")
Back to Journals » Drug Design, Development and Therapy » Volume 12
Pharmacokinetic and bioequivalence study of a telmisartan/S-amlodipine fixed-dose combination (CKD-828) formulation and coadministered telmisartan and S-amlodipine in healthy subjects
Authors Kang WY , Seong SJ, Ohk B , Gwon M, Kim BK , La S , Kim HJ, Cho S , Yoon Y , Yang DH, Lee HW
Received 8 November 2017
Accepted for publication 29 December 2017
Published 14 March 2018 Volume 2018:12 Pages 545—553
DOI https://doi.org/10.2147/DDDT.S156492
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Manfred Ogris
Woo Youl Kang,1,2,* Sook Jin Seong,1,* Boram Ohk,1,2 Mi-Ri Gwon,1,3 Bo Kyung Kim,1,2 Sookie La,4 Hyun-Ju Kim,3 Seungil Cho,1 Young-Ran Yoon,1,2 Dong Heon Yang,5 Hae Won Lee1
1Clinical Trial Center, Kyungpook National University Hospital, Daegu, Republic of Korea; 2Department of Biomedical Science, BK21 Plus KNU Bio-Medical Convergence Program for Creative Talent, Kyungpook National University Graduate School, Daegu, Republic of Korea; 3Department of Molecular Medicine, Cell and Matrix Research Institute, Kyungpook National University School of Medicine, Daegu, Republic of Korea; 4Analytical Research Division, Biocore Co Ltd, Seoul, Republic of Korea; 5Division of Cardiology, Department of Internal Medicine, Kyungpook National University School of Medicine & Hospital, Daegu, Republic of Korea
*These authors contributed equally to this work
Purpose: A new fixed-dose combination (FDC) formulation of telmisartan 80 mg and S-amlodipine 5 mg (CKD-828) has been developed to increase convenience (as only one tablet is required per day) and improve treatment compliance.
Methods: The pharmacokinetic characteristics and tolerability of an FDC of telmisartan and S-amlodipine were compared to those after coadministration of the individual agents in this randomized, open-label, single-dose, two-way, four-period, crossover study. To analyze the telmisartan and S-amlodipine plasma concentrations using a validated liquid chromatography–tandem mass spectrometry method, serial blood samples were collected up to 48 hours post-dose for telmisartan and 144 hours post-dose for S-amlodipine, in each period.
Results: Forty-eight healthy subjects were enrolled, and 43 completed the study. The mean peak plasma concentration (Cmax) and the area under the plasma concentration–time curve from time 0 to the last measurement (AUC0–t) values of telmisartan were 522.29 ng/mL and 2,475.16 ng⋅h/mL for the FDC, and 540.45 ng/mL and 2,559.57 ng⋅h/mL for the individual agents concomitantly administered, respectively. The mean Cmax and AUC0–t values of S-amlodipine were 2.71 ng/mL and 130.69 ng⋅h/mL for the FDC, and 2.74 ng/mL and 129.81 ng⋅h/mL for the individual agents concomitantly administered, respectively. The geometric mean ratio (GMR) and 90% confidence interval (CI) for the telmisartan Cmax and AUC0–t (FDC of telmisartan and S-amlodipine/concomitant administration) were 0.8509 (0.7353–0.9846) and 0.9431 (0.8698–1.0226), respectively. The GMR and 90% CI for the S-amlodipine Cmax and AUC0–t (FDC/concomitant administration) were 0.9829 (0.9143–1.0567) and 0.9632 (0.8798–1.0546), respectively. As the intrasubject variability of the Cmax for telmisartan administered individually was 42.94%, all 90% CIs of the GMRs fell within the predetermined acceptance range. Both treatments were well tolerated in this study.
Conclusion: CKD-828 FDC tablets were shown to be bioequivalent to coadministration of the individual agents with the respective strength, in healthy subjects under fasting conditions. There was no significant difference in safety profile between the two treatments.
Keywords: fixed-dose combination, pharmacokinetics, S-amlodipine, telmisartan, safety
Introduction
Hypertension is one of the major risk factors for the development of cardiovascular disease (CVD).1 After a reduction of 10 mmHg in systolic blood pressure (SBP) or 5 mmHg in diastolic blood pressure (DBP), incidence of coronary heart disease and stroke have been shown to decrease by 20% and 32%, respectively, in 1 year.2 Accordingly, a reduction in blood pressure (BP) through effective pharmacotherapy significantly reduces the risk of CVD and death, irrespective of the initial BP.3
Four different classes of antihypertensive drugs are recommended as first-line therapy for hypertension: calcium-channel blockers (CCBs), angiotensin-converting enzyme inhibitors, angiotensin II receptor blockers (ARBs), and thiazide-type diuretics.4,5 Combination therapy with two or more agents from different drug classes is required to achieve BP control in the majority of patients, as hypertension is not easily controlled by monotherapy unless it is mild.6,7 Several clinical trials have demonstrated that combination therapy using two or more antihypertensive agents with complementary mechanisms exhibited greater efficacy in reducing BP, with no increase in adverse events (AEs), compared to high-dose monotherapy.8–12 Furthermore, fixed-dose combinations (FDCs) of two or more antihypertensive agents in a single pill can improve medication compliance by reducing the number of concomitant medications, and consequently have beneficial effects on BP control, compared to free combinations or monotherapy.13–16 In retrospective database analyses, FDCs can decrease the cost of cardiovascular care in the hypertensive population.17,18 Among the preferred combinations of classes of antihypertensive drugs, ARB/CCB combination therapy is a particularly useful option, as the addition of ARBs to CCBs has been found to reduce the incidence of peripheral edema, the most common CCB-related adverse effect.19–21
Telmisartan is an ARB, with a selective type 1 receptor blockade effect.22 The antihypertensive efficacy of telmisartan, administered once daily at doses of 20–160 mg in patients with mild-to-moderate essential hypertension, has been shown to be superior to that of placebo and comparable to that of other common antihypertensive agents, such as losartan, amlodipine, and lisinopril.23 The peak plasma concentration (Cmax) of telmisartan has been observed to be reached between 0.5 and 1 hour after oral administration, with a bioavailability of 30%–60%.22,24 Telmisartan is highly bound (>99%) to plasma proteins, with a very high volume of distribution (~500 L), and an elimination half-life (t1/2) of 21–38 hours.22,24 It is not metabolized by the cytochrome P450 system, and most of the oral dose (98%) is excreted unaltered in feces, mainly via bile.22
Amlodipine, a third-generation dihydropyridine CCB, is a racemic mixture consisting of the R-enantiomer and S-enantiomer in a 1:1 ratio.25 The antihypertensive effect of amlodipine through calcium-channel blocking properties is ascribed to S-amlodipine, while R-amlodipine may be responsible for the side effects of racemic amlodipine, as it has 1,000-fold lower CCB activity.26,27 Accordingly, an amlodipine formulation containing only S-amlodipine is more favorable, with fewer side effects than racemic amlodipine.27,28 Following oral administration, amlodipine shows different pharmacokinetic (PK) characteristics (high oral bioavailability of 60%–65%, a longer time to Cmax [tmax] of 6–12 hours, and a longer t1/2 of 30–50 hours) compared to other dihydropyridine CCBs.29,30 CYP3A4 plays an important role in the metabolic clearance of amlodipine.31
The drug–drug interactions between telmisartan and S-amlodipine have not been reported following multiple-dose coadministration of high doses of telmisartan and S-amlodipine.27,32 FDC formulations of telmisartan (40 or 80 mg) and S-amlodipine besylate (2.5 mg) have been developed as antihypertensive drugs (CKD-828 40/2.5 mg or 80/2.5 mg), by Chong Kun Dang Pharmaceutical Co, Ltd (Seoul, Republic of Korea). Recently, a new FDC formulation of telmisartan (80 mg) and S-amlodipine besylate (5 mg) was developed. The aim of this study was to investigate the PK profile, bioequivalence (BE), and safety of telmisartan and S-amlodipine, administered as a CKD-828 80/5 mg FDC formulation or in corresponding doses as individual tablets.
Methods
Study subjects
The study protocol was approved by the Institutional Review Board of Kyungpook National University Hospital (KNUH, Daegu, Republic of Korea). The study (ClinicalTrials.gov registry no: NCT02358824) was conducted at the KNUH Clinical Trial Center, in accordance with the ethical standards of the Declaration of Helsinki and the applicable Good Clinical Practice guidelines. Written informed consent was obtained from all subjects before their participation in this study.
Healthy Korean male volunteers aged ≥19 years, who weighed ≥50 kg and were within ±20% of their ideal body weight, were eligible to participate in this study. All subjects had no clinically significant abnormalities as judged by clinical history, detailed physical examination, routine clinical laboratory tests (hematology, biochemistry, and urinalysis), serology tests (hepatitis B surface antigens, anti-hepatitis C virus antibody, anti-human immunodeficiency virus antibody, and the venereal disease research laboratory test), and 12-lead electrocardiography, conducted within 4 weeks prior to study drug administration.
Exclusion criteria included a history of hypersensitivity to any drug including telmisartan and amlodipine; history or evidence of cardiovascular, hepatobiliary, renal, endocrine, hematological, respiratory, gastrointestinal, central nervous system, psychiatric, neuromuscular, or malignant disease; presence of hyperkalemia; presence of active hepatitis or serum aspartate aminotransferase, alanine aminotransferase, or total bilirubin levels >1.5× the upper limit of normal; SBP of >150 or <90 mmHg, or DBP of >100 or <50 mmHg; creatinine clearance (estimated by the Cockcroft–Gault equation using serum creatinine concentrations) <80 mL/min; presence of gastrointestinal disease (eg, Crohn’s disease or peptic ulcer) or history of resection surgery (excluding simple appendectomy or herniorrhaphy); presence or history of major injury, operation, or acute disease within 4 weeks prior to the first administration of the study drug; history of alcohol abuse (>21 units/week), or subjects who could not abstain from drinking during the study period, regular intake of caffeine (>5 cups per day), or excessive smoking (>10 cigarettes per day); use of any prescription medication or herbal remedies within the previous 2 weeks, or use of any over-the-counter remedies within 1 week prior to the first administration of the study drug, as this could have affected the study or the safety of the subjects in the opinion of the investigator; use of any other investigational drug within 3 months prior to the first administration of the study drug; donation of whole blood within 2 months or any blood products within 1 month prior to the first administration of the study drug; abnormal diet that could affect absorption, distribution, metabolism, and excretion of a given drug within 7 days prior to study medication administration; positive serologic tests; or subjects who were not eligible to participate in this study at the discretion of the study investigator.
Study design and procedure
This randomized, open-label, single-dose, two-sequence, four-period, two-treatment crossover study was conducted at the KNUH Clinical Trial Center. The FDC formulation of CKD-828 (batch no QQ001; expiration date, August 2017; Chong Kun Dang Pharmaceutical Co, Ltd) was used as the test treatment (T), and the coadministration of telmisartan 80 mg (Micardis®; batch no 304342; expiration date, May 2017; Boehringer Ingelheim, Ingelheim, Germany) and S-amlodipine 5 mg (Anydipine S®; batch no QE001; expiration date, March 2016; Chong Kun Dang Pharmaceutical Co, Ltd) was used as the reference treatment (R). Based on a computer-generated table of randomization according to a randomization schedule generated using SAS software (version 9.4; SAS Institute Inc, Cary, NC, USA), a total of 48 enrolled subjects were allocated to one of two treatment sequence groups in a 1:1 ratio: sequence A (RTRT) and sequence B (TRTR). In each study period, the subjects were administered a single oral dose of either T or R with a 14-day washout period between treatments. A schematic diagram of the study design is shown in Figure 1.
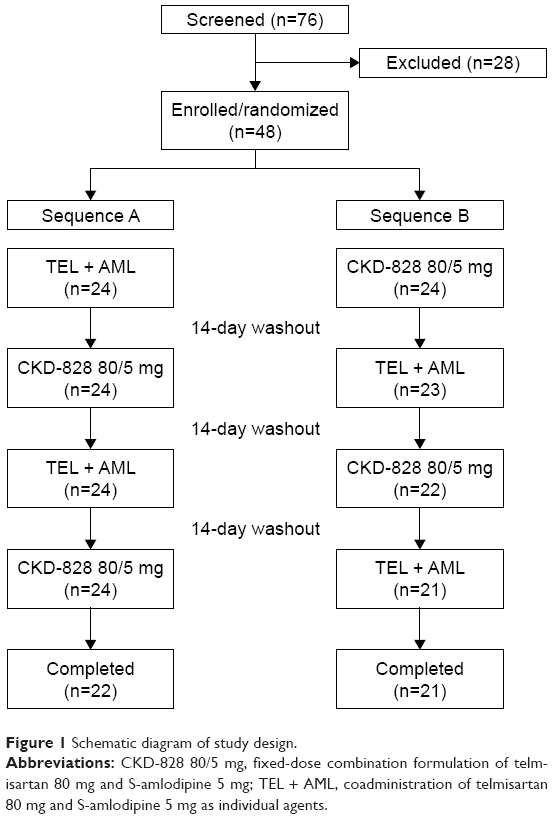 | Figure 1 Schematic diagram of study design. |
The subjects were admitted to the study center at 8 pm, the day before dosing. Each study drug was administered under fasting conditions along with 150 mL of water. Food was restricted from 10 hours before dosing until 4 hours after dosing. Standard meals were provided 4 and 10 hours after dosing. Additional water intake was not permitted for 2 hours before and 2 hours after dosing during each period.
Blood samples for the determination of telmisartan concentrations were collected before dosing and at 0.25, 0.5, 0.75, 1, 1.5, 2, 4, 8, 10, 12, 24, 36, and 48 hours after dosing. Blood samples for the determination of S-amlodipine plasma concentrations were collected before dosing and at 1, 2, 3, 4, 5, 6, 7, 8, 10, 12, 24, 48, 96, and 144 hours after dosing. A 20 G, 0.75 inch indwelling intravenous catheter was placed in either the forearm or dorsum of the hand of each subject. After 1 mL of blood was discarded from the catheter, 6 mL of blood was drawn into a tube containing dipotassium (K2) EDTA and centrifuged at 3,000 rpm for 10 minutes at 4°C to separate plasma. Following centrifugation, plasma samples were transferred to three tubes and stored at −70°C until analysis by the analytical laboratory (Biocore Co Ltd, Seoul, Republic of Korea).
Analysis of telmisartan and S-amlodipine concentrations in plasma
Telmisartan plasma concentrations were determined by a validated method using high performance liquid chromatography (2795 Alliance HT HPLC system, Waters Corporation, Milford, MA, USA) coupled to tandem mass spectrometry (MS/MS, Quattro Premier XE, Waters Corporation). Chromatographic separations were performed on a Unison UK-C18 column (2.0×75 mm, 3 μm particle size) at a flow rate of 0.2 mL/min. The mobile phase consisted of a 30:70:0.1 (v/v/v) mixture of 10 mM ammonium formate, acetonitrile, and formic acid. Multiple reaction monitoring transitions were performed at mass-to-charge ratios (m/z) of 515.10→276.10 and 518.15→279.15 for telmisartan and telmisartan-d3, the internal standard, respectively. Frozen plasma was thawed at room temperature and vortexed for 10 seconds. After 10 μL of telmisartan-d3 (600 ng/mL) was added to 100 μL of plasma in a polypropylene tube, 1,250 μL of acetonitrile was then added. After mixing for 1 minute, the tube was centrifuged at 13,000 rpm for 5 minutes. The supernatant (100 μL) was mixed with 200 μL of 50% acetonitrile, and a 10 μL aliquot of this solution was injected into the LC-MS/MS system for analysis.
S-amlodipine plasma concentrations were determined using liquid chromatography (Shiseido Nanospace SI-2; Shiseido, Tokyo, Japan) coupled to 4000 QTRAP® tandem mass spectrometer operated with Analyst 1.6.2 (AB SCIEX, Foster City, CA, USA). Chromatographic separations were performed on an Atlantis-C18 column (2.1×100 mm, 3 μm particle size) at a flow rate of 0.2 mL/min. The mobile phase consisted of a 50:50:0.1 (v/v/v) mixture of acetonitrile, deionized water, and formic acid. Multiple reaction monitoring transitions were performed at mass-to-charge ratios (m/z) of 409.2→238.2 and 413.2→238.2 for S-amlodipine and (S)-amlodipine-d4, the internal standard, respectively. Frozen plasma was thawed at room temperature and vortexed for 10 seconds. After 20 μL of (S)-amlodipine-d4 (100 ng/mL) was added to 300 μL of plasma in a polypropylene tube, 30 μL of 1 M sodium hydroxide was then added and mixed. After 2 mL of methyl tert-butyl ether was added, the tube was extracted for 20 minutes and then centrifuged at 2,500 rpm for 5 minutes. The organic layer was transferred and dried with a stream of nitrogen gas. The residue was reconstituted with 150 μL of the mobile phase, and filtrated with a 0.2 μm filter. A 10 μL aliquot of this solution was then injected into the LC-MS/MS system for analysis.
Linear calibration curves ranged between 1 and 2,000 ng/mL for telmisartan (r≥0.9962), and between 50 and 20,000 pg/mL for S-amlodipine (r≥0.9999). The overall intraday accuracy ranged from 91.0% to 106.8% for telmisartan, and from 94.0% to 111.6% for S-amlodipine. The overall inter-day accuracy ranged from 92.2% to 101.4% for telmisartan, and from 100.5% to 102.8% for S-amlodipine. The intraday precision (% coefficient of variation, CV) ranged from 0.9% to 5.4% for telmisartan and from 0.5% to 8.0% for S-amlodipine. The inter-day precision (%CV) ranged from 1.5% to 7.1% for telmisartan and from 1.6% to 8.5% for S-amlodipine. The lower limit of quantification was 1 ng/mL for telmisartan and 50 pg/mL for S-amlodipine.
PK analysis
PK parameters for telmisartan and S-amlodipine in plasma were estimated by a non-compartmental method using Phoenix WinNonlin version 6.4 software (Pharsight Corporation, St Louis, MO, USA). Cmax and tmax were obtained directly from the observed plasma concentration–time data. The area under the plasma concentration–time curve from time 0 to the last measurement (AUC0–t) was calculated using the linear trapezoidal method. The AUC from time 0 to infinity (AUC0–∞) was calculated using the following formula: AUC0–∞ = AUC0–t + Ct/ke, where Ct is the last measurable concentration, and ke is the terminal elimination rate constant determined from a linear regression line of the log-transformed plasma concentrations versus time over the terminal log-linear portion (at least three final data points). The terminal t1/2 was calculated to be 0.693/ke.
Statistical analysis
The sample size for the present study was calculated based on the intrasubject variability of telmisartan Cmax (47.5%), the largest value among AUC0–t values, and Cmax values of telmisartan and S-amlodipine in earlier PK studies. In the four-period replicate design, 14 subjects per group were required to detect a difference of ≥20% in the log-transformed PK parameters between the two different treatments (ie, FDC vs individual tablets) with a geometric mean ratio (GMR) of 0.9, a significance level of 0.05, and a power of 80%.32 Therefore, a total of 48 subjects were to be enrolled, assuming an estimated 40% dropout rate.
Demographics, safety data, and PK parameters were summarized using descriptive statistics. All PK parameters are presented as means ± standard deviation (SD), except for tmax values, which are expressed as the median, maximum, and minimum values. To assess the bioequivalence between T and R, the GMR and 90% confidence interval (CI) of Cmax and AUC0–t of telmisartan and S-amlodipine were calculated after natural logarithm transformation. The FDC formulation was considered bioequivalent if the 90% CI of Cmax and AUC0–t for S-amlodipine fell within a predetermined range of 0.800–1.250, according to the standard used by the Korea Ministry of Food and Drug Safety. However, telmisartan has been reported to be a highly variable drug; therefore, the widening of the acceptance criteria for Cmax of telmisartan was prospectively defined in the study protocol. This was based on the intrasubject variability of the reference product obtained from this replicate design study as follows: 1) if the intrasubject variability for Cmax of the reference compound in the study is <30%, the acceptance criteria of the 90% CI for BE would be the conventional BE range of 0.800–1.250; 2) if the intrasubject variability for Cmax of the reference compound is ≥30%, the acceptance criteria for Cmax can be widened to 0.732–1.367 (the criteria were calculated using the following formula presented in the European Medicines Agency and Korean BE study guidelines: [upper limit, lower limit] = exp[±k*sWR], where k is the regulatory constant set to 0.760, and sWR is the intrasubject SD of the log-transformed values of Cmax of the reference product).33–35 The conventional acceptance limit range of 0.800–1.250 was applied to the AUC0–t for telmisartan. All statistical analyses for GMRs with 90% CIs were performed using Phoenix WinNonlin version 6.4 software.
Safety and tolerability assessments
Subjects who received at least one dose of the study drugs throughout the study period were evaluated in the safety and tolerability analysis, based on clinical and laboratory AEs collected after dosing, including all subjective symptoms reported by subjects and objective signs observed by investigators. Vital signs (BP and pulse rate) of the participants were monitored at screening, before and after administration of study drugs for 2, 4, 6, 12, and 24 hours, and at the follow-up visit (14±2 days after administration of the last dose). Body temperature was assessed at screening and at the follow-up visit.
Twelve-lead electrocardiograms were obtained and routine laboratory tests (hematology, urinalysis, and serum chemistry) were conducted at screening, before administration of the study drugs in periods 2 and 4, and at the follow-up visit. All laboratory tests were performed at the Department of Laboratory Medicine, KNUH.
Results
Subject characteristics
A total of 48 healthy male subjects were enrolled in the study and randomly assigned into each of the treatment groups. The means (ranges) for subject age, height, and weight were 25.5 years (19–45 years), 173.7 cm (165.0–183.8 cm), and 67.8 kg (54.2–89.0 kg), respectively. Baseline demographics showed no statistical difference between the sequence groups (Table 1). Five subjects withdrew consent during the study. Accordingly, 43 subjects completed this study and were included in the PK analysis. All 48 subjects who received a study drug at least once were included in the safety assessment.
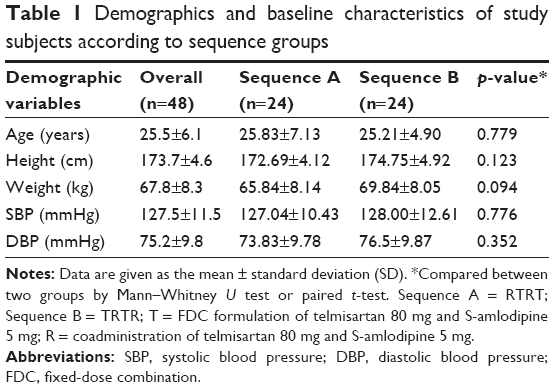 | Table 1 Demographics and baseline characteristics of study subjects according to sequence groups |
PK data
The mean (SD) plasma concentration versus time profiles for telmisartan and S-amlodipine, following a single oral administration of T or R, are illustrated in Figure 2. The differences in PK parameters for telmisartan and S-amlodipine between T and R are summarized in Table 2. All 90% CIs of the GMRs fell within the predetermined acceptance range, including the CI value of 0.7353–0.9846 for telmisartan Cmax, as the intrasubject variability for telmisartan Cmax of the single agents in this study was 42.94% (Table 3).
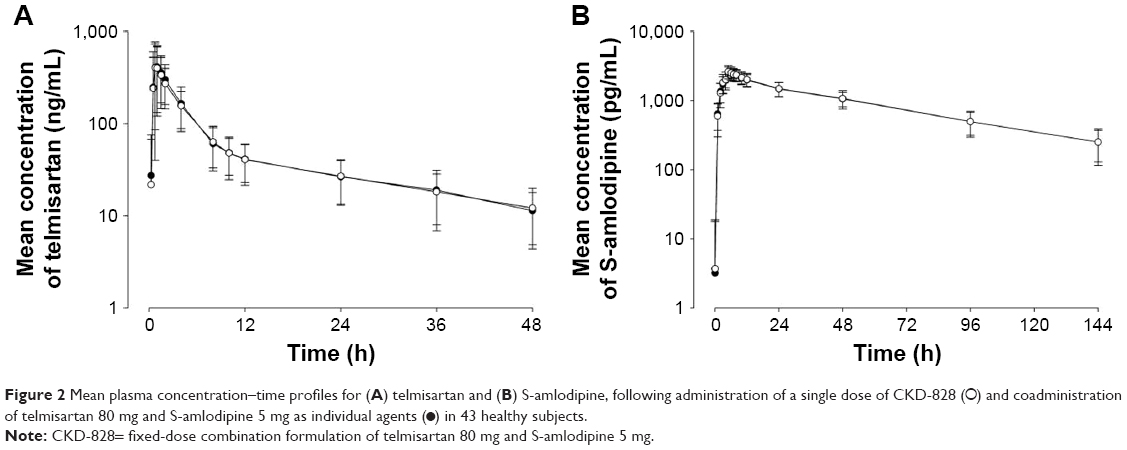 | Figure 2 Mean plasma concentration–time profiles for (A) telmisartan and (B) S-amlodipine, following administration of a single dose of CKD-828 (○) and coadministration of telmisartan 80 mg and S-amlodipine 5 mg as individual agents (•) in 43 healthy subjects. |
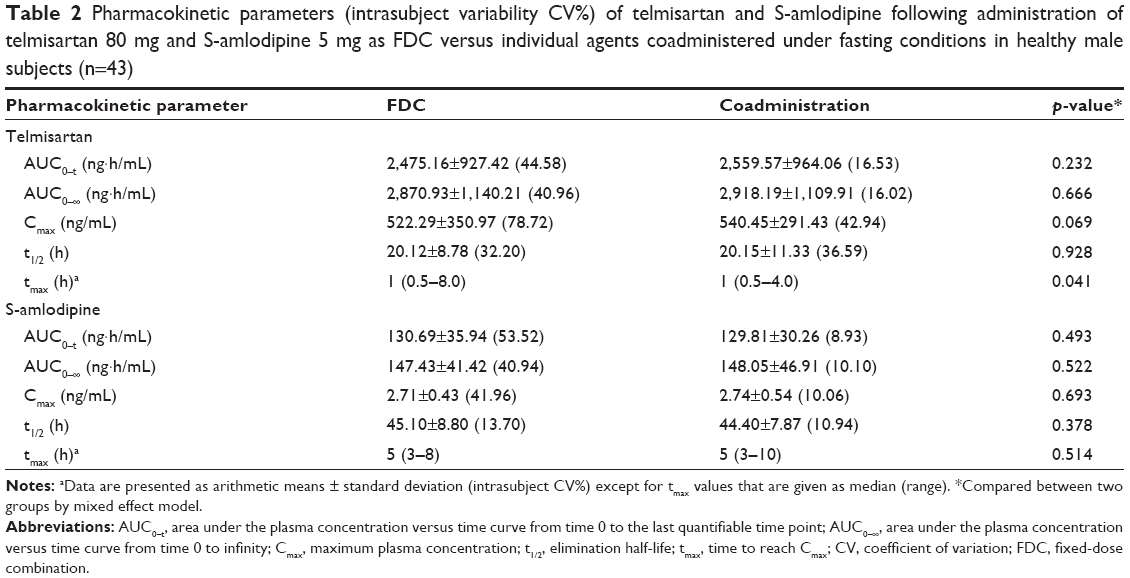 | Table 2 Pharmacokinetic parameters (intrasubject variability CV%) of telmisartan and S-amlodipine following administration of telmisartan 80 mg and S-amlodipine 5 mg as FDC versus individual agents coadministered under fasting conditions in healthy male subjects (n=43) |
 | Table 3 Geometric mean ratios and 90% CIs for Cmax, AUC0–t, and AUC0–∞ following administration of telmisartan 80 mg and S-amlodipine 5 mg as a fixed-dose combination versus individual agents in healthy subjects |
Safety and tolerability assessments
Single oral doses of telmisartan 80 mg and S-amlodipine 5 mg, as an FDC tablet or as individual agents, were generally well tolerated in healthy adult subjects in this study. In total, 11 subjects (22.9% of 48 subjects) experienced at least one of 12 reported AEs during this study. Of all 12 AEs, four events were determined to be possibly related to the study drugs (one headache, one instance of decreased BP, and one instance of myalgia after T treatment; and one upper respiratory infection after R treatment), and three events were determined to be probably related (two instances of increased creatine phosphokinase [CPK] after T treatment; and one instance of increased CPK after R treatment). All AEs were transient and spontaneously resolved without specific treatment, with no severe or serious AEs. No subjects withdrew from the study due to AEs.
Figure 3 illustrates the changes in mean SBP, DBP, and pulse rate in the two groups from baseline (0 hours) to 24 hours after administration of a single oral dose of the FDC or the individual agents concomitantly administered. The changes in vital signs showed no statistical difference between the two groups.
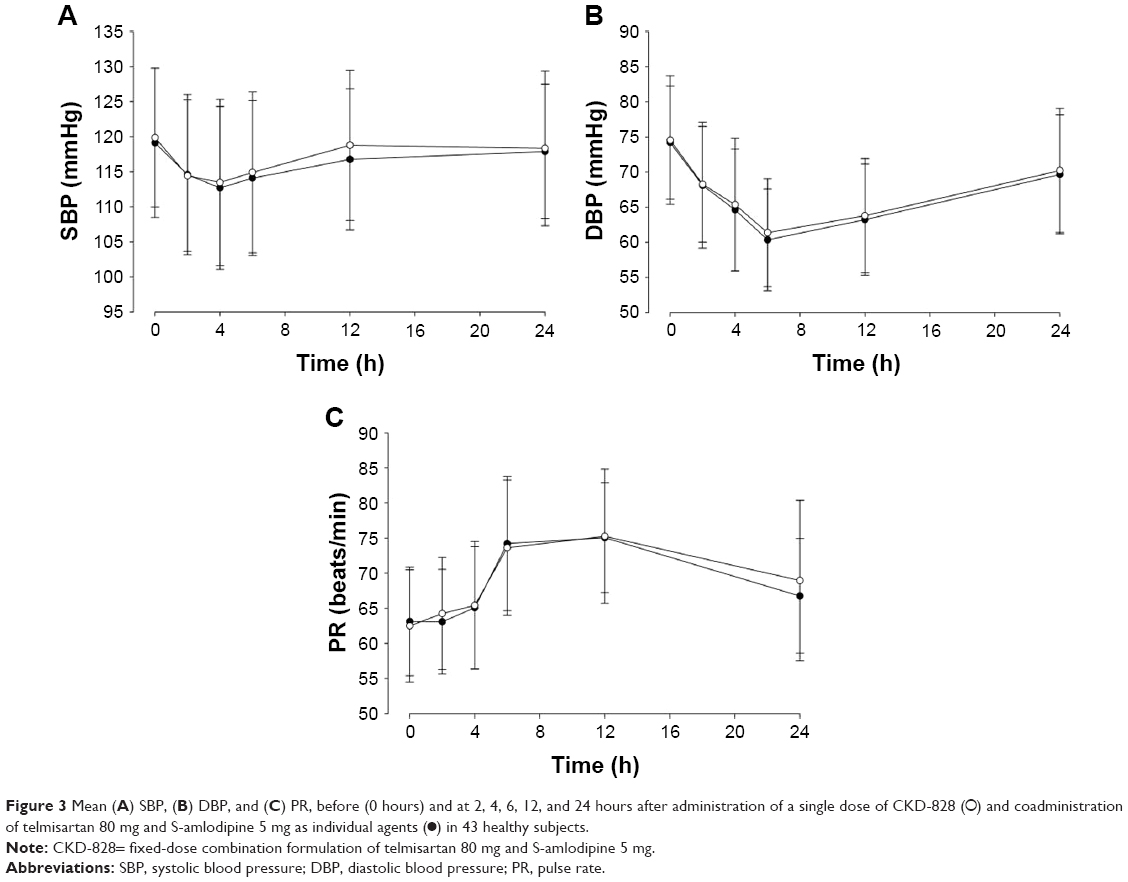 | Figure 3 Mean (A) SBP, (B) DBP, and (C) PR, before (0 hours) and at 2, 4, 6, 12, and 24 hours after administration of a single dose of CKD-828 (○) and coadministration of telmisartan 80 mg and S-amlodipine 5 mg as individual agents (•) in 43 healthy subjects. |
Discussion
This randomized, open-label, two-sequence, four-period, two-treatment crossover study demonstrated that the FDC formulation of telmisartan 80 mg and S-amlodipine 5 mg is bioequivalent to coadministration of the individual tablets. In addition, both the FDC formulation and separate tablets of telmisartan and S-amlodipine were well tolerated in this study.
The mean ratio of AUC0–t for telmisartan and S-amlodipine in our study accounted for 87.0%–89.0% of the total AUC0–∞, indicating that the sampling schedule was appropriate to provide a reliable estimate of the extent of exposure (AUC0–t/AUC0–∞ ratio >80%). The washout period of 14 days in this study based on the long t1/2 of amlodipine (30–50 hours) from earlier PK studies was enough to ensure complete elimination of the study drugs from the blood, as the S-amlodipine and telmisartan plasma concentrations in the pre-dose samples for periods 2–4 were not detectable.
The intrasubject variability %CV of telmisartan Cmax for FDC tablets in this study was 78.72%, higher than the value we had used to calculate the sample size (47.5%). However, the sample size adjusted from the intrasubject variability in our study (about 45 subjects in total) is not much different from the number of subjects who completed this study, suggesting that the sample size was not insufficient to demonstrate the bioequivalence of telmisartan in this study.33
This four-period replicate study design enabled us to obtain repeated measures to improve the precision of the comparisons between the two treatments by using each subject as their own control, and to determine the true intrasubject variability for the T and R treatments independently. Our calculated intrasubject variability of telmisartan Cmax (42.94%) was comparable with the value (47.5%) that had been estimated from our earlier two-way, two-period, crossover PK study and used for the sample size calculation in this study, and was higher than the value (30%) predefined in this study protocol according to the Korean BE study guideline for highly variable drug products. Accordingly, 90% CIs of the GMR for telmisartan Cmax from our study (0.7353–0.9846) fell within the widened acceptance limit of 0.732–1.367.34–36 Consequently, it was verified that all GMR and 90% CI values for both telmisartan and S-amlodipine fell within the predetermined bioequivalence range for both Cmax and AUC0–t.
The source of the larger intrasubject variability %CV values of telmisartan and S-amlodipine for FDC tablets than those for individual agents coadministered in this study is unclear, but the formulation complexities might be a plausible explanation. Mitra and Wu concluded that the biopharmaceutical and PK behavior could be complicated by combining multiple active ingredients in a single FDC formulation.37 High drug loading is required in the FDC tablet due to restrictions on its final size, leading to dissolution slowdown.
There were several limitations in this study. First, the results were obtained from healthy male subjects, who may not represent the target patients. Second, even though long-term antihypertensive treatment is required in clinical settings, only a single dose was administered in this study.
In conclusion, telmisartan/S-amlodipine FDC tablets were bioequivalent to coadministration of the individual agents with the respective strength in healthy subjects under fasting conditions. There was no significant difference in safety profile between the two treatments.
Acknowledgments
This study was sponsored by Chong Kun Dang Pharmaceutical Co, Ltd, and was supported by grants from the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health and Welfare, Republic of Korea (HI14C2750); the Bio & Medical Technology Development Program of the National Research Foundation (NRF), funded by the Ministry of Science, ICT and Future Planning, Republic of Korea (NRF-2013M3A9B6046416); and the Industrial Core Technology Development Program (10051129; development of the system for ADME assessment using radiolabeled compounds), funded by the Ministry of Trade, Industry and Energy (MOTIE, Korea).
Disclosure
The authors report no conflicts of interest in this work.
References
Chobanian AV, Bakris GL, Black HR, et al; Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure; National Heart, Lung, and Blood Institute; National High Blood Pressure Education Program Coordinating Committee. Seventh report of the Joint National Committee on prevention, detection, evaluation, and treatment of high blood pressure. Hypertension. 2003;42(6):1206–1252. | ||
Law MR, Morris JK, Wald NJ. Use of blood pressure lowering drugs in the prevention of cardiovascular disease: meta-analysis of 147 randomized trials in the context of expectations from prospective epidemiological studies. BMJ. 2009;338:b1665. | ||
Ettehad D, Emdin CA, Kiran A, et al. Blood pressure lowering for prevention of cardiovascular disease and death: a systematic review and meta-analysis. Lancet. 2016;387(10022):957–967. | ||
James PA, Oparil S, Carter BL, et al. 2014 evidence-based guideline for the management of high blood pressure in adults: report from the panel members appointed to the Eighth Joint National Committee (JNC 8). JAMA. 2014;311(5):507–520. | ||
Zhang PY. Review of new hypertension guidelines. Eur Rev Med Pharmacol Sci. 2015;19(2):312–315. | ||
Mancia G, De Backer G, Dominiczak A, et al. 2007 ESH-ESC practice guidelines for the management of arterial hypertension: ESH-ESC Task Force on the management of arterial hypertension. J Hypertens. 2007;25(9):1751–1762. | ||
Düsing R. Optimizing blood pressure control through the use of fixed combinations. Vasc Health Risk Manag. 2010;6:321–325. | ||
Neutel JM. The role of combination therapy in the management of hypertension. Nephrol Dial Transplant. 2006;21(6):1469–1473. | ||
Ke YN, Dong YG, Ma SP, et al. Improved blood pressure control with nifedipine GITS/valsartan combination versus high-dose valsartan monotherapy in mild-to-moderate hypertensive patients from Asia: results from the ADVISE study, a randomized trial. Cardiovasc Ther. 2012;30(6):326–332. | ||
Kim KI, Shin MS, Ihm SH, et al. A randomized, double-blind, multicenter, phase III study to evaluate the efficacy and safety of fimasartan/amlodipine combined therapy versus fimasartan monotherapy in patients with essential hypertension unresponsive to fimasartan monotherapy. Clin Ther. 2016;38(10):2159–2170. | ||
Sohn IS, Kim CJ, Ahn T, et al. Efficacy and tolerability of combination therapy versus monotherapy with candesartan and/or amlodipine for dose finding in essential hypertension: a phase II multicenter, randomized, double-blind clinical trial. Clin Ther. 2017;39(8):1628–1638. | ||
Sung J, Jeong JO, Kwon SU, et al. Valsartan 160 mg/amlodipine 5 mg combination therapy versus amlodipine 10 mg in hypertensive patients with inadequate response to amlodipine 5 mg monotherapy. Korean Circ J. 2016;46(2):222–228. | ||
Gupta AK, Arshad S, Poulter NR. Compliance, safety, and effectiveness of fixed-dose combinations of antihypertensive agents: a meta-analysis. Hypertension. 2010;55(2):399–407. | ||
Egan BM, Bandyopadhyay D, Shaftman SR, Wagner CS, Zhao Y, Yu-Isenberg KS. Initial monotherapy and combination therapy and hypertension control the first year. Hypertension. 2012;59(6):1124–1131. | ||
Erding S. Compliance with the treatment of hypertension: the potential of combination therapy. J Clin Hypertens (Greenwich). 2010;12(1):40–46. | ||
da Silva PM. Efficacy of fixed-dose combination therapy in the treatment of patients with hypertension: focus on amlodipine/valsartan. Clin Drug Investig. 2010;30(9):625–641. | ||
Sherrill B, Halpern M, Khan S, Zhang J, Panjabi S. Single-pill vs free-equivalent combination therapies for hypertension: a meta-analysis of health care costs and adherence. J Clin Hypertens (Greenwich). 2011;13(12):898–909. | ||
Taylor AA, Shoheiber O. Adherence to antihypertensive therapy with fixed-dose amlodipine besylate/benazepril HCl versus comparable component-based therapy. Congest Heart Fail. 2003;9(6):324–332. | ||
Mancia G, Fagard R, Narkiewicz K, et al. 2013 ESH/ESC guidelines for the management of arterial hypertension: the Task Force for the management of arterial hypertension of the European Society of Hypertension (ESH) and of the European Society of Cardiology (ESC). J Hypertens. 2013;31(7):1281–1357. | ||
Canbakan B. Rational approaches to the treatment of hypertension: drug therapy – monotherapy, combination, or fixed-dose combination? Kidney Int Suppl (2011). 2013;3(4):349–351. | ||
Ihm SH, Jeon HK, Cha TJ, et al. Efficacy and safety of two fixed-dose combinations of S-amlodipine and telmisartan (CKD-828) versus S-amlodipine monotherapy in patients with hypertension inadequately controlled using S-amlodipine monotherapy: an 8-week, multicenter, randomized, double-blind, phase III clinical study. Drug Des Devel Ther. 2016;10:3817–3826. | ||
Israili ZH. Clinical pharmacokinetics of angiotensin II (AT1) receptor blockers in hypertension. J Hum Hypertens. 2000;14(Suppl 1):S73–S86. | ||
Smith DH, Matzek KM, Kempthorne-Rawson J. Dose response and safety of telmisartan in patients with mild to moderate hypertension. J Clin Pharmacol. 2000;40(12 Pt 1):1380–1390. | ||
Hernández-Hernández R, Sosa-Canache B, Velasco M, Armas-Hernández MJ, Armas-Padilla MC, Cammarata R. Angiotensin II receptor antagonists role in arterial hypertension. J Hum Hypertens. 2002;16(Suppl 1):S93–S99. | ||
Mason RP, Marche P, Hintze TH. Novel vascular biology of third-generation L-type calcium channel antagonists: ancillary actions of amlodipine. Arterioscler Thromb Vasc Biol. 2003;23(12):2155–2163. | ||
Oh MJ, Hwang HH, Kim HG, et al. Bioequivalence study of a new fixed-dose combination tablet containing S-amlodipine nicotinate and olmesartan medoxomil in healthy Korean male subjects. Clin Ther. 2017;39(7):1371–1379. | ||
Noh YH, Lim HS, Kim MJ, et al. Pharmacokinetic interaction of telmisartan with S-amlodipine: an open-label, two-period crossover study in healthy Korean male volunteers. Clin Ther. 2012;34(7):1625–1635. | ||
Goldmann S, Stoltefuss J, Born L. Determination of the absolute configuration of the active amlodipine enantiomer as (−)-S: a correction. J Med Chem. 1992;35(18):3341–3344. | ||
Abernethy DR. The pharmacokinetic profile of amlodipine. Am Heart J. 1989;118(5 Pt 2):1100–1103. | ||
Meredith PA, Elliott HL. Clinical pharmacokinetics of amlodipine. Clin Pharmacokinet. 1992;22(1):22–31. | ||
Zhu Y, Wang F, Li Q, et al. Amlodipine metabolism in human liver microsomes and roles of CYP3A4/5 in the dihydropyridine dehydrogenation. Drug Metab Dispos. 2014;42(2):245–249. | ||
Stangier J, Su CA. Pharmacokinetics of repeated oral doses of amlodipine and amlodipine plus telmisartan in healthy volunteers. J Clin Pharmacol. 2000;40(12 Pt 1):1347–1354. | ||
Tothfalusi L, Endrenyi L. Sample sizes for designing bioequivalence studies for highly variable drugs. J Pharm Pharm Sci. 2012;15(1):73–84. | ||
European Medicines Agency (EMA), Committee for Medicinal Products for Human Use. Guideline on the Investigation of Bioequivalence. London: EMA, Committee for Medicinal Products for Human Use; 2010. | ||
Korea Ministry of Food and Drug Safety (MFDS). [Guideline on Bioequivalence Studies for Orally Administered Drug Products (No. 2014–188)]. Available from: http://www.mfds.go.kr/index.do?mid=1013&seq=8562&cmd=v. Accessed November 12, 2016. Korean. | ||
Karalis V, Symillides M, Macheras P. Bioequivalence of highly variable drugs: a comparison of the newly proposed regulatory approaches by FDA and EMA. Pharm Res. 2012;29(4):1066–1077. | ||
Mitra A, Wu Y. Challenge and opportunities in achieving bioequivalence for fixed-dose combination products. AAPS J. 2012;14(3):646–655. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.