")
Back to Journals » Drug Design, Development and Therapy » Volume 13
Pharmacokinetic and bioequivalence study between two formulations of S-1 in Korean gastric cancer patients
Authors Lee HW , Seong SJ, Kang WY , Ohk B , Gwon MR, Kim BK , Cho S , Cho K, Sung YK , Yoon YR , Kim JG
Received 17 June 2019
Accepted for publication 3 August 2019
Published 3 September 2019 Volume 2019:13 Pages 3127—3136
DOI https://doi.org/10.2147/DDDT.S219822
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Anastasios Lymperopoulos
Hae Won Lee*,1,2, Sook Jin Seong*,1,2, Woo Youl Kang1,2, Boram Ohk1,2, Mi-Ri Gwon1,2, Bo Kyung Kim1,2, Seungil Cho1,2, Kyunghee Cho3, Yong Kyung Sung4, Young-Ran Yoon1,2, Jong Gwang Kim5
1Department of Molecular Medicine, School of Medicine, Kyungpook National University, Daegu, Republic of Korea; 2Department of Clinical Pharmacology, Kyungpook National University Hospital, Daegu, Republic of Korea; 3Analytical Research Division, Biocore Co. Ltd., Seoul, Republic of Korea; 4Department of R&D, Myungmoon Pharm. Co., Ltd., Seoul, Republic of Korea; 5Department of Oncology/Hematology, Kyungpook National University Hospital, Kyungpook National University School of Medicine, Kyungpook National University Cancer Research Institute, Kyungpook National University, Daegu, Republic of Korea
*These authors contributed equally to this work
Correspondence: Young-Ran Yoon
Department of Molecular Medicine, School of Medicine, Kyungpook National University, 130 Dongduk-Ro, Jung-gu, Daegu 41944, Republic of Korea
Tel +82 53 420 4950
Fax +82 53 426 4944
Email [email protected]
Jong Gwang Kim
Department of Oncology/Hematology, Kyungpook National University Hospital, Kyungpook National University School of Medicine, Kyungpook National University Cancer Research Institute, Kyungpook National University, 130 Dongduk-Ro, Jung-gu, Daegu 41944, Republic of Korea
Tel +82 53 200 6522
Fax +82 53 420 5218
Email [email protected]
Purpose: S-1 is an oral fluoropyrimidine anticancer drug consisting of the 5-fluorouracil prodrug tegafur combined with gimeracil and oteracil. The purpose of this study was to evaluate the pharmacokinetic (PK), bioequivalence, and safety of a newly developed generic formulation of S-1 compared with the branded reference formulation, in Korean gastric cancer patients.
Methods: This was a single-center, randomized, open-label, single-dose, two-treatment, two-way crossover study. Eligible subjects were randomly assigned in a 1:1 ratio to receive the test formulation or reference formulation, followed by a one-week washout period and administration of the alternate formulation. Serial blood samples were collected at 0 hrs (predose), 0.25, 0.5, 1, 2, 3, 4, 5, 6, 8, 10, 12, 24, 36, and 48 hrs after dosing in each period. The plasma concentrations of tegafur, 5-FU, gimeracil, and oteracil were analyzed using a validated liquid chromatography-tandem mass spectrometry method. The PK parameters were calculated using a non-compartmental method.
Results: In total, 29 subjects completed the study. All of the 90% confidence intervals (CIs) of the geometric mean ratios (GMRs) fell within the predetermined acceptance range. No serious adverse events were reported during the study.
Conclusion: The new S-1 formulation met the Korean regulatory requirement for bioequivalence. Both S-1 formulations were well tolerated in all subjects.
Clinical trial registry: https://cris.nih.go.kr CRIS KCT0003855.
Keywords: S-1, pharmacokinetics, bioequivalence, tegafur, gimeracil, oteracil
Introduction
The oral fluoropyrimidine S-1 (TS-1, Taiho Pharmaceutical) is an orally active triple-drug mixture of tegafur/gimeracil/oteracil in a molar ratio of 1:0.4:1.1,2 S-1 was developed to overcome the drawbacks of 5-fluorouracil (5-FU), 90% of which is actively catabolized in the liver to inactive metabolites by the enzyme dihydropyrimidine dehydrogenase (DPD).3 S-1 has been demonstrated to be effective not only in gastric cancer, but also in a variety of malignancies, including head and neck cancer, breast cancer, colorectal cancer, and pancreatic cancer.4–8 According to the Adjuvant Chemotherapy Trial of TS-1 for Gastric Cancer (ACTS-GC), S-1-based adjuvant chemotherapy for one year after curative gastrectomy has become a standard treatment in stage II or stage III gastric cancer patients.4,9
Tegafur, a 5-fluorouracil (5-FU) prodrug, is gradually converted, primarily by cytochrome P450 2A6 (CYP2A6), to 5-FU. According to several studies on the correlation of CYP2A6 polymorphisms with the pharmacokinetics and efficacy of S-1 in patients with gastric cancer, exposure to 5-FU and relapse-free survival differed significantly between the patients with CYP2A6 variant alleles and those with wild-type alleles.10–12 In the case of genetic polymorphism in DPYD, the gene encoding for DPD, reduced activity of DPD can increase the risk of developing severe fluoropyrimidine-related toxicity.13,14 Following single-dose oral administration of S-1, the maximum plasma concentration (Cmax) of tegafur achieved was reached 1.0–3.6 hrs after dosing, with a terminal half-life (t1/2) of 8.2–13.1 hrs, and the Cmax and t1/2 of 5-FU were 3.0–4.0 hrs and 1.9–3.4 hrs, respectively.15–17
Gimeracil [5-chloro-2,4-dihydroxypyridine (CDHP)], a competitive inhibitor of DPD, reduces the degradation of 5-FU, resulting in prolonged effective concentrations of 5-FU in plasma and tumor tissue.1,18 The Cmax of gimeracil has been observed to be reached between 1.5–3.3 hrs after oral administration, with a mean t1/2 of 3.3–5.8 hrs.17,18 Oteracil, a competitive inhibitor of orotate phosphoribosyltransferase, inhibits the phosphorylation of 5-FU in the gastrointestinal tract, thereby decreasing the gastrointestinal toxicity of 5-FU.19 Following oral administration, oteracil reaches Cmax (tmax) in 2.5–4.0 hrs and t1/2 in 4.0–7.8 hrs.17,18
The objective of the present study was to evaluate the pharmacokinetic (PK) characteristics and bioequivalence between a test formulation and branded reference formulations of S-1 capsules in cancer patients.
Methods
Study subjects
This study was conducted at the Clinical Trial Center, Kyungpook National University Hospital (KNUH, Daegu, Republic of Korea), in accordance with the ethical principles of the Declaration of Helsinki, International Conference on Harmonization Good Clinical Practice Guideline, and local laws and regulations. The protocol was approved by the Institutional Review Board of KNUH. Written informed consent was obtained from all of the subjects prior to their participation in this study.
All of the subjects enrolled in this study met the following conditions: (1) between 20 and 70 years of age; (2) gastric cancer or head and neck cancer patients who had been followed up after curative surgery (follow-up period ≤5 years), with no tegafur/gimeracil/oteracil potassium administration required; (3) subjects who were considered eligible for participating in the study by an investigator, based on clinical laboratory tests including hematology, clinical chemistry, and urinalysis (hemoglobin ≥9.0 g/dL; neutrophil count ≥1500 mm3; platelets ≥100,000/ mm3; total bilirubin ≤ three times the upper limit of the normal range (ULN); aspartate aminotransferase (AST), alanine aminotransferase (ALT), and alkaline phosphatase (ALP) ≤2.5 times the ULN; serum creatinine ≤ the ULN; creatinine clearance (estimated using the Cockcroft-Gault equation using serum creatinine concentrations) ≥60 mL/min)); patients capable of oral administration of TeGO 25 or TS-1® 25 capsules; having a body mass index (BMI) between 17.6 kg/m2 and 26.4 kg/m2 (BMI = body weight (kg)/{height (m)}2); having a body surface area (BSA) ≥1.25 m2 (BSA (m2) = height (cm)0.663 X body weight (kg)0.444 X 0.008883); having histologically or cytologically proven cancer; Eastern Cooperative Oncology Group (ECOG) performance status (PS) of 0–2; negative pregnancy test at screening, in the case of female patients; patients who voluntarily signed a written informed consent and consent on private information utilization, approved by the Institutional Review Board of KNUH. National University Hospital (KNUH, Daegu, Republic of Korea).
Exclusion criteria were as follows: use of any drug that could induce or inhibit drug metabolizing enzymes such as barbiturates, or history of alcohol abuse within one month before the start of the trial; patients who could not take S-1; use of S-1 within four weeks prior to the first administration of the study drug; patients within six months after subtotal gastrectomy, or patients with total gastrectomy, or patients with frequent relapse of peptic ulcer; participation in any other clinical trial within 12 weeks prior to the first administration of the study drug, or patients on concomitant medications taken within 10 days before the start of the trial, that could affect the trial (excluding amlodipine, lecanidipine, losartan, valsartan, olmesartan, irbesartan, atenolol, or ramipril); history of other surgery within four weeks prior to the first administration of the study drug; history of any other chemotherapy within five weeks prior to the first administration of the study drug; history of radiotherapy within six weeks prior to the first administration of the study drug; evidence of metastases to other organs (brain or bone, etc.) other than gastric cancer; active infectious disease (any febrile disease with fever >38°C); serious concurrent disease such as intestinal palsy, bowel obstruction, interstitial pneumonia, pulmonary fibrosis, gastrointestinal bleeding, uncontrollable diabetes mellitus, heart failure, myocardial infarction, angina pectoris, renal failure, hepatic failure, psychiatric disorder, cerebrovascular disease, or peptic ulcer in need of transfusion, or patients judged inappropriate for bioequivalence study by their physicians; patients with diarrhea in need of treatment (watery diarrhea); women who were pregnant or breast-feeding, or patients with reproductive potential who were unwilling to use an effective method of contraception, or patients with reproductive potential; patients who were taking phenytoin, warfarin, flucytosine, allopurinol, idoxuridine, leucovorin, dipyridamole, cimetidine, methoxsalen, leflunomide, letrozole, fluoropyrimidine, anti-cancer agents (fluorouracil, tegafur–uracil combination drug, tegafur doxifluridine, capecitabine, carmofur, Horinato–tegafur–uracil therapy, Rebohorinato–fluorouracil therapy), pilocarpine, or proton pump inhibitors; patients judged inappropriate for the study by their investigators; a medical history of serious hypersensitivity to the study drug; patients with serious myelosuppression, serious renal disease, or serious hepatic disease; patients who were taking other fluoropyrimidine anticancer drugs, or fluoropyrimidine antifungals (flucytosine); evidence of hereditary disease, including galactose intolerance, Lapp lactase deficiency, or glucose–galactose malabsorption.
Study design and procedure
This was a randomized, open-label, single-dose, two-period, two-way crossover study conducted at the KNUH Clinical Trial Center. Thirty patients with gastric cancer were enrolled and randomly assigned to one of the two treatment sequences of test-reference or reference-test in a 1:1 ratio. TeGO capsules 25 mg (lot no 4012P1; expiration date, March 2017; Myungmoon Pharm Co., Ltd., Seoul, Republic of Korea) were used as the test formulation, and TS-1® capsules 25 mg (lot no TZO202; expiration date, October 2016; Jeil Pharmaceutical Co., Ltd., Seoul, Republic of Korea) were used as the branded reference formulation. Both formulations contained 25 mg tegafur, 7.25 mg gimeracil, and 24.5 mg oteracil. A single oral dose of test or branded reference formulation of S-1 capsules was administered in each period. The washout period was seven days, which was five times longer than the terminal half-lives of tegafur, gimeracil, and oteracil as reported in previous PK studies.15–18
The subjects were admitted to the study center at 8 pm the day prior to dosing. Each study drug was orally administered under fasting conditions along with 240 mL of water. The subjects fasted for 10 hrs prior to dosing and the fasting was continued until four hours after dosing. Standard meals were provided at 4 and 10 hrs after dosing. No additional water intake was allowed for two hours before and after dosing during each period.
For PK analysis of tegafur, 5-FU, gimeracil, and oteracil, blood samples were collected at 0 hrs (pre-dose), 0.25, 0.5, 1, 2, 3, 4, 5, 6, 8, 10, 12, 24, 36, and 48 hrs post-administration of the drug. An indwelling intravenous catheter was placed in either the forearm or dorsum of the hand of each subject. After discarding 1 mL of blood from the catheter, 10 mL of blood was collected into an EDTA vacutainer and was centrifuged at 3200 rpm for 6 mins, in order to obtain plasma. Following centrifugation, the plasma samples were transferred to three different tubes and stored at −70°C until analyzed by the analytical laboratory, Biocore Co. Ltd. (Seoul, Republic of Korea).
Analysis of the plasma concentrations of tegafur, gimeracil, oteracil, and 5-FU
The plasma concentrations of tegafur, gimeracil, oteracil, and 5-FU were determined by ultra-fast liquid chromatography (UFLC, Shimadzu UFLC system, Shimadzu Corp., Kyoto, Japan) coupled with tandem mass spectrometry (MS/MS, API 5000, AB Sciex, Foster City, CA, USA), with some modifications of a validated method.20
For gimeracil and tegafur, chromatographic separation was performed on a C18 column (4.6 i.d. ×50 mm, 3.0 µm particle size; Imtakt Corp., Tokyo, Japan), at a flow rate of 0.64 mL/min (A pump) and 0.16 mL/min (B pump). The mobile phase consisted of acetonitrile (A pump) and 0.1% formic acid in deionized water (B pump). Multiple reaction monitoring transitions were performed at mass-to-charge (m/z) ratios of 145.9 → 128.0 and 149.0 → 130.9 for gimeracil and gimeracil-13C3 (the internal standard (IS)), and 201.1 → 130.9 and 204.1 → 133.9 for tegafur and tegafur-13C, 15N2, respectively. The frozen plasma was thawed at room temperature. Following the addition of 10 μL of the IS (5000 ng/mL; gimeracil-13C3: tegafur-13C, 15N2=1:1, v/v) to 200 μL of plasma in a polypropylene tube, 1 mL of acetonitrile was added and vortexed for 5 mins. After the mixture was centrifuged at 13,000 rpm for 5 mins, the upper layer was evaporated to dryness under a stream of nitrogen. The residue was reconstituted with 100 μL of 50% acetonitrile solution, and centrifuged at 13,000 rpm for 5 mins. A 3 μL aliquot of this solution was injected into the LC-MS/MS system for analysis. The lower limit of quantification was 1 ng/mL for gimeracil and 2 ng/mL for tegafur, and the linear calibration curves ranged between 1 and 500 ng/mL for gimeracil (r≥0.9950), and between 2 and 2000 ng/mL for tegafur (r≥0.9950). The overall intra-day accuracy ranged from 88.0% to 108.0%, and inter-day accuracy ranged from 90.4% to 105.7%, at concentrations of 1, 3, 30, and 400 ng/mL for gimeracil. Intra-day accuracy ranged from 89.1% to 107.0%, while inter-day accuracy ranged from 99.0% to 103.7%, at concentrations of 2, 6, 60, and 1,600 ng/mL for tegafur. The intra-day and inter-day precision (% coefficient of variation (CV)) ranged from 0.6% to 10.4%, and from 1.8% to 7.4%, respectively, for gimeracil and from 1.0% to 6.2%, and from 2.2% to 7.4%, respectively, for tegafur.
For oteracil, chromatographic separation was performed on a C18 column (2.1 i.d. ×100 mm, 3.0 µm particle size; Waters Corp., Milford, MA, USA), at a flow rate of 0.09 mL/min (A pump) and 0.01 mL/min (B pump). The mobile phase consisted of acetonitrile (A pump) and 0.1% formic acid in deionized water (B pump). Multiple reaction monitoring transitions were performed at mass-to-charge (m/z) ratios of 156.0 → 112.0 and 161.0 → 117.0 for oteracil and oxonic acid-13C2, 15N3 (the IS), respectively. The frozen plasma was thawed at room temperature, and 10 μL of the IS (3000 ng/mL) was added to 200 μL of plasma in a polypropylene tube, and mixed. After 500 μL of methanol and 500 μL of deionized water were added to an SPE cartridge, the sample was loaded, washed with 1.5 mL of deionized water, eluted with 2 mL of methanol, and evaporated to dryness under a stream of nitrogen gas. The residue was reconstituted with 150 μL in 50% acetonitrile solution, and centrifuged at 13,000 rpm for 5 mins. A 1 μL aliquot of this solution was injected into the LC-MS/MS system for analysis. The lower limit of quantification was 2 ng/mL, and the linear calibration curves ranged between 2 and 500 ng/mL for oteracil (r≥0.9950). The overall intra-day and inter-day accuracy ranged from 93.4% to 109.0%, and from 97.7% to 106.3%, respectively, at concentrations of 2, 6, 30, and 400 ng/mL for oteracil. The intra-day and inter-day precision (%CV) ranged from 0.9% to 15.6%, and from 2.1% to 13.0%, respectively.
For 5-FU, chromatographic separation was performed on a C18 column (2.1 i.d. × 100 mm, 3.0 µm particle size; Waters Corp., Milford, MA, USA), at a flow rate of 0.18 mL/min (A pump) and 0.02 mL/min (B pump). The mobile phase consisted of a 0.1% formic acid in deionized water (A pump) and acetonitrile (B pump). Multiple reaction monitoring transitions were performed at mass-to-charge (m/z) ratios of 129.0 → 42.0 and 132.0 → 44.0 for 5-fluorouracil and 5-fluorouracil-13C,15N2 (the IS), respectively. The frozen plasma was thawed at room temperature. Following the addition of 10 μL of the IS (2500 ng/mL) to 200 μL of plasma in a polypropylene tube, 600 μL of acetonitrile was added and vortexed for 5 mins. After the mixture was centrifuged at 13,000 rpm for 5 mins, the upper layer was evaporated to dryness under a stream of nitrogen. The residue was reconstituted as 100 μL in 50% acetonitrile solution, and centrifuged at 13,000 rpm for 5 mins. A 1 μL aliquot of this solution was injected into the LC-MS/MS system for analysis. The lower limit of quantification was 1 ng/mL, and the linear calibration curves ranged between 1 and 200 ng/mL for 5-FU (r≥0.9950). The overall intra-day and inter-day accuracy ranged from 89.8% to 103.6%, and from 97.3% to 99.8%, respectively, at concentrations of 1, 3, 16, and 160 ng/mL for 5-FU. The intra-day and inter-day precision (% CV) ranged from 0.7% to 14.4%, and from 1.9% to 11.8%, respectively.
The extraction recoveries of tegafur, 5-FU, gimeracil, and oteracil in the low, medium, high QC samples ranged from 66.3% to 94.4% with CV of <3.0%. The matrix effect ranged from 88.9% to 104.9% with CV of <2.6%, indicating that no significant interference occurred. The precision and accuracy of QC samples consisting of a mixture of the four analytes were within 8.0% and within 103.7% for short-term stability, and within 6.7% and within 108.0% for three freeze-thaw cycle stability, and within 6.9% and within 107.1% for post-preparative stability, respectively. Accordingly, tegafur, 5-FU, gimeracil, and oteracil in plasma samples were found to exhibit no problems in these three stability tests. The method reproducibility was checked by reanalysis of 10% of the samples (87 incurred samples each for tegafur, 5-FU, and gimeracil, and 70 samples for oteracil) near the Cmax and the elimination phase in the PK profile of the drug. The % change between the initial concentration and the concentration during the repeat analysis were within 20% of their mean for all the repeats of the four analytes.
PK analysis
The PK parameters for tegafur, gimeracil, oteracil, and 5-FU were calculated by non-compartmental methods using the Phoenix WinNonlin software, version 6.4 (Pharsight, Sunnyvale, CA, USA). The Cmax and tmax were obtained directly from the observed plasma concentration-time data. The area under the plasma concentration-time curve from time 0 to the last measurement (AUC0-t) was calculated using the linear trapezoidal method for ascending concentrations and the log trapezoidal method for descending concentrations. The AUC from time 0 to infinity (AUC0-∞) was calculated using the following formula: AUC0-∞ = AUC0-t+ Ct/λz, where Ct is the last measurable concentration and λz is the terminal elimination rate constant estimated from a linear regression line of the log-transformed plasma concentrations versus time over the terminal log-linear portion (at least three final data points). The t1/2 was calculated to be 0.693/λz.
Statistical analyses
Descriptive statistics were used to summarize the baseline demographics, PK parameters, and safety data. The differences in baseline demographics between the two treatment groups were determined by the Mann-Whitney U test or independent t-test for the age, height, body weight, and BMI of the individuals, using the SPSS software for Windows OS (ver. 18.0; SPSS Korea, Seoul, Republic of Korea). The differences in the PK parameters between the two groups were compared using a mixed-effects model analysis of variance (ANOVA) model, with subject-within-sequence as a random effect, and sequence, period, and treatment as fixed effects. The results were presented as the mean ± standard deviation (SD), except for the tmax values, which were expressed as the median, maximum, and minimum values. A p-value below 0.05 was taken to indicate statistical significance.
The bioequivalence between the test and reference formulations was evaluated based on the primary PK parameters (Cmax and AUC0-t) of tegafur, gimeracil, and oteracil after natural logarithm (ln) transformation. The test formulation was considered bioequivalent according to the standard used by the Korea Ministry of Food and Drug Safety as follows: 1) if the 90% confidence interval (CI) of the geometric mean ratios (GMRs) (test/reference formulations) for those parameters of tegafur, gimeracil, and oteracil fell within the conventional BE range of 0.8000–1.2500; or 2) if the GMR was within the range of 0.9–1.11, and the total number of subjects was greater than or equal to 24, with the similarity of in vitro dissolution profiles demonstrated at all conditions in in vitro dissolution tests conducted according to the standard.21 All statistical analyses for GMRs with 90% CIs were performed using the SAS software (ver. 9.2.; SAS Institute Inc., Cary, NC, USA).
Assessment of safety and tolerability
Safety and tolerability were evaluated for all the subjects who received at least one dose of the study drugs throughout the study period, by monitoring clinical adverse events (AEs) or AEs identified in the laboratory, which were observed after dosing, and included all subjective symptoms reported by the subjects and objective signs observed by the investigators. Vital signs (blood pressure, pulse rate, body temperature) of the participants were monitored at screening, on days one and eight (predose and at 4, 8, 12 and 24 hrs after dosing), and at the follow-up visit. Physical examination was performed at screening, before dosing in each period at days one and eight, and at the follow-up visit. Electrocardiograms were conducted at screening. Routine laboratory tests (hematology, urinalysis and serum chemistry) were conducted at screening, before dosing in period I, and at the follow-up visit. The AEs were monitored and recorded using the Medical Dictionary for Regulatory Activities (version 16.0), categorized per system organ class and preferred term, and summarized according to the number of events, number of subjects, severity, seriousness, and causality. All laboratory tests were performed at the Department of Laboratory Medicine, KNUH.
Results
Demographic characteristics
A total of thirty subjects (26 males and four females) were enrolled in this study, and were randomly assigned to one of two different groups in a 1:1 ratio. One subject withdrew consent before drug administration in period I. Accordingly, 29 subjects (group A, n=15; group B, n=14) who completed the study were considered for the PK analyses and for the safety assessment.
The means ± SD (ranges) for the age, height, and weight of the subjects were 56.6±9.1 years (38–69 years), 167.5±7.6 cm (149.7–180.0 cm), and 59.3±8.2 kg (46.0–77.2 kg). The baseline demographics showed no statistical difference between the two groups (Table 1). All of the patients had gastric cancers.
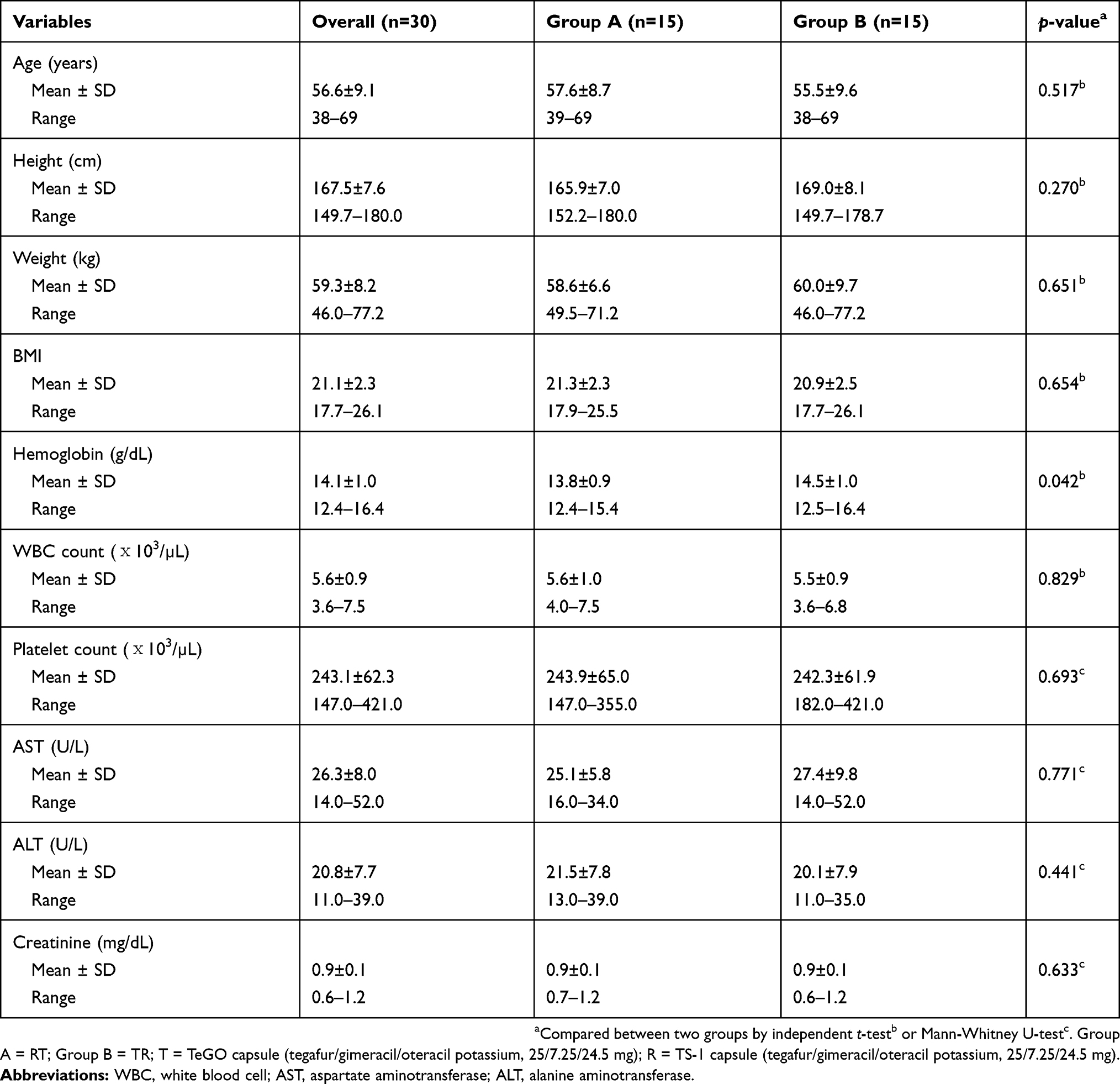 |
Table 1 Demographic and baseline characteristics of study subjects enrolled in this study |
PK data
The mean (SD) plasma concentration versus time profiles for tegafur, 5-FU, gimeracil, and oteracil following a single oral administration of test or reference formulation are illustrated in Figure 1A–D, respectively. The main PK parameters (AUC0-t, AUC0-∞, Cmax, t1/2, tmax) for both formulations are summarized in Table 2. All 90% CIs for the GMRs of test/reference formulations for Cmax, AUC0-t, and AUC0-∞ fell within the predetermined acceptance range for bioequivalence (0.8000–1.2500) (Table 3), except the 90% CI for the oteracil Cmax value of 0.7518–1.1049 (Table 3). However, the GMR of the oteracil Cmax value was 0.9114, which is within the range of 0.9–1.11, the total number of subjects was greater than or equal to 24, and the in vitro dissolution profiles were similar under all conditions in in vitro dissolution tests conducted according to the standard.21 Accordingly, the test formulation was considered bioequivalent.
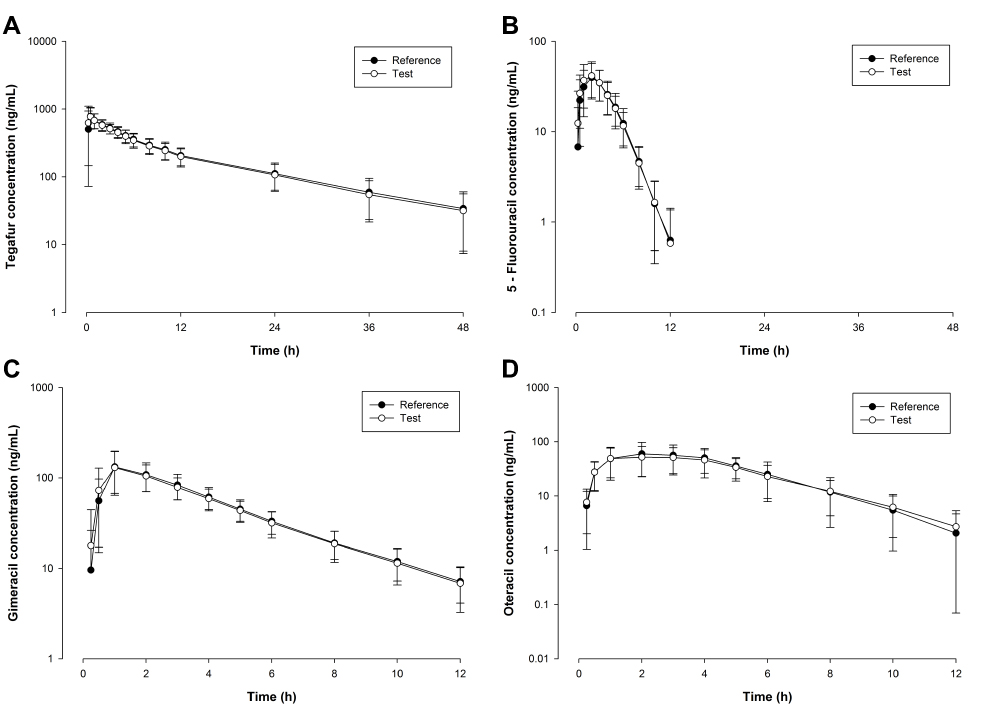 |
Figure 1 Mean plasma concentration-time profiles for (A) tegafur, (B) 5-fluorouracil, (C) gimeracil, and (D) oteracil, following a single-dose administration of tegafur/gimeracil/oteracil potassium (25/7.25/24.5 mg) of the test (○) and as reference (●) formulations in 29 gastric cancer patients. |
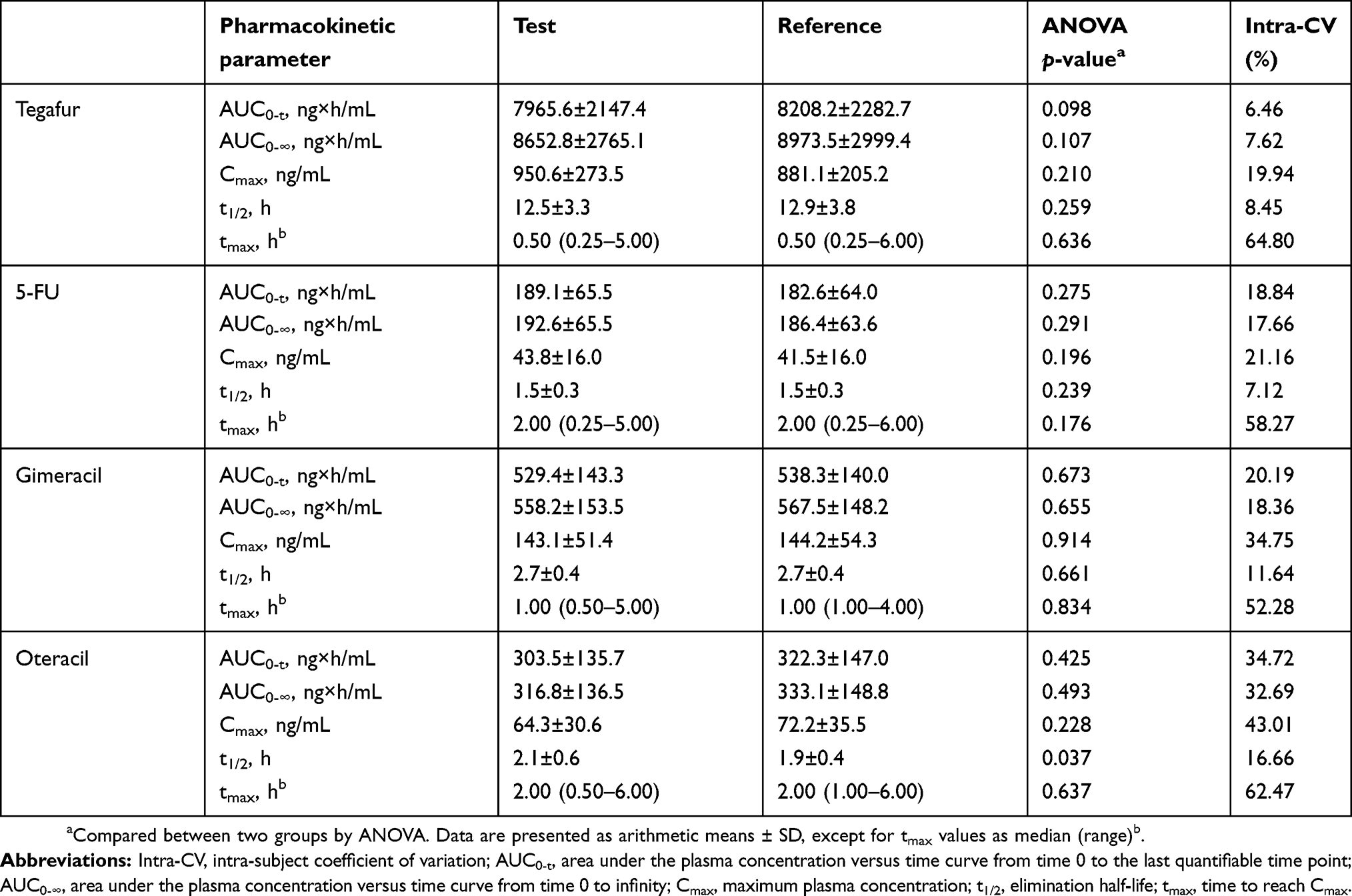 |
Table 2 Pharmacokinetic parameters of tegafur, 5-fluorouracil (5-FU), gimeracil, and oteracil following administration of a single tegafur/gimeracil/oteracil potassium (25/7.25/24.5 mg) dose of the test and reference formulations in 29 cancer patients |
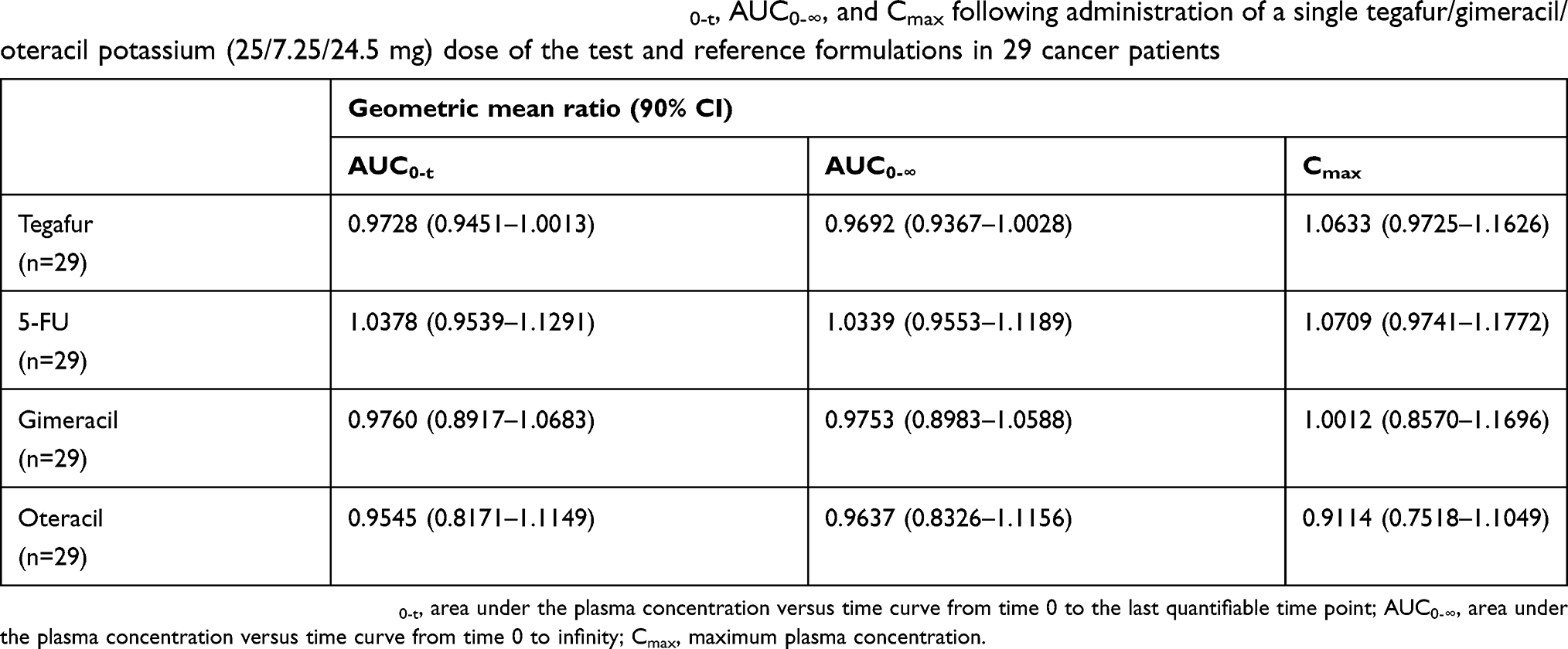 |
Table 3 Geometric mean ratios and 90% CIs for the AUC0-t, AUC0-∞, and Cmax following administration of a single tegafur/gimeracil/oteracil potassium (25/7.25/24.5 mg) dose of the test and reference formulations in 29 cancer patients |
Safety and tolerability assessments
A total of 12 subjects (41.4% of 29 subjects) experienced at least one of the 20 reported AEs. After the administration of the test formulation, eight subjects (27.6% of 29 subjects) experienced at least one of the 11 reported AEs: four incidences of diarrhea; and one incidence each of cold sweating, vomiting, increased AST, increased ALT, increased urinary occult blood, increased urinary RBC, and increased urinary WBC. In total, nine subjects (31.0% of 29 subjects) experienced nine reported AEs (three incidences of hyperkalemia, two incidences of diarrhea, and one incidence each of chest discomfort, blurred vision, bloating, and increased urinary RBC) after the administration of the reference formulation. Of all the 20 AEs, 18 were determined to be possibly related to the study medication. Two AEs—increased urinary WBC and cold sweating—were unlikely to be related to the medication. All AEs were transient and spontaneously resolved without specific treatment, with no severe or serious AEs. No subjects withdrew from the study due to AEs.
Discussion
This randomized, open-label, two-period, two-way crossover study demonstrated that the test formulation of S-1 (25 mg tegafur, 7.25 mg gimeracil, and 24.5 mg oteracil) was bioequivalent to the reference formulation, after administration of a single oral dose in Korean cancer patients. Both formulations were tolerated well in this study.
As recommended by the guidelines for bioavailability and bioequivalence studies, blood samples were collected for up to 48 hrs after dosing (at least three or more times the terminal t1/2s of tegafur, the longest value among t1/2 values of tegafur, 5-FU, gimeracil, and oteracil in earlier PK studies) in this study, in order to capture 90% of the relevant AUCs.15–18,21 The mean AUC0-t/AUC0-∞ ratios for tegafur, 5-FU, gimeracil, and oteracil ranged from 92.9% to 95.8% in our study. The sampling schedule was thus appropriate for providing a reliable estimate of the extent of exposure. Tegafur, 5-FU, gimeracil, and oteracil were not detectable in the pre-dose plasma samples in period II of our study, indicating that the washout period of seven days in this study based on the t1/2 values obtained from earlier PK studies was adequate for ensuring the complete elimination of the study medication from the blood after period I.
In this study, ANOVA showed no significant differences in 29 subjects between the two formulations on the PK parameters tested for tegafur, 5-FU, gimeracil, and oteracil (p>0.05), except for the t1/2 of oteracil (p=0.037). In our study, both formulations contained 25 mg tegafur, 7.25 mg gimeracil, and 24.5 mg oteracil potassium per capsule. As the mean body surface area (BSA) of the subjects who completed this study was 1.63 m2, the BSA-normalized dosage of tegafur administered in our study was 15.3 mg/m2. After dose-normalization to 40 mg/m2, the mean Cmax and AUC0-t values of tegafur from our study (2485.2–2303.5 ng/mL and 20.8–21.5 µg×h/mL, respectively) were comparable with those reported by Zhuang et al, after administration of a single oral dose of 40 mg per square meter of body surface (BSA) of test and reference formulations in 30 Chinese patients with cancer. They reported the mean tmax, Cmax, AUC0-t, and t1/2 values for tegafur of 1.9–3.6 hrs, 1869.7–1901.0 ng/mL, 21.0 −22.7 µg×h/mL, and 10.8–10.9 hrs, respectively.
The 90% CIs of the GMR of the log-transformed AUC0-t, AUC0-∞, and Cmax values for tegafur, 5-FU, gimeracil, and oteracil were within the acceptable range for bioequivalence predetermined according to the guidelines of the Ministry of Food and Drug Safety (MFDS) of Republic of Korea.21 Although the 90% CI of oteracil Cmax (0.7518–1.1049) was not within the conventional BE range of 0.8000–1.2500, the GMR of the oteracil Cmax value was within the range of 0.9–1.11, with the total number of subjects greater than or equal to 24, and similar in vitro dissolution tests conducted for all conditions. It therefore also met the acceptance criteria predetermined in the study protocol, according to the guidelines of the MFDS.
The intra-subject variability (%CV) values of Cmax, AUC0-t, AUC0-∞ of tegafur, 5-FU, gimeracil, and oteracil in our study ranged from 6.46% to 43.01%. The %CV values of gimeracil Cmax, and oteracil AUC0-t and Cmax were ≥30%, compatible with or less than those reported by Chu et al after once-daily-for-28-day 50 mg/m2/day administration of S-1 in patients with advanced malignancies.
The AEs of all grades occurring in at least 30% of patients in a randomized study of postoperative adjuvant chemotherapy with S-1 for gastric cancer were leukopenia, decreased hemoglobin, increased AST, increased ALT, increased bilirubin, stomatitis, anorexia, nausea, diarrhea, rash, pigmentation, and fatigue.22 However, the frequency of grade three or four AEs was less than 5%, except for neutropenia and anorexia. According to several clinical studies, S-1 and combination therapy were demonstrated not to be inferior to 5-FU continuous infusion, with fewer AEs and lower incidence of grade 3–4 toxicity. Of the 20 AEs in our study, the two most frequent were diarrhea (six of 20 AEs) and hyperkalemia (three of 20 AEs). All of the AEs were transient and resolved spontaneously without any specific treatment, and there were no severe or serious AEs. Both formulations were well tolerated in this study.
In conclusion, the PK profiles of the two S-1 formulations evaluated in this study met the regulatory requirements for bioequivalence. Both formulations were generally well tolerated.
Data sharing statement
We, the authors, intend to share individual de-identified participant data. However, there must be a limit on our data sharing, because this study was sponsored by a pharmaceutical company.
Acknowledgment
This study was sponsored by Myungmoon Pharm. Co., Ltd., Seoul, Republic of Korea, and was supported by grants from the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (HI14C2750, HI15C0001), as well as by the Industrial Core Technology Development Program (10051129, Development of the system for ADME assessment using radiolabeled compounds), funded by the Ministry of Trade, Industry & Energy (MOTIE, Korea).
Disclosure
At the time of the study, KC was employed by Biocore Co. Ltd. and YKS was employed by Myungmoon Pharm. Co., Ltd. The authors report no other conflicts of interest in this work.
References
1. Diasio RB. Clinical implications of dihydropyrimidine dehydrogenase inhibition. Oncology (Williston Park). 1999;13(Suppl):17–21.
2. Namikawa T, Maeda H, Kitagawa H, et al. Treatment using oxaliplatin and S-1 adjuvant chemotherapy for pathological stage III gastric cancer: a multicenter phase II study (TOSA trial) protocol. BMC Cancer. 2018;18(1):186. doi:10.1186/s12885-018-4242-8
3. Chhetri P, Giri A, Shakya S, Shakya S, Sapkota B, Pramod KC. Current development of anti-cancer drug S-1. J Clin Diagn Res. 2016;10(11):XE01–XE05. doi:10.7860/JCDR/2016/19345.8776
4. Sakuramoto S, Sasako M, Yamaguchi T, et al. Adjuvant chemotherapy for gastric cancer with S-1, an oral fluoropyrimidine. N Engl J Med. 2007;357(18):1810–1820. doi:10.1056/NEJMoa072252
5. Yoon DH, Kim SB. S-1 plus cisplatin: another option in the treatment of advanced head and neck cancer? Expert Rev Anticancer Ther. 2010;10(5):659–662. doi:10.1586/era.10.45
6. Fujii T, Horiguchi J, Yanagita Y, et al. Phase II study of S-1 plus trastuzumab for HER2-positive metastatic breast cancer (GBCCSG-01). Anticancer Res. 2018;38(2):905–909. doi:10.21873/anticanres.12301
7. Li J, Xu R, Xu J, et al. Phase II study of S-1 plus leucovorin in patients with metastatic colorectal cancer: regimen of 1 week on, 1 week off. Cancer Sci. 2017;108(10):2045–2051. doi:10.1111/cas.13335
8. Zhu X, Ju X, Cao F, et al. Safety and efficacy of stereotactic body radiation therapy combined with S-1 simultaneously followed by sequential S-1 as an initial treatment for locally advanced pancreatic cancer (SILAPANC) trial: study design and rationale of a phase II clinical trial. BMC Open. 2016;6(12):e013220. doi:10.1136/bmjopen-2016-013220
9. Sasako M, Sakuramoto S, Katai H, et al. Five-year outcomes of a randomized phase III trial comparing adjuvant chemotherapy with S-1 versus surgery alone in stage II or III gastric cancer. J Clin Oncol. 2011;29(33):4387–4393. doi:10.1200/JCO.2011.36.5908
10. Jeong JH, Park SR, Ahn Y, et al. Associations between CYP2A6 polymorphisms and outcomes of adjuvant S-1 chemotherapy in patients with curatively resected gastric cancer. Gastric Cancer. 2017;20(1):146–155. doi:10.1007/s10120-015-0586-9
11. Tanner JA, Tyndale RF. Variation in CYP2A6 activity and personalized medicine. J Pers Med. 2017;7(4):E18. doi:10.3390/jpm7040018
12. Kim MJ, Kong SY, Nam BH, Kim S, Park YI, Park SR. A randomized phase II study of S-1 versus capecitabine as first-line chemotherapy in elderly metastatic gastric cancer patients with or without poor performance status: clnical and pharmacogenetics results. Pharmacogenet Genomics. 2018;28(1):23–30. doi:10.1097/FPC.0000000000000320
13. Meulendijks D, Henricks LM, van Kuilenburg AB, et al. Patients homozygous for DPYD c.1129-5923C>G/haplotype B3 have partial DPD deficiency and require a dose reduction when treated with fluoropyrimidines. Cancer Chemother Pharmacol. 2016;78(4):875–880. doi:10.1007/s00280-016-3137-0
14. Amstutz U, Henricks LM, Offer SM, et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) guideline for dihydropyrimidine dehydrogenase genotype and fluoropyrimidine dosing: 2017 update. Clin Pharmacol Ther. 2018;103(2):210–216. doi:10.1002/cpt.911
15. Ueno H, Okusaka T, Ikeda M, Takezako Y, Morizane C. Phase II study of S-1 in patients with advanced biliary tract cancer. Br J Cancer. 2004;91(10):1769–1774. doi:10.1038/sj.bjc.6602208
16. Hirata K, Horikoshi N, Aiba K, et al. Pharmacokinetic study of S-1, a novel oral fluorouracil antitumor drug. Clin Cancer Res. 1999;5(8):2000–2005.
17. Chu QS, Hammond LA, Schwartz G, et al. Phase I and pharmacokinetic study of the oral fluoropyrimidine S-1 on a once-daily-for-28-day schedule in patients with advanced malignancies. Clin Cancer Res. 2004;10(15):4913–4921. doi:10.1158/1078-0432.CCR-04-0469
18. Furuse J, Okusaka T, Kaneko S, et al. Phase I/II study of the pharmacokinetics, safety, and efficacy of S-1 in patients with advanced hepatocellular carcinoma. Cancer Sci. 2010;101(12):2606–2611. doi:10.1111/j.1349-7006.2010.01730.x
19. Shirasaka T, Shimamoto Y, Fukushima M. Inhibition of oxonic acid of gastrointestinal toxicity of 5-fluorouracil without loss of its antitumor activity in rats. Cancer Res. 1993;53(17):4004–4009.
20. Liu K, Zhong D, Zou H, Chen X. Determination of tegafur, 5-fluorouracil, gimeracil and oxonic acid in human plasma using liquid chromatography-tandem mass spectrometry. J Pharm Biomed Anal. 2010;52(4):550–556. doi:10.1016/j.jpba.2010.01.026
21. Korea Ministry of Food and Drug Safety (MFDS). [Guideline on Bioequivalence Studies for Orally Administered Drug Products (No. 2017-28)]. Korean. Available from: https://www.mfds.go.kr/brd/m_211/view.do?seq=11835.
22. TS-ONE®. Capsule 20 [package Insert]. Tokyo, Japan: Taiho Pharmaceutical Co. Ltd.; 2017.
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.