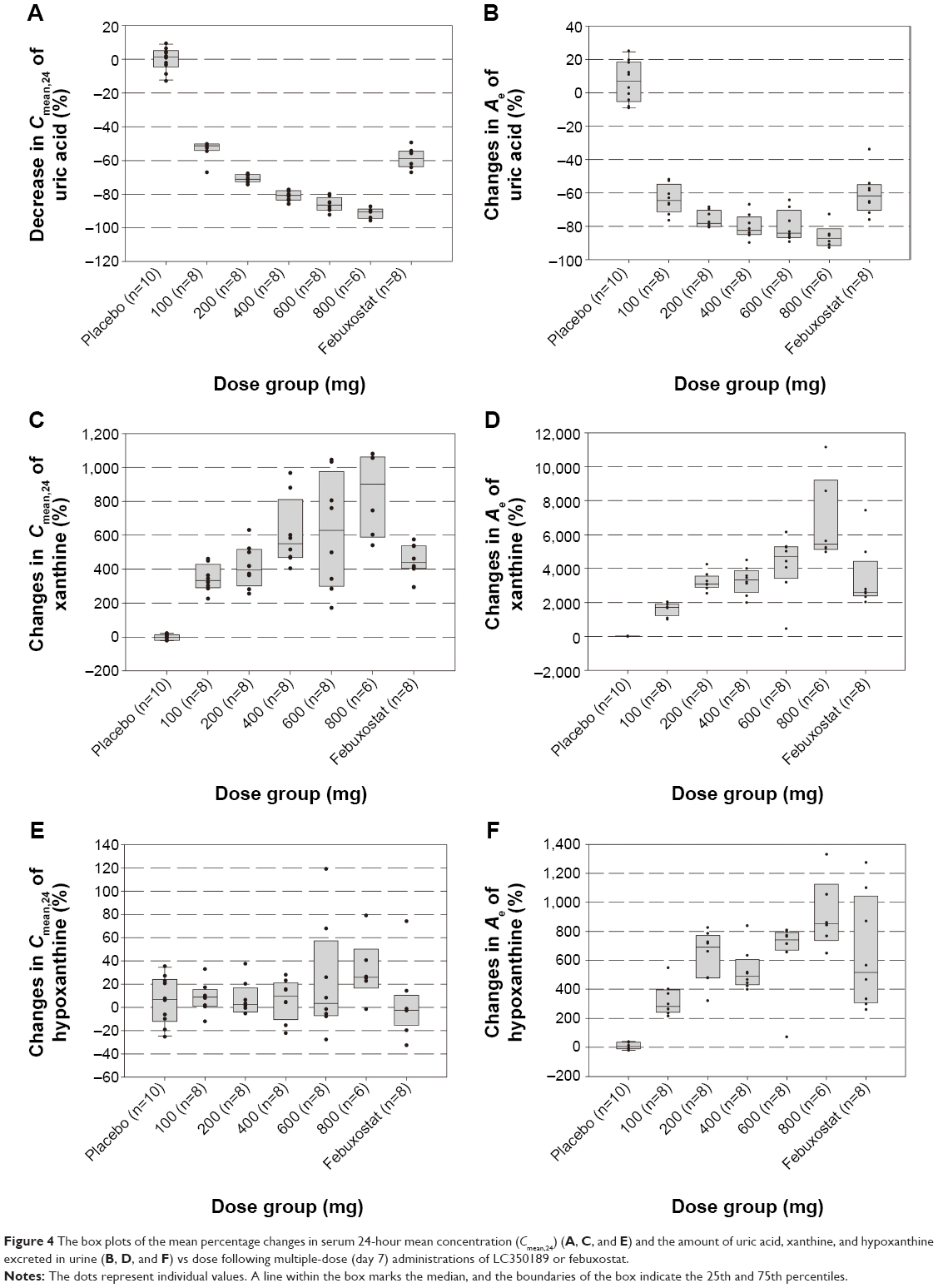 | Figure 4 The box plots of the mean percentage changes in serum 24-hour mean concentration (Cmean,24) (A, C, and E) and the amount of uric acid, xanthine, and hypoxanthine excreted in urine (B, D, and F) vs dose following multiple-dose (day 7) administrations of LC350189 or febuxostat. |
After a single and multiple oral administrations of LC350189, the Cmean,24 of serum xanthine increased drastically up to greater than seven times from the baseline in a dose-dependent manner (Table 2 and Figures 3C and 4C), while the amount of xanthine excreted in the urine increased at a much greater level than the increase in serum xanthine concentrations (Figures 3D and 4D). The renal clearance of xanthine was also increased in all of the dose groups (Table 2). Although the Cmean,24 of serum hypoxanthine increased in all of the dose groups except for the 10 mg single-dose group, the pattern and extent of increase were not so apparent as that seen with serum xanthine (Table 3 and Figures 3E and 4E). However, the amount of hypoxanthine excreted in urine also increased as the dose was increased (Table 3 and Figures 3F and 4F). The renal clearance of hypoxanthine also increased after the administration of LC350189.
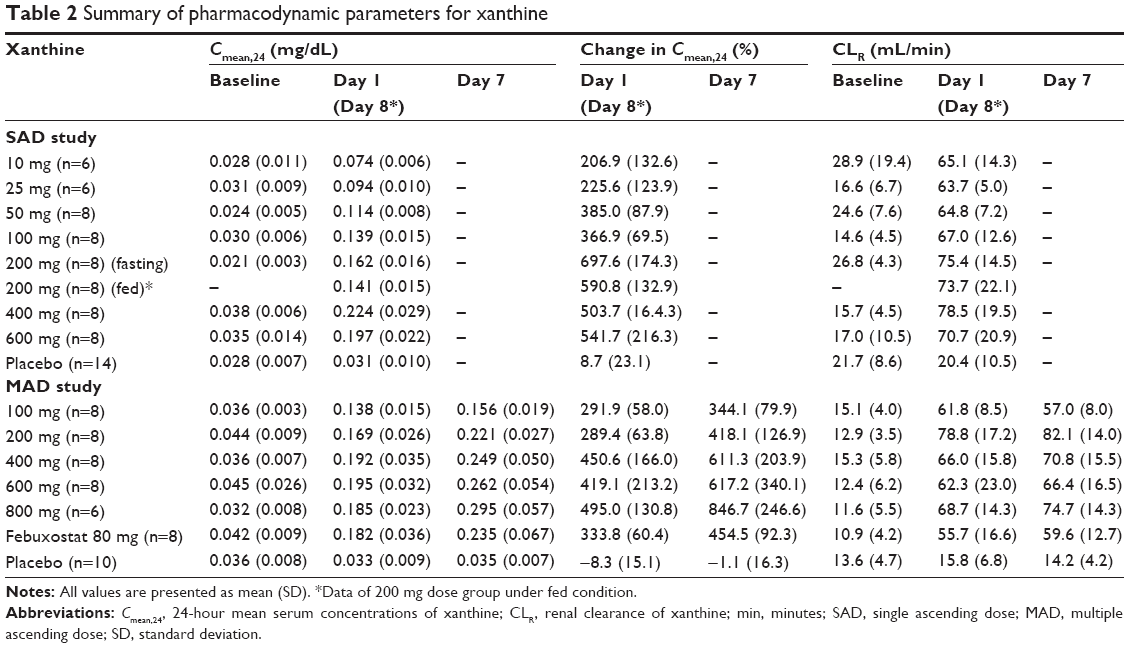 | Table 2 Summary of pharmacodynamic parameters for xanthine |
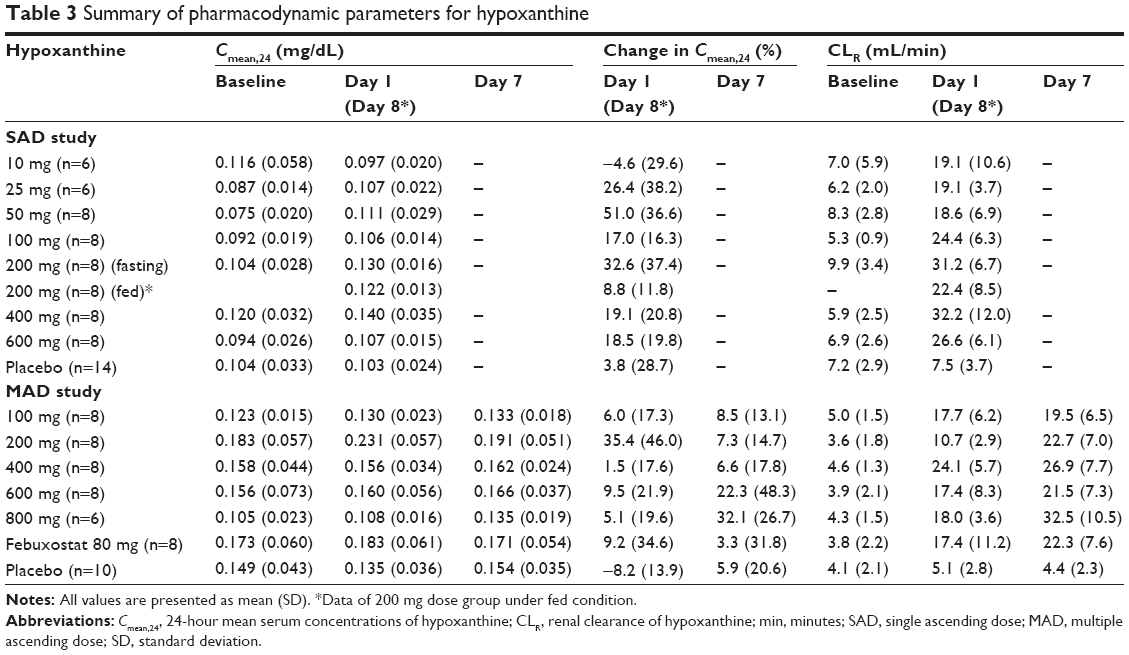 | Table 3 Summary of pharmacodynamic parameters for hypoxanthine |
Pharmacokinetics
LC350189 was rapidly absorbed after both a single and multiple oral administrations, reaching the peak plasma concentration approximately 3 hours post dose (Figure 5 and Table 4). Cmax and AUClast were judged to have increased in a dose-proportional manner based on the finding that the 95% CIs of the slope of the log-transformed Cmax and AUClast included 1.0 (Cmax 0.97–1.08, AUClast 0.99–1.11). Approximately, 20%–30% of LC350189 was excreted in the urine. Food delayed the absorption of LC350189 (~2 hours), and it also decreased Cmax by 38% (Figure 6); the geometric men ratio (90% CI) of Cmax (fed vs fasting status) was 0.624 (0.485–0.802). However, the overall exposure to LC350189 was comparable between the fasted and fed states, ie, the geometric mean ratio (90% CI) of AUClast with and without food was 0.973 (0.837–1.131). Steady state was reached within 2–3 days after multiple administrations of LC350189 (Figure 6 and Table 5). The 95% CI of the slope of the log-transformed Cmax and AUCtau,ss was 0.64–0.89 and 0.78–1.06, respectively, meaning that exposure to LC350189 was dose proportional. The fraction of the drug excreted unchanged in urine was similar to that in the SAD study.
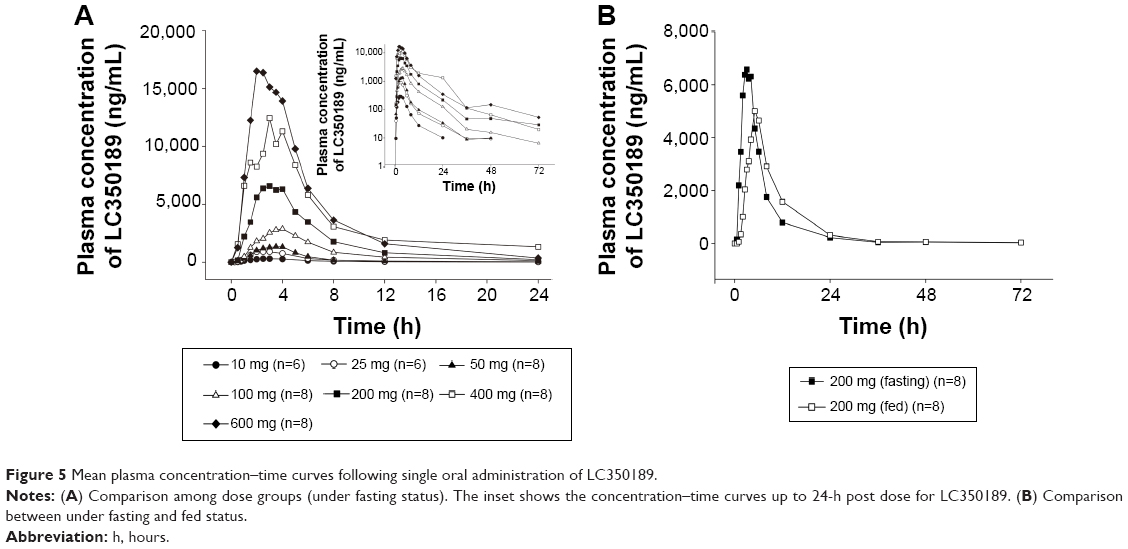 | Figure 5 Mean plasma concentration–time curves following single oral administration of LC350189. |
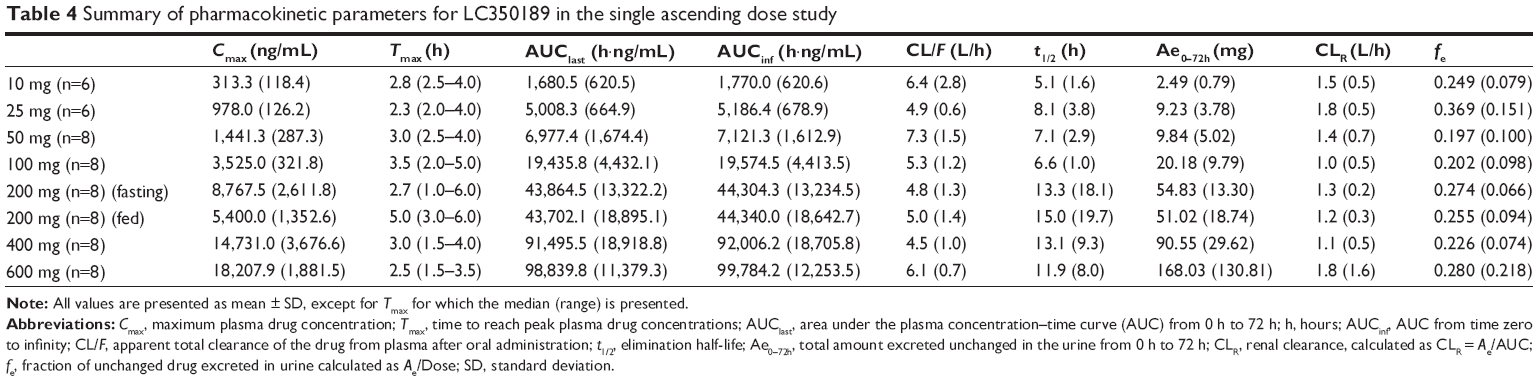 | Table 4 Summary of pharmacokinetic parameters for LC350189 in the single ascending dose study |
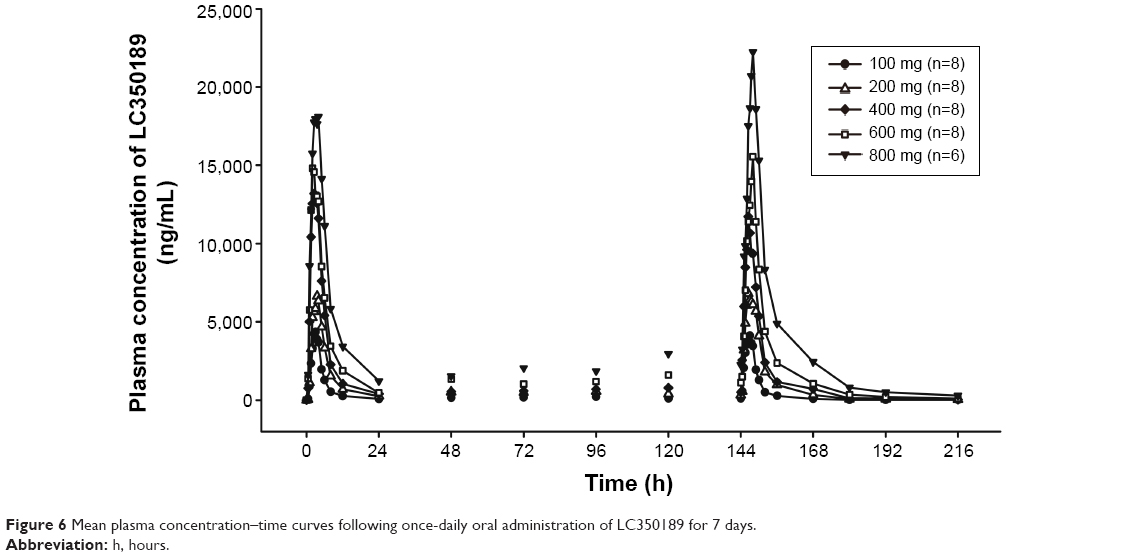 | Figure 6 Mean plasma concentration–time curves following once-daily oral administration of LC350189 for 7 days. |
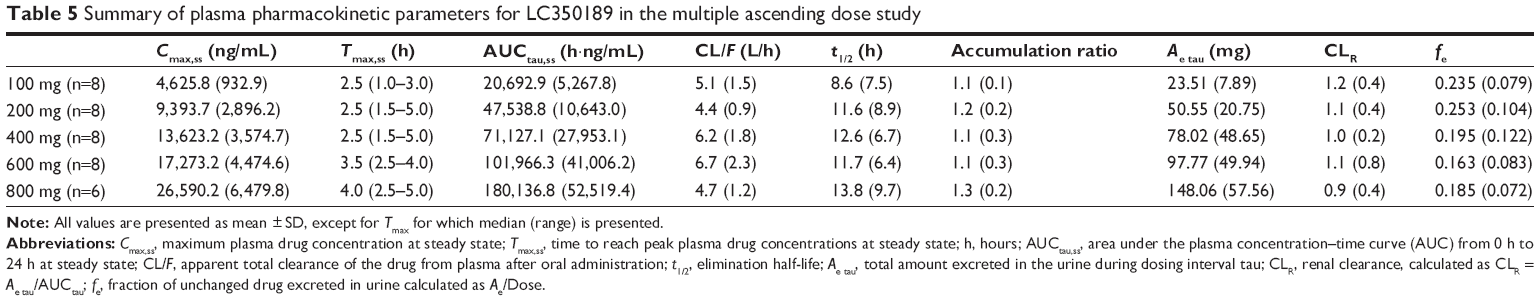 | Table 5 Summary of plasma pharmacokinetic parameters for LC350189 in the multiple ascending dose study |
PK–PD relationship of LC350189
A simple Emax model well described the relationship between the AUCtau,ss of LC350189 on day 7 after multiple administrations and the percentage decrease in the Cmean,24 of SUA (Figure 7), where Emax and EAUC50 were estimated to be 100.1% (95% CI: 94.8%–105.4%) and 16,942.2 h·ng/mL (13,029.6–20,854.8 h·ng/mL), respectively.
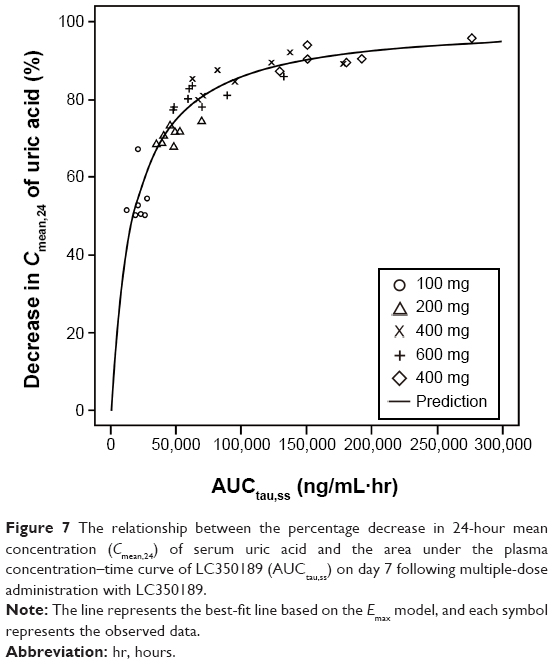 | Figure 7 The relationship between the percentage decrease in 24-hour mean concentration (Cmean,24) of serum uric acid and the area under the plasma concentration–time curve of LC350189 (AUCtau,ss) on day 7 following multiple-dose administration with LC350189. |
Safety
A total of 17 and 20 AEs were reported by eleven (out of 67, 16.4%) and 15 (out of 58, 25.9%) subjects in parts I and II, respectively, of which eleven and six cases were considered as drug related, respectively (Table 6). The most common organ class affected was the gastrointestinal system, where four cases of diarrhea were reported (two cases in the LC350189 dose groups). The frequency of AEs was comparable among the dose groups. There was no serious AE reported, and all AEs were mild in intensity. There were no clinically significant changes in vital signs, ECGs, physical examination, or clinical laboratory tests, including the NGAL test.
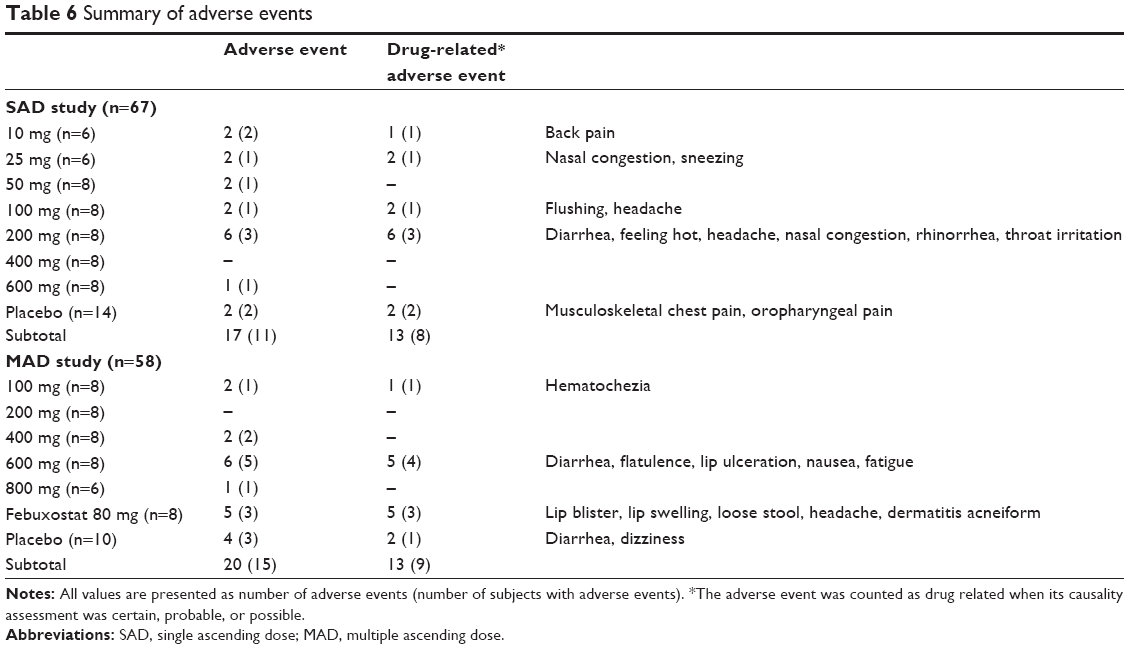 | Table 6 Summary of adverse events |
Discussion
Our results showed that the level of SUA decreased significantly from baseline in a dose-dependent manner after LC350189 administrations.
The level of uric acid decreased until 12 hours or 24 hours post administration, depending on the dose strength in the SAD study, even though the concentration of LC350189 decreased rapidly after Tmax, reaching concentrations that are less than 10% of Cmax in each dose group. A similar phenomenon occurred in the MAD study, where the uric acid level decreased until 12 hours post administration. It is hypothesized that LC350189 binds to XO for a few hours before release, during which LC350189 inhibits the enzyme’s activity. Inhibition reaches steady state after five doses of LC350189. After the last dose of LC350189, the SUA level increased at a relatively consistent rate regardless of the doses (Figure 2), suggesting zero-order kinetics for XO in this dose range.
After multiple administrations of LC350189, the maximum percentage decrease from baseline in Cmean,24 for SUA was 91.2%, while it was 58.8% in the febuxostat group. In a previously reported multiple-dose study of febuxostat, the maximum percentage decrease from baseline in Cmean,24 was 27%–76% in the dose range of 10 mg–240 mg, and it was ~40% in the 40 mg dose group and 60% in the 90 mg dose group.25 Considering that the approved doses of febuxostat are 40 mg and 80 mg in the USA, it is expected that LC350189 in the range of 100 mg and 200 mg would have similar effects to those of febuxostat at 80 mg on SUA concentrations (Figures 2 and 4).
In this study, exposure to LC350189 under the fed state remained similar to that of the fasted state, and the changes in SUA concentration were also similar between the fasted and fed states (Figure 2B). Even though the percentage decrease from baseline in the Cmean,24 for SUA was a little greater after food intake (Table 1), it may be because the SUA concentrations at baseline (ie, on days 1 and 8) were different within subjects, ie, decrease in SUA concentration did not return to the pretreatment level after the washout period. It is one of the limitations of this study, which states that there was no separate baseline evaluation for Cmean,24 right before day 8. Therefore, although it is expected that LC350189 would have similar SUA lowering effect regardless of food intake, further studies are warranted to adequately investigate the food effect on the PDs of LC350189.
Because LC350189 inhibits XO, the serum level of xanthine increased as expected. While the Cmean,24 of SUA decreased from baseline by 8.7%–31.7% after a single dose of LC350189, the Cmean,24 of xanthine increased two- to seven-fold. On the other hand, the Cmean,24 of SUA decreased from baseline by 53.5%–91.2% after multiple administrations of LC350189, whereas the Cmean,24 of xanthine increased three- to eight-fold on day 7. Thus, the change in xanthine appears to be greater than that in SUA, which may be because the concentration of xanthine itself is normally approximately one-hundredth that of SUA. While the amount of uric acid excreted in the urine decreased slightly after a single LC350189 administration, it decreased to one-third to one-tenth of the baseline level after multiple administrations. In contrast, the amount of xanthine excreted in the urine increased from baseline after a single administration and multiple administrations of LC351089 (Table S2). Increased urinary excretion of xanthine can cause acute renal failure when treating tumor lysis syndrome in cancer patients.26–29 The amount of xanthine excreted in the urine in the present study was similar to that reported previously in the febuxostat study.25 Furthermore, no AE was considered related to an increased xanthine level in the present study. However, the level of increase in serum xanthine concentration by XO inhibition, including LC350189, should be taken into account when selecting their optimal dose.
Neither a single dose nor multiple doses of LC350189 change the renal clearance of uric acid, whereas the renal clearance of xanthine and hypoxanthine was increased by LC350189 (Tables 1–3). It was possibly because the extent of increase in the amount excreted in the urine was greater than that of Cmean,24 for xanthine. This phenomenon was also observed with allopurinol.30 It is known that xanthine and hypoxanthine filtered in the glomerulus is reabsorbed and secreted into the renal tubule.31 If reabsorption involves a transporter, the increase in the renal clearance of xanthine and hypoxanthine could be caused by saturated reabsorption.
The increase in serum xanthine concentration was greater in the higher dose groups, whereas the increase in serum hypoxanthine concentration was not only unapparent and inconsistent but also its extent was small compared with xanthine. Xanthine is produced not only from hypoxanthine but also from guanine. Thus, even if the production of xanthine is inhibited by the XO inhibitor, xanthine could still be produced from guanine (Figure 1). The unapparent increase in serum hypoxanthine concentrations by LC350189 in the present study could be due to the combination of the increased renal clearance of hypoxanthine and the decreased de novo synthesis of hypoxanthine. Another possibility is that the binding affinity of xanthine to XO could be different than that of hypoxanthine, which renders the extent of inhibition by the XO inhibitor variable.
Based on the Emax model developed in the present study, the inhibition of XO by LC350189 approaches 100%, whereas febuxostat inhibited 81.4% of the enzyme.25 Multiple doses of LC350189 at 100 mg appeared to inhibit 50% of the XO activity because the AUC at steady is close to the EAUC50 of 16,942.2 h·ng/mL.
Because the range of fluctuation in uric acid level after multiple dosing with once-daily dosing was small, the use of LC350189 with once-daily dosing regimen would be suitable, and it could be confirmed in the later phase of the drug development.
In the low-dose (10 mg, 25 mg, and 50 mg) groups of the SAD study, the concentrations of LC350189 were detected only up to 48 hours and the terminal slopes were steeper when compared to higher doses. The 100 mg dose group also had very low concentrations, close to the lower limit of quantification (LLOQ) (5 ng/mL) around the last sampling time point (72 hours post dose). This led to truncation of the terminal elimination phase, such that the range of the mean terminal half-life of the lower dose groups (10–100 mg) was smaller (5.18.1 hours) compared to that of higher doses (11.9–13.3 hours). In the MAD study, the same phenomenon was observed.
LC350189 was the major circulating component of the plasma and urine in the preclinical study, and only the concentrations of LC350189 were measured in this study, not its metabolites. Approximately 20%–30% of orally administered LC350189 was excreted into the urine in an unchanged form. A double peak was observed in some of the individual plasma profiles of LC350189, suggesting that LC350189 was present in the enterohepatic circulation. LC350189 was found to be metabolically stable in the in vitro liver microsome study and also when incubated with CYP isoenzymes, indicating that CYP enzymes were not responsible for LC350189 metabolism.
The Tmax of LC350189 (~3 hours) was slightly longer than that of febuxostat (~1 hour).25 The t1/2 of LC350189 (~10 hours) was similar to that of febuxostat (mean ± SD, 40 mg: 10.3±7.4 hours; 70 mg: 12.5±7.7 hours). Febuxostat can be taken with or without food, because there is no clinically significant change in the percentage decrease in SUA concentration, with a slight decrease in AUC following multiple 80 mg once-daily doses after a high-fat meal.32
Orally administered LC351089 was well tolerated. There was no allergic reaction similar to that reported in the previous febuxostat multiple-dose study (six out of 118 subjects who were treated with febuxostat).25 The vasodilatory symptoms reported in the previous febuxostat study at higher doses were reported in two subjects in the present study (flushing and feeling hot after a single dose). There were no signals of renal toxicity according to the results of the NGAL test.
Conclusion
In this study, LC350189 was well tolerated after a single oral administration and multiple oral administrations in the dose range of 10–800 mg. LC350189 decreased SUA concentrations and reduced the amount of uric acid excreted in the urine. Particularly, the decrease in SUA concentration after multiple oral doses of LC350189 200 mg was comparable to that of febuxostat at 80 mg. Once-daily LC350189 can be developed as an effective treatment option for patients with hyperuricemia or gout.
Acknowledgments
This study was sponsored by a research grant from LG Life Sciences, Seoul, Korea. The authors thank Tae Hun Kim and Jin-ah Hwang at LG Life Sciences for their invaluable comments on the manuscript. This work has been presented as a poster at the 2014 Annual Meeting of American Society for Clinical Pharmacology and Therapeutics.
Disclosure
The authors declare no conflicts of interest.
References
Roddy E, Zhang W, Doherty M. Is gout associated with reduced quality of life? A case–control study. Rheumatology. 2007;46(9):1441–1444. | ||
Singh JA, Strand V. Gout is associated with more comorbidities, poorer health-related quality of life and higher healthcare utilisation in US veterans. Ann Rheum Dis. 2008;67(9):1310–1316. | ||
Chandratre P, Roddy E, Clarson L, Richardson J, Hider SL, Mallen CD. Health-related quality of life in gout: a systematic review. Rheumatology. 2013;52(11):2031–2040. | ||
Neogi T. Clinical practice. Gout. N Engl J Med. 2011;364(5):443–452. | ||
Roddy E, Choi HK. Epidemiology of gout. Rheum Dis Clin North Am. 2014;40(2):155–175. | ||
Lee CH, Sung NY. The prevalence and features of Korean gout patients using the national health insurance corporation database. J Rheum Dis. 2011;18(2):94–100. | ||
Shields GE, Beard SM. A systematic review of the economic and humanistic burden of gout. Pharmacoeconomics. 2015. Epub 2015 May 13. | ||
Wu EQ, Forsythe A, Guerin A, Yu AP, Latremouille-Viau D, Tsaneva M. Comorbidity burden, healthcare resource utilization, and costs in chronic gout patients refractory to conventional urate-lowering therapy. Am J Ther. 2012;19(6):e157–e166. | ||
Brook RA, Kleinman NL, Patel PA, et al. The economic burden of gout on an employed population. Curr Med Res Opin. 2006;22(7):1381–1389. | ||
Wertheimer A, Morlock R, Becker MA. A revised estimate of the burden of illness of gout. Curr Ther Res Clin Exp. 2013;75:1–4. | ||
Pluim HJ, van Deuren M, Wetzels JF. The allopurinol hypersensitivity syndrome. Neth J Med. 1998;52(3):107–110. | ||
Lee HY, Ariyasinghe JT, Thirumoorthy T. Allopurinol hypersensitivity syndrome: a preventable severe cutaneous adverse reaction? Singapore Med J. 2008;49(5):384–387. | ||
Hande KR, Noone RM, Stone WJ. Severe allopurinol toxicity. Description and guidelines for prevention in patients with renal insufficiency. Am J Med. 1984;76(1):47–56. | ||
Perez-Ruiz F, Calabozo M, Fernandez-Lopez MJ, et al. Treatment of chronic gout in patients with renal function impairment: an open, randomized, actively controlled study. J Clin Rheumatol. 1999;5(2):49–55. | ||
Vazquez-Mellado J, Morales EM, Pacheco-Tena C, Burgos-Vargas R. Relation between adverse events associated with allopurinol and renal function in patients with gout. Ann Rheum Dis. 2001;60(10):981–983. | ||
Edwards NL. Treatment-failure gout: a moving target. Arthritis Rheum. 2008;58(9):2587–2590. | ||
Chao J, Terkeltaub R. A critical reappraisal of allopurinol dosing, safety, and efficacy for hyperuricemia in gout. Curr Rheumatol Rep. 2009;11(2):135–140. | ||
Jackson RL, Hunt B, MacDonald PA. The efficacy and safety of febuxostat for urate lowering in gout patients >/=65 years of age. BMC Geriatr. 2012;12:11. | ||
Becker MA, Schumacher HR Jr, Wortmann RL, et al. Febuxostat compared with allopurinol in patients with hyperuricemia and gout. N Engl J Med. 2005;353(23):2450–2461. | ||
Chohan S, Becker MA, MacDonald PA, Chefo S, Jackson RL. Women with gout: efficacy and safety of urate-lowering with febuxostat and allopurinol. Arthritis Care Res (Hoboken). 2012;64(2):256–261. | ||
Garcia-Valladares I, Khan T, Espinoza LR. Efficacy and safety of febuxostat in patients with hyperuricemia and gout. Ther Adv Musculoskelet Dis. 2011;3(5):245–253. | ||
Gallin JI. Principles and Practice of Clinical Research. San Diego, CA: Academic Press; 2002. | ||
Hummel J, McKendrick S, Brindley C, French R. Exploratory assessment of dose proportionality: review of current approaches and proposal for a practical criterion. Pharm Stat. 2009;8(1):38–49. | ||
Smith BP, Vandenhende FR, DeSante KA, et al. Confidence interval criteria for assessment of dose proportionality. Pharm Res. 2000;17(10):1278–1283. | ||
Khosravan R, Grabowski BA, Wu JT, Joseph-Ridge N, Vernillet L. Pharmacokinetics, pharmacodynamics and safety of febuxostat, a non-purine selective inhibitor of xanthine oxidase, in a dose escalation study in healthy subjects. Clin Pharmacokinet. 2006;45(8):821–841. | ||
Wyngaarden JB. Allopurinol and xanthine nephropathy. N Engl J Med. 1970;283(7):371–372. | ||
Band PR, Silverberg DS, Henderson JF, et al. Xanthine nephropathy in a patient with lymphosarcoma treated with allopurinol. N Engl J Med. 1970;283(7):354–357. | ||
Gomez GA, Stutzman L, Chu TM. Xanthine nephropathy during chemotherapy in deficiency of hypoxanthine-guanine phosphoribosyltransferase. Arch Intern Med. 1978;138(6):1017–1019. | ||
LaRosa C, McMullen L, Bakdash S, et al. Acute renal failure from xanthine nephropathy during management of acute leukemia. Pediatr Nephrol. 2007;22(1):132–135. | ||
Turnheim K, Krivanek P, Oberbauer R. Pharmacokinetics and pharmacodynamics of allopurinol in elderly and young subjects. Br J Clin Pharmacol. 1999;48(4):501–509. | ||
Goldfinger S, Klinenberg JR, Seegmiller JE. The renal excretion of oxypurines. J Clin Invest. 1965;44:623–628. | ||
Khosravan R, Grabowski B, Wu JT, Joseph-Ridge N, Vernillet L. Effect of food or antacid on pharmacokinetics and pharmacodynamics of febuxostat in healthy subjects. Br J Clin Pharmacol. 2008;65(3):355–363. |
Supplementary materials
Analytical methods
Concentrations of LC350189 in the plasma and urine were determined by a validated method using high-performance liquid chromatography (SCL-10AVP; Shimadzu, Kyoto, Japan) coupled with tandem mass spectrometry (4000 QTRAP®; AB Sciex, Darmstadt, Germany). The internal standard for LC350189 was LC35-0303. The plasma samples containing the internal standard were deproteinated using acetonitrile. A Luna C8 column (3 μm, 30 mm ×2 mm; Phenomenex, Torrance, CA, USA) was used with a gradient mobile phase consisting of 0.1% formic acid in distilled water (DW) and 0.1% formic acid in acetonitrile. The tandem mass spectrometry (MS/MS) system was operated in positive ionization mode along with a turbo spray mode. The lower limit of quantification (LLOQ) for LC350189 was 5 ng/mL. The linear range of the calibration curves for plasma and urine was 5–2,000 ng/mL (r2≥0.9954 for plasma and r2≥0.9936 for urine). In plasma, the interassay accuracy ranged from 97.1% to 103.4%, and the precision ranged from 4.4% to 10.5%. In urine, the interassay accuracy ranged from 92.7% to 103.9% and the precision ranged from 2.2% to 9.8%.
Serum concentrations of uric acid, xanthine, and hypoxanthine were determined by a validated method similar to the method used for the determination of LC350189 concentrations. The internal standards were uric acid, xanthine, and hypoxanthine. The plasma samples containing the internal standard were deproteinated using methanol. A Hypersil GOLD column (5 μm, 100 mm ×2.1 mm, Thermo Scientific, Waltham, MA, USA) was used with a gradient mobile phase consisting of 0.1% formic acid in DW and 0.1% formic acid in methanol. The MS/MS system was operated in negative ionization mode along with a turbo spray mode. The LLOQs were 1 μg/mL for uric acid and 30 ng/mL for xanthine and hypoxanthine. The linear ranges of the calibration curves were as follows: 1–100 μg/mL for uric acid (r2≥0.997) and 30–3,000 ng/mL for xanthine and hypoxanthine (r2≥0.9978 for xanthine and r2≥0.9949 for hypoxanthine). The interassay accuracy ranged from 97.9% to 104.4% for uric acid, from 96.4% to 100.1% for xanthine, and from 97.5% to 100.9% for hypoxanthine. The interassay precision ranged from 4.9% to 6.7% for uric acid, from 5.1% to 12.2% for xanthine, and from 5.7% to 8.0% for hypoxanthine.
Pharmacokinetic and pharmacodynamic assessments
The individual pharmacokinetic (PK) parameters were obtained by noncompartmental methods using Phoenix™ WinNonlin® (Version 6.2; Certara, Princeton, NJ, USA). The actual collection times were used in the PK analysis. The maximum drug concentrations in plasma (Cmax) and the time at which Cmax was reached (Tmax) were determined directly from the observed values. The terminal half-life (t1/2) was calculated as follows: t1/2= ln(2)/λz. The area under the time–concentration curves (AUC) (from time 0 to the last measurable time (AUClast) and within a dosing interval in a steady state (AUCtau,ss)) were calculated using the linear trapezoidal rule for ascending concentrations and the log trapezoidal method for descending concentrations. The AUC extrapolated to infinity (AUCinf) was estimated as the sum of AUClast and the extrapolated area given by the quotient of the last measured concentration and λz (Clast/λz). The apparent clearance (CL/F) was calculated as dose/AUCinf, and the apparent clearance in a steady state (CLss/F) was calculated as dose/AUCtau,ss. The cumulative amount of drug excreted in urine (Ae) was computed from the urine volume and urine drug concentration. The fraction of unchanged drug excreted in urine (fe) was calculated as follows: fe = Ae/Dose. The individual renal clearance (CLR) was calculated as follows: CLR = Ae/AUC. For the multiple ascending dose study, additional parameters determined in the steady state included the accumulation ratio, defined as the ratio of AUCtau,ss (day 7, from 144 hours to 168 hours)/AUCtau (day 1, from 0 hours to 24 hours).
Pharmacodynamic (PD) parameters were estimated for uric acid, xanthine, and hypoxanthine. The AUC0–24 was calculated by noncompartmental methods using Phoenix™ WinNonlin® (Version 6.2; Pharsight Corporation). The 24-hour mean serum concentrations (Cmean,24) were calculated by dividing AUC0–24 by 24. The cumulative amount of uric acid, xanthine, and hypoxanthine excreted in urine (Ae) was computed using the urine volume and urine compound concentration. The individual CLR was calculated as follows: CLR = Ae/AUC. The % change from baseline (day 1) in Cmean,24 for uric acid was calculated as follows: (Cmean,base − Cmean,1d(or 7d))/Cmean,base ×100.
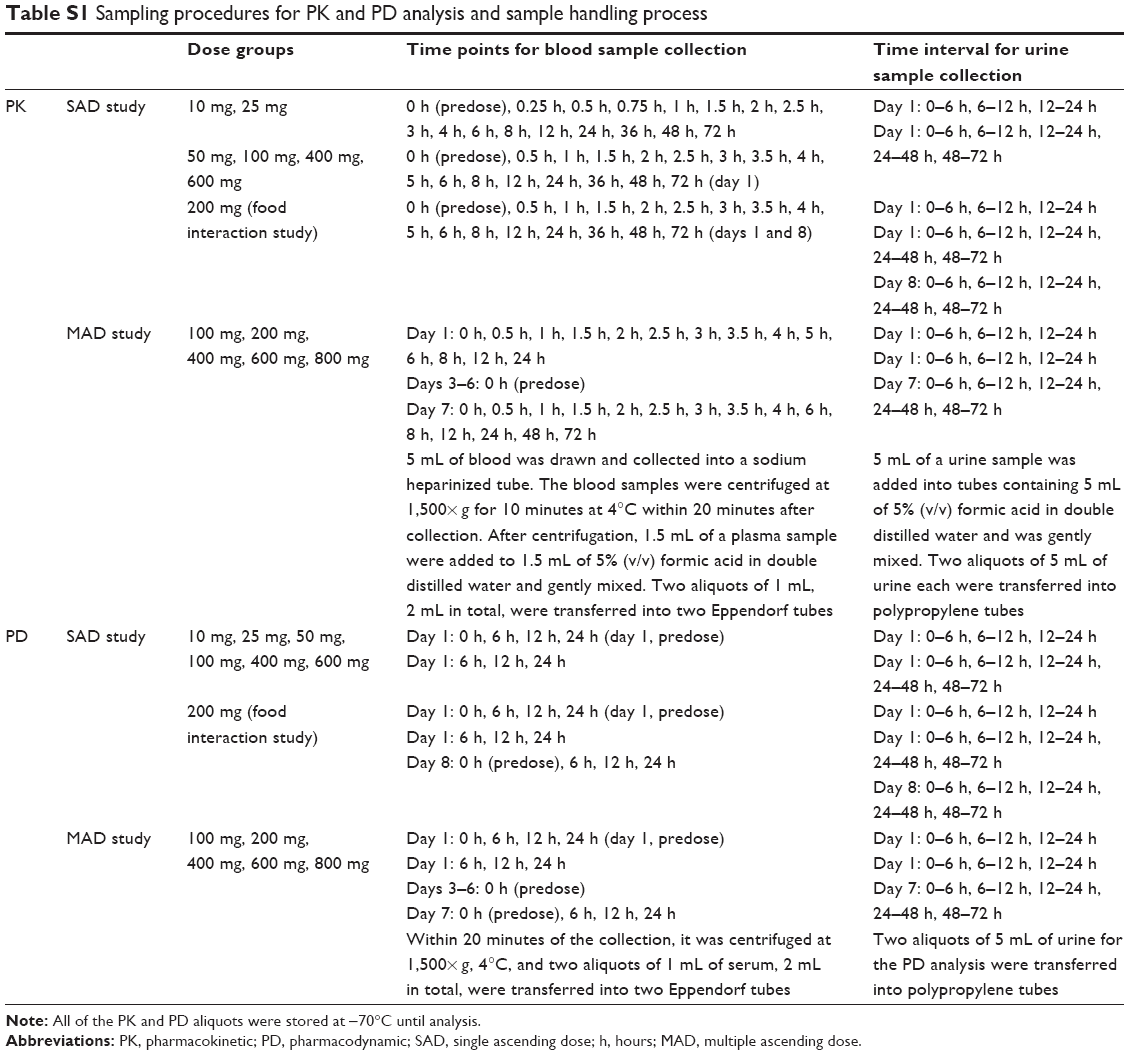 | Table S1 Sampling procedures for PK and PD analysis and sample handling process |
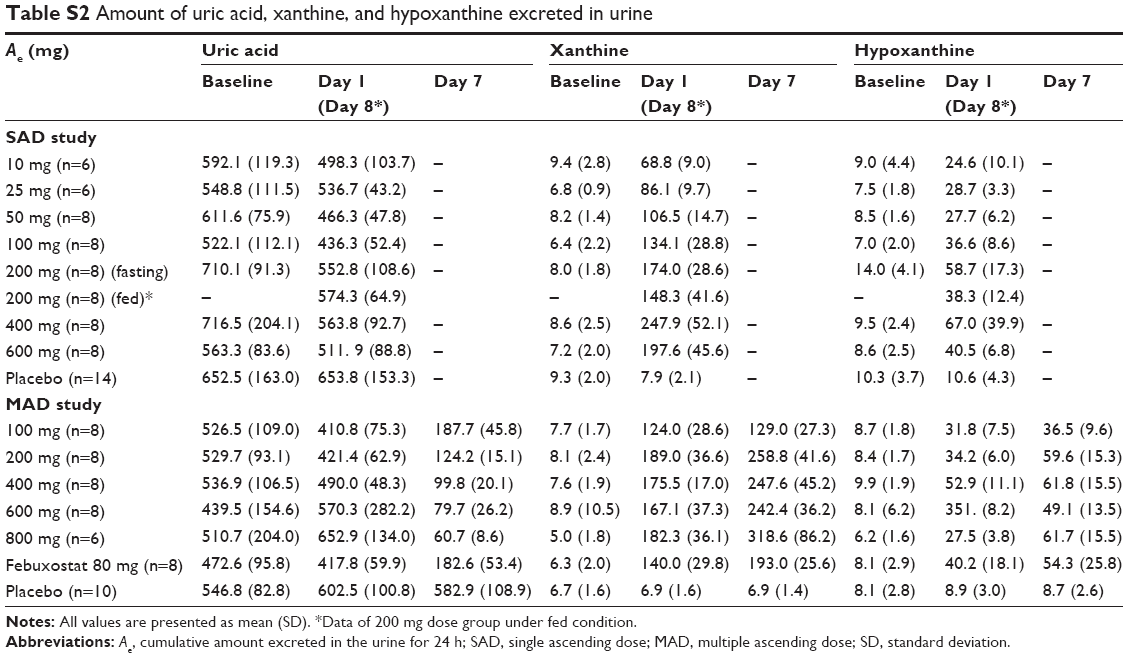 | Table S2 Amount of uric acid, xanthine, and hypoxanthine excreted in urine |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.