")
Back to Journals » Journal of Blood Medicine » Volume 9
Pharmacokinetics-based clinical management of acquired von Willebrand syndrome: a case report
Authors Baldeo CM, Rivera CE, Tun HW , Vishnu P
Received 27 September 2017
Accepted for publication 12 December 2017
Published 19 January 2018 Volume 2018:9 Pages 9—13
DOI https://doi.org/10.2147/JBM.S152663
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Martin H Bluth
Candice M Baldeo, Candido E Rivera, Han W Tun, Prakash Vishnu
Division of Hematology and Medical Oncology, Mayo Clinic, Jacksonville, FL, USA
Abstract: von Willebrand disease (VWD) is a common bleeding disorder caused by defective or low levels of von Willebrand factor (VWF). Although most cases of VWD are caused by genetic mutations, some are acquired due to various disease states. In managing VWD, the aim is to normalize plasma levels of both VWF and factor VIII (FVIII), as this aids in hemostasis. Desmopressin usually corrects VWF level in type 1 VWD by inducing the release of endogenous VWF. In cases where desmopressin is ineffective or cannot be used, transfusion of virally inactivated, plasma-derived VWF/FVIII concentrate or infusion of recombinant VWF (Vonvendi) is indicated. Treatment of acquired von Willebrand syndrome (AVWS) aims to control the underlying disease while regulating life-threatening hemorrhages with infusions of VWF/FVIII concentrate. Wide intrasubject variability in VWF and FVIII levels, particularly in AVWS, necessitates verification of response to treatment by frequent monitoring of the plasmatic VWF level. Clinical pharmacokinetics of VWF may facilitate calculation of the necessary loading and maintenance doses of VWF/FVIII concentrate in the management of AVWS patients undergoing surgery, thereby avoiding unnecessary infusion of coagulation factor concentrate.
Keywords: acquired von Willebrand syndrome, bleeding disorder, pharmacokinetics, surgical hemostasis
Introduction
von Willebrand disease (VWD) is a heterogeneous bleeding disorder characterized by quantitative (types 1 and 3) and qualitative (types 2A, 2B, 2M, and 2N) defects in von Willebrand factor (VWF).1 Acquired von Willebrand syndrome (AVWS) is a rare bleeding disorder caused by absence or a structural or functional defect in VWF that can be associated with autoimmune conditions, malignancy, and lymphoproliferative, myeloproliferative, or cardiovascular disorders (Table 1).2,3 AVWS usually occurs at a later age, with a median age of 62 years, in individuals without any personal or family history of bleeding disorders.
 | Table 1 Causes of AVWS Abbreviations: AVWS, acquired von Willebrand syndrome; MGUS, monoclonal gammopathy of undetermined significance. |
In VWD, defective hemostasis involves abnormal platelet adhesion and aggregation and decreased factor VIII (FVIII) levels. Therapy for AVWS is directed at the underlying disorder, while also controlling hemorrhage. Some management options include desmopressin, VWF/FVIII concentrates, cryoprecipitate, extracorporeal immunoadsorption, and chemotherapy for the underlying malignancy-based disorders.4 Plasma-derived, virus-inactivated VWF/FVIII concentrate can also be used to effectively treat VWD.5,6 Approved in 1981, human antihemophilic factor/VWF complex (Humate-P) has become the most widely used factor replacement therapy in VWD. More recently, a recombinant VWF (Vonvendi) was approved by the US Food and Drug Administration for on-demand treatment and control of bleeding episodes in adults with VWD.7 While Humate-P and Vonvendi have been used extensively, their pharmacokinetic profiles can help to manage VWD and other hemostatic disorders more comprehensively.8
The precise activity level of VWF needed for sufficient hemostasis in VWD is still largely unknown. Essentially, the goal is to maintain FVIII levels between 50 and 150 U/dL by adjusting doses and modifying the timing of administration of VWF/FVIII concentrate;9 however, caution must be practiced since this can elevate FVIII levels, increasing the risk of venous thrombosis.10 Conversely, underdosing replacement therapy can hinder adequate control of hemorrhage. Without a doubt, the pharmacokinetic profile of VWF can maximize dosing to adequately control bleeding while minimizing the risk of thrombotic complications. This is particularly important in patients with AVWS, in whom ascertaining an adequate response to infusion of VWF/FVIII concentrate following therapeutic interventions for the underlying disorder is vitally important.
Case report
A 74-year-old man with a history of VWD (non-congenital), diagnosed 1 year prior to presentation, was referred to our institution for surgical management of papillary thyroid cancer. His symptoms of VWD included excessive bleeding following minor trauma and elective surgical procedures (intra-articular hematoma after an arthroscopic right knee surgery and prolonged bleeding after removal of a basal cell carcinoma on the nose and after undergoing fine-needle aspiration of the thyroid nodule) and episodes of spontaneous epistaxis, which manifested 3 years prior to the diagnosis of VWD. He did not have any congenital or acquired cardiac defects, and there was no family history of bleeding disorders. He was not taking any medications that could increase the risk of bleeding.
On examination, the patient appeared well, without any signs of mucocutaneous bleeding, lymphadenopathy, or splenomegaly. Coagulation tests revealed prolonged activated partial thromboplastin time of 53.9 seconds (normal range: 22.7–36.1 seconds), normal prothrombin time of 14.5 seconds, and low FVIII procoagulant activity and VWF antigen levels of 7% and 8%, respectively. There was no evidence of a specific inhibitor to FVIII. VWF ristocetin cofactor activity (VWF:Ab) was 6% (the assay that was used for testing was the HemosIL VWF activity assay), and VWF propeptide antigen level was normal (101 IU/dL). Thus, VWF propeptide antigen to VWF antigen ratio was very high, suggesting increased clearance of VWF. Multimer analysis showed presence of low multimers but essentially near-complete absence of large and intermediate multimers, consistent with acquired type 2A VWD (Figure 1). Serum protein electrophoresis showed a stable level of monoclonal protein (0.2 g/dL), and serum immunoglobulins A, G, and M and free light chain levels were in the normal range. These findings were consistent with monoclonal gammopathy of undetermined significance (MGUS) in the absence of evidence for plasma cell proliferative or lymphoproliferative disorders, leading to differential diagnoses of AVWS secondary to MGUS or adsorption of VWF on thyroid cancer cells (Table 2).
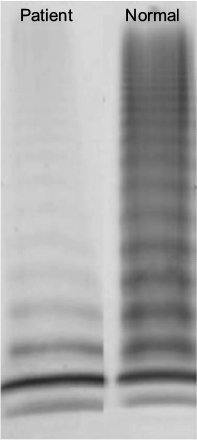 | Figure 1 VWF multimer analysis. Note: Multimer analysis showed presence of low multimers but essentially near-complete absence of large and intermediate multimers, consistent with acquired type 2A VWD. Abbreviations: VWF, von Willebrand factor; VWD, von Willebrand disease. |
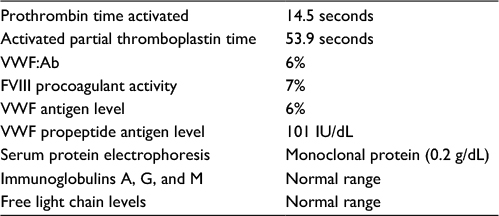 | Table 2 Laboratory testing done in the patient Abbreviations: VWF, von Willebrand factor; FVIII, factor VIII. |
Plasma VWF:Ab was low (17%) prior to elective thyroidectomy. The patient was treated with intravenous immunoglobulin (IVIG) 1 g/kg daily for 2 days (80 g daily, dose based on ideal body weight) as the literature indicates that IVIG is effective in MGUS-induced AVWS and in other cases with immunoglobulin G autoantibodies.11 Plasma VWF:Ab levels tested 1 hour after completion of IVIG had improved to 82%, confirming the diagnosis of MGUS-induced AVWS and indicating that adsorption of VWF on thyroid cancer cells was not a major factor. With a preoperative plasma VWF:Ab level of 79%, the patient received a bolus infusion of 80 ristocetin cofactor units/kg of Humate-P 1 hour prior to surgery. Plasma VWF:Ab 30 minutes after Humate-P infusion had increased to 214%. A total thyroidectomy was performed successfully with very minimal blood loss and without any perioperative hemostatic complications. Since the preoperative plasma VWF:Ab was 214%, the planned postoperative maintenance dose of Humate-P was not administered. Daily plasma VWF:Ab and FVIII levels were checked for 3 days postoperatively, with levels remaining >100% (Figure 2). Our patient demonstrated an excellent response to IVIG therapy with notable improvement in VWF:Ab levels. Clinical pharmacokinetic analysis by serial estimation of plasmatic levels of VWF activity and FVIII suggested that IVIG transiently reversed MGUS-mediated accelerated clearance of VWF.11 Pharmacokinetic monitoring following IVIG was beneficial in confirming the diagnosis of MGUS-induced AVWS and guiding appropriate perioperative dosing of infusion of VWF/FVIII concentrate.
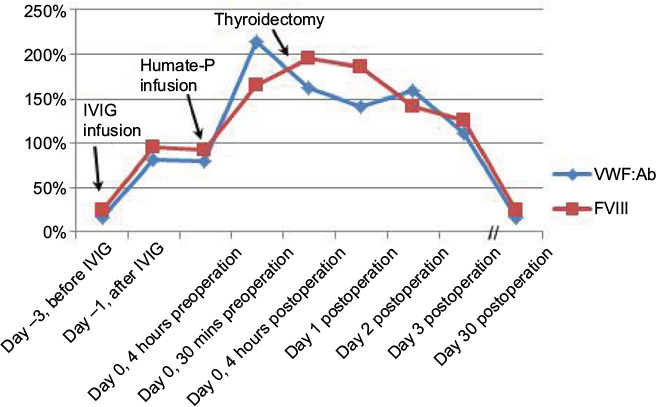 | Figure 2 Clinical pharmacokinetics of VWF and FVIII before and after surgery. Notes: VWF activity (VWF:Ab) and FVIII levels improved with IVIG infusion but were <80% preoperatively. With a bolus infusion of Humate-P®, both VWF:Ab and FVIII levels improved to >100% and remained stable for ~3 days after the thyroidectomy. The patient did not have any intra- or postoperative bleeding and recovered well from surgery. Abbreviations: VWF, von Willebrand factor; FVIII, factor VIII; IVIG, intravenous immunoglobulin. |
Approximately 1 month after surgery, VWF antigen, VWF:Ab, and FVIII levels became low, but the patient did not have any signs or symptoms of bleeding. We recommended IVIG and Humate-P infusions only for any major bleeding episodes or electively before surgical procedures.
Ethics approval and consent to participate were not applicable in this study as it is a single case report. Consent for publication was obtained from the patient.
Discussion
There are multiple mechanisms that contribute to AVWS, including antibody-mediated clearance of VWF, adsorption of VWF to surfaces of cancer cells or platelets, and proteolysis of VWF. By treating the underlying contributing disorder, AVWS is managed simultaneously. However, it sometimes can be difficult to treat the underlying disorder, so additional treatment options for AVWS are needed to control bleeding.
Determining optimal dosing of VWF/FVIII concentrate in AVWS is challenging. The probability of wide intrasubject variability in FVIII and VWF:Ab levels necessitates verification of response to treatment of the underlying disorder and frequent monitoring of these levels to adjust the dose of infusion of VWF/FVIII concentrate.8,12,13 Additionally, there are often inconsistencies between presurgical and postsurgical values. Monitoring individual plasmatic VWF:Ab levels and FVIII levels subsequently determines the precise dosing needed to tailor management. Regardless of the way dose adjustments are made, peak and trough levels of coagulation factors need to be followed with infusions of VWF/FVIII concentrate adjusted appropriately.
Although occurrence of thromboembolic events is not common in VWD treated with VWF/FVIII concentrate, those patients who are at an increased risk for venous thromboembolic events should be monitored closely. FVIII levels should be assayed every 12 hours on the day VWF/FVIII concentrate is infused and every 24 hours thereafter.1 For those who are at a considerable risk for venous thromboembolic events, prophylactic anticoagulation is advocated, similar to what is used in non-VWD patients.
In our case, performing clinical pharmacokinetic analysis confirmed the diagnosis of MGUS-induced AVWS and enabled better dosing of Humate-P. It avoided the unnecessary administration of coagulation factor concentrate in the setting of major surgery without compromising hemostasis. We propose that the practice of performing clinical pharmacokinetic analysis by serial estimation of plasmatic levels of VWF activity and FVIII can help not only save cost of avoidable treatment but also limit the possible adverse effects of Humate-P and, thus, should be considered the standard of care in perioperative management of patients with AVWS. This tailored management promotes the individualized care of these patients. Our case also demonstrates that IVIG can correct VWF and FVIII activities for several days. Prospective studies are required to evaluate the efficacy and safety of IVIG in AVWS.
Author contributions
All authors contributed equally to this work, discussed the results and implications, and commented on the manuscript at all stages. All authors contributed toward data analysis, drafting and critically revising the paper and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Nichols WL, Hultin MB, James AH, et al. von Willebrand disease (VWD): evidence-based diagnosis and management guidelines, the National Heart, Lung, and Blood Institute (NHLBI) Expert Panel report (USA). Haemophilia. 2008;14:171–232. | ||
Curnow J, Pasalic L, Favaloro EJ. Treatment of von Willebrand disease. Semin Thromb Hemost. 2016;42(2):133–146. | ||
Federici AB, Rand JH, Bucciarelli P, et al. Acquired von Willebrand syndrome: data from an international registry. Thromb Haemost. 2000;84:345–349. | ||
Jakway JL. Acquired von Willebrand’s disease. Hematol Oncol Clin North Am. 1992;6:1409–1419. | ||
Favaloro EJ, Pasalic L, Curnow J. Monitoring therapy during treatment of von Willebrand disease. Semin Thromb Hemost. 2017;43(3):338–354. | ||
Federici AB. Management of von Willebrand disease with factor VIII/von Willebrand factor concentrates: results from current studies and surveys. Blood Coagul Fibrinolysis. 2005;16(suppl 1):S17–S21. | ||
Gill JC, Castaman G, Windyga J, et al. Hemostatic efficacy, safety, and pharmacokinetics of a recombinant von Willebrand factor in severe von Willebrand disease. Blood. 2015;126:2038–2046. | ||
Lethagen S, Kyrle PA, Castaman G, Haertel S, Mannucci PM; HAEMATE P Surgical Study Group. von Willebrand factor/factor VIII concentrate (Haemate P) dosing based on pharmacokinetics: a prospective multicenter trial in elective surgery. J Thromb Haemost. 2007;5:1420–1430. | ||
Franchini M. Surgical prophylaxis in von Willebrand’s disease: a difficult balance to manage. Blood Transfus. 2008;6(suppl 2):s33–s38. | ||
Kyrle PA, Minar E, Hirschl M, et al. High plasma levels of factor VIII and the risk of recurrent venous thromboembolism. N Engl J Med. 2000;343:457–462. | ||
Federici AB. Use of intravenous immunoglobulin in patients with acquired von Willebrand syndrome. Hum Immunol. 2005;66:422–430. | ||
Di Paola J, Lethagen S, Gill J, et al. Presurgical pharmacokinetic analysis of a von Willebrand factor/factor VIII (VWF/FVIII) concentrate in patients with von Willebrand’s disease (VWD) has limited value in dosing for surgery. Haemophilia. 2011;17:752–758. | ||
McEneny-King A, Iorio A, Foster G, Edginton AN. The use of pharmacokinetics in dose individualization of factor VIII in the treatment of hemophilia A. Expert Opin Drug Metab Toxicol. Epub 2016 Aug 19:1–9. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.