")
Back to Journals » Drug Design, Development and Therapy » Volume 14
Pharmacokinetic-Based Drug–Drug Interactions with Anaplastic Lymphoma Kinase Inhibitors: A Review
Authors Zhao D, Chen J, Chu M, Long X, Wang J
Received 10 February 2020
Accepted for publication 2 April 2020
Published 30 April 2020 Volume 2020:14 Pages 1663—1681
DOI https://doi.org/10.2147/DDDT.S249098
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Jianbo Sun
Dehua Zhao,1 Jing Chen,1 Mingming Chu,2 Xiaoqing Long,1 Jisheng Wang1
1Department of Clinical Pharmacy, The Third Hospital of Mianyang (Sichuan Mental Health Center), Mianyang 621000, People’s Republic of China; 2Department of Clinical Pharmacy, The Second Affiliated Hospital of Army Medical University, Chongqing 400037, People’s Republic of China
Correspondence: Jisheng Wang; Dehua Zhao Email [email protected]; [email protected]
Abstract: Anaplastic lymphoma kinase (ALK) inhibitors are important treatment options for non-small-cell lung cancer (NSCLC), associated with ALK gene rearrangement. Patients with ALK gene rearrangement show sensitivity to and benefit clinically from treatment with ALK tyrosine kinase inhibitors (ALK-TKIs). To date, crizotinib, ceritinib, alectinib, brigatinib, lorlatinib, and entrectinib have received approval from the US Food and Drug Administration and/or the European Medicines Agency for use during the treatment of ALK-gene-rearrangement forms of NSCLC. Although the oral route of administration is convenient and results in good compliance among patients, oral administration can be affected by many factors, such as food, intragastric pH, cytochrome P450 enzymes, transporters, and p-glycoprotein. These factors can result in increased risks for serious adverse events or can lead to reduced therapeutic effects of ALK-TKIs. This review characterizes and summarizes the pharmacokinetic parameters and drug–-drug interactions associated with ALK-TKIs to provide specific recommendations for oncologists and clinical pharmacists when prescribing ALK-TKIs.
Keywords: ALK, TKIs, NSCLC, PK, drug–drug interactions
Introduction
Lung cancer is one of the most common and lethal malignancies worldwide, and non-small cell lung cancer (NSCLC) is the most frequently occurring form of lung cancer.1 NSCLC has been shown to be driven by various activated oncogenes.2 NSCLC was first associated with activating mutations in the epidermal growth factor receptor (EGFR).3 Because of the high clinical response rates to EGFR inhibitors among patients with NSCLC associated with EGFR mutations, the detection of activating mutations in EGFR and the utilization of EGFR inhibitors introduced a new therapeutic strategy to combat NSCLC.4 In addition to mutations in EGFR, mutations in anaplastic lymphoma kinase (ALK) have been associated with NSCLC.5 ALK gene rearrangements occur in approximately 5% of NSCLC patients, indicating that ALK may represent a new and promising molecular target for NSCLC treatment.6 To date, several ALK tyrosine kinase inhibitors (ALK-TKIs) have been developed and are widely available in clinical practice, some of which have received approval by the US Food and Drug Administration (FDA) and/or the European Medicines Agency (EMA),7 such as crizotinib, ceritinib, alectinib, brigatinib, lorlatinib, and entrectinib.
All ALK-TKIs are administered orally, which makes administration flexible and convenient and improves quality of life. However, despite these advantages, the oral route of administration also increases the risk of potential drug–drug interactions (DDIs), leading to high interpatient variability and subsequent risks for increased toxicity and/or reduced treatment efficacy.
DDIs can be classified into pharmacodynamic DDIs and pharmacokinetic (PK) DDIs.8 PK DDIs are defined as drug interactions that affect absorption, distribution, metabolism, and excretion, leading to the altered bioavailability of a drug and possible unfavorable outcomes.9 Pharmacodynamic DDIs refer to interactions during which active compounds alter pharmacological effects, which can be additive, antagonistic, or synergistic.10,11
The primary objective of this review article is to present an overview of existing PK and DDI data for each of the FDA- and EMA-approved ALK-TKIs. In addition, we will provide specific recommendations designed to guide oncologists and clinical pharmacists through the process of managing DDIs during treatment with ALK-TKIs.
PK Parameters of ALK-TKIs
Crizotinib is a first-generation ALK-TKI, ceritinib, alectinib, and brigatinib are second-generation ALK-TKIs, and lorlatinib is a third-generation ALK-TKI. For entrectinib, it is a potent oral inhibitor of the tyrosine kinases tropomyosin receptor kinases (TRK) A/B/C, c-ros oncogene 1 (ROS1), and ALK. After oral intake, the median maximum plasma concentration (Cmax) of crizotinib, ceritinib, alectinib, and entrectinib are reached from 4 to 6 hours, whereas the median times to achieve Cmax for brigatinib and lorlatinib are 1 to 4 hours and 1.2 to 2 hours, respectively.12–24 Among the six existing ALK-TKIs, lorlatinib has been shown to have the highest bioavailability (81%).21 For lorlatinib,23 66% is bound to plasma proteins, whereas greater than 90% of the other five ALK-TKIs are bound to plasma proteins. All six ALK-TKIs have been demonstrated to be well-distributed in tissues. Crizotinib and ceritinib have been associated with poor central nervous system (CNS) penetration, whereas alectinib, brigatinib, lorlatinib, and entrectinib have been shown to cross the blood-brain barrier (BBB), allowing their distribution to CNS tissues. Crizotinib, ceritinib, alectinib, brigatinib, and entrectinib have been shown to be predominantly metabolized by the cytochrome P450 (CYP450) pathway,12–24 whereas lorlatinib has been shown to be predominantly metabolized by both CYP3A4 and UDP-glucuronosyltransferase (UGT1A4).22 For all six agents, the mean plasma elimination half-lives (t1/2) are longer than 20 hours. Apart from lorlatinib,22 these agents are predominantly excreted through the feces.12–24 The PK parameters of the six agents are listed in Table 1.
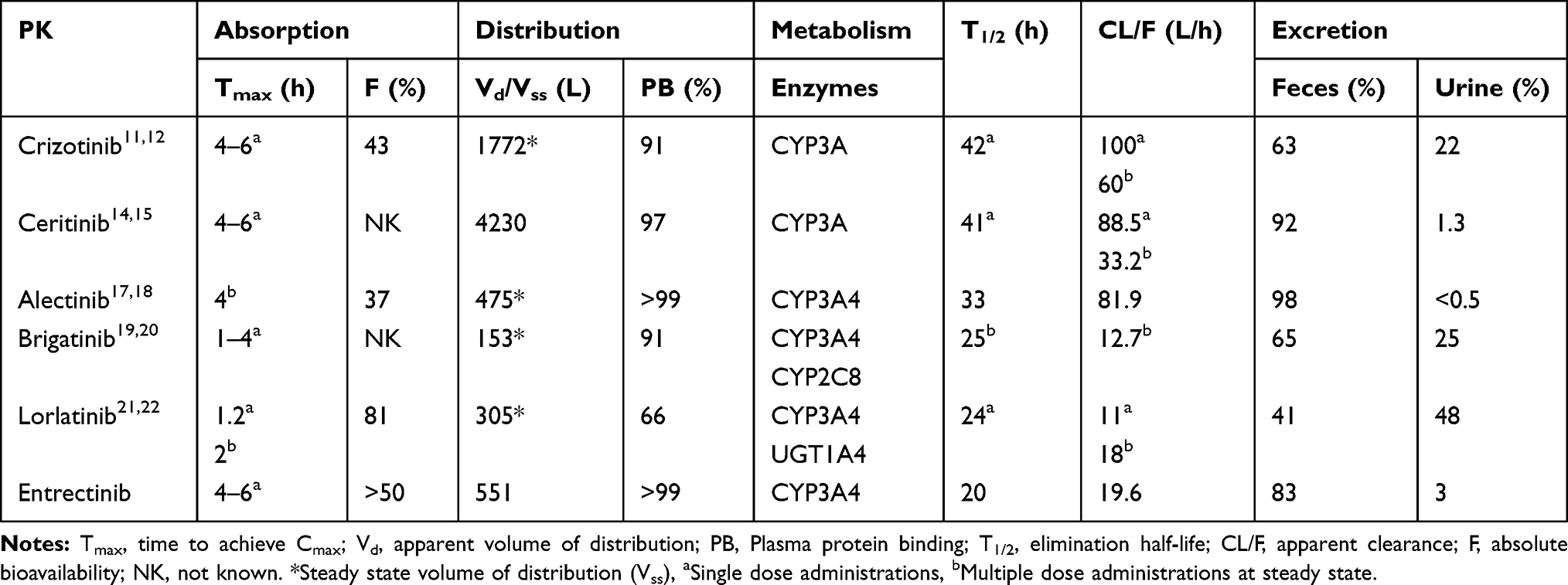 |
Table 1 PK Parameters for ALK-TKIs |
Crizotinib
Absorption
Following a single-dose administration of crizotinib, the median time to achieve Cmax for crizotinib ranges from 4 to 6 hours, and a steady-state concentration can be achieved within 15 days of repeated administration, with a median accumulation ratio of 4.8.12–14 The mean absolute bioavailability of crizotinib is 43%.12,13
Distribution
The Steady sate volume of distribution (Vss) for crizotinib is 1772 L, following a single, intravenous dose. The protein binding percentage of crizotinib is 91%, and the blood-to-plasma concentration ratio is approximately.12,13
Elimination
After a single dose, the mean t1/2 of crizotinib is 42 hours, with a mean apparent clearance (CL/F) of 100 L/h. When administered at an oral dosage of 250 mg, twice daily, the t1/2 is approximately 43–51 hours, with a mean CL/F of 60 L/h.12,13 Crizotinib has been shown to be primarily metabolized by CYP3A and was also shown to be a CYP3A inhibitor.12,13 This auto-inhibition of CYP3A could explain why the mean CL/F of crizotinib is lower under steady-state conditions than after a single, oral dose.13,14
Metabolism
Crizotinib is predominantly metabolized by CYP3A, which also significantly mediated the formation of the crizotinib lactam and O-desalkyl metabolites, to a significant extent.12,13
Excretion
Following the administration of a single dose of radiolabeled crizotinib, 63% (53% of which was unchanged) of the administered dose was recovered in feces and 22% (2.3% of which was unchanged) was recovered in urine.12,13
Ceritinib
Absorption
After a single oral administration of ceritinib to patients, the Cmax of ceritinib is achieved after approximately 4 to 6 hours.15,16 A steady-state concentration can be achieved within 15 days of repeated administration, and the accumulation ratio is 6.2 after 3 weeks.15,16 The absolute bioavailability of ceritinib has not been established but, based on the proportion of metabolites that have been excreted in mass balance studies, the lower limit of oral absorption has been estimated to be approximately 25%.15,16
Distribution
Ceritinib is 97% bound to plasma proteins. The Vd is 4230 L, following a single dose in patients. The mean blood to-plasma ratio in humans is 1.35.15,16
Elimination
Following a single dose, under fasted conditions, the t1/2 of ceritinib is 41 hours.15 The geometric mean CL/F under steady-state conditions is 33.2 L/h, which is lower than after a single dose in patients (88.5 L/h).15
Metabolism
CYP3A is the major enzyme involved in the metabolism of ceritinib.15 Following a single dose of radiolabeled ceritinib, unchanged ceritinib was the major circulating component (82%) in plasma, whereas 11 metabolites were identified at low levels (each accounting for ≤ 2.3% of the initial amount).15,16 Ceritinib has also been shown to be a CYP3A inhibitor, suggesting that the non-linearity pharmacokinetics of ceritinib may be attributable to the auto-inhibition of CYP3A.
Excretion
After a single dose of radiolabeled ceritinib, 92% of the administered dose was recovered in the feces (68% of which was unchanged) and 1.3% was recovered in the urine.15,16
Alectinib
Absorption
Alectinib reaches Cmax within 4 to 6 hours, after the administration of multiple doses of alectinib, under fed conditions in patients.17,18 A steady-state concentration of alectinib can be achieved within 7 days, after which it remains stable, with a median accumulation ratio of 5.6.18,19 The absolute bioavailability of alectinib is 37%.19
Distribution
The VSS for alectinib is 475 L.19 Protein binding percentages for both alectinib and M4 are greater than 99%.18,19 The mean blood-to-plasma ratios for alectinib and M4 are 2.64 and 2.50, respectively.18,19 As a result of its lipophilic properties, alectinib exhibits good penetration through the BBB, which leads to high concentrations in the cerebrospinal fluid (CSF).19
Elimination
In patients, the CL/F is 81.9 L/h for alectinib, and 217 L/h for M4, and the t1/2 is 33 hours for alectinib, and 31 hours for M4.19
Metabolism
Alectinib is predominantly metabolized into M4 by CYP3A4.18 Subsequently, M4 is metabolized into M6, also by CYP3A4.18 Following the administration of a single radiolabeled dose, unchanged alectinib is the primary circulating moiety found in plasma, constituting 61% of total radioactivity, whereas M4 constituted 15% of the total radioactivity in plasma.18,19
Excretion
Following a single radiolabeled dose of alectinib, 97.8% of the radioactivity is recovered in the feces and 0.46% is recovered in the urine.17–19 In the feces, 84% of alectinib is unchanged, whereas 5.8% of M4 is unchanged.17–19
Brigatinib
Absorption
Following a single dose of brigatinib, the median time to Cmax ranged between 1 and 4 hours.20,21 The absolute bioavailability of brigatinib has not been established. The mean accumulation ratio after repeated dosing is 1.9 to 2.4.20,21
Distribution
Brigatinib is 91% bound to plasma proteins, and its blood-to-plasma concentration ratio is 0.69.20,21 The Vss for brigatinib is 153 L.21
Elimination
The mean CL/F of brigatinib under steady-state conditions is 12.7 L/h, and its mean t1/2 is 25 hours after multiple daily doses of 180 mg brigatinib.20,21
Metabolism
Brigatinib is metabolized primarily by CYP2C8 and CYP3A4.20 Following a single radiolabeled dose, unchanged brigatinib represents the major circulating radioactive component, accounted for 92% of radioactivity, whereas its primary metabolite, AP26123, accounted for 3.5% of the radioactivity.20,21
Excretion
Following a single dose of radiolabeled brigatinib, the percent recovery of the administered dose was 65% in feces and 25% in urine.20,21 Unchanged brigatinib represented 41% and 86% of the total radioactivity in feces and urine, respectively.20,21
Lorlatinib
Absorption
The median time to achieve Cmax for lorlatinib is 1.2 hours, following a single oral dose, and 2 hours, following multiple daily doses.22 The steady-state concentration of lorlatinib can be reached within 23 hours.23 The mean absolute bioavailability is 81%.22,23
Distribution
Lorlatinib is 66% bound to plasma proteins. The blood-to-plasma ratio is 0.99 and the CSF-to-plasma ratio is 0.75.22 The Vss is 305 L, after a single intravenous dose.22,23
Elimination
The mean t1/2 of lorlatinib is 24 hours.22 The mean oral CL/F is 11 L/h (35%), following a single oral dose, and 18 L/h (39%) under steady-state conditions, suggesting autoinduction.22,23
Metabolism
Lorlatinib is metabolized primarily by CYP3A4 and UGT1A4, with minor contributions from CYP2C8, CYP2C19, CYP3A5, and UGT1A3.23 Following a single radiolabeled dose, a benzoic acid metabolite (M8) of lorlatinib, which is pharmacologically inactive, accounted for 21% of the circulating radioactivity in plasma.22,23
Excretion
Following a single dose of radiolabeled lorlatinib, 41% of the radioactivity was excreted in feces (less than 1% was unchanged) and 48% was excreted in urine (approximately 9% was unchanged).22,23
Entrectinib
Absorption
The time to achieve Cmax for entrectinib ranges from 4 to 6 hours after oral administration of a 600 mg dose.24 The oral bioavailability of entrectinib is estimated to be at least 50% based on total radioactivity recovered in urine and in feces.24
Distribution
Entrectinib and its active major metabolite M5 are both > 99% bound to plasma proteins.24 The Vd/F is 551 L and 81.1 L for entrectinib and M5, respectively.24 The blood‐to-plasma ratio is 1.3 for entrectinib and 1.0 for M5.25
Elimination
The CL/F is 19.6 L/h and 52.4 L/h for entrectinib and M5, respectively.24 The t1/2 of entrectinib and M5 were estimated to be 20 and 40 hours, respectively.24
Metabolism
Entrectinib is metabolized primarily by CYP3A4.24 The active metabolite M5 is the only major active circulating metabolite and circulating M5 exposures at steady-state in patients were 40% of the corresponding entrectinib exposure.24
Excretion
Following oral administration of a single oral dose of radiolabeled entrectinib, 83% of radioactivity was excreted in feces (36% was unchanged and 22% was M5) and 3% was excreted in excretion in urine.24
PK DDIs
PK DDIs: Absorption
Absorption-related drug interactions are commonly associated with drugs that undergo incomplete absorption (eg, have low bioavailability, first-pass effects, or are dependent on drug transporters).26 Important factors that can affect drug absorption include acid-reducing agents (ARAs), food, drug transporter inhibitors or inducers, and intestinal CYP inhibitors or inducers.8
Intragastric pH
Oral bioavailability is determined by absorption and first-pass effect. Dissolution in stomach is the first step in drug absorption, and only when a drug dissolves into small molecules, it can pass through the intestinal mucosa to the portal circulation. The effect of pH on the absorption of an oral drug depends on its chemical nature. Weak-base drugs may show decreased absorption as a result of decreased solubility at higher pH values.27,28
Patients with cancer frequently use ARAs, such as proton pump inhibitors (PPIs), H2-receptor antagonists (H2RAs), and antacids, to treat diseases caused by the hypersecretion of gastric acid. For drugs that exhibit pH-dependent solubility (eg, weak-base drugs), the elevation of gastric pH by ARAs may decrease solubility and absorption and, subsequently, reduce treatment efficacy.27 Effects of ARAs on the absorption of ALK-TKIs are listed in Table 2.
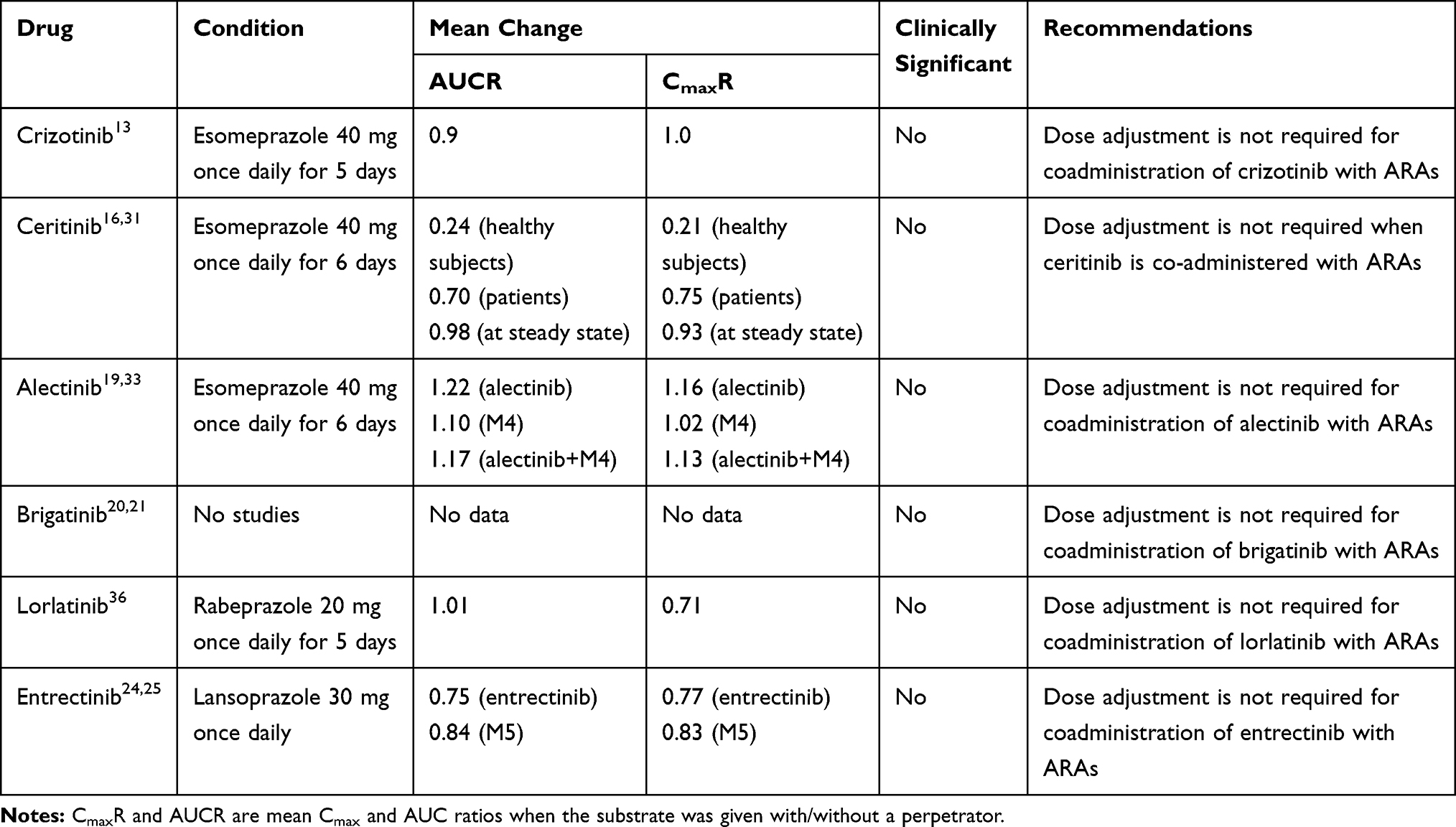 |
Table 2 Effect of ARAs on the Absorption of ALK-TKIs |
Crizotinib exhibits pH-dependent aqueous solubility, with solubility decreasing from more than 10 mg/mL at pH 1.6 to less than 0.1 mg/mL at pH 8.2.29 In a pharmacokinetic study, the administration of crizotinib following 5 days of treatment with 40 mg esomeprazole, once daily, resulted in a 10% decrease in the area under the curve (AUC), which is not considered to be clinically meaningful.13
Ceritinib is a weak-basic agent and displays pH-dependent solubility, with solubility decreasing from 11.9 to 0.01 mg/mL as pH values increase from 1 to 6.8.30 In clinical practice, the coadministration of a single dose of ceritinib with 40 mg esomeprazole, once daily, for 6 days, in healthy subjects, resulted in decreases in the AUC and Cmax of ceritinib, by 76% and 79%, respectively.31 However, the coadministration of ceritinib with PPIs for 6 days in ALK-positive cancer patients, the AUC and Cmax of ceritinib decreased by 30% and 25%, respectively.31 Consistent with the steady-state PK results, the effects of PPIs on the steady-state exposure of ceritinib is not considered to be clinically meaningful.16,31
Alectinib shows pH-dependent solubility, the solubility of alectinib ranging from 7.45µg/mL at pH1 to 0.013 at pH 6 in aqueous buffers.32 A pharmacokinetic study showed that administration of alectinib following multiple doses of esomeprazole (40 mg once daily for 6 days), the AUC and Cmax increased by 22% and 16% for alectinib, and increased by 10% and 2% for M4, respectively.33 Due to the low solubility of alectinib in the stomach at all pH values, the impact of gastric pH changing on its solubility and oral absorption seems with no clinically relevant.19
Brigatinib is classified as a high-solubility agent, with solubility decreasing from 157.1 to 2.5 mg/mL as pH values increase from 1.6 to 7.5.34 Thus far, no studies have been performed to assess the effects of ARAs on the absorption of brigatinib; however, considering its high solubility, the absorption of brigatinib is not expected to be significantly altered by the presence of ARAs. Therefore, dose adjustment is not required when brigatinib is co-administered with ARAs.20,21
Lorlatinib exhibits pH-dependent aqueous solubility that decreases over the pH range from 2.55 to 8.02, from 32.38 mg/mL to 0.17 mg/mL, respectively.35 Drug-interaction studies have demonstrated that the co-administered of rabeprazole (20 mg, once daily), resulted in a 29% decrease in Cmax, and 1% decrease in AUC, which was not considered to be clinically meaningful.36
Entrectinib has low and pH-dependent solubility, and its solubility decreases with increasing pH.25 When entrectinib was co-administered with lansoprazole (30 mg qd) under fasting condition, the AUC and Cmax decreased by 25% and 23% for entrectinib, and decreased by 16% and 17% for M5, respectively.25 Based on the results, coadministration of entrectinib with a PPI did not significantly alter the exposure of entrectinib.25 Thus, no dose adjustment is required for coadministration of PPI with entrectinib.24,25
Food
Food–drug interactions can have one of the following four pharmacokinetic effects on the absorption of an orally administered drug: delay, increase, decrease, or no effect.37 The mechanisms of food effects on oral drug absorption include increased gastrointestinal pH values, increased bile salt secretion, delayed gastric emptying, increased hepatic blood and gastrointestinal fluid flow, changes in the activity of drug transporters and intestinal enzymes, and binding to the drug.38 During the daily lives of patients, the oral administration of drugs, either with or without food, can have several advantages and disadvantages. Therefore, understanding whether a specific orally administered drug should be taken with or without food is essential. Effects of food on the absorption of ALK-TKIs are listed in Table 3.
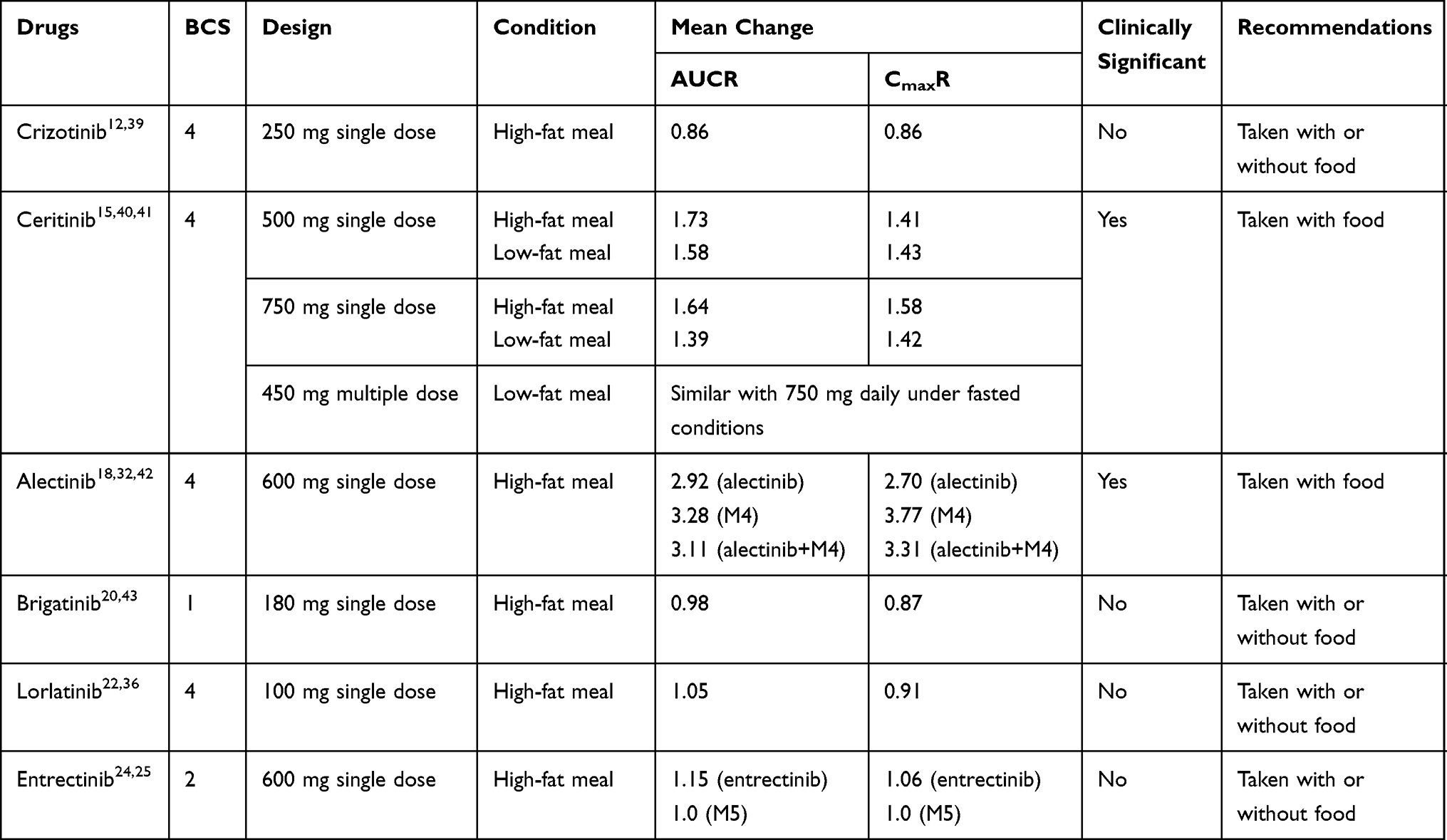 |
Table 3 Effects of Food on the Absorption of ALK-TKIs |
Crizotinib is classified as a class 4 compound (low solubility and low permeability) by the Biopharmaceutics Classification System (BCS).29 According to pharmacokinetic studies, the coadministration of crizotinib with a high-fat meal resulted in slight reductions (approximately 14%) in both the AUC and Cmax.39 Thus, crizotinib may be taken either with or without food.12,39
Ceritinib is classified as a BCS class 4 compound.30 In a food-effect trial, conducted in healthy subjects, a 500-mg dose of ceritinib administered with a high-fat meal (1000 calories and 58 grams of fat) increased the ceritinib AUC by 73% and the Cmax by 41%, whereas a low-fat meal (330 calories and 9 grams of fat) increased the ceritinib AUC by 58% and the Cmax by 43%.15 When the dose of ceritinib increased to 750 mg, the AUC and Cmax increased by 64% and 58%, with a high-fat meal, and by 39% and 42%, with a low-fat meal, respectively, compared with fasted conditions.40,41 In a dose-optimization study performed in patients, receiving a 450-mg dose of ceritinib daily, with food (100 to 500 calories and 1.5 to 15 grams of fat), resulted in similar steady-state exposure to ceritinib and more favorable gastrointestinal tolerability than receiving 750 mg, daily, under fasted conditions.41 Therefore, the recommended dosage of ceritinib is 450 mg, orally, once daily, with food.15
Alectinib is classified as a BCS class 4 agent.32 A study examining the effects of food on the pharmacokinetics of alectinib showed that a high-fat meal markedly increased the combined exposure to alectinib and M4, by 211% for AUC and 231% for Cmax, after the oral administration of a single dose of alectinib, with toxicities being well-tolerated.32,42 As a result, alectinib is recommended to be administered with food.18
Brigatinib is considered as a BCS class 1 substance (high solubility and high permeability).34 During food-effect studies, the brigatinib AUC and Cmax were reduced by 2% and 13%, respectively, in healthy subjects when administered with a high-fat meal (920 calories, 58 grams carbohydrate, 59 grams fat and 40 grams protein).43 Therefore, patients were instructed to take brigatinib without regard to meals.20
Lorlatinib meets the criteria for a BCS class 4 compound.35 The administration of lorlatinib with a high-fat meal (1000 calories, with 150 calories from protein, 250 calories from carbohydrates, and 500 to 600 calories from fat) resulted in an 5% increase in the AUC and an 9% decrease in the Cmax compared with fasted conditions.36 Food did not have a clinically meaningful effect on lorlatinib exposure. Thus, lorlatinib may be administered without regard to food.22
Entrectinib is considered as a BCS class 2 compound.25 When a single dose 600 mg entrectinib was administered with a high‐fat, high‐calorie meal, the entrectinib AUC and Cmax were increased by 15% and 6%, respectively.25 Moreover, food has no effect on the exposure of M5, indicating that food did not have a significant effect on entrectinib exposure.25 Therefore, entrectinib is recommended to be administered with or without food.24
Drug Transporters
Drug transporters are located throughout the body, especially in the intestinal gut, liver, kidney, and the BBB, where they play important roles in the membrane transport of many drugs.44,45 Drug transporters may be divided into two large families, the ATP-binding cassette transporters (ABCs) and the solute carrier transporters (SLCs). The ABCs are efflux drug transporters, including P-gp, multidrug resistance-associated protein 2 (MRP2), and breast cancer resistance protein (BCRP). Conversely, the SLCs are influx drug transporters, including organic anion transporting peptides (OATPs), organic anion transporters (OATs), and organic cation transporters (OCTs). The inhibition and induction of drug transporters can have significant impacts on the pharmacokinetics of drugs by altering the absorption, distribution, metabolism, and elimination.46 Likewise, elacridar, a P-gp and ABCG2 inhibitor, can increase the oral bioavailability of crizotinib.47 Therefore, more attention should be paid to the DDIs mediated by drug transporters. The potential DDIs between drug transporters and ALK-TKIs are mentioned in Table 4. The potential DDIs between ALK-TKIs and other transporter subsrates are mentioned in Table 5.
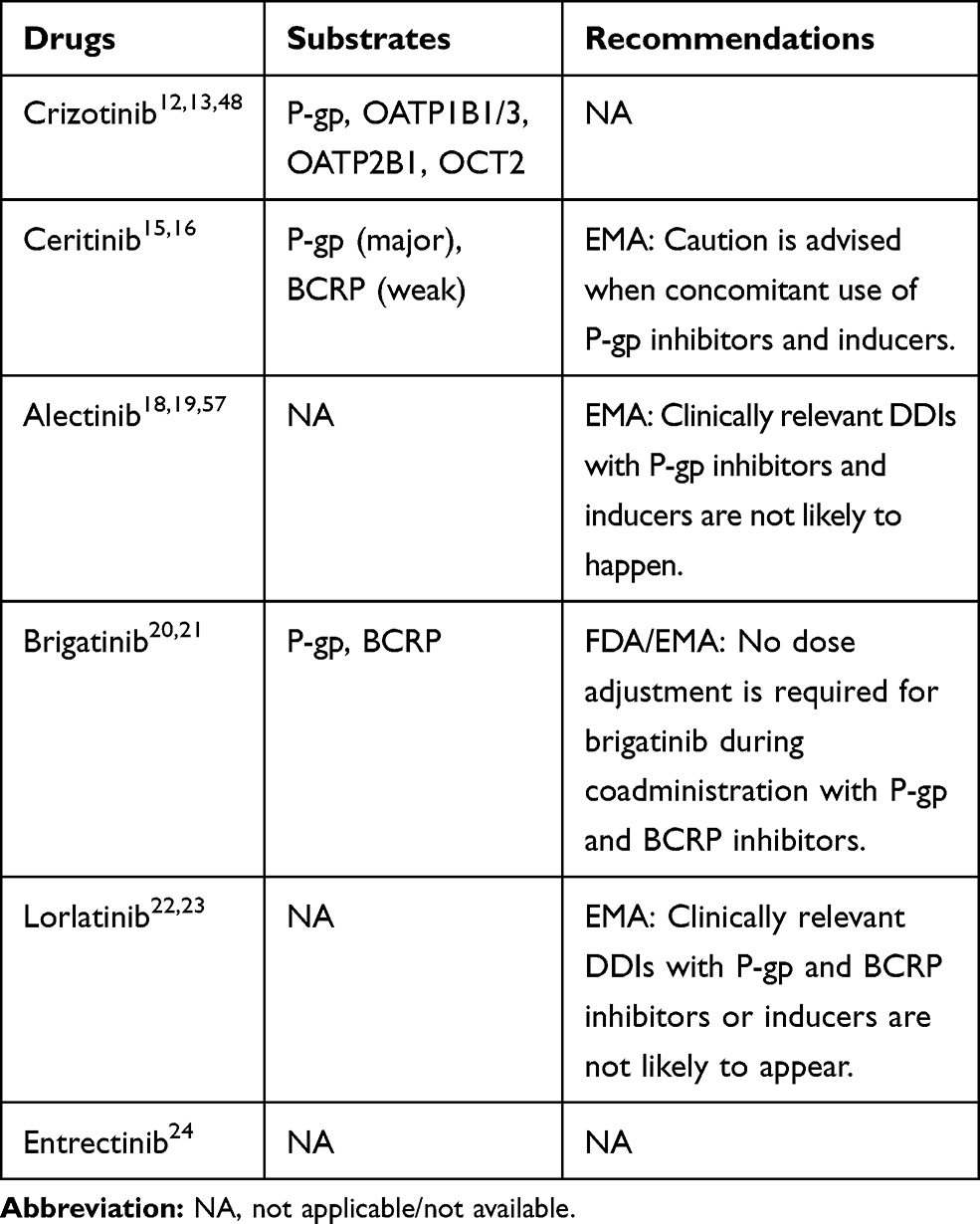 |
Table 4 The Potential DDIs Between Drug Transporters and ALK-TKIs |
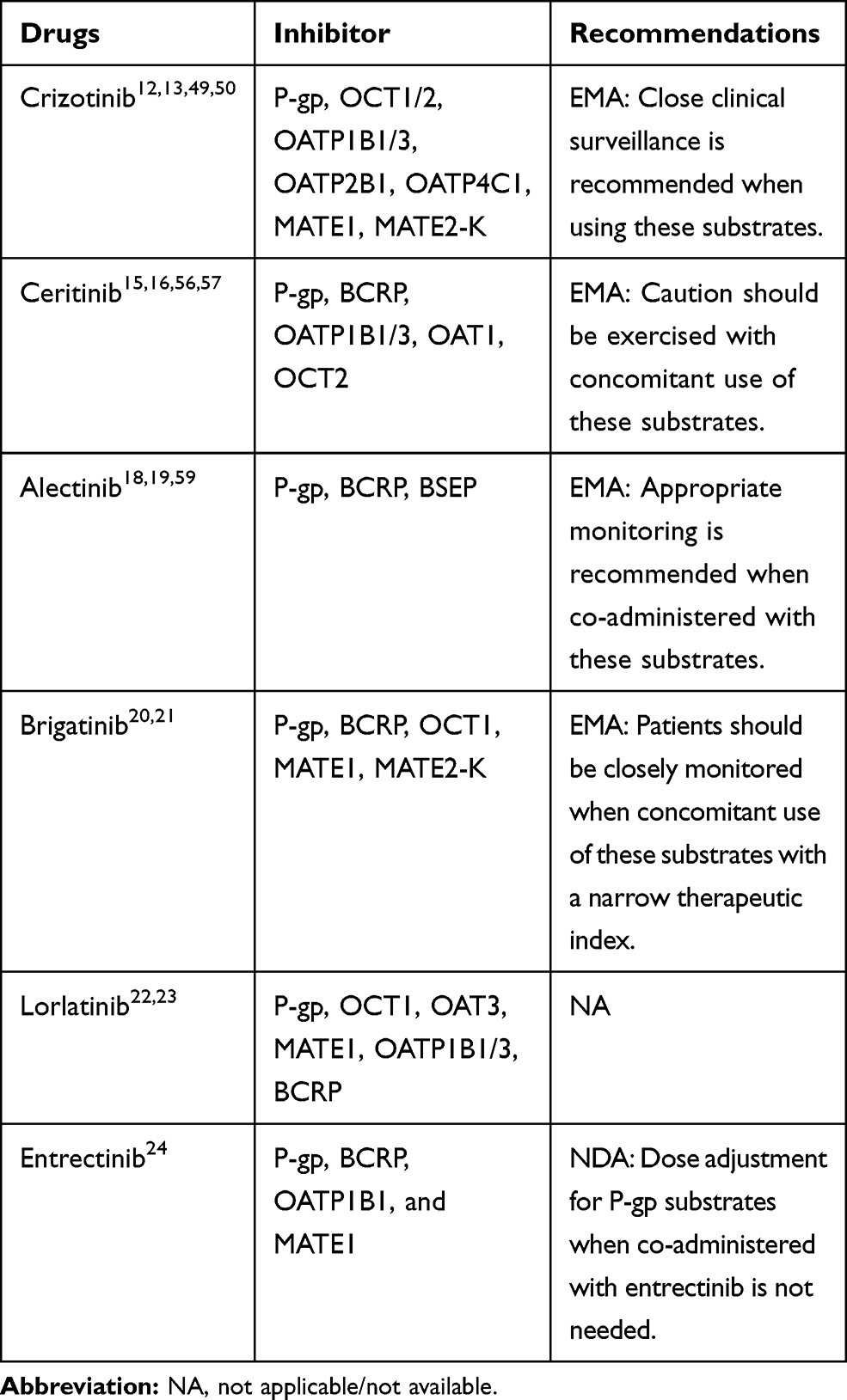 |
Table 5 The Potential DDIs Between ALK-TKIs and Other Transporter Substrates |
Based on in vitro data, crizotinib is transported by the P-gp, OATP1B1/2, OATP2B1, and OCT2.12,13 A mouse model study, conducted by Tang,43 demonstrated that the oral availability and brain accumulation of crizotinib were primarily restricted by P-gp, at a non-saturating dose, and coadministration with elacridar was able to substantially increase the oral availability of crizotinib. Shu et al48 reported the effect of the co-administered of entecavir on the PK of crizotinib in NSCLC patients. The crizotinib AUCss increased by 22% but Cmax, ss decreased by 7%. Moreover, the incidence of adverse reaction of crizotinib was significantly increased.
Crizotinib inhibits the P-gp, OCT1/2, OATPB1/3, OATP2B1, OATP4C1, multidrug and toxin extrusion protein (MATE1), and MATE2-K, but not OAT1/3, or bile salt export pump transporter (BSEP).11,12,49,50 Razaet al51 reported that crizotinib significantly increased the sensitivity of P-gp over-expressing cells in response to doxorubicin and paclitaxel. Therefore, crizotinib may have the potential to increase the plasma concentrations of co-administered P-gp substrates, and close clinical surveillance is recommended when crizotinib is administered with these agents.
A significant association between ABCB1 single nucleotide polymorphisms (SNPs) and the functionality of P-gp has been demonstrated by previous studies.52–54 Because crizotinib is a substrate of P-gp, ABCB1 SNPs may lead to differences in the intracellular versus extracellular concentrations of crizotinib. Fujiwaraet al55 recently reported that one patient, with the 1236TT-2677TT-3435TT genotype, represented a distinct outlier, with crizotinib AUC and Cmax values on day 15 that were 2.8- and 2.6-fold greater than the mean, respectively, compared with other genotypes.
Ceritinib is a good substrate for P-gp and a weak substrate for BCRP.15,16 If ceritinib is administered with P-gp inhibitors, an increase in ceritinib concentrations is likely.16 Caution is necessary with the concomitant use of P-gp inhibitors or inducers. Based on in vitro data, ceritinib is a weak inhibitor of OATP1B1/3, OAT1, OCT2, and is predicted to inhibit P-gp and BCRP, but not MRP2, OAT3, or OCT1.16,56 Ceritinib may have the potential to increase the plasma concentrations of co-administered drugs that are transported by P-gp and BCRP.56,57 Caution should be exercised with the concomitant use of P-gp and BCRP substrates.16
Alectinib is not a substrate for P-gp, BCRP, or OATP1B1/3, but M4 is a substrate for P-gp.18,19,58 Because alectinib inhibits P-gp, that coadministration of P-gp inhibitors is not expected to have clinically relevant effects on M4 exposure.19 In vitro, alectinib and M4 are inhibitors of P-gp, BSEP, and BCRP. Therefore, alectinib and M4 may have the potential to increase the plasma concentrations of co-administered P-gp, BSEP, and BCRP substrates.19,59 Thus, appropriate monitoring is recommended when alectinib is co-administered with these substrates.19
Brigatinib is a substrate for P-gp and BCRP.20,21 Due to the high solubility and high permeability of brigatinib, in vitro, P-gp and BCRP inhibitors are unlikely to alter systemic exposure, and no dose adjustments are necessary for brigatinib when coadministered with P-gp and BCRP inhibitors.21 Brigatinib is an inhibitor of P-gp, BCRP, OCT1, MATE1, and MATE2K, in vitro.20,21 Therefore, brigatinib may have the potential to increase the concentrations of co-administered substrates for these transporters.21 Patients should be closely monitored when brigatinib is concomitantly used with those substrates with narrow therapeutic indexes (eg, digoxin, dabigatran, and methotrexate).21
Based on in vitro data, lorlatinib is not a substrate for P-gp or BCRP, and these efflux mechanisms are expected to have minimal effects on its absorption.22,23 Lorlatinib may have the potential to inhibit P-gp, OCT1, OAT3, MATE1, OATP1B1/3, and intestinal BCRP, and mediate DDIs by inhibiting these drug transporters, at clinically relevant concentrations.23 The DDIs between drug transporter substrates and lorlatinib still require investigating in in vivo studies.23
Entrectinib is not a substrate of P-gp, BCPR, or OATP1B1/3. However, M5 is a substrate of P-gp and BCPR.24 Based on in vitro dada, entrectinib and M5 have a potential inhibitory effect on P-gp, BCRP, OATP1B1, and MATE1.24 A clinical DDI study showed that coadministration of entrectinib single dose 600 mg increased digoxin AUC and Cmax by 18% and 28%, respectively.24 Therefore, dose adjustment for P-gp substrates when co-administered with entrectinib is not needed.24
Intestinal CYP Enzymes
CYP enzymes metabolize a wide variety of drugs. CYPs are localized in both the liver and intestine. Thus, for many orally administered drugs, the first-pass drug clearance is contributed to by both liver and intestine.60 CYP3A (CYP3A4 and 3A5) and CYP2C9 represent the major intestinal CYPs, accounting for 80% and 14%, respectively, of total CYP enzymes.61 The activities of hepatic and intestinal CYP3A share commonalities, but also exhibit differences. For example, rifampicin is an inducer of hepatic and intestinal CYP3A, but the effects of grapefruit juice appeared to be selective for intestinal CYP3A and are not sensitive to hepatic CYP3A.60
Because intestinal CYP enzymes and P-gp share common locations and common inhibitors and inducers, they might act as a coordinated absorption barrier against oral drugs. Without efflux transporters, the high concentration of orally administered drugs in the gut might easily saturate CYP enzymes in enterocytes, resulting in fewer metabolites and more unchanged drug being released into the blood.60 Therefore, the interplay between intestinal CYP enzymes and P-gp can enable highly efficient intestinal metabolism, which could have a substantial effect on the first-pass elimination of orally administered drugs.
For some orally administered drugs, intestinal CYP metabolism can eliminate a large proportion of the drugs before they can reach the systemic circulation. Drugs that are intestinal CYP substrates not only suffer from low bioavailability but they are also more likely to be susceptible to DDIs with other CYP substrates, inhibitors, or inducers.61 However, because many drugs undergo extensive first-pass and are dependent on hepatic and intestinal CYP metabolism and transporters, distinguishing their individual contributions to altered drug bioavailability can be difficult.
PK DDIs: Distribution
Distribution is largely determined by blood flow and the binding properties of drugs with plasma proteins.62 ALK-TKIs can bind to several plasma proteins, such as albumin, α1-acid glycoprotein, lipoproteins, and immunoglobulins. The unbound drugs can cross the membrane, distribute to tissues, and exert biological activity. In theory, if two highly protein-bound drugs are combined, one drug can displace the other from its protein binding site, increasing the concentration of the unbound drug and altering its apparent distribution volume.63 Crizotinib, ceritinib, alectinib, brigatinib, and entrectinib are highly bound to plasma proteins (≥ 90%),12,15,18,20,24 and could theoretically interact with other highly protein-bound drugs, such as phenytoin and warfarin; however, little evidence exists to support clinically relevant interactions due to displacement from protein binding sites, likely because, although changes in protein binding can influence the pharmacokinetic parameters of a drug, they rarely alter the overall exposure to a drug.64
Drug distribution into tissues is mediated by transporters, and inhibition or induction of transporters can also influence the distribution of drugs.46 For instance, crizotinib is a substrate of P-gp, which is consistent with crizotinib being relatively ineffective for cancer associated with brain metastasis. However, when co-administered with elacridar, the brain accumulation of crizotinib was increased.47 Similar with crizotinib, brain accumulation of the ceritinib is restricted by P-gp and BRCP, and coadministration of P-gp and BRCP inhibitor can increase its brain accumulation.65 For alectinib, lorlatinib, and entrectinib, they are not a substrate of P-gp and BRCP, this may be an important reason why they exhibit good penetration through the BBB.
PK DDIs: Metabolism
Most PK DDIs involve the metabolism of drugs in vivo, especially those drugs metabolized by CYP enzymes.64 Although some drugs are also metabolized at the small intestine, the primary site for drug metabolism is the liver.66 Potent CYP inhibitors and inducers can modify the exposure of CYP substrates. Moreover, the competitive binding of two substrates at the same CYP enzyme-binding site can also mediate DDIs. The five ALK-TKIs are CYP enzyme substrates; thus, the DDIs may occur when ALK-TKIs are co-administered with CYP enzyme inhibitors, inducers, and substrates.66 The effects of CYP inhibitors and inducers on the exposure of ALK-TKIs are mentioned in Table 6. The effect of ALK-TKIs on the exposure of other CYP3A substrates are mentioned in Table 7.
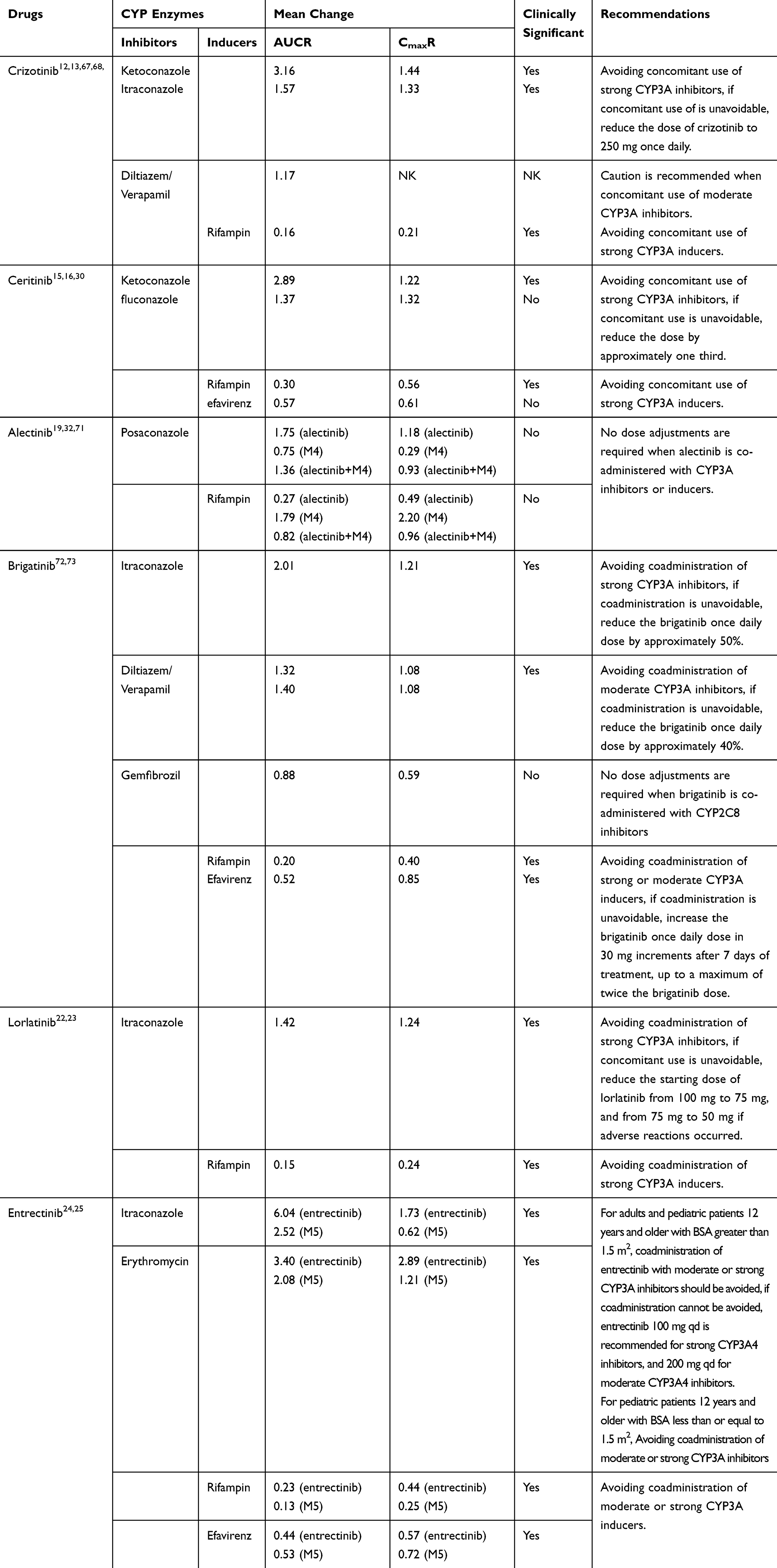 |
Table 6 Effects of CYP Inhibitors and Inducers on the Exposure of ALK-TKIs |
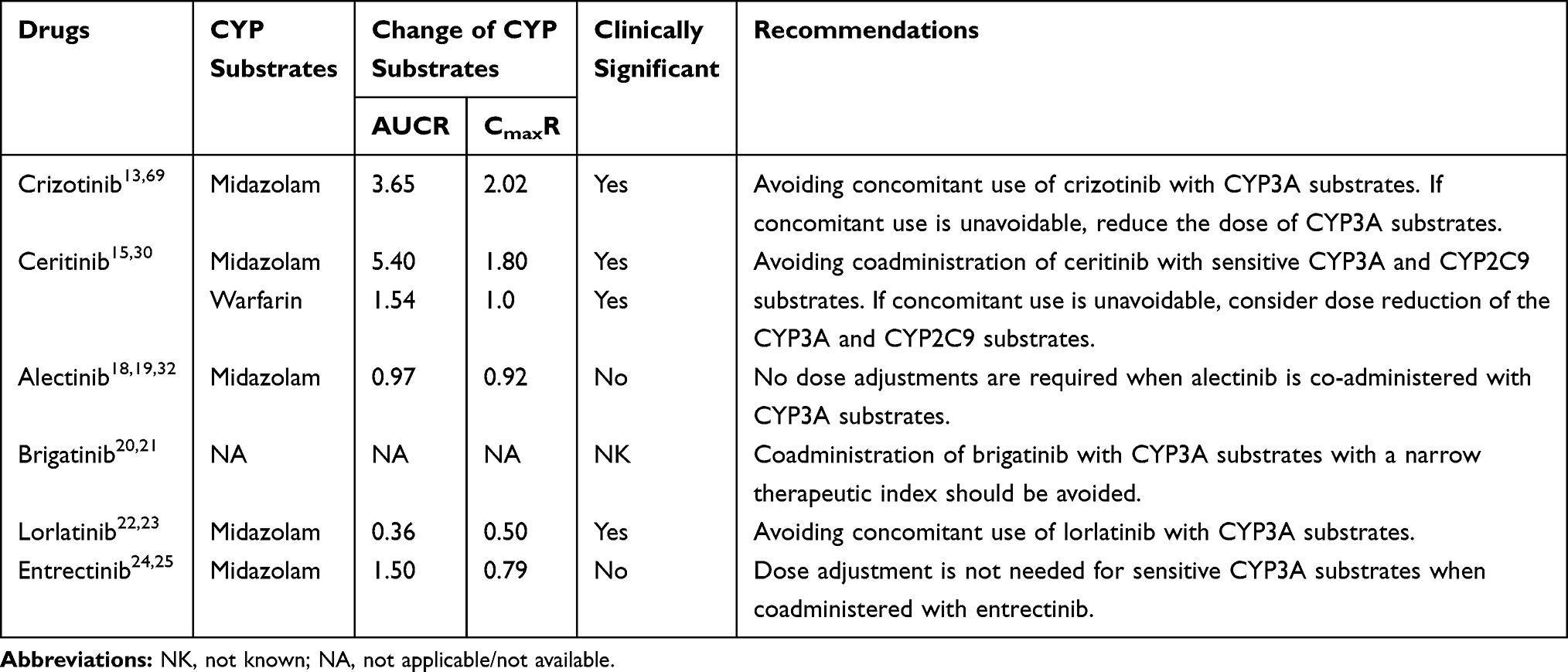 |
Table 7 Effects of ALK-TKIs on the Exposure of Other Drugs |
Crizotinib
The coadministration of a single 150-mg orally administered dose of crizotinib with ketoconazole, increased the crizotinib AUC and Cmax by 216% and 44%, respectively.67 When crizotinib was co-administered with another strong CYP3A inhibitor, itraconazole, the AUC and Cmax of crizotinib increased by 57% and by 33%, respectively.13 Therefore, the concomitant use of strong CYP3A inhibitors and crizotinib should be avoided. If the concomitant use of CYP3A inhibitors with crizotinib is unavoidable, the crizotinib dose should be reduced to 250 mg, once daily.12,13 The effects of moderate CYP3A inhibitors on crizotinib exposure might also be clinically relevant, and the physiologically based pharmacokinetic (PBPK) model predicted a 17% increase in crizotinib AUC after treatment with moderate CYP3A inhibitors (diltiazem or verapamil).13 Caution is recommended when co-administering crizotinib with moderate CYP3A inhibitors.
During the concomitant use of crizotinib with rifampin, a strong CYP3A inducer, the AUC and Cmax of crizotinib decreased by 84% and 79%, respectively.67 Therefore, the concomitant use of strong CYP3A inducers with crizotinib should be avoided. A clinical study was conduced to assess the effect of dexamethasone on the exposure and efficacy of crizotinib.68 The results showed that dexamethasone has no effect on crizotinib exposure or efficacy.68
Crizotinib is a reversible and time-dependent inhibitor of CYP3A.13,69 Coadministration of crizotinib increases the AUC and Cmax values for midazolam (a sensitive CYP3A substrate), by 116% and 32%, 265% and 102%, and 250% and 139%, on average, at crizotinib dose levels of 100 mg daily, 250 mg twice daily, and 300 mg twice daily, respectively, suggesting that crizotinib is a moderate CYP3A inhibitor.29,69 The warfarin (R) enantiomer is metabolized by CYP3A4, kubomura et al70 reported a case of increased prothrombin time-international normalized ratio when crizotinib and warfarin were co-administered. Therefore, the concomitant use of crizotinib with sensitive CYP3A substrates should be avoided, if the concomitant use of crizotinib is unavoidable, reducing the dose of CYP3A substrates is recommended.
Ceritinib
The coadministration of ceritinib with ketoconazole, under fasted conditions, for 14 days, increased the ceritinib AUC by 189% and Cmax by 22%.15,16 Thus, we recommend avoiding the concomitant use of strong CYP3A inhibitors with ceritinib, if the concomitant use of CYP3A inhibitors is unavoidable, the dose of ceritinib should be reduced by approximately one third.15 The coadministration of ceritinib with rifampin for 14 days decreased the AUC and Cmax of ceritinib by 70% and 44%, respectively.15 Therefore, the concomitant use of crizotinib with strong CYP3A inducers should be avoided.15
PBPK modeling predicted that fluconazole (a moderate CYP3A4 inhibitor) can increase ceritinib AUC by 37% and Cmax by 32%, while efavirenz (a moderate CYP3A4 inducer) can decrease ceritinib AUC by 43% and Cmax by 39%.16,30 A clinical trial to evaluate the effect of a moderate CYP3A4 inhibitor or inducer on ceritinib PK is considered unnecessary given the magnitude of predicted changes in steady-state exposures after coadministration with a moderate CYP3A4 inhibitor and inducer.16 Restricting the concomitant use of moderate CYP3A4 inhibitors and inducers when using ceritinib is not recommended.16
Ceritinib has been classified as a strong CYP3A4 inhibitor and a weak CYP2C9 inhibitor. The concomitant use of midazolam with ceritinib increased the midazolam AUC by 440% and the Cmax by 80%.15 When co-administered with warfarin (a CYP2C9 substrate), the AUC of S-warfarin increased by 54%, whereas no statistically significant effect was observed for Cmax.15,16 These findings suggested that the coadministration of ceritinib with CYP3A4 or CYP2C9 substrates should be avoided, if concomitant use of such substrates is unavoidable, a dose reduction for the sensitive CYP substrates should be considered.15
Alectinib
A clinical study was conducted to assess the DDIs between alectinib and CYP inhibitors, inducers, and substrates.19 The coadministration of alectinib with posaconazole (a strong CYP3A inhibitor) increased the alectinib AUC and Cmax by 75% and 18%, respectively, and reduced the M4 AUC and Cmax by 25% and 71%, respectively.32,71 The effects on the combined exposure to alectinib and M4 were minor, reducing the total Cmax by 7% and increasing the total AUC by 36%.32,71 The concomitant use of rifampicin with alectinib reduced the alectinib AUC and Cmax by 73% and 51%, respectively, and increased the M4 AUC and Cmax by 79% and 120%, respectively.32,71 The effects on the combined exposure to alectinib and M4 were minor, reducing the total AUC and Cmax by 18% and 4%, respectively.32,71 Based on these results, no dose adjustments are required when alectinib is co-administered with CYP3A inhibitors or inducers.19
In vitro, alectinib and M4 show weak, time-dependent inhibition of CYP3A4, and alectinib exhibits the weak induction of CYP3A4 and CYP2B6.18,19 A study has shown that multiple doses of alectinib had no influence on the exposure to midazolam.32 Alectinib is a weak inhibitor of CYP2C8, and a PBPK model study showed that alectinib had no clinically significant effects of on repaglinide (a substrate of CYP2C8) metabolism in vivo.32 Therefore, no dose adjustment is required for co-administered CYP3A substrates.19
Brigatinib
The coadministration of brigatinib with itraconazole increased the brigatinib AUC and Cmax by 101% and 21%, respectively.72,73 Compared with strong CYP3A4 inhibitors, moderate CYP3A inhibitors (eg, diltiazem and verapamil) may increase the AUC of brigatinib by approximately 40%.73 Therefore, the coadministration of strong or moderate CYP3A inhibitors should be avoided, if the coadministration of a strong or moderate CYP3A inhibitor cannot be avoided, reduce the brigatinib once-daily dose by approximately 50% for strong inhibitors and by 40%, for moderate inhibitors.20
Brigatinib is also a substrate for CYP2C8.20,21 In healthy subjects, the coadministration of brigatinib with gemfibrozil (a strong CYP2C8 inhibitor) reduced the brigatinib AUC and Cmax by 12% and 41%, respectively; however, these effects are not clinically meaningful, as total systemic exposure of brigatinib was not meaningfully altered.72 The effect of gemfibrozil on the PK of brigatinib was unexpected, and the underlying mechanism for decreased AUC and Cmax in the presence of gemfibrozil is unknown, it may be related to the inhibition of transporters that may be important for brigatinib uptake.
When co-administered with rifampicin, the AUC and Cmax of brigatinib were decreased by 80% and 60%, respectively.72,73 Therefore, the concomitant use of strong CYP3A inducers with brigatinib should be avoided.20,21 Compared with strong CYP3A4 inducers, moderate CYP3A inducers may decrease the AUC of brigatinib by approximately 50%, based on PBPK model.73 The concomitant use of moderate CYP3A inducers with brigatinib should also be avoided, if the coadministration of moderate or strong CYP3A4 inducers is unavoidable, increase the brigatinib once-daily dose by 30-mg increments for 7 days of treatment, up to a maximum of twice the starting brigatinib dose.21
Brigatinib is an inducer for CYP3A4.21 Clinical DDI studies examining CYP3A sensitive substrates have not yet been conducted; however, brigatinib may decrease the exposure to CYP3A substrates.21 Therefore, the coadministration of brigatinib with CYP3A substrates with narrow therapeutic indexes should be avoided, as their effectiveness may be reduced.21
Lorlatinib
When given concomitantly with itraconazole, the AUC of lorlatinib increased by 42% and the Cmax increased by 24%.22 Therefore, the coadministration of strong or moderate CYP3A inhibitors should be avoided, if a strong CYP3A4/5 inhibitor must be concomitantly administered, a dose reduction for lorlatinib is recommended.22
In DDI studies, rifampin reduced the mean lorlatinib AUC by 85% and reduced the Cmax by 76%.22 Thus, the concomitant administration of lorlatinib with strong CYP3A4/5 inducers should be avoided.22 The effect of moderate CYP3A4/5 inducers on lorlatinib pharmacokinetics has not yet been clearly established; therefore, they may reduce lorlatinib plasma concentrations.23 Thus, the concomitant use with moderate CYP3A4/5 inducers should also be avoided.22,23
In in vitro studies, lorlatinib is a time-dependent inhibitor and an inducer of CYP3A4/5.22 The coadministration of lorlatinib with midazolam decreased the midazolam AUC and Cmax by 64% and 50%, respectively.22 Thus, the concurrent administration of lorlatinib with CYP3A4/5 substrates that have narrow therapeutic indices should be avoided.22 Lorlatinib may have the potential to inhibit CYP2C9 and CYP1A2, based on in vitro studies, and may have the weak potential to induce CYP2B6.23 Caution is recommended when co-administering lorlatinib with these CYP substrates.23
Entrectinib
Entrectinib is metabolized primarily by CYP3A4 to form the major active metabolite M5.24 Similarly, M5 is also primarily metabolized by CYP3A4.24 Coadministration of itraconazole with entrectinib increased entrectinib AUC and Cmax by 504% and 73%, respectively, and increased M5 AUC by 152% but decreased the M5 Cmax by 38%.25 Coadministration of erythromycin with entrectinib is predicted to increase entrectinib AUC by 240% and Cmax by 189%, and increase M5 AUC by 108% and Cmax by 21%.25 Tus, for adults and pediatric patients 12 years and older with BSA greater than 1.5 m2, coadministration of entrectinib with moderate or strong CYP3A inhibitors should be avoided, if coadministration cannot be avoided, entrectinib 100 mg once daily is recommended for coadministration of strong CYP3A4 inhibitors, and 200 mg once daily is recommended for coadministration of moderate CYP3A4 inhibitors.24 For pediatric patients 12 years and older with BSA less than or equal to 1.5 m2, coadministration of entrectinib with moderate or strong CYP3A inhibitors should be avoided.24
When co-administered with rifampicin, the AUC and Cmax were decreased by 77% and 56% for entrectinib, and decreased by 87% and 75% for M5, respectively.25 Concomitant use of efavirenz is expected to decrease entrectinib AUC and Cmax by 56% and 43%, and decrease M5 AUC and Cmax by 47% and 28%, respectively.25 Based on these data, coadministration of entrectinib with moderate or strong CYP3A inducers should be avoided.24
In vitro, entrectinib and M5 exhibited inhibitory potential toward CYP3A4/5, CYP2D6, and CYP2C8/9.24,25 A clinical DDI study showed that coadministration of entrectinib once daily with midazolam increase the midazolam AUC by 50% but decreased Cmax by 21%.25 The results suggest that dose adjustment is not needed for sensitive CYP3A substrates when co-administered with entrectinib.25
PK DDIs: Elimination
Drug transporters, located in the kidney and liver, associated with hepatic CYP enzymes are important for the elimination of drugs.45,74,75 For the six ALK-TKIs, liver elimination represents the major clearance pathway, with minor contributions from renal elimination. Thus, the inhibition or induction of hepatic transporters and CYP enzymes may increase or decrease the excretion of affected drugs, resulting in clinically relevant DDIs. However, clinical data supporting these possibilities are very limited, and more research is necessary to assess the effects of clinically relevant changes in hepatic transporters on the pharmacokinetics of crizotinib, ceritinib, brigatinib, and entrectinib. Because alectinib and lorlatinib are not substrates for transporters, coadministration with transporter inhibitors or inducers are not expected to have relevant effects on the elimination of these two drugs.19,23 In addition, ALK-TKIs also can affect the PK of other drugs by altering the expression and activity of enzymes and transporters, the effects of ALK-TKIs on the elimination of other substrates also should be examined.
Liver and kidney are the two major organs for the elimination of drugs. Thus, DDI related to elimination can occur due to the renal impairment (RI) or hepatic impairment (HI).76 For drugs are primarily eliminated by the liver, HI may significantly impact their PK and cause DDIs.77 Similarly, for drugs are primarily eliminated by the kidney, RI may significantly impact their PK and cause DDIs.78 Moreover, RI can also affect the non- renal disposition of drugs that are eliminated by the liver.76,78 Therefore, it is important to know the effect of RI and HI on the exposure of drugs. The effect of RI and HI on the exposure of ALK-TKIs are illustrated in Table 8.
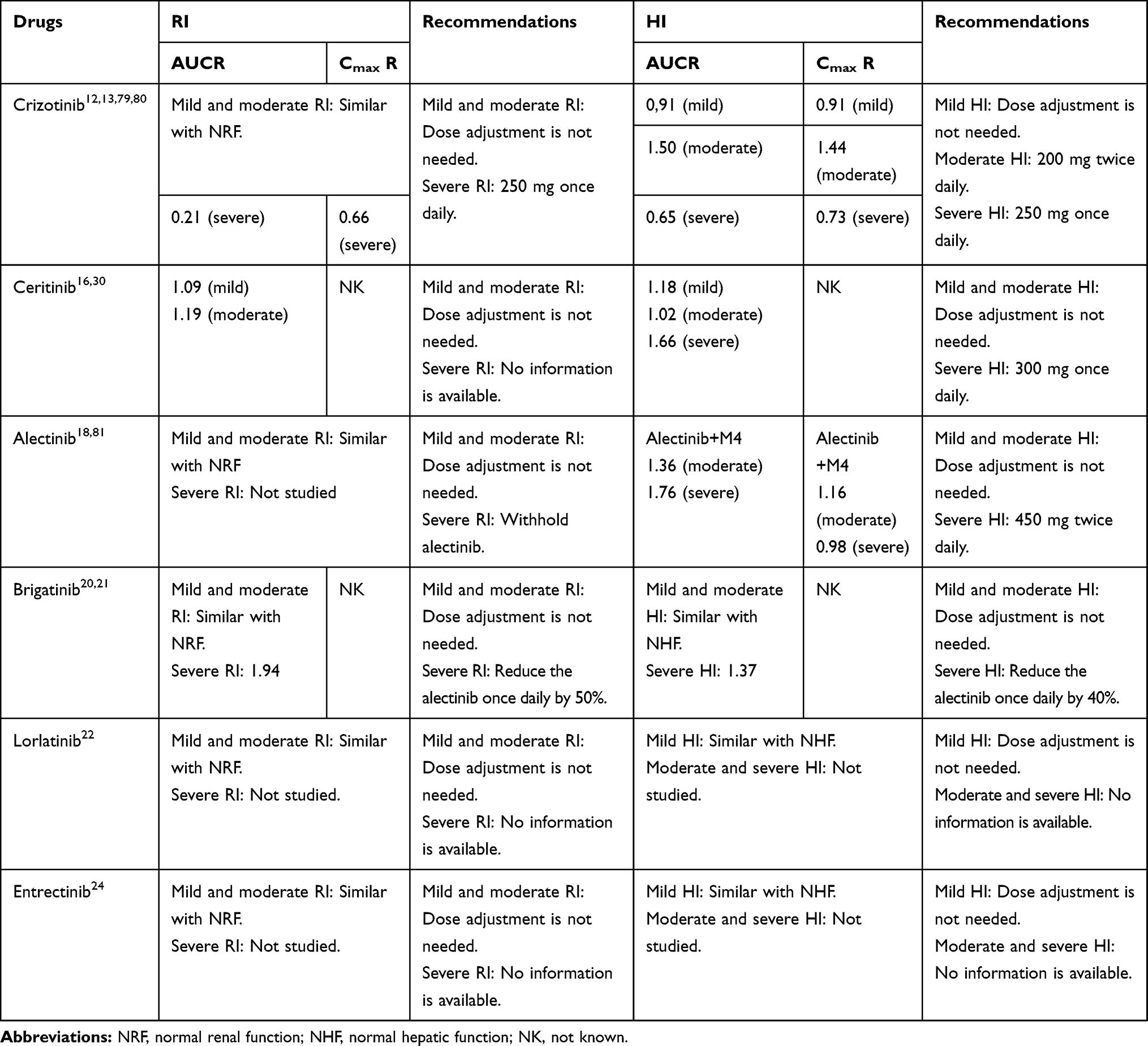 |
Table 8 The Effect of RI and HI on the Exposure of ALK-TKIs |
Conclusions
ALK-TKIs have rapidly become an established component of regular oncology practice in the past few years.6,82 This review of the PK of crizotinib, ceritinib, alectinib, brigatinib, lorlatinib, and entrectinib highlighted the differences in absorption, metabolism, distribution, and excretion among these ALK-TKIs, which can influence their potential for DDIs.
Multiple clinically significant DDIs have been reported due to the inhibition or induction of hepatic CYP enzymes. Except for alectinib, CYP inhibitors and inducers can significantly alter the exposure of ALK-TKIs and can lead to clinically relevant DDIs.12,15,20,22,24 Thus, for crizotinib, ceritinib, brigatinib, lorlatinib, and entrectinib, the concomitant use of strong CYP3A inhibitors or inducers should be avoided. If concomitant use is unavoidable, the doses of these five ALK-TKIs should be reduced. In addition, all six ALK-TKIs are CYP3A inhibitors, and crizotinib, ceritinib, brigatinib, and lorlatinib can significantly influence the exposure of midazolam, indicating that clinical intervention is necessary when these four ALK-TKIs are co-administered with other CYP3A substrates.13,16,21,23 In addition to hepatic CYP enzymes, intestinal CYP enzymes are also involved in drug metabolism, and the inhibition or induction of intestinal CYP enzymes may change the absorption of a drug.60 For instance, grapefruit is an intestinal CYP inhibitor, which may increase the bioavailability of ALK-TKIs.13,16,19,21,23,24,83 Thus, grapefruit juice or grapefruit should be avoided during treatment with AKL-TKIs.
PK interactions between food and orally administered drugs primarily affect drug absorption, which may have clinically relevant effects. For ceritinib and alectinib, food can significantly increase absorption, and the toxicities are well-tolerated.33,40,41 As a result, they are recommended to be taken with food. In contrast, for crizotinib, brigatinib, lorlatinib, and entrectinib, the effects of food on their absorption are considered to have no clinical implications;22,24,39,43 therefore, they can be taken either with or without food. ARAs can also affect the absorption of many orally administered drugs, however, based on the results of clinical trials, the effects of ARAs on the absorption of ALK-TKIs are not clinically significant.25,29-33,36 Therefore, the concomitant use of ARAs is not contraindicated.
Transporters play an important role in the membrane transport of many drugs and are involved in intestinal tissue distribution, hepatic uptake, and biliary and renal excretion. The inhibition or induction of transporters by co-administered drugs can alter the PK of victim drugs, leading to transporter-mediated DDIs. Crizotinib, ceritinib, and brigatinib are substrates for P-gp and other transporters.13,16,21 Thus, caution should be used when the concomitant use of these ALK-TKIs with strong transporter inhibitors or inducers. Furthermore, all six ALK-TKIs are transporter inhibitors (eg, P-gp), and close clinical surveillance is recommended when the coadministration of ALK-TKIs with P-gp or other transporter substrates is necessary.
In summary, PK DDIs can occur in every step during which a drug interacts with the body, including during absorption, distribution, metabolism, and excretion, and many agents (eg, food, ARAs, transporter inhibitors/inducers, and CYP inhibitors/inducers) can lead to DDIs.9,84 In this review, we presented the PK and DDIs of crizotinib, ceritinib, alectinib, brigatinib, lorlatinib, and entrectinib in clinical practice, to help clinicians maximize efficacy and minimize the incidence of adverse events when prescribing ALK-TKIs.
Disclosure
The authors report no conflict of interest regarding the publication of this paper.
References
1. Ferlay J, Steliarova-Foucher E, Lortet-Tieulent J, et al. Cancer incidence and mortality patterns in Europe: estimates for 40 countries in 2012. Eur J Cancer. 2013;49(6):1374–1403. doi:10.1016/j.ejca.2012.12.027
2. Tabchi S, Kourie HR, Klastersky J. Concurrent driver mutations/rearrangements in non-small-cell lung cancer. Curr Opin Oncol. 2017;29(2):118–122. doi:10.1097/CCO.0000000000000353
3. da Cunha Santos G, Shepherd FA, Tsao MS. EGFR mutations and lung cancer. Annu Rev Pathol. 2011;6(1):49–69. doi:10.1146/annurev-pathol-011110-130206
4. Lee DH. Treatments for EGFR-mutant non-small cell lung cancer (NSCLC): the road to a success, paved with failures. Pharmacol Ther. 2017;174:1–21. doi:10.1016/j.pharmthera.2017.02.001
5. Mori M, Hayashi H, Fukuda M, et al. Clinical and computed tomography characteristics of non-small cell lung cancer with ALK gene rearrangement: comparison with EGFR mutation and ALK/EGFR-negative lung cancer. Thorac Cancer. 2019;10(4):872–879. doi:10.1111/1759-7714.13017
6. Roskoski R
7. Spagnuolo A1, Maione P1, Gridelli C. Evolution in the treatment landscape of non-small cell lung cancer with ALK gene alterations: from the first- to third-generation of ALK inhibitors. Expert Opin Emerg Drugs. 2018;23(3):231–241. doi:10.1080/14728214.2018.1527902
8. Tannenbaum C, Sheehan NL. Understanding and preventing drug-drug and drug-gene interactions. Expert Rev Clin Pharmacol. 2014;7(4):533–544. doi:10.1586/17512433.2014.910111
9. Kucharczuk CR, Ganetsky A, Vozniak JM. Drug-drug interactions, safety, and pharmacokinetics of EGFR tyrosine kinase inhibitors for the treatment of non-small cell lung cancer. J Adv Pract Oncol. 2018;9(2):189–200.
10. van Leeuwen RW, van Gelder T, Mathijssen RH, et al. Drug-drug interactions with tyrosine-kinase inhibitors: a clinical perspective. Lancet Oncol. 2014;15(8):e315–e326. doi:10.1016/S1470-2045(13)70579-5
11. Scripture CD, Figg WD. Drug interactions in cancer therapy. Nat Rev Cancer. 2006;6(7):546–558. doi:10.1038/nrc1887
12. US Food and Drug Administration. Label. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2011/202570s000lbl.pdf.
13. European Medicines Agency. Product information. Available from: https://www.ema.europa.eu/en/documents/product-information/xalkori-epar-product-information_en.pdf. Accessed December 1, 2019.
14. Hamilton G1, Rath B, Burghuber O. Pharmacokinetics of crizotinib in NSCLC patients. Expert Opin Drug Metab Toxicol. 2015;11(5):835–842. doi:10.1517/17425255.2015.1021685
15. US Food and Drug Administration. Label. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/205755s016lbl.pdf.
16. European Medicines Agency. Product information. Available from: https://www.ema.europa.eu/en/documents/product-information/zykadia-epar-product-information_en.pdf.
17. Morcos PN, Yu L, Bogman K, et al. Absorption, distribution, metabolism and excretion (ADME) of the ALK inhibitor alectinib: results from an absolute bioavailability and mass balance study in healthy subjects. Xenobiotica. 2017;47(3):217–229. doi:10.1080/00498254.2016.1179821
18. US Food and Drug Administration. Label. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/208434s004lbl.pdf. Accessed December 1, 2019.
19. European Medicines Agency. Product information. Available from: https://www.ema.europa.eu/en/documents/product-information/alecensa-epar-product-information_en.pdf.
20. US Food and Drug Administration. Label. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/208772s004lbl.pdf.
21. European Medicines Agency. Product information. Available from: https://www.ema.europa.eu/en/documents/product-information/alunbrig-epar-product-information_en.pdf.
22. US Food and Drug Administration. Label. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/210868s000lbl.pdf. Accessed December 1, 2019.
23. European Medicines Agency. Product information. Available from: https://www.ema.europa.eu/en/documents/product-information/lorviqua-epar-product-information_en.pdf. Accessed December 1, 2019.
24. US Food and Drug Administration. Label. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/212725s000lbl.pdf.
25. European Medicines Agency. Multi-discipline review. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2019/212725Orig1s000,%20212726Orig1s000MultidisciplineR.pdf. Accessed December 1, 2019.
26. Roberts AG, Gibbs ME. Mechanisms and the clinical relevance of complex drug-drug interactions. Clin Pharmacol. 2018;10:123–134. doi:10.2147/CPAA.S146115
27. Budha NR, Frymoyer A, Smelick GS, et al. Drug absorption interactions between oral targeted anticancer agents and PPIs: is pH-dependent solubility the Achilles heel of targeted therapy? Clin Pharmacol Ther. 2012;92(2):203–213. doi:10.1038/clpt.2012.73
28. Willemsen AE, Lubberman FJ, Tol J, et al. Effect of food and acid-reducing agents on the absorption of oral targeted therapies in solid tumors. Drug Discov Today. 2016;21(6):962–976. doi:10.1016/j.drudis.2016.03.002
29. US Food and Drug Administration. Clinical pharmacology and biopharmaceutics reviews. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2011/202570Orig1s000harmR.pdf. Accessed December 1, 2019.
30. US Food and Drug Administration. Clinical pharmacology and biopharmaceutics reviews. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2014/205755Orig1s000ClinPharmR.pdf.
31. Lau YY, Gu W, Lin T, et al. Assessment of drug-drug interaction potential between ceritinib and proton pump inhibitors in healthy subjects and in patients with ALK-positive non-small cell lung cancer. Cancer Chemother Pharmacol. 2017;79(6):1119–1128. doi:10.1007/s00280-017-3308-7
32. US Food and Drug Administration. Clinical pharmacology and biopharmaceutics reviews. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2015/208434Orig1s000ClinPharmR.pdf.
33. Parrott NJ, Yu LJ, Takano R, et al. Physiologically based absorption modeling to explore the impact of food and gastric pH changes on the pharmacokinetics of alectinib. AAPS J. 2016;18(6):1464–1474. doi:10.1208/s12248-016-9957-3
34. US Food and Drug Administration. Chemistry Reviews. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2017/208772Orig1s000ChemR.pdf.
35. US Food and Drug Administration. Chemistry reviews. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2018/210868Orig1s000ChemR.pdf.
36. European Medicines Agency. Multi-discipline review. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2018/210868Orig1s000MultidisciplineR.pdf.
37. Koziolek M, Alcaro S, Augustijns P, et al. The mechanisms of pharmacokinetic food-drug interactions - A perspective from the UNGAP group. Eur J Pharm Sci. 2019;134:31–59. doi:10.1016/j.ejps.2019.04.003
38. Deng J, Zhu X, Chen Z, et al. A Review of food-drug interactions on oral drug absorption. Drugs. 2017;77(17):1833–1855. doi:10.1007/s40265-017-0832-z
39. Xu H, O’Gorman M, Boutros T, et al. Evaluation of crizotinib absolute bioavailability, the bioequivalence of three oral formulations, and the effect of food on crizotinib pharmacokinetics in healthy subjects. J Clin Pharmacol. 2015;55(1):104–113. doi:10.1002/jcph.356
40. Otoukesh S, Sanchez T, Mirshahidi S, et al. ASCEND-8 pharmacokinetic, safety, and efficacy data for ceritinib 450 mg with food in patients with anaplastic lymphoma kinase-positive non-small cell lung Cancer: a clinical perspective. Cancer Treat Res Commun. 2019;20:100149. doi:10.1016/j.ctarc.2019.100149
41. Cho BC, Kim DW, Bearz A, et al. ASCEND-8: a randomized phase 1 Study of Ceritinib, 450 mg or 600 mg, taken with a low-fat meal versus 750 mg in fasted state in patients with anaplastic lymphoma kinase (ALK)-Rearranged Metastatic Non-Small Cell Lung Cancer (NSCLC). J Thorac Oncol. 2017;12(9):1357–1367. doi:10.1016/j.jtho.2017.07.005
42. European Medicines Agency. Assessment report. Available from: https://www.ema.europa.eu/en/documents/assessment-report/alecensa-epar-public-assessment-report_en.pdf.
43. Tugnait M, Gupta N, Hanley MJ, et al. The Effect of a high-fat meal on the pharmacokinetics of brigatinib, an oral anaplastic lymphoma kinase inhibitor, in healthy volunteers. Clin Pharmacol Drug Dev. 2019;8(6):734–741. doi:10.1002/cpdd.641
44. Yu J, Petrie ID, Levy RH, et al. Mechanisms and clinical significance of pharmacokinetic-based drug-drug interactions with drugs approved by the U.S. food and drug administration in 2017. Drug Metab Dispos. 2019;47(2):135–144. doi:10.1124/dmd.118.084905
45. Nakanishi T, Tamai I. Interaction of drug or food with drug transporters in intestine and liver. Curr Drug Metab. 2015;16(9):753–764. doi:10.2174/138920021609151201113537
46. König J, Müller F, Fromm MF. Transporters and drug-drug interactions: important determinants of drug disposition and effects. Pharmacol Rev. 2013;65(3):944–966. doi:10.1124/pr.113.007518
47. Tang SC, Nguyen LN, Sparidans RW, et al. Increased oral availability and brain accumulation of the ALK inhibitor crizotinib by coadministration of the P-glycoprotein (ABCB1) and breast cancer resistance protein (ABCG2) inhibitor elacridar. Int J Cancer. 2014;134(6):1484–1494. doi:10.1002/ijc.28475
48. Shu W, Ma L, Hu X, et al. Drug-drug interaction between crizotinib and entecavir via renal secretory transporter OCT2. Eur J Pharm Sci. 2020;142:105153. doi:10.1016/j.ejps.2019.105153
49. Omote S, Matsuoka N, Arakawa H, et al. Effect of tyrosine kinase inhibitors on renal handling of creatinine by MATE1. Sci Rep. 2018;8(1):9237. doi:10.1038/s41598-018-27672-y
50. Sato T, Mishima E, Mano N, et al. Potential drug interactions mediated by renal organic anion transporter OATP4C1. J Pharmacol Exp Ther. 2017;362(2):271–277. doi:10.1124/jpet.117.241703
51. Raza A, Kopp SR, Kotze AC. Synergism between ivermectin and the tyrosine kinase/P-glycoprotein inhibitor crizotinib against Haemonchus contortus larvae in vitro. Vet Parasitol. 2016;227:64–68. doi:10.1016/j.vetpar.2016.07.026
52. Kurata Y, Ieiri I, Kimura M, et al. Role of human MDR1 gene polymorphism in bioavailability and interaction of digoxin, a substrate of P-glycoprotein. Clin Pharmacol. 2002;72:209–219.
53. Sakaeda T, Nakamura T, Okumura K. Pharmacogenetics of MDR1 and its impact on the pharmacokinetics and pharmacodynamics of drugs. Pharmacogenomics. 2003;4(4):397–410. doi:10.1517/phgs.4.4.397.22747
54. Dessilly G, Elens L, Panin N, et al. ABCB1 1199G>A polymorphism (rs2229109) affects the transport of imatinib, nilotinib and dasatinib. Pharmacogenomics. 2016;17(8):883–890. doi:10.2217/pgs-2016-0012
55. Fujiwara Y, Hamada A, Mizugaki H, et al. Pharmacokinetic profiles of significant adverse events with crizotinib in Japanese patients with ABCB 1 polymorphism. Cancer Sci. 2016;107(8):1117–1123. doi:10.1111/cas.12983
56. Hu J, Zhang X, Wang F, et al. Effect of ceritinib (LDK378) on enhancement of chemotherapeutic agents in ABCB1 and ABCG2overexpressing cells in vitro and in vivo. Oncotarget. 2015;6(42):44643–44659. doi:10.18632/oncotarget.5989
57. Yang L, Li M, Wang F, et al. Ceritinib enhances the efficacy of substrate chemotherapeutic agent in human ABCB1-overexpressing leukemia cells in vitro,in vivo and ex-vivo. Cell Physiol Biochem. 2018;46(6):2487–2499. doi:10.1159/000489655
58. Hofman J, Sorf A, Vagiannis D, et al. Interactions of alectinib with human ATP-binding cassette drug efflux transporters and cytochrome P450 biotransformation enzymes: effect on pharmacokinetic multidrug resistance. Drug Metab Dispos. 2019;47(7):699–709. doi:10.1124/dmd.119.086975
59. Yang K, Chen Y, To KK, et al. Alectinib (CH5424802) antagonizes ABCB1- and ABCG2-mediated multidrug resistance in vitro,in vivo and ex vivo. Exp Mol Med. 2017;49(3):e303. doi:10.1038/emm.2016.168
60. Thelen K1, Dressman JB. Cytochrome P450-mediated metabolism in the human gut wall. J Pharm Pharmacol. 2009;61(5):541–558. doi:10.1211/jpp.61.05.0002
61. Xie F, Ding X, Zhang QY. An update on the role of intestinal cytochrome P450 enzymes in drug disposition. Acta Pharm Sin B. 2016;6(5):374–383. doi:10.1016/j.apsb.2016.07.012
62. Roberts JA1, Pea F, Lipman J. The clinical relevance of plasma protein binding changes. Clin Pharmacokinet. 2013;52(1):1–8. doi:10.1007/s40262-012-0018-5
63. Benet LZ, Hoener BA. Changes in plasma protein binding have little clinical relevance. Clin Pharmacol Ther. 2002;71(3):115–121. doi:10.1067/mcp.2002.121829
64. Haddad A, Davis M, Lagman R. The pharmacological importance of cytochrome CYP3A4 in the palliation of symptoms: review and recommendations for avoiding adverse drug interactions. Support Care Cancer. 2007;15(3):251. doi:10.1007/s00520-006-0127-5
65. Kort A, Sparidans RW, Wagenaar E, et al. Brain accumulation of the EML4-ALK inhibitor ceritinib is restricted by P-glycoprotein (P-GP/ABCB1) and breast cancer resistance protein (BCRP/ABCG2). Pharmacol Res. 2015;102:200–207. doi:10.1016/j.phrs.2015.09.003
66. Manikandan P, Nagini S. Cytochrome P450 structure, function and clinical significance: a review. Curr Drug Targets. 2018;19(1):38–54. doi:10.2174/1389450118666170125144557
67. Xu H, O’Gorman M, Tan W, et al. The effects of ketoconazole and rifampin on the single-dose pharmacokinetics of crizotinib in healthy subjects. Eur J Clin Pharmacol. 2015;71(12):1441–1449. doi:10.1007/s00228-015-1945-5
68. Lin S, Nickens DJ, Patel M, et al. Clinical implications of an analysis of pharmacokinetics of crizotinib coadministered with dexamethasone in patients with non-small cell lung cancer. Cancer Chemother Pharmacol. 2019;84(1):203–211. doi:10.1007/s00280-019-03861-y
69. Mao J, Johnson TR, Shen Z, et al. Prediction of crizotinib-midazolam interaction using the Simcyp population-based simulator: comparison of CYP3A time dependent inhibition between human liver microsomes versus hepatocytes. Drug Metab Dispos. 2013;41(2):343–352. doi:10.1124/dmd.112.049114
70. Kubomura Y, Ise Y, Wako T, et al. A drug interaction between crizotinib and warfarin in non-small-cell lung cancer: a case report. J Nippon Med Sch. 2017;84(6):291–293. doi:10.1272/jnms.84.291
71. Morcos PN, Cleary Y, Guerini E, et al. Clinical drug-drug interactions through cytochrome P450 3A (CYP3A) for the selective ALK inhibitor alectinib. Clin Pharmacol Drug Dev. 2017;6(3):280–291. doi:10.1002/cpdd.298
72. Tugnait M, Gupta N, Hanley MJ, et al. Effects of strong CYP2C8 or CYP3A Inhibition and CYP3A induction on the pharmacokinetics of brigatinib, an oral anaplastic lymphoma kinase inhibitor, in healthy volunteers. Clin Pharmacol Drug Dev. 2019;8(6):734–741. doi:10.1002/cpdd.723
73. European Medicines Agency. Assessment report. Available from: https://www.ema.europa.eu/en/documents/assessment-report/alunbrig-epar-public-assessment-report_en.pdf.
74. Feng B, Varma MV. Evaluation and quantitative prediction of renal transporter-mediated drug-drug interactions. J Clin Pharmacol. 2016;56(7):S110–S121. doi:10.1002/jcph.702
75. Patel M, Taskar KS, Zamek-Gliszczynski MJ. Importance of hepatic transporters in clinical disposition of drugs and their metabolites. J Clin Pharmacol. 2016;56(7):S23–S39. doi:10.1002/jcph.671
76. Krens SD, Lassche G, Jansman FGA, et al. Dose recommendations for anticancer drugs in patients with renal or hepatic impairment. Lancet Oncol. 2019;20(4):e200–e207. doi:10.1016/S1470-2045(19)30145-7
77. Verbeeck RK. Pharmacokinetics and dosage adjustment in patients with hepatic dysfunction. Eur J Clin Pharmacol. 2008;64(12):1147–1161. doi:10.1007/s00228-008-0553-z
78. Verbeeck RK, Musuamba FT. Pharmacokinetics and dosage adjustment in patients with renal dysfunction. Eur J Clin Pharmacol. 2009;65(8):757–773. doi:10.1007/s00228-009-0678-8
79. El-Khoueiry AB, Sarantopoulos J, O’Bryant CL, et al. Evaluation of hepatic impairment on pharmacokinetics and safety of crizotinib in patients with advanced cancer. Cancer Chemother Pharmacol. 2018;81(4):659–670. doi:10.1007/s00280-018-3517-8
80. Tan W, Yamazaki S, Johnson TR, et al. Effects of renal function on crizotinib pharmacokinetics: dose recommendations for patients with ALK-positive non-small cell lung cancer. Clin Drug Investig. 2017;37(4):363–373. doi:10.1007/s40261-016-0490-z
81. Morcos PN, Cleary Y, Sturm-Pellanda C, et al. Effect of hepatic impairment on the pharmacokinetics of alectinib. J Clin Pharmacol. 2018;58(12):1618–1628. doi:10.1002/jcph.1286
82. Rocco D, Battiloro C, Della Gravara L, et al. Safety and tolerability of anaplastic lymphoma kinase inhibitors in non-small-cell lung cancer. Drug Saf. 2019;42(2):199–209. doi:10.1007/s40264-018-0771-y
83. Cholewka-Stafińska M, Polaniak R, Kardas M, et al. Interaction of oral form anticancer drugs with grapefruit juice. Pol Merkur Lekarski. 2017;42(247):30–33.
84. Levêque D, Lemachatti J, Nivoix Y, et al. Mechanisms of pharmacokinetic drug-drug interactions. Rev Med Interne. 2010;31(2):170–179. doi:10.1016/j.revmed.2009.07.009
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.