")
Back to Journals » Drug Design, Development and Therapy » Volume 9
Pharmacokinetics and bioavailability study of two ondansetron oral soluble film formulations in fasting healthy male Chinese volunteers
Authors Zhu Y, Zhang Q, Zou J, Wan M, Zhao Z, Zhu J
Received 10 April 2015
Accepted for publication 19 May 2015
Published 12 August 2015 Volume 2015:9 Pages 4621—4629
DOI https://doi.org/10.2147/DDDT.S86415
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Wei Duan
Yubing Zhu,1 Qian Zhang,1 Jianjun Zou,2 Meng Wan,1 Zheng Zhao,1 Junrong Zhu1
1Department of Pharmacy, 2Laboratory of Clinical Pharmacology, Nanjing First Hospital, Nanjing Medical University, Nanjing, Jiangsu, People’s Republic of China
Background: Ondansetron oral soluble film is designed to be applied on top of the tongue without requiring water to aid dissolution or swallowing, which is especially fitting for nausea and vomiting patients.
Purpose: This study was conducted to compare the bioavailability of two 8 mg ondansetron oral soluble film formulations.
Patients and methods: This randomized, open-label, two-period crossover study was performed under fasting conditions. A total of ten eligible subjects were randomly assigned at a 1:1 ratio to receive a single 8 mg dose of the test and reference ondansetron oral soluble film formulations, followed by a 1-week washout period and administration of the alternate formulation. The concentrations of ondansetron were assayed using an liquid chromatograph-mass spectrometer/mass spectrometer (LC-MS/MS) method. For analysis of pharmacokinetic properties, including the peak concentration of Tmax (Cmax), AUC from time 0 (baseline) to t hours (AUC0–t), and AUC from baseline to infinity (AUC0–∞), blood samples were obtained at intervals over the 24-hour period after studying drug administration. Tolerability was assessed by monitoring vital signs and laboratory tests (hematology, blood biochemistry, hepatic function, and urinalysis) and by questioning subjects about adverse events.
Results: The mean (standard derivation [SD]) relative bioavailability was 96.5 (23.7%). The 90% confidence intervals (CIs) for the log-transformed ratios of Cmax and AUC0–t were 84.71%–103.28% and 91.38%–108.60%, respectively (P>0.05). Similar results were found for the data without log-transformation. No statistically significant differences were found based on analysis of variance. No significant adverse events occurred or were reported during the study.
Conclusion: As the 90% CIs based on the differences between the test and reference formulation were within the 80%–125% range for both the Cmax and AUC0–t, we concluded that the two formulations were bioequivalent with respect to the rate or the extent of absorption. Both formulations are well tolerated.
Keywords: ondansetron, oral soluble film, LC-MS/MS, bioequivalence
Introduction
Ondansetron, an available selective serotonin-blocking agent, has been widely used in the treatment of preventing nausea and vomiting caused by cancer chemotherapy, radiation therapy, and surgery.1 The approved formulations of ondansetron in the market include intramuscular, intravenous, and oral.2 Intramuscular administration of medications can be painful and may be associated with risks for infection, bleeding, and abscess formation. Intravenous administration of medications was often limited with complications such as phlebitis, infiltration, extravasation, and infections.3 Although traditional oral formulations can be self-administered, it is not always possible if a patient has difficulties in swallowing tablets. Oral soluble film is a novel formulation that can dissolve in seconds and can be swallowed without liquid, which is very fitting for the antiemetic drugs such as ondansetron.4
But the pharmacokinetic (PK) properties of oral soluble film ondansetron in Chinese population have not been well reported in the previous studies. Based on the literature search, the PK parameters of oral soluble film ondansetron were reported in the previous study,5 but they are not well documented in Chinese population. Considering the effect of racial and genetic differences on drug interactions, the study of the PK properties of oral soluble film ondansetron is necessary.
Therefore, the aim of this study was to investigate the PK properties of ondansetron and the bioequivalence of a test ondansetron soluble film 8 mg (Jiangsu Hengrui Pharmaceutical Co. Ltd., Nanjing, People’s Republic of China) and a reference ondansetron soluble film 8 mg (Zuplenz®; Galena Biopharma, Inc., Portland, OR, USA) in a healthy Chinese adult male population to obtain regulatory approval for the test formulation. We also established a validated LC-MS/MS method for the determination and quantification of ondansetron in the human plasma.
Materials and methods
Subjects and study design
This open-label, randomized-sequence, single-dose, two-way crossover study involved ten healthy subjects under fasting condition, with 1-week washout period.
Healthy male volunteers aged 18–40 years were eligible for recruitment (body mass index, 20.1–24.8 kg/m2). Inclusion criteria included a healthy status confirmed by medical documentation (complete medical history, physical examination, chest radiography, serum laboratory evaluation, and electrocardiography). Other inclusion criteria included abstinence from all drug as well as alcoholic or caffeinated beverages for 2 weeks prior to study entry. Subjects were excluded from the study if they had a history or evidence of a renal, gastrointestinal, hepatic, or hematologic abnormality or any acute or chronic disease. This was done to ensure that the existing degree of variation would not be due to an influence of illness or other medications. Eligible subjects were informed of the aim and risks of the study by the clinical investigators and provided written informed consent prior to study initiation.
This study was performed in accordance with the current revision of the Declaration of Helsinki,6 Good Clinical Practice,7 and Good Laboratory Practice.8 The study and the informed consent form were approved prior to the start of the study by the First Hospital of Nanjing Medical University ethnics committee.
Subjects were admitted to the hospital at 9 pm the day before the study and fasted 8 hours before each drug administration. A computer-generated random number table was used to assign subjects in a 1:1 ratio to receive a single 8 mg dose of the test or reference formulation of ondansetron. Subjects and investigators were blinded to sequence. As the t1/2 of ondansetron oral soluble film is 4.6 hours, a 1-week washout period followed administration of the initial formulation, after which the alternate formulation was administered.
Chemicals and reagents
Ondansetron (lot: RS1002100510, the reference standard; purity: 100.3%) and lacosamide (lot: 20120316-2, the internal standard [IS]; purity: 99.51%) were provided by Hangzhou Pharm & Chem Co., Ltd. (Hangzhou, People’s Republic of China). High-performance liquid chromatography (HPLC) grade of methanol and methyl tert-butyl ether was purchased from Tedia Company (Fairfield, OH, USA). Other reagents were all of analytical grade and were purchased from Nanjing Chemical Reagent Co., Ltd. (Nanjing, People’s Republic of China).
The test formulation (8 mg ondansetron oral soluble film) was manufactured by Jiangsu Hengrui Pharmaceutical Co. Ltd. The reference formulation was the leading product in the international market (Zuplenz®, Galena Biopharma, Inc.) and provided by Jiangsu Hengrui Pharmaceutical Co. Ltd.
Blood sampling and assaying
After the 8-hour overnight fast and before administration of the study drug, blood was drawn for baseline (time 0) measurements. A 20 G catheter (WeiGao Company, Shandong, People’s Republic of China) was placed in a forearm vein, and a 5 mL blood sample was drawn into a vacuum tube with heparin sodium (Shanghai Biochemistry Co., Ltd., Shanghai, People’s Republic of China) as an anticoagulant. Additional blood samples were drawn at 20 minutes, 40 minutes, 60 minutes, 80 minutes, 100 minutes, 120 minutes, 140 minutes, and 160 minutes and 3 hours, 4 hours, 6 hours, 8 hours, 10 hours, 12 hours, 14 hours, and 24 hours after dosing. Subjects were provided with a standard meal (60% carbohydrate, 30% protein, and 10% fat; ~900 kcal) at 4 hours and 10 hours after studying drug administration in each treatment group. Plasma was immediately separated by nonrefrigerated centrifuge (Jiangsu Jintan Huanyu Instrument Company, Changzhou, People’s Republic of China) at 1,072 g (r=0.03 m) for 10 minutes and then stored at −65°C until analyzed by Agilent 6460A Triple Quad LC-MS/MS systems with multiple reaction monitoring (Agilent Technologies, Santa Clara, CA, USA). After washout period and administration of the alternate formulation, blood samples were drawn and analyzed in the same way.
Determination of ondansetron
An HPLC system, Agilent 1200 series LC equipped with an autosampler, an online degasser, a binary pump, a thermostatted column compartment, and a UV detector connected with a Zorbax SB-C18 column (3.5 μm, 2.1 mm ×100 mm, Agilent Technologies), was used for chromatographic separation. MassLynx 4.0 analysis workstation (Waters Corporation) was used for system control, data acquisition, and quantification.
All the plasma was thawed at room temperature. In all, 100 μL of thawed plasma was placed with 500 μL of methyl tert-butyl ether (containing 10 μL of 100 ng/mL IS lacosamide) into clean Eppendorf tubes, vortexed for 60 seconds, and centrifuged at 1,072 g for 5 minutes at 20°C. Then 450 μL of the supernatants were transferred to clean vials, evaporated to dryness at 37°C under nitrogen, and resolved with 200 μL mobile phase.
For a complete separation of all the compounds, various concentrations of mobile phases were tested. The mobile phase of 0.1% methanoic acid aqueous solution/methanol (65:35, vol/vol) proved to show the best separation efficiency.
Then the column effluent was directly introduced into the MS detector Agilent Technologies 6460 Triple Quadrupole LC-MS/MS operated in a positive electrospray ionization (ESI) mode. Nitrogen was used as both the sheath gas and collision gas. The ESI source parameters were as follows: gas temperature 320°C, gas flow 10 L/min, nebulizer gas pressure 45 psi, sheath gas temperature 300°C, sheath gas flow 11 L/min, and capillary voltage 3,500 V. Fragmentor voltages were selected for ondansetron and IS ranging from 80 V to 140 V and 60 V to 120 V, respectively. The parameters of collision energy were selected from ondansetron and are ranging from 16 eV to 22 eV and 0 eV to 6 eV, respectively. The optimized conditions are as follows: fragmentor voltages of 120 V (ondansetron) and 60 V (IS), respectively. The parameters of collision energy are 22 eV (ondansetron) and 2 eV (IS). Samples were analyzed by LC-MS/MS in the multiple reaction monitoring mode to maximize sensitivity. Characteristic transitions (precursor ion → product ion) are [M+H]+ (ondansetron) m/z 294.2→170.2 and [M+H]+ (IS) m/z 251.2→108.2 (Figure 1).
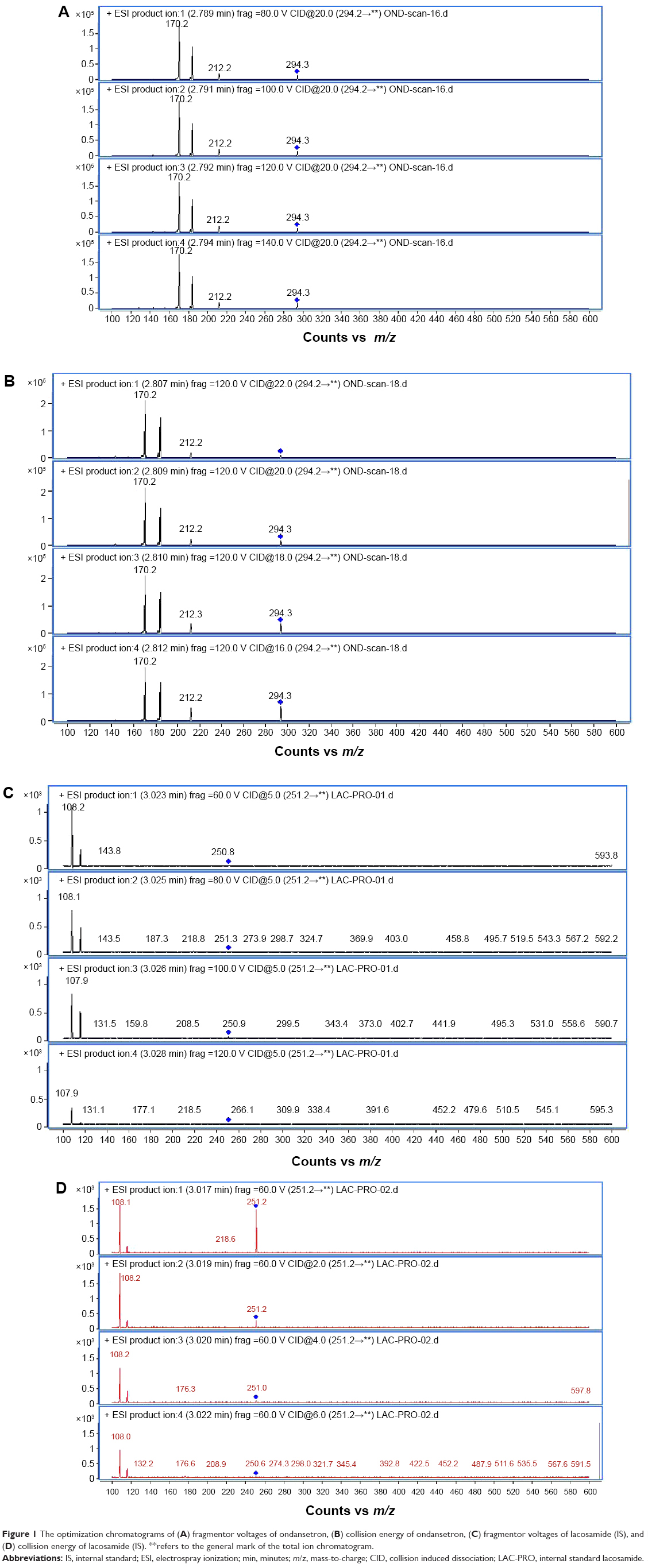 | Figure 1 The optimization chromatograms of (A) fragmentor voltages of ondansetron, (B) collision energy of ondansetron, (C) fragmentor voltages of lacosamide (IS), and (D) collision energy of lacosamide (IS). **refers to the general mark of the total ion chromatogram. |
Method validation
The validation of methods is characterized by sensitivity, specificity, linearity, recovery, accuracy, and interday and intraday precisions. The lower limit of quantification for this method was 0.2 ng/mL, and the signal-to-noise ratio was >5. The samples were measured at three levels of 0.4 ng/mL, 4.0 ng/mL, and 64.0 ng/mL of quality control (QC) concentrations in all cases, indicating the acceptable precision and accuracy of the method. No endogenous peaks interfering with quantification were observed throughout the validation process (Figure 2). The validation data, as presented in Table 1, were taken from our validation report. The values for intraday and interday precisions and accuracy (all <7% in all cases) meet with the requirements of China Food and Drug Administration (CFDA) and US Food and Drug Administration (FDA) guidelines (Table 1).9,10
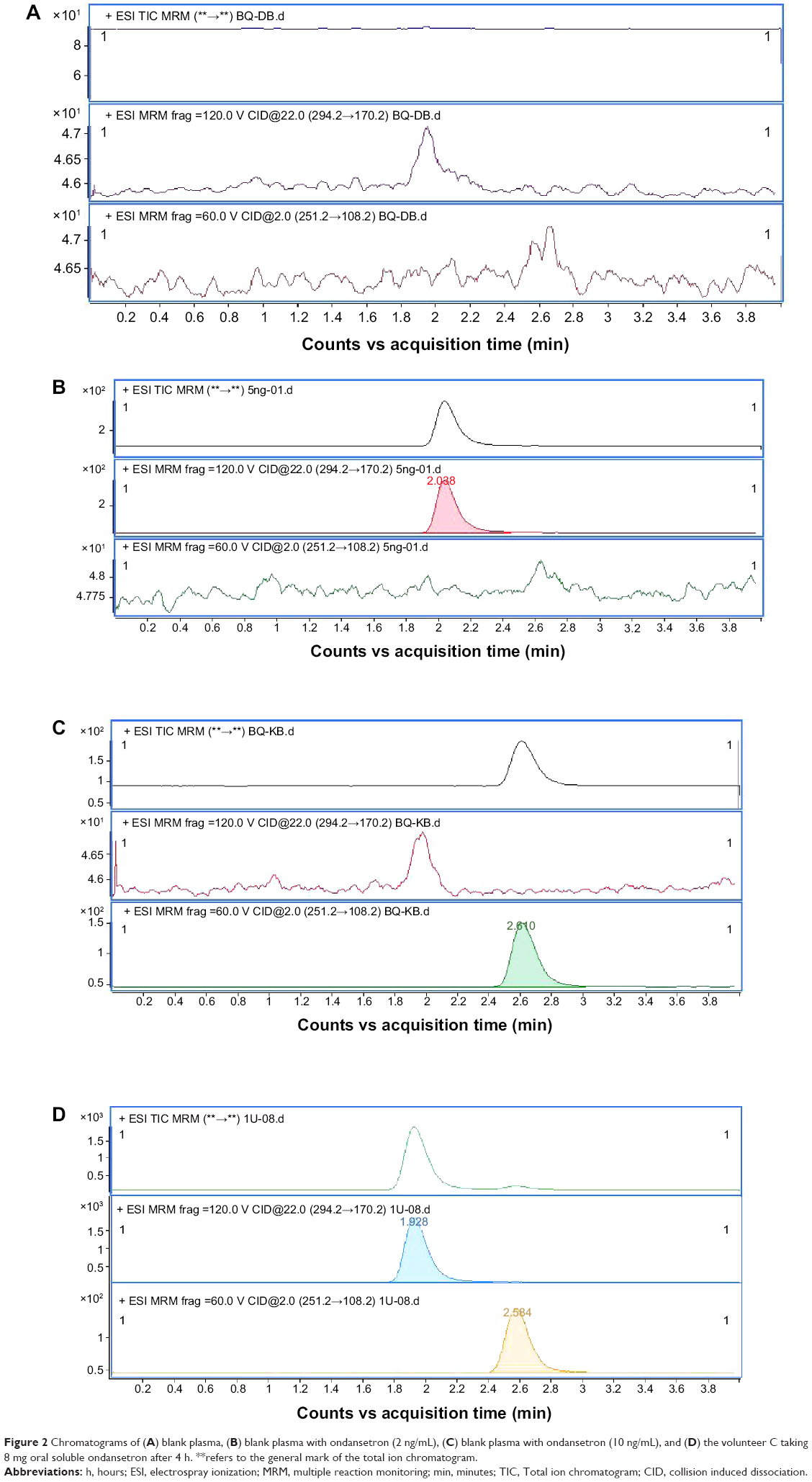 | Figure 2 Chromatograms of (A) blank plasma, (B) blank plasma with ondansetron (2 ng/mL), (C) blank plasma with ondansetron (10 ng/mL), and (D) the volunteer C taking 8 mg oral soluble ondansetron after 4 h. **refers to the general mark of the total ion chromatogram. |
 | Table 1 Accuracy and interday and intraday precisions of ondansetron |
Matrix effects
The matrix effects were evaluated by the QC samples of three different concentrations (4.0 ng/mL, 40.0 ng/mL, and 64.0 ng/mL). In all, 100 μL drug-free plasma or 100 μL blank matrix (distilled water) samples were prepared by the procedures of determination of ondansetron. Then we evaluated matrix effects by comparing the peak area of ondansetron and lacosamide (IS) of drug-free plasma (n=5) with them in blank matrix (n=5). Mean (SD) of the ratios of ondansetron and lacosamide (IS) were 95.44 (3.24) and 101.22 (3.11), respectively, which suggested that there were no matrix effects that affect the determination of ondansetron.
PK and statistical analysis
Using a power analysis (expected value, ≥1−β=0.8), it was determined that the power of the analysis of variance (ANOVA) was >0.8 at a 90% confidence interval (CI), according to the FDA11 guidelines on bioequivalence testing, indicating that ten subjects would be sufficient for the purposes of the study.
A noncompartmental PK method was employed to determine the PK properties of ondansetron. Cmax and Tmax were obtained directly from the plasma concentration–time curves of ondansetron. Other PK parameters (AUC0–∞, elimination rate constant (ke), and t1/2) were calculated on noncompartmental analysis using Drug and Statistics Software version 2.1 (University of Science and Technology, Hefei, People’s Republic of China). AUC0–24 was calculated using the trapezoidal rule.12 AUC0–∞ was calculated as follows:
AUC0−∞ = AUC0−24+C24/ke |
where C24 represented the last measurable concentration. ke was determined from the slope of the natural logarithm (ln)-linear portion of the plasma concentration–time curve using least-squares regression analysis, and t1/2 was estimated using the following equation:
|
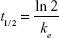
The relative bioavailability (F) of the test was calculated as follows:
|
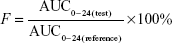
To test the bioequivalence of the formulations, ANOVA for a two-way crossover design was performed on log-transformed Cmax, AUC0–24, and AUC0–∞. The ratios of the log-transformed Cmax, AUC0–24, and AUC0–∞ were obtained for both formulations, and ANOVA was performed using the F-score. The probability of exceeding the limits of acceptance (80%–125%) was obtained using two-sided t-tests, as described by Schuirmann13 and the FDA.11 The 90% CIs of the geometric means of the individual test/reference (T/R) ratios for Cmax, AUC0–24, and AUC0–∞ were obtained.
Results
In all, ten male subjects (mean [range] age, 28.4 years [18–40 years]; weight, 62.4 kg [55–75 kg]; height, 172 cm [163–184 cm]) were enrolled in the study. Five subjects each received the test or reference formulations first changed after 1-week period. All volunteers completed the study. The mean ondansetron concentration–time profiles after administration of the two formulations are shown in Figure 3.
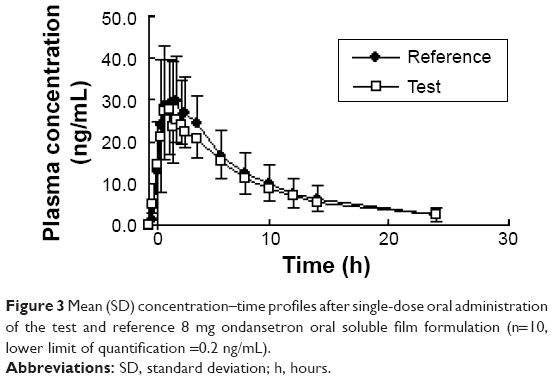 | Figure 3 Mean (SD) concentration–time profiles after single-dose oral administration of the test and reference 8 mg ondansetron oral soluble film formulation (n=10, lower limit of quantification =0.2 ng/mL). |
The values of the PK parameters (AUC0–24, AUC0–∞, Cmax, t1/2, and Tmax) and the geometric mean ratios (90% CI) of AUC0–24, AUC0–∞, and Cmax of ondansetron are presented in detail in Table 2.
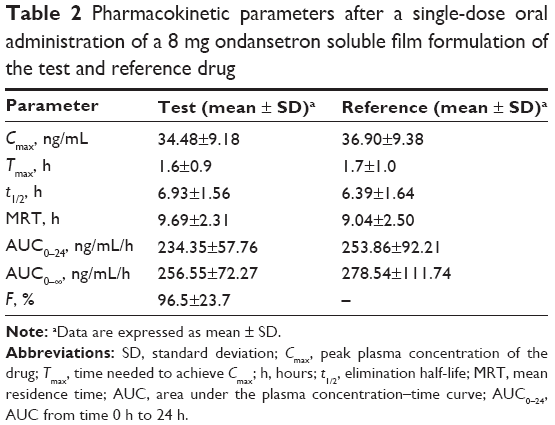 | Table 2 Pharmacokinetic parameters after a single-dose oral administration of a 8 mg ondansetron soluble film formulation of the test and reference drug |
Out of three volunteers, two experienced abdominal pain after administration of the test and reference formulation and one experienced skin rash after administration of the reference formulation. All the symptoms resolved spontaneously within 1 hour. Physical examination revealed no abnormalities attributable to ondansetron administration. The studied biochemical, hematological, and electrocardiography (ECG) parameters revealed no abnormalities. The subjects were closely monitored after the symptoms resolved, and no other effects were reported. The adverse events (AEs) were considered by three independent investigators as mild. None of the volunteers withdrew from the study. No serious AEs were reported. At the end of the study, all vital signs were considered to be normal in all subjects on the basis of physical examination and results of blood chemistry analysis. There were no significant AEs encountered during this bioequivalence study.
Discussion
The aim of this study was to investigate the PK properties of two 8 mg oral soluble film formulations of ondansetron in a healthy Chinese adult male population to obtain regulatory approval for the test formulation.
Previous studies reported many methods, such as radioimmunoassay,14 spectrophotometric,15 HPLC–UV,16 high-performance thin layer chromatography,17 and capillary electrophoresis,18 to determine the plasma concentrations of ondansetron. However, these methods have some drawbacks such as laborious sample preparation procedures, time-consuming gradient elution, lower sensitivity, and comparatively longer runtime, which limited their application to large amounts of quantification in biological fluids. LC-MS/MS19,20 was recently the most commonly used method to determine the plasma, but it required expensive and time-consuming solid phase extraction columns, which are not fit for using in developing countries. As ondansetron is a medium polar compound, the determination of biological fluids always suffered with residues in the chromatography systems, especially for the concentration of low limitation of quantification.
So it is necessary to develop a rapid, accurate, precise, and sensitive method to overcome the drawbacks of reported methods. We created and validated a LC-MS/MS method for the determination and the quantification of ondansetron in human plasma. Compared to previous studies, gradient elution was always used in the determination of ondansetron for better results in chromatographic separation. We used isocratic elution to save analytical times and the mobile phase with good sensitivity. According to the results of specificity, the assay is suitable for the PK research of ondansetron in humans.
Quimby et al studied the PK parameters of ondansetron in healthy cats with approximately a single 0.4 mg/kg dose of oral, subcutaneous, and intravenous formulation.21 The t1/2 of ondansetron was 1.84±0.58 hours (intravenous), 1.18±0.27 hours (oral), and 3.17±0.53 hours (subcutaneous), suggesting that subcutaneous administration of ondansetron to healthy cats is a more prolonged exposure than oral and intravenous administration. Dychter et al studied the PK properties with a single 8 mg dose of ondansetron. The PK data of ondansetron were AUC0–∞ 287.19 ng/mL/h and Cmax 33.5 hours.2 Dadey reported bioequivalence study in healthy Indian populations.4 The results of a single 8 mg dose ondansetron were AUC0–∞ 293.73 ng/mL/h and Cmax 39.05 hours, which had similar PK properties with the orally disintegrating formulations in their study.
The mean (SD) data for the test and reference drug ondansetron in our study were t1/2 6.93 hours (1.56 hours) and 6.39 hours (1.64 hours), Cmax 34.48 ng/mL (9.18 ng/mL) and 36.90 ng/mL (9.38 ng/mL), and AUC0–∞ 256.55 ng/mL/h (72.27 ng/mL/h) and 278.54 ng/mL/h (111.74 ng/mL/h), respectively. These values were similar to the results in the previous literature.4 The 90% CIs of the T/R AUC0–24 and Cmax ratios of ondansetron were 91.38%–108.60% and 84.71%–103.28%, respectively. The differences between the test and reference products for Cmax and AUC values were not found to be statistically significant (P<0.05), which can be assumed to be bioequivalent according to FDA and CFDA guidelines.9,10 There were no significant AEs encountered during this study, which suggested that all the two formulations are well tolerated.
Conclusion
Based on the PK results of this study, it was concluded that the two formulations of the 8 mg ondansetron oral soluble formulations were bioequivalent.
Study limitation
This study was a single-dose, open-label design in a small group of young healthy male volunteers; therefore, the study results cannot be extrapolated to female, older population or to patients. Additionally, although sex differences were reported on the PK parameters of ondansetron, they are not discussed in this study as our study aims to estimate the bioequivalence of the test and reference formulations.
Acknowledgment
The authors would like to acknowledge Jiangsu Hengrui Pharmaceutical Co. Ltd. for providing the test formulation of ondansetron and the reference standards used in this research.
Disclosure
The authors have indicated that they have no conflicts of interest in this work.
References
Mujtaba A, Kohli K, Ali J, Baboota S. Development of HPTLC method for the estimation of ondansetron hydrochloride in bulk drug and sublingual tablets. Drug Test Anal. 2013;5:122–125. | ||
Dychter SS, Harrigan R, Bahn JD, et al. Tolerability and pharmacokinetic properties of ondansetron administered subcutaneously with recombinant human hyaluronidase in minipigs and healthy volunteers. Clin Ther. 2014;36(2):211–224. | ||
Gabriel J. Subcutaneous fluid administration and the hydration of older people. Br J Nurs. 2014;23(14):S12–S14. | ||
Dadey E. Bioequivalence of ondansetron oral soluble film 8 mg (ZUPLENZ) and ondansetron orally disintegrating tablets 8 mg (ZOFRAN) in healthy adults. Am J Ther. 2015;22(2):90–97. | ||
Choudhary DR, Patel VA, Chhalotiya UK, Patel HV, Kundawala AJ. Comparative pharmacokinetic studies of fast dissolving film and oral solution of ondansetron in rats. Curr Drug Deliv. 2013;10(6):696–700. | ||
World Medical Association. Declaration of Helsinki. Ethical principles for Medical Research Involving Human Subjects. Adopted by the 18th WMA General Assembly, Helsinki, Finland, June 1964 and Amended by the 64th WMA General Assembly, Fortaleza, Brazil, October, 2013; 2015. Available from: http://www.wma.net/en/30publications/10policies/b3/index.html | ||
International Conference on Harmonisation Expert Working Group. ICH Harmonised Tripartite Guideline: Guideline for Good Clinical Practice E6 (R1). Geneva, Switzerland: International Conference on Harmonisation; 1996. | ||
Organization for Economic Co-operation and Development. OECD Series on Principles of Good Laboratory Practice and Compliance Monitoring Number 1: OECD Principles on Good Laboratory Practice. Paris, France: Organization for Economic Co-operation and Development; 1997. | ||
China Food and Drug Administration (CFDA), Center for Drug Evaluation. Guideline for Bioavailability and Bioequivalence Studies of Generic Drug Products [in Chinese]. Available from: http://www.sda.gov.cn/gsz05106/08.pdf. Accessed July 31, 2015. | ||
Center for Drug Evaluation and Research, US Food and Drug Administration. Guidance for Industry. Human Drugs In Vivo Bioequivalence Compliance Program. Available from: http://www.fda.gov/downloads/ICECI/EnforcementActions/BioresearchMonitoring/UCM133760.pdf. Accessed July 31, 2015. | ||
Center for Drug Evaluation and Research, US Food and Drug Administration. Guidance for Industry: Human Drugs In Vivo Bioequivalence Compliance Program 7348.001. Available from: http://www.fda.gov/ora/complianceref/bimo/7348_001/default.htm. Accessed July 31, 2015. | ||
Center for Drug Evaluation and Research, US Food and Drug Administration, Guidance for Industry. Bioanalytical Method Validation. Available from: http://www.fda.gov/cder/guidance/4252fnl.htm | ||
Schuirmann DJ. A comparison of the two one-sided tests procedure and the power approach for assessing the equivalence of average bioavailability. J Pharmacokinet Biopharm. 1987;15:657–680. | ||
Wring SA, Rooney RM, Goddard CP, Waterhouse I, Jenner WN. A sensitive radio-immunoassay, combined with solid-phase extraction, for the sub-nanogram per ml determination of ondansetron in human plasma. J Pharm Biomed Anal. 1994;12(3):361–371. | ||
Bourdon F, Lecoeur M, Odou P, Vaccher C, Foulon C. Complementarity of UV-PLS and HPLC for the simultaneous evaluation of antiemetic drugs. Talanta. 2014;120:274–282. | ||
Chandrasekar D, Ramakrishna S, Diwan PV. A rapid, sensitive and validated method for the determination of ondansetron in human plasma by reversed-phase high-pressure liquid chromatography. Arzneimittelforschung. 2004;54(10):655–659. | ||
Raval PB, Puranik M, Wadher SJ, Yeole PG. A validated HPTLC method for determination of ondansetron in combination with omeprazole or rabeprazole in solid dosage form. Indian J Pharm Sci. 2008;70(3):386–390. | ||
Li L, Xu L, Huang J, You T. On-line focusing of 5-hydroxy-tryptamine type 3 receptor antagonists via the combination of field-enhanced sample injection and dynamic pH junction in capillary electrophoresis with amperometric detection. J Chromatogr A. 2014;1331:117–122. | ||
Dotsikas Y, Kousoulos C, Tsatsou G, Loukas YL. Development and validation of a rapid 96-well format based liquid-liquid extraction and liquid chromatography-tandem mass spectrometry analysis method for ondansetron in human plasma. J Chromatogr B Analyt Technol Biomed Life Sci. 2006;836(1–2):79–82. | ||
Xu X, Bartlett MG, Stewart JT. Determination of ondansetron and its hydroxy metabolites in human serum using solid-phase extraction and liquid chromatography/positive ion electrospray tandem mass spectrometry. J Mass Spectrom. 2000;35(11):1329–1334. | ||
Quimby JM, Lake RC, Hansen RJ, Lunghofer PJ, Gustafson DL. Oral, subcutaneous, and intravenous pharmacokinetics of ondansetron in healthy cats. J Vet Pharmacol Ther. 2014;37(4):348–353. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.