")
Back to Journals » Drug Design, Development and Therapy » Volume 14
Pharmacokinetics and Bioequivalence of Two Formulations of Valsartan 80 mg Capsules: A Randomized, Single Dose, 4-Period Crossover Study in Healthy Chinese Volunteers Under Fasting and Fed Conditions
Authors Wu Q, Wang X, Chen Q, Zou Y, Xu X, Li T, Yu C, Zhu F, Zhang KE, Jia J , Liu Y
Received 9 March 2020
Accepted for publication 7 September 2020
Published 12 October 2020 Volume 2020:14 Pages 4221—4230
DOI https://doi.org/10.2147/DDDT.S253078
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Manfred Ogris
Qingqing Wu,1,2 Xiaodong Wang,3 Qian Chen,1,2 Yang Zou,1,2 Xiaoyan Xu,1,2 Tingting Li,1,2 Chen Yu,1,2 Fu Zhu,1,2 Kanyin E Zhang,4 Jingying Jia,1,2 Yanmei Liu1,2
1Center Laboratory, Shanghai Xuhui Central Hospital, Shanghai, People’s Republic of China; 2Shanghai Engineering Research Center of Phase I Clinical Research & Quality Consistency Evaluation for Drugs, Shanghai, People’s Republic of China; 3Changzhou Siyao Pharmaceuticals Co., Ltd, Jiangsu, People’s Republic of China; 4ViaClinical Ltd, Shanghai, People’s Republic of China
Correspondence: Yanmei Liu
Center Laboratory, Shanghai Xuhui Central Hospital, No. 966, Huaihai Road(M), Shanghai, People’s Republic of China
Tel/Fax +86-21-54030254
Email [email protected]
Purpose: To compare the bioequivalence of two formulations of valsartan (80 mg capsules) under fasting and fed conditions in healthy Chinese volunteers using a full-replicate study design.
Methods: A total of 78 Subjects were randomly assigned to fasting cohort (n = 48) or fed cohort (n = 30). Each cohort includes 4 single-dose observation periods and 3-day washout periods. Blood samples were collected at designed time point. Plasma concentration of valsartan was analyzed by a validated LC-MS/MS method. Noncompartmental analysis method was employed to determine the pharmacokinetic parameters. Based on the within-subject standard deviation (SWR) of the reference formulation, either reference-scaled average bioequivalence (RSABE) or average bioequivalence (ABE) method was used to evaluate the bioequivalence of the two formulations.
Results: Under fasting conditions, the RSABE method was used to evaluate the bioequivalence of Cmax (SWR> 0.294), while ABE method was used to evaluate the bioequivalence of AUC0-t and AUC0-∞. The geometric mean ratio (GMR) of the test/reference for Cmax was 99.52%, and the 95% upper confidence bound was < 0. For AUC0-t and AUC0-∞ comparisons, GMRs were 102.07% and 101.92%, and the 90% CIs of the test/reference were 96.28%– 108.21%, 96.28%– 107.88%, respectively. Under fed conditions, the SWR value of Cmax, AUC0-t and AUC0-∞ all exceeded the cutoff value of 0.294 and therefore, the RSABE method was used. The GMRs for Cmax, AUC0-t and AUC0-∞ were 98.78%, 103.33% and 103.08%, respectively, while the 95% upper confidence bound values were all < 0. These results all met the bioequivalence criteria for highly variable drugs. All adverse events were mild and transient.
Conclusion: In this study, the generic formulation of valsartan 80 mg capsule was considered to be bioequivalent to the reference product under both fasting and fed conditions, and satisfied the requirements for marketing in China.
NMPA Registration No: CTR20181422.
Keywords: valsartan, bioequivalence, replicate, pharmacokinetics, safety
Introduction
Valsartan is an orally active angiotensin II type 1 receptor blocker for the treatment of hypertension alone or in combination with other antihypertensive agents.1,2 Angiotensin receptor blockers (ARBs) are recommended to both hypertension treatment and cardiovascular prevention in different settings.3,4 It was patented in 1990 and came into medical use in 1996 known as Diovan which was initially used for the treatment for hypertension. It became available as a generic medication in 2012 worldwide. Bioequivalence studies comparing generic to innovator products are required for marketing a new generic product by the National Medical Products Administration (NMPA) of China. A systemically active generic drug is considered to be bioequivalent to the reference drug if the rate and extent of absorption of the two products do not show any significant difference, by conducting bioequivalence (BE) studies in human subjects to compare its pharmacokinetic characteristics.5
Valsartan is mainly administrated at a dose of 80 mg or 160 mg/d.6 The absorption of Valsartan is rapid with the Tmax of 2–4 hours. Intravenous administration showed a double exponential decay of pharmacokinetics with the T1/2 of 6 hours. The absolute bioavailability of Diovan is reported to be 25% (range 10% to 35%).7 The solubility of valsartan is pH dependent and thus may cause subject variability in drug absorption and failure in bioequivalence studies.8 The within-subject variabilities for valsartan in Cmax (CV~30%) and AUC (CV~44%) are considerable.9,10 In light of these findings to indicate the high intra-subject variability of valsartan, we applied a two-sequence and four-period crossover (2 x 4), full replicate study design, such that the true intra-subject variability for the test and reference products can be determined independently, such that the reference scaled average bioequivalence (RSABE) method may be applied to assess bioequivalence with widened acceptance limit, according to NMPA Guideline for human bioavailability and bioequivalence studies for pharmaceuticals.11
Orally administered valsartan is excreted mainly in the feces (~83%) and minor portion in the urine (~13%).10 Valsartan is highly bound to serum proteins (95%), mainly serum albumin. The volume of distribution valsartan after IV (20 mg in 12 volunteers) administration was small (17 L), indicating that valsartan was not widely distributed in tissues.12,13 After intravenous administration, the plasma clearance was approximately 2 L/h and the renal clearance was approximately 0.62 L/h (approximately 30% of total clearance).7
Clinical trials for Valsartan treatment for hypertension versus placebo demonstrated its side effects like viral infection (3% vs 2%), fatigue (2% vs 1%) and abdominal pain (2% vs 1%).7 Compared with ACEI, the incidence of dry cough is significantly less. A bioequivalence trial (randomized, single-dose, 2-period crossover study) of 2 formulations of valsartan 160-mg tablets was conducted in 60 healthy Korean male volunteers. The results indicated that the test preparation and the reference preparation were bioequivalent and both formulations were well tolerated, with no serious adverse events reported.14 Although there are reports in the literature regarding the pharmacokinetic (PK) characteristics of valsartan,15–19 but few have fully addressed its intra-subject variability, especially under fed conditions. Thus, the current study was designed to compare the pharmacokinetic properties and bioequivalence of two formulations of valsartan (80 mg capsules) under fasting and fed conditions in healthy Chinese volunteers, using a full-replicate study design, and seek regulatory approval for the generic formulation to be marketed in China.
Subjects and Methods
Subjects
This study was conducted at the Phase I clinical research center of Shanghai Xuhui Central Hospital from August to October 2018. All subjects had been informed about the study and had provided written informed consent before participating in the study. Also, before starting any screening or research-related procedures, the researcher had explained to the subjects the nature of the study and had answered all questions related to the study. The subject had been asked to provide informed consent. All clinical laboratory tests were performed by masked bio-analysis at the Clinical Laboratory of Shanghai Xuhui Central Hospital, which was accredited by Shanghai Center for Clinical Laboratory of China. All QC samples, which were purchased from the National Center for Clinical Laboratories of China or Shanghai Center for Clinical Laboratory of China, will be detected together with the samples from patients at the laboratory every day. All participants were free to withdraw from the study at any time.
The inclusion criteria comprised healthy Chinese volunteers aged 18 to 45 years, with the body mass index between 19.0 to 28.0 kg/m2, in good health and physical condition as determined by medical history, vital signs, physical examinations, 12-lead ECG, chest X-ray examination and laboratory tests (blood chemistry, hematology, urinalysis, hepatitis B surface antigen, hepatitis C antibody, HIV antibody and syphilis antibody).
The exclusion criteria included a history of heavy smoking (more than 10 cigarettes per day) and/or drug dependence and/or alcohol abuse. Those who had participated in other clinical trials within 3 months prior to this study, or who had blood loss of >400 mL within 3 months, or who had blood transfusions or donated blood >200 mL within 1 month before this study were also excluded. Subjects must not use any medications, vitamins and/or herbal supplements, within 2-week prior to study drug administration. For female subjects, their weight should be more than 45 kg besides the body mass index between 19.0 to 28.0 kg/m2. Positive pregnancy test or lactation women should be excluded.
Study Design
This study was a single-dose, open-label, randomized, two-sequence, four-Period crossover bioequivalence study. The study was performed in accordance with the ethical standards for studies in humans of the Declaration of Helsinki and its amendments,20 the International Conference on Harmonisation Guideline for Good Clinical Practice,21 and the Guideline for Good Clinical Principles recommended by NMPA of China.22 The study protocol and informed-consent form were approved by the ethic committee of Shanghai Xuhui Central Hospital. The study was also approved by NMPA of China (B201800338-01). According to NMPA guideline on the investigation of bioequivalence,11 this study was conducted in two cohorts under fasting and fed conditions. The sample size for fasting and fed conditions were calculated based on preliminary studies by using SAS (version 9.4) software.
In cohort 1, 48 subjects were randomly assigned to sequence A (TRTR) and sequence B (RTRT), and received valsartan under fasting conditions; in cohort 2, 30 subjects were randomly assigned to sequence C and (TRTR) sequence D (RTRT), and received valsartan under fed conditions. Two randomization tables were generated for the cohort 1 and 2, by SAS version 9.4 (SAS Institute Inc., Cary, North Carolina) (Supplementary Table 1). The test product (T) was 80 mg valsartan capsule (lot # 201,706,082, expiration date 06/2019) was manufactured by Changzhou Siyao Pharmaceutical Co., Ltd.) and the reference product (R) was DIOVAN 80 mg capsule (lot# X2125, expiration date 12/2019), manufactured by Beijing Novartis Pharmaceutical Co., Ltd.).
All participants were required to undergo an overnight (10-hour) fast prior to dosing. Subjects who participated in the fed study must take a high-calorie, high-fat breakfast (which contains 148 calories of protein, 260 calories of carbohydrate, and 525 calories of fat) 30 minutes before dosing.11 Investigational drugs were administered with 240 mL of water under supervision of a qualified pharmacist. Subjects were not allowed to drink water 1 hour before and 1 hour after dosing, except the water used for drug administration. Food intake was strictly controlled and standardized lunch and dinner were provided approximately 4 and 10 hours postdose, respectively. Alcoholic beverages, coffee, xanthine-containing drinks, intense physical activity, and smoking were not allowed during the study period.
All participating volunteers were under medical supervision by a physician throughout the study. Blood samples (2 mL) were drawn to K2-EDTA containing vacuum blood collection tubes at 0 (pre-dosing), 0.5, 1, 1.5, 2, 2.5 3, 3.5, 4, 5, 6, 8, 12, 24, and 36 hours post-dose in the fasting cohort, and at 0 (pre-dosing), 0.5, 1, 2, 3, 3.5, 4.4.5, 5.5.5, 6, 8, 12, 24, and 36 hours post-dose in the fed cohort of the study. After sample collection, plasma was separated by centrifugation (2000 g ×10 minutes, 4°C), transferred into polypropylene tube within 1 hour, and was stored at −80°C until analyzed by liquid chromatography tandem mass spectrometry (LC-MS/MS).
Safety
Safety assessments included physical examinations, vital signs (oral body temperature, pulse rate, and sitting blood pressure), 12-lead ECGs, clinical laboratory tests and adverse event (AE) monitoring.
Determination of Plasma Valsartan Concentrations
In this study, the concentration of valsartan in plasma was determined by a validated LC-MS/MS method.23,24 A deuterium labeled analytical internal standard (valsartan-d9) was used for the quantitative determination of valsartan. In order to ensure the reliability of the analytical method for the determination, validation of this analytical method was performed to evaluate its selectivity, linearity, precision, accuracy and stability in solution according to the technical guideline for clinical pharmacokinetics of chemical drugs published by NMPA.25
The bioanalytical method and validation results are briefly described as follows: Protein precipitation method was used to extract valsartan from human plasma samples: An aliquot (10 μL) of valsartan-d9 (20 μg/mL in 50% [vol/vol] methanol) was added as internal standard to100 μL of plasma and mixed well, then 200 μL of acetonitrile was added into each sample and mixed well (vortex 10 s). After centrifuging the mixture at 15,000 rpm× 3 min at 4°C, the supernatant was transferred into auto-sampler vials and 20 μL was injected into the LC-MS/MS system for analysis.
Chromatographic conditions were as follows: The column was ACQUITY UPLC BEH C18 2.1× 50 mm, 1.7 μm, and the column temperature was 40°C, with a flow rate of 0.600 mL/min. The mobile phase A was 100% water containing 5 mM ammonium acetate and 1.0% formic acid, mobile phase B was 100% acetonitrile containing 0.1% formic acid. The starting gradient was 40% mobile phase B, and increased to 55% mobile phase B at 1 min applying a linear gradient elution program, then increased to 90% at 1.01 min and held until 1.80 min, then return to 40% B at 1.81 min and stop at 2.50 min.
The valsartan and internal standard valsartan-d9 were detected by a triple-quadruple mass detector (API4000; AB Sciex) in positive-ion mode with electrospray ionization (ESI) in multiple-reaction-monitoring (MRM) mode (m/z 436.2/235.2 for valsartan, m/z 445.2/235.2 for valsartan-d9).
The lower limit of quantitation (LLOQ) of valsartan was 7.00 ng/mL with dynamic range of 7.00 to 7000 ng/mL. And quality control (QC) samples (low QC, 21 ng/mL; Geometric Mean QC, 210 ng/Ml; medium QC, 3500 ng/mL; high QC, 5250 ng/mL; dilution QC, 14,000 ng/mL) were analyzed to assess the accuracy and precision of the method. All QC samples are evenly distributed among the samples to be analyzed.
Pharmacokinetic and Statistical Analysis
Pharmacokinetic analysis was conducted with noncompartmental model using SAS version 9.4 (SAS Institute Inc., Cary, North Carolina). Cmax and Tmax values were obtained directly from individual concentration-time curves (C-T) of valsartan. AUC0−t was calculated using the linear trapezoidal rule. AUC0−∞ was calculated as AUC0−t + Ct/z, where Ct was the concentration at the last point and λz was the slope of the linear regression of the log-transformed C-T curve. Valsartan plasma t1/2 was calculated as 0.693/z.25
Primary pharmacokinetic parameters (AUC0-t, AUC0-∞, Cmax) and the secondary pharmacokinetic parameters (Tmax, t1/2, λz) were summarized to their mean, median, standard deviation, maximum, minimum values and coefficient of variation, etc.
Statistical analysis was also performed using SAS version 9.4 on Ln-transformed primary pharmacokinetic parameters..
Due to the anticipated high intra-individual variation of valsartan, this study used a full-replicate design based on the within-subject variability of the reference formulation, according to the FDA-recommended reference-scaled average bioequivalence (RSABE) method.26
Prior to bioequivalence evaluation, the within-subject standard deviation (SWR) of the reference formulation was calculated and if SWR < 0.294 for any primary PK parameters (Cmax, AUC0-t, AUC0-∞), which corresponds to within-subject variability (CVW%) <30%, bioequivalence was evaluated by the standard average bioequivalence (ABE). Bioequivalence was claimed if the 90% confidence interval (CI) for the test/reference GMR of the above pharmacokinetic parameter(s) falls within the range of 80.00% −125.00%.
If SWR≥0.294 (CVW% ≥30%) for any primary PK parameter(s), RSABE method was used for bioequivalence evaluation. In this case, the 95% upper confidence bound for (YT−YR)2–θS2WR was calculated based on Howe’s Approximation I.27
Where YT and YR are the natural log transformed AUC or Cmax mean values for test and reference formulation, respectively.
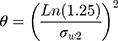 is the BE limit and σw0 = 0.25 is the regulatory constant set by US FDA and China NMPA.
is the BE limit and σw0 = 0.25 is the regulatory constant set by US FDA and China NMPA.
For any given primary PK parameter(s), bioequivalence was concluded if the 95% upper confidence bound for (YT−YR)2–θS2WR is ≤0 and the test/reference GMR falls within 80.00% −125.00%.
Results
Volunteers Characteristics
A total of 78 healthy Chinese volunteers were enrolled and all completed the study (Figure 1). In the fasting study cohort, 48 (45 males, 3 females) healthy Chinese volunteers were enrolled (Mean ± SD age, 27.0 ± 4.5 years [range, 18–39]; height, 167.2 ± 6.4 cm [range, 148.0 −178.5]; weight, 63.2 ± 7.6 kg [range, 50.2–79.0]; and BMI, 22.6 ± 2.1 kg/m2 [range, 19.1–27.7]). In the fed study cohort, 30 (24 males, 6 females) healthy Chinese volunteers were enrolled (Mean ± SD age, 27.0 ± 4.9 years [range, 19–39]; height, 166.9 ± 7.3 cm [range, 149.0 −180.0]; weight, 63.5 ± 7.0 kg [range, 50.2–79.3]; and BMI, 22.8 ± 1.8 kg/m2 [range, 19.5–26.1]).
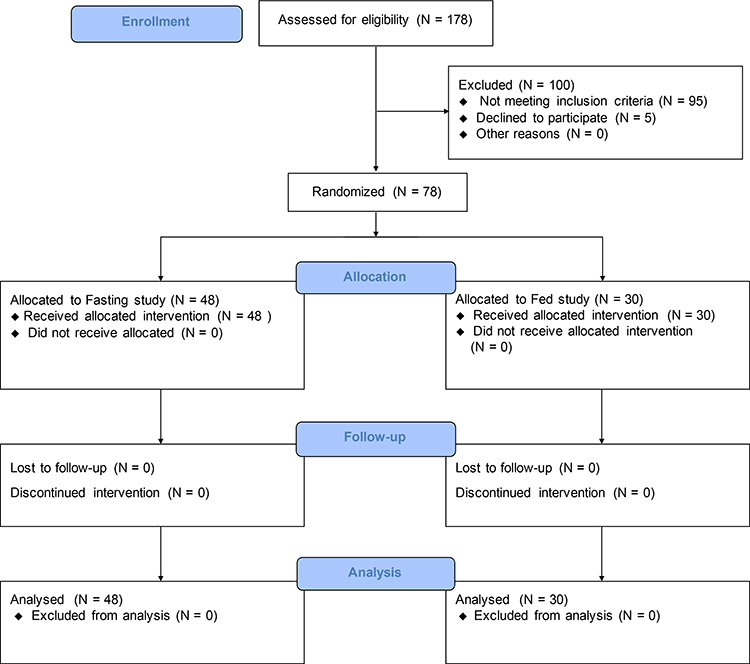 |
Figure 1 Study flow diagram according to CONSORT recommendations. |
Safety
Under fasting conditions, 6 subjects (12.5%) who received test formulation and 7 subjects (14.6%) who received reference formulation were reported to experience drug-related treatment-emergent AEs (TEAEs). Under fed conditions, 4 (13.3%) who received test formulation and 2 subjects (6.7%) who received reference formulation were reported to experience TEAEs (Table 1). The most common TEAEs in test formulation was hypotension, whereas in reference formulation the most common TEAEs were diarrhea. All AEs were considered to be mild intensity by the investigators and were transient as the subjects recovered by the end of the study. No serious AEs were reported and none of the subjects were withdrawn from the study due to AEs.
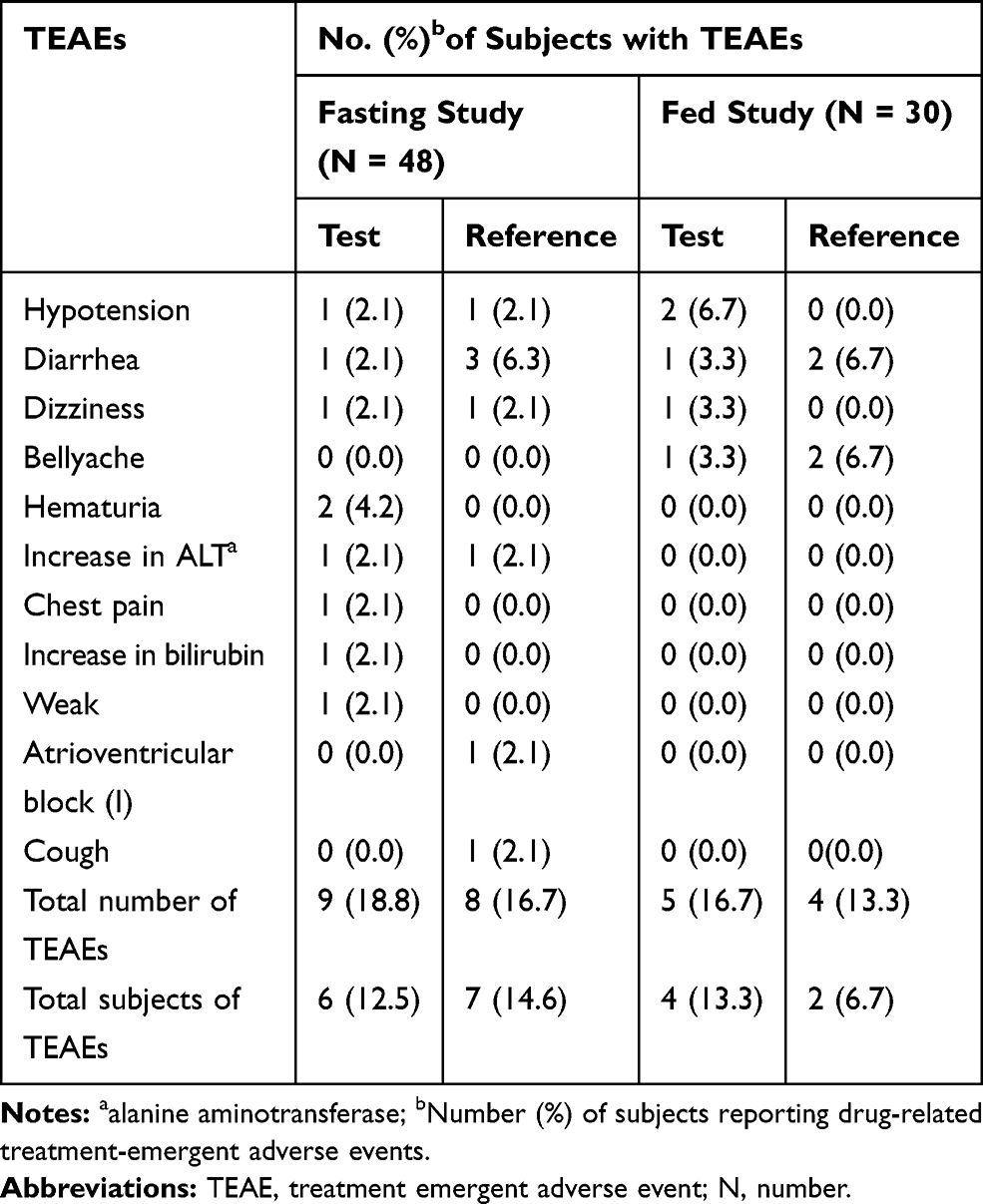 |
Table 1 All Drug-Related Treatment-Emergent AEs of Test and Reference Formulations of Valsartan 80-Mg Capsules After Oral Administration Under Fasting and Fed Conditions in Healthy Chinese Volunteers |
Pharmacokinetics
The mean (± SD) plasma concentration-time (C-T) curves of valsartan following single-dose oral administration of four individual sequence (T1, T2, R1, R2) under fasting and fed conditions are shown in Figure 2. The primary PK parameters under fasting and fed conditions are summarized in Tables 2 and 3.
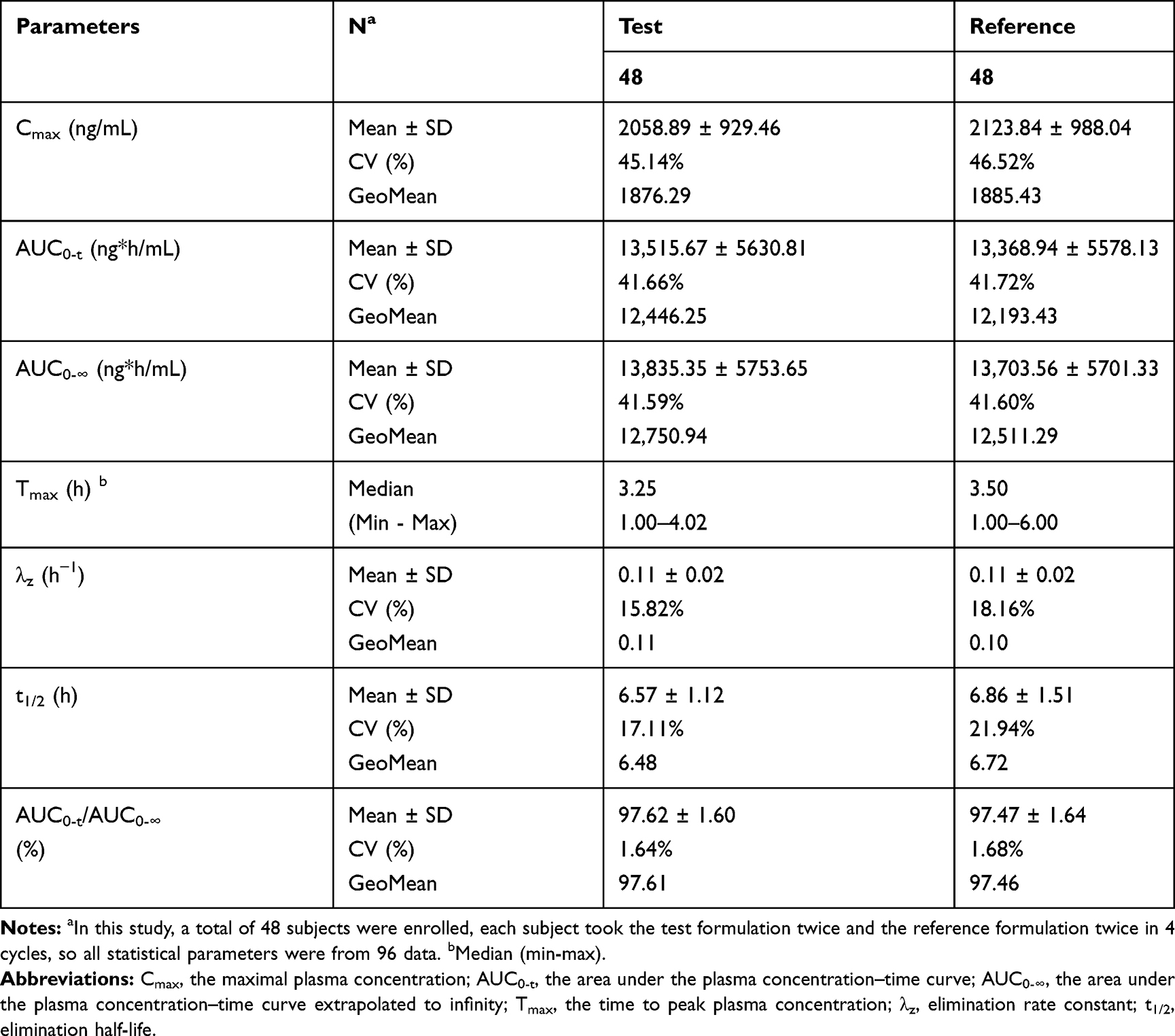 |
Table 2 Pharmacokinetic Properties of Test and Reference Formulations of Valsartan 80-Mg Capsules After Single-Dose Administration in Healthy Chinese Volunteers Under Fasting Condition |
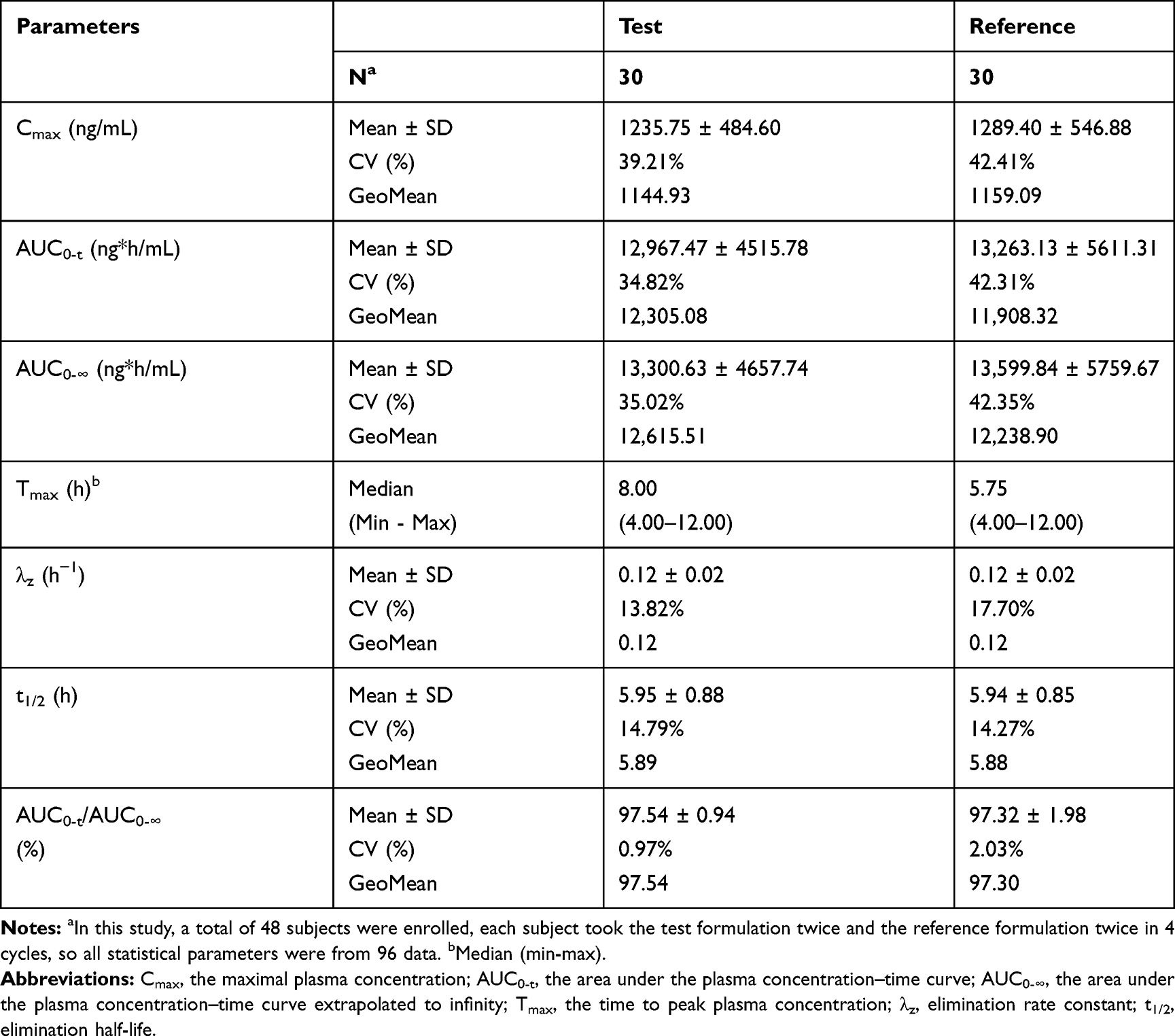 |
Table 3 Pharmacokinetic Properties of Test and Reference Formulations of Valsartan 80-Mg Capsules After Single-Dose Administration in Healthy Chinese Volunteers Under Fed Condition |
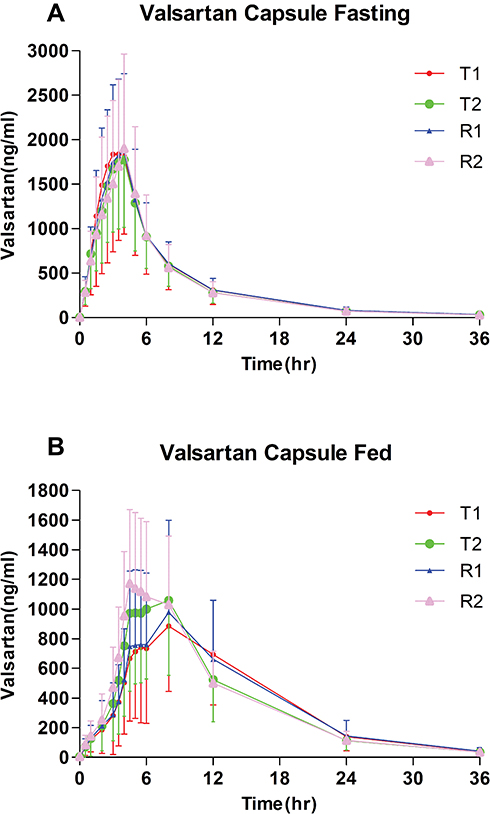 |
Figure 2 Mean (SD) plasma concentration-time curves of valsartan following single-dose oral administration of four individual sequence (T1, T2, R1, R2) under fasting (A, N = 48) and fed (B, N = 30) conditions in healthy Chinese volunteers. |
Bioequivalence
The bioequivalence evaluation between the test and reference formulations of valsartan 80 mg capsules in healthy volunteers under fasting and fed conditions are presented in Tables 4 and 5.
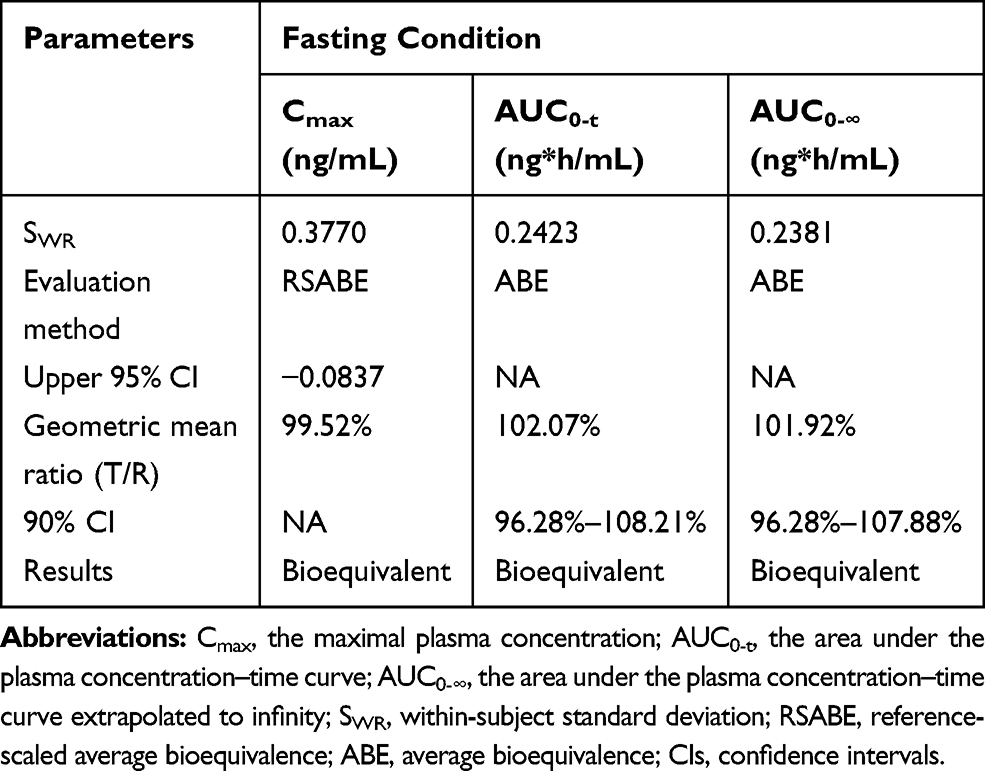 |
Table 4 Bioequivalence Evaluation of Test and Reference Formulations of Valsartan 80-Mg Capsules in Healthy Volunteers Under Fasting Condition |
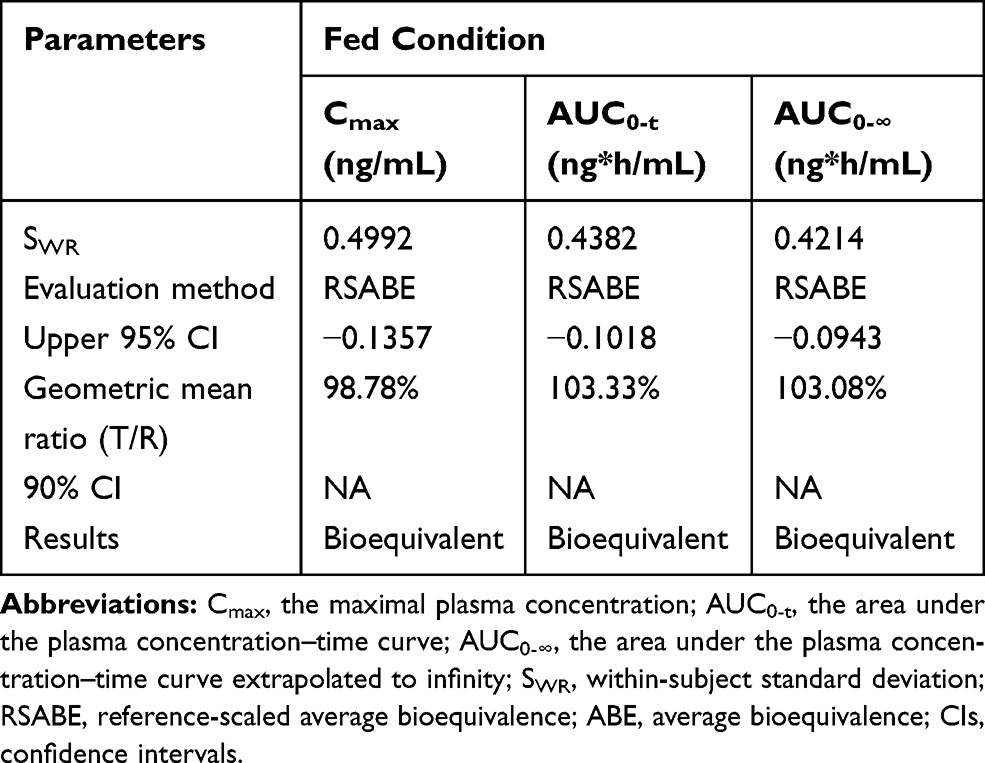 |
Table 5 Bioequivalence Evaluation of Test and Reference Formulations of Valsartan 80-Mg Capsules in Healthy Volunteers Under Fed Condition |
As shown in Table 4, under fasting conditions, the SWR for Cmax exceeded the regulatory cutoff of 0.294, and therefore, RSABE method was used for equivalence evaluation. The test/reference GMR for Cmax was 99.52%, falls within 80.00% −125.00%, and the 95% upper confidence bound was −0.0837<0, which demonstrated that Cmax comparison has met bioequivalence criteria for highly variable drugs. Since SWR for AUC0-t and AUC0-∞ were less than 0.294, ABE method was used to evaluate the bioequivalence for these PK parameters. The 90% CIs of GMR were 96.28%–108.21%, 96.28%–107.88%, all fell within the range of 80.00% −125.00%, also met the bioequivalence criteria for ABE. In summary, the test and reference formulations of valsartan 80 mg capsules were bioequivalent under fasting conditions.
Statistical analysis of pharmacokinetic parameters under fed conditions (Table 5) showed that the SWR of the primary pharmacokinetic parameters (Cmax, AUC0-t, AUC0-∞) all exceeded the cutoff value 0.294, which means CVW% >30%, the bioequivalence evaluation for all three PK parameters were performed using RSABE method.
The results showed that the GMR of the test and the reference formulations for Cmax, AUC0-t and AUC0-∞ all fell within the range of 80.00% −125.00%, and the 95% upper confidence bound values were less than 0, which met the bioequivalence criteria for highly variable drugs and therefore, the test and reference formulation were concluded to be bioequivalent under fed conditions.
On ANOVA, using logarithmic-transformed data, no sequence or formulation effects were observed for any PK parameters.
Discussion
This study evaluated the bioequivalence of two formulations of valsartan (80 mg capsules) in two study cohorts under fasting and fed conditions, both cohorts were open label, randomized, single-dose, two sequence, four-period, full replicate crossover study in healthy Chinese volunteers. As Changzhou Siyao Pharmaceuticals Co., Ltd produced two kinds of valsartan, 4080 mg, 80 mg capsules were chosen to be evaluated in our study, which was in consistent with the guideline that recommended to consider highest dose for bioequivalence study. The results of this study provide support for the marketing of the new generic valsartan capsule (80 mg) in China.
A total of 78 healthy subjects were enrolled in this study under fasting and fed conditions and all completed the study. The drug administration and blood collection process were well followed. The subjects and numbers of AEs in the test and reference formulations were similar. In the fed study cohort, 2 subjects developed local skin infections, which were considered to be caused by the implanted catheters and successfully treated with topical drugs by clinical investigator. All other subjects who experienced AEs were recovered without any treatment.
We compared the results of pharmacokinetic parameters from our study to those reported in drug product prescribing information and literatures. Under fasting conditions, the Tmax and t1/2 values are close to the range described in the reference drug instructions (Tmax, 2–4 hours, t1/2 6 hours).5 The peak plasma concentration (Cmax) and total exposure (AUC0-t, AUC0-∞) values in our study were also comparable to those reported in the literatures10,11 after dose adjustment (Supplementary Table 2).
After taking high-calory and high-fat meal, the t1/2 value is close to the reported range, whereas the Tmax was significantly prolonged and peak concentration (Cmax) was also significantly decreased by ~40%, but the total exposure (AUC0-t, AUC0-∞) did not decrease significantly, which were equivalent to the results under fasting condition after dose adjustment (Supplementary Table 2). According to the drug product prescribing information, food intake with valsartan capsules reduces the exposure (as measured by AUC) of valsartan by about 48% and Cmax by about 50%.5 The results of our study partially support the food effects reported for this drug.
For bioequivalence evaluation under fasting conditions, RSABE method was used to evaluate Cmax, while ABE method was used to evaluate AUC0-t and AUC0-∞, for all three primary PK parameters, the test and reference valsartan capsules were determined to be bioequivalent.
This study clearly demonstrated, under fasting conditions, the reference product valsartan capsule is highly variable only in terms of Cmax (>30%), but not in terms of AUC0-t and AUC0-∞. Moreover, the full-replicate study design allowed us to obtain within-subject variability for the test and reference product separately, and the results showed the CVw% for Cmax for the test formulation was 30.5%, which was significantly less than that for the reference formulation with CVw% of 39.0%. The CVw% values for AUC0-t and AUC0-∞ were similar between the test and reference formulations (Supplementary Table 3).
Under fed conditions, bioequivalence evaluation for the test and reference formulations of valsartan were all assessed using RSABE method, since the SWR for all primary PK parameters (Cmax, AUC0-t, AUC0-∞) exceeded the cutoff value of 0.294 and all met the corresponding bioequivalence criteria for highly variable drugs in terms of 1) the 95% upper confidence bound and 2) GMR falls within 80–125%.
Once again, this study demonstrated, under fed conditions, the reference product valsartan capsule is highly variable in all primary PK parameters. Similar to the observation made under fasting conditions, the CVw% value of Cmax for the test formulation was 29.6%, significantly less than that for the reference formulation of 50.0%, and the CVw% values of AUC0-t and AUC0-∞ were 19.0%, 18.9% for the test formulation, significantly less than those of the reference formulation of 44.5%, 42.6%, respectively (Supplementary Table 3).
Conclusions
The results from this study suggest that the test formulation of valsartan 80 mg capsule was bioequivalent to the reference formulation under both fasting and fed conditions. It meets the requirements of bioequivalence according to NMPA Guidelines for the Study of Bioequivalence of Highly Variable Drugs and the Guiding Principles for the Investigation of Human Bioequivalence for Pharmaceuticals Based on Pharmacokinetic Parameters. Both formulations were generally well tolerated.
Data Sharing Statement
All of the individual participant data collected during the trial, after deidentification, can be shared. Study Protocol, Statistical Analysis Plan, Informed Consent Form, and Clinical Study Report can also be available. All shared and available data besides the information presented in the article can be obtained by contacting the corresponding author. To gain access, data requestors will need to sign a data access agreement. Data are available indefinitely.
Acknowledgments
The authors would like to thank the subjects and staffs who participated in the study. The authors acknowledge Shanghai Xihua Scientific Co., Ltd. for bioanalytical method validation and sample analysis of the study. The authors also acknowledge ViaClinical Ltd. (Shanghai, China) for statistical analysis of the study.
Author Contributions
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; gave final approval of the version to be published; and agree to be accountable for all aspects of the work.
Funding
This study was sponsored by Changzhou Siyao Pharmaceutical Co., Ltd. (Changzhou, China). And it did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors. The study sponsor had a role in study design, data analysis, and data interpretation.
Disclosure
Xiaodong Wang is the employee from Changzhou Siyao Pharmaceutical Co., Ltd. Kanyin E Zhang reports that ViaClinical Ltd received service fees from Changzhou Siyao Pharmaceutical Co. to perform this study. The authors report no other conflicts of interest in this work.
References
1. Criscione L, de Gasparo M, Buhlmayer P, Whitebread S, Ramjoue HP, Wood J. Pharmacological profile of valsartan: a potent, orally active, nonpeptide antagonist of the angiotensin II AT1-receptor subtype. Br J Pharmacol. 1993;110(2):761–771.
2. Markham A, Goa KL. Valsartan. A review of its pharmacology and therapeutic use in essential hypertension. Drugs. 1997;54(2):299–311. doi:10.2165/00003495-199754020-00009
3. Spannella F, Giulietti F, Balietti P, et al. Renin-angiotensin system blockers and statins are associated with lower in-hospital mortality in very elderly hypertensives. J Am Med Dir Assoc. 2018;19(4):342–347. doi:10.1016/j.jamda.2017.09.023
4. Thomopoulos C, Parati G, Zanchetti A. Effects of blood-pressure-lowering treatment on outcome incidence in hypertension: 10 - Should blood pressure management differ in hypertensive patients with and without diabetes mellitus? Overview and meta-analyses of randomized trials. J Hypertens. 2017;35(5):922–944. doi:10.1097/HJH.0000000000001276
5. Federal food, drug, and cosmetic act, chapter v, subchapter a, drugs and devices section 355(j)(8)(B)(i).
6. Zaid AN, Cortesi R, Qaddomi A, Khammash S. Formulation and bioequivalence of two valsartan tablets after a single oral administration. Sci Pharm. 2011;79(1):123–135. doi:10.3797/scipharm.1009-01
7. DIOVAN®(Valsartan) Capsules [Prescribing Information]. East Hanover, NJ: Novartis Pharmaceuticals Corporation; 2020.
8. Yamada I, Goda T, Kawata M, Shiotuki T, Ogawa K. Gastric acidity-dependent bioavailability of commercial sustained release preparations of indomethacin, evaluated by gastric acidity-controlled beagle dogs. Chem Pharm Bull. 1990;38(11):3112–3115. doi:10.1248/cpb.38.3112
9. Mansouri K, Behloul S, Cherait I, Nekhoul K, Hadjaz IM, Mansouri MB. Bioequivalence of two brands of valsartan 80 mg coated breakable tablets in 15 healthy algerian volunteers: a pilot study. J Pharm Pharmacol. 2017;661–667.
10. Saydam M, Takka S. Bioavailability File: valsartan, FABAD. J Pharm Sci. 2007;32(4):185–196.
11. National Medical Products Administration of China, Center for Drug Evaluation. Guideline for bioavailability and bioequivalence studies of generic drug products [in Chinese].
12. Colussi DM, Parisot C, Rossolino ML, Brunner LA, Lefevre GY. Protein binding in plasma of valsartan, a new angiotensin II receptor antagonist. J Clin Pharmacol. 1997;37(3):214–221. doi:10.1002/j.1552-4604.1997.tb04783.x
13. Flesch G, Muller P, Lloyd P. Absolute bioavailability and pharmacokinetics of valsartan, an angiotensin II receptor antagonist, in man. Eur J Clin Pharmacol. 1997;52(2):115–120. doi:10.1007/s002280050259
14. Kim JE, Ki MH, Yoon IS, et al. Pharmacokinetic properties and bioequivalence of 2 formulations of valsartan 160-mg tablets: a randomized, single-dose, 2-period crossover study in healthy Korean male volunteers. Clin Ther. 2014;36(2):273–279. doi:10.1016/j.clinthera.2014.01.004
15. Iqbal M, Khuroo A, Batolar LS, Tandon M, Monif T, Sharma PL. Pharmacokinetics and bioequivalence study of three oral formulations of valsartan 160 mg: a single-dose, randomized, open-label, three-period crossover comparison in healthy Indian male volunteers. Clin Ther. 2010;32(3):588–596. doi:10.1016/j.clinthera.2010.03.004
16. Muller P, Flesch G, de Gasparo M, Gasparini M, Howald H. Pharmacokinetics and pharmacodynamic effects of the angiotensin II antagonist valsartan at steady state in healthy, normotensive subjects. Eur J Clin Pharmacol. 1997;52(6):441–449.
17. Sioufi A, Marfil F, Jaouen A, et al. The effect of age on the pharmacokinetics of valsartan. Biopharm Drug Dispos. 1998;19(4):237–244. doi:10.1002/(SICI)1099-081X(199805)19:4<237::AID-BDD100>3.0.CO;2-7
18. Sechaud R, Graf P, Bigler H, Gruendl E, Letzkus M, Merz M. Bioequivalence study of a valsartan tablet and a capsule formulation after single dosing in healthy volunteers using a replicated crossover design. Int J Clin Pharmacol Ther. 2002;40(1):35–40.
19. Spinola AC, Almeida S, Filipe A, Neves R, Trabelsi F, Farre A. Results of a single-center, single-dose, randomized-sequence, open-label, two-way crossover bioequivalence study of two formulations of valsartan 160-mg tablets in healthy volunteers under fasting conditions. Clin Ther. 2009;31(9):1992–2001. doi:10.1016/j.clinthera.2009.09.002
20. World Medical Association Declaration of Helsinki (WMA). Ethical Principles for Medical Research Involving Human Subjects. Adopted by the 18th WMA General Assembly, Helsinki, Finland, June 1964, and amended by the 52nd WMA General Assembly, Edinburgh, Scotland, October 7, 2000.
21. International Council for Harmonization of technical requirements for registration of pharmaceuticals for human use.
22. National Medical Products Administration of China. Guideline for good clinical practice [in Chinese].
23. Hanpithakpong W, Kamanikom B, Dondorp AM, et al. A liquid chromatographic-tandem mass spectrometric method for determination of artesunate and its metabolite dihydroartemisinin in human plasma. J Chromatogr B Analyt Technol Biomed Life Sci. 2008;876(1):61–68. doi:10.1016/j.jchromb.2008.10.018
24. Hodel EM, Zanolari B, Mercier T, et al. A single LC-tandem mass spectrometry method for the simultaneous determination of 14 antimalarial drugs and their metabolites in human plasma. J Chromatogr B Analyt Technol Biomed Life Sci. 2009;877(10):867–886. doi:10.1016/j.jchromb.2009.02.006
25. National Medical Products Administration of China. Technical guideline for clinical pharmacokinetics of chemical drugs [in Chinese].
26. Davit BM, Chen ML, Conner DP, et al. Implementation of a reference-scaled average bioequivalence approach for highly variable generic drug products by the US Food and Drug Administration. AAPS J. 2012;14(4):915–924. doi:10.1208/s12248-012-9406-x
27. Howe WG. Approximate confidence limits on the mean of X+Y where X and Y are two tabled independent variables. J Amer Statist Assoc. 1974;69:789–794.
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.