")
Back to Journals » Drug Design, Development and Therapy » Volume 14
Pexidartinib, a Novel Small Molecule CSF-1R Inhibitor in Use for Tenosynovial Giant Cell Tumor: A Systematic Review of Pre-Clinical and Clinical Development
Authors Benner B, Good L , Quiroga D , Schultz TE, Kassem M , Carson WE, Cherian MA , Sardesai S , Wesolowski R
Received 10 March 2020
Accepted for publication 15 April 2020
Published 4 May 2020 Volume 2020:14 Pages 1693—1704
DOI https://doi.org/10.2147/DDDT.S253232
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Georgios Panos
Brooke Benner,1,* Logan Good,1,* Dionisia Quiroga,2 Thomas E Schultz,3 Mahmoud Kassem,2 William E Carson,1 Mathew A Cherian,2 Sagar Sardesai,2 Robert Wesolowski2
1Department of Surgery, The Ohio State University Comprehensive Cancer Center, Columbus, OH, USA; 2Division of Medical Oncology, The Ohio State University Comprehensive Cancer Center, Columbus, OH, USA; 3Department of Pharmacy, The Ohio State University Comprehensive Cancer Center, Columbus, OH, USA
*These authors contributed equally to this work
Correspondence: Robert Wesolowski
Division of Medical Oncology, The Ohio State University Comprehensive Cancer Center, 1800 Cannon Drive, 1250 Lincoln Tower, Columbus, OH 43210 Tel +1 614 366 5125
Fax +1 614 293 7264
Email [email protected]
Abstract: Tenosynovial giant cell tumor (TGCT) is a rare benign tumor that involves the synovium, bursa, and tendon sheath, resulting in reduced mobility of the affected joint or limb. The current standard of care for TGCT is surgical resection. However, some patients have tumor recurrence, present with unresectable tumors, or have tumors that are in locations where resection could result in amputations or significant debility. Therefore, the development of systemic agents with activity against TGCT to expand treatment options is a highly unmet medical need. Pathologically, TGCT is characterized by the overexpression of colony-stimulating factor 1 (CSF-1), which leads to the recruitment of colony-stimulating factor-1 receptor (CSF-1R) expressing macrophages that make up the primary cell type within these giant cell tumors. The binding of CSF-1 and CSF-1R controls cell survival and proliferation of monocytes and the switch from a monocytic to macrophage phenotype contributing to the growth and inflammation within these tumors. Therefore, molecules that target CSF-1/CSF-1R have emerged as potential systemic agents for the treatment of TGCT. Given the role of macrophages in regulating tumorigenesis, CSF1/CSF1R-targeting agents have emerged as attractive therapeutic targets for solid tumors. Pexidartinib is an orally bioavailable and potent inhibitor of CSF-1R which is one of the most clinically used agents. In this review, we discuss the biology of TGCT and review the pre-clinical and clinical development of pexidartinib which ultimately led to the FDA approval of this agent for the treatment of TGCT as well as ongoing clinical studies utilizing pexidartinib in the setting of cancer.
Keywords: pexidartinib, colony-stimulating factor 1 receptor, tenosynovial giant cell tumors, pigmented villonodular synovitis
Introduction
Tenosynovial Giant Cell Tumor
Tenosynovial giant cell tumor (TGCT), also referred to as giant cell tumor of the tendon sheath (GCT-TS), is a rare, commonly non-malignant tumor that typically affects small and large joints.1 These are locally aggressive mesenchymal neoplasms arising primarily in the synovium-lined joints, bursa, or tendon sheaths. A majority of cases are classified by their growth pattern as nodular or localized.2,3 A minority of cases are malignant and only 10% of cases are classified as diffuse (known as pigmented villonodular synovitis, PVNS).1–3 These tumors can also be characterized by their site (intra- or extra-articular). TGCT primarily affects people between the ages of 20–50.14 The presentation of TGCT can vary but affected patients frequently present with a painless joint swelling or with a firm, slow-growing joint mass. The most common primary sites of TGCT are the digits of the hands and the knees.3,4 Limited range of motion, pain, hemorrhagic joint effusions, and cartilage destruction occurs in more advanced disease and can result in significant morbidity and loss of functionality.2,3
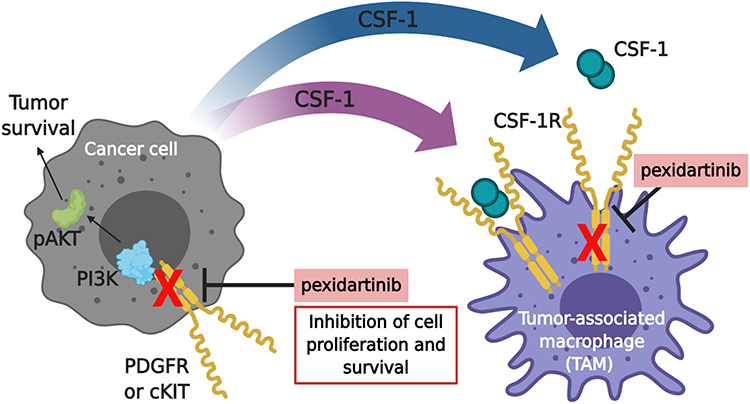 |
Figure 1 Pexidartinib mechanism of actiona. The proliferation and survival of cancer cells and macrophages are regulated by colony-stimulating factor-1 (CSF-1). Pexidartinib is a selective CSF-1R inhibitor that stimulates the auto-inhibited state of CSF-1R by interacting with the juxtamembrane region of CSF-1R, responsible for folding and inactivating the kinase domain, and prevents the binding of CSF-1 and ATP to the region. Without the binding of CSF-1 to the receptor, CSF1-R cannot undergo ligand-induced auto-phosphorylation. Thus, inhibiting the CSF-1/CSF-1R pathway, resulting in the inhibition of tumor cell proliferation and other cells populations such as macrophages. Pexidartinib has also been shown to inhibit cKIT, FMS-like tyrosine kinase 3 (FLT3), and platelet-derived growth factor receptor (PDGFR)-β, all receptor tyrosine kinases that regulate cellular processes such as cell proliferation and survival. This illustration was created with BioRender.com. aData from Plexxikon Inc.45 Abbreviations: PI3K, phosphatidylinositol 3-kinase; pAKT, phospho-protein kinase B. |
The current standard of care for localized TGCT is surgical resection.5 Radiotherapy is only seldom used because of late sequela and most often used in TGCT in large joints such as the knee.6 In patients with diffuse disease, surgery is often not feasible.2 Many patients with diffuse disease require repeat surgeries due to disease recurrence resulting in increased morbidity, including pain and reduced function of the affected joints sometimes requiring joint replacement or even amputation.2 Moreover, conventional cytotoxic chemotherapy has shown limited clinical benefit. Given the number of unsuccessful treatment options, novel, effective systemic therapies for TGCT are needed.
TGCT cells are characterized by the overexpression of colony-stimulating factor 1 (CSF-1) and in most cases, CSF-1 overexpression is driven by a chromosomal translocation of chromosome 1p13 to chromosome 2q35. This leads to the fusion of the coding region of CSF-1 to that of collagen 6A3, resulting in constitutive expression of CSF-1 under the control of collagen 6A3 regulatory elements.5,7 In a study by Cupp et al, the frequency of TGCT patients with the CSF-1 translocation was 61%.8 However, cells overexpressing CSF-1 only make up a small portion of cells found in the tumor (2–16%). Yet, the overproduction of CSF-1 recruits cells expressing colony-stimulating factor 1 receptor (CSF-1R), such as monocytes and macrophages, which make up the bulk of a TGCT. CSF-1 binding to CSF-1R likely accounts for the inflammatory changes and disease burden associated with these tumors, highlighting the CSF-1/CSF-1R pathway as an attractive therapeutic strategy for TGCT.3,7 CSF-1R signaling has been well described in the setting of TGCT. Herein, we provide a comprehensive review of the role of CSF-1R signaling in TGCT as well as other solid tumors.
CSF-1R Signaling
CSF-1 is known to stimulate the formation of colonies of macrophages by binding to its receptor CSF-1R. CSF-1R belongs to the platelet-derived growth factor (PDGF) receptor family. It possesses a highly glycosylated extracellular region comprised of five immunoglobulin domains, a transmembrane domain, and an intracellular domain. In turn, the intracellular domain is comprised of a juxtamembrane domain (JMD) and an intracellular tyrosine kinase domain that is interrupted by a kinase insert domain.9 Binding of CSF-1R to its ligands, CSF-1 or interleukin 34 (IL-34), induces homo-dimerization of CSF-1R and subsequently activates receptor signaling and tyrosine phosphorylation of CSF-1R.10 This results in the activation of downstream signaling pathways including the phosphatidylinositol 3-kinase/phosphorylated AKT (PI3K/pAKT) pathway which regulates cell survival (Figure 1).9 The differentiation, proliferation, and survival of several cell types is dependent on CSF-1R-mediated signaling. The activation of CSF-1R directly induces a monocytic cell fate in hematopoietic stem cells. Furthermore, it also instructs monocytes to differentiate into macrophages.9 Macrophage proliferation is associated with CSF-1R-mediated activation of the mitogen-activated protein kinase (MEK) kinase and PI3K pathways.11 Finally, CSF-1 is thought to promote the survival of macrophages by stimulating glucose uptake via glucose transporter 1 (Glut1) as well as through activation of the PI3K and phospholipase C (PLC) signaling pathways.11
CSF-1R is primarily expressed on the surface of circulating monocytes and macrophages (the result of the migration of monocytes to tissues and differentiation to resident immune cells), but can also be found on other cell types such as neutrophils, dendritic cells, myeloid-derived suppressor cells (MDSC), and tumor cells.9,12 Macrophages are known for their ability to be highly plastic and respond to cues from their stromal environment such as tissue necrosis, low oxygen levels, and high concentrations of lactate and pyruvate.10 Broadly, macrophages are categorized as being pro-inflammatory or anti-inflammatory. The pro-inflammatory macrophages are also termed classically activated or M1 macrophages, while anti-inflammatory are often referred to as alternatively activated or M2 macrophages. Tumor-associated macrophages (TAMs) are considered to be M2-like and have been reported to contribute to tumor growth, angiogenesis, metastasis, and resistance to cancer therapies.13,14 This phenotype is the result of cytokines present in the stromal environment, including CSF-1, IL-4, IL-13, and IL-10.10 Furthermore, CSF-1R signaling is involved in various diseases. Inappropriate expression of CSF-1R can lead to the development of certain leukemias and lymphomas, while autocrine and paracrine regulation of the CSF-1R has been linked to the progression and metastasis of solid tumors.15 Moreover, CSF-1R can also contribute to chronic inflammatory diseases, such as systemic lupus erythematosus, arthritis, atherosclerosis, and obesity.16,17 While there are multiple functions of CSF-1 signaling, this review will focus on CSF-1R-mediated signaling in monocytes and macrophages.
CSF-1R Inhibitors
Targeting the CSF-1/CSF-1R pathway has emerged as an attractive therapeutic strategy in cancer and other diseases. Of the components involved in the CSF-1/CSF-1R pathway, only molecules that directly target CSF-1 or CSF-1R are in clinical development.2 To the best of our knowledge, there are no strategies to target IL-34 (the second known ligand for CSF-1R). A variety of small molecules and monoclonal antibodies (mAbs) targeting CSF-1R or its ligand CSF-1 are currently in clinical development as monotherapy or in combination with other tumor-targeting agents such as chemotherapy or immunotherapy. A small-molecule CSF-1R inhibitor, pexidartinib (PLX3397), is the highlighted CSF-1R inhibitor in this review and casts the broadest net of clinical studies using monotherapy CSF-1R inhibition in the setting of c-KIT-mutated melanoma, prostate cancer, glioblastoma, classical Hodgkin lymphoma, neurofibroma, sarcoma, and leukemia.33 In addition to pexidartinib, other small molecule CSF-1R inhibitors include ARRY-382 (Array BioPharma), PLX7486 (Plexxikon), BLZ945 (Novartis), and JNJ-40346527 (Johnson& Johnson). Current anti-CSF-1R mAbs include emactuzumab (Roche), AMG820 (Amgen), IMC-CS4 (also referred to as LY3022855; Eli Lilly), cabiralizumab (Five Prime Therapeutics), MCS110 (Novartis; CSF-1), and PD-0360324 (Pfizer, CSF-1).10
Pexidartinib
Pexidartinib or PLX3397 was first discovered by Plexxikon Inc., the small molecule structure-guided research and development center of Daiichi Sankyo. Pexidartinib (brand name Turalio) is an orally bioavailable small-molecule tyrosine kinase inhibitor that inhibits CSF-1R and gained Food and Drug Administration (FDA) approval for the treatment of TGCT in August 2019 (Figure 2). Pexidartinib was designed to stabilize CSF-1R in the auto-inhibited state by interacting with the CSF-1R juxtamembrane region, resulting in inactivation of the kinase domain and prevention of CSF-1 and adenosine triphosphate (ATP) binding (Figure 2). Pexidartinib effectively inhibits CSF-1R at a half-maximal inhibitory concentration (IC50) of 17 nanomolar (nM) and is also able to inhibit the proto-oncogene c-KIT, (IC50 12 nM) and FMS-like tyrosine kinase 3- internal tandem duplication (FLT3-ITD) (IC50 9 nM).18
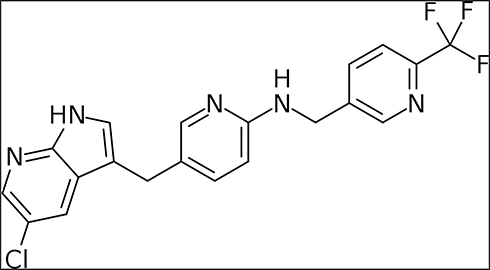 |
Figure 2 Chemical structure of pexidartinib (Turalio). |
The overexpression of CSF-1 by TGCT and the importance of CSF-1R signaling in the pathogenesis of this disease was one of the reasons for the development of pexidartinib and other CSF-1R inhibitors. Pexidartinib has also been shown to slow tumor growth through a decrease in immune suppression and angiogenesis resulting in a reduction in TAMs and an increase of intratumoral CD4+ and CD8+ T lymphocytes.20,21 These findings provide a rationale to study CSF-1R inhibition in multiple other cancers. Other than TGCT, pexidartinib has been studied pre-clinically and clinically in gastrointestinal stromal tumors, colorectal cancer, pancreatic cancer, melanoma, glioblastoma, and hematologic malignancies. These studies are reviewed in the following sections.
Preclinical Data
Previous preclinical data have demonstrated the ability of cytotoxic therapies such as chemotherapy and radiation to induce macrophage recruitment and furthermore increase CSF-1 expression within the tumor.19 These preclinical in vitro studies utilizing microvascular endothelial cells (MECs) from both murine mammary and human breast tumors found that exposure to paclitaxel, cisplatin, and radiation increased the levels of CSF-1 mRNA.19 Furthermore, in vivo data from mammary tumors of MMTV-PyMT mice had a higher expression of CSF-1 mRNA and increased density of macrophages in the tumor stroma following exposure to paclitaxel and other cytotoxic therapies. However, when MMTV-PyMT mice were treated with paclitaxel and pexidartinib the combination demonstrated a decrease in macrophage infiltration to the tumor, significant reduction in tumor growth, and lower amounts of pulmonary metastases compared to single-agent paclitaxel.19 Interestingly, mice treated with paclitaxel and pexidartinib also had an increase in intratumoral CD4+ T cells, CD8+ T cells, and dendritic cells with enhanced production of functional cytokines within MMTV-PyMT tumors. A reduction of the immunosuppressive factor Arginase1 was also found in mice treated with pexidartinib, suggesting that it can modulate the immune landscape within tumors.19
The preclinical safety of pexidartinib was evaluated in single and multiple-dose studies. In one toxicity study, Sprague-Dawley rats were treated with pexidartinib for 7 days at doses of 30, 100, and 300 mg/kg/day, which demonstrated modest toxicities including leukopenia, anemia, increased liver enzymes, hepatomegaly, bone marrow hematopoietic atrophy, and cystic corpora lutea (Investigator’s Brochure). In a subsequent 28-day study with a 14-day recovery period, Sprague-Dawley rats were dosed at 20, 60, and 200 mg/kg/day with exposure increased as the dosage was increased over the 20 to 200 mg/kg/day range. Hepatocellular hypertrophy was observed and correlated with higher liver enzyme levels, liver weights, and incidence and/or severity of chronic progressive nephropathy in a dose-dependent manner. However, these findings were also observed in the vehicle control groups and therefore are difficult to attribute to pexidartinib alone. Further animal toxicity studies were performed in dogs where they were dosed twice daily for 28 consecutive days at doses of 100 and 300 mg/kg/day. This study revealed toxicities of emesis, body weight loss, anorexia, and pathologic tissue changes in the testes, bone marrow, kidneys, spleen, and lymphoid depletion in the thymus for animals (Investigator’s Brochure).
Finally, preclinical animal studies have demonstrated an anti-tumor effect of pexidartinib. The ability of pexidartinib to decrease levels of tumor-infiltrating myeloid cells (TIMs) and inhibit tumor growth was described in a mouse model of prostate cancer.20 In this study, RM-1 prostate tumor-bearing mice were treated with control chow, local radiation (3Gy×5 days), pexidartinib chow, or both radiation and pexidartinib. Their results demonstrated that pexidartinib alone had little effect on tumor growth compared with the control group. However, radiation alone reduced tumor size by 43% at day 10.21 Tumors were stabilized for a short duration but resumed an aggressive growth rate shortly after the treatment had been completed. Interestingly, the combined radiation and pexidartinib treated group maintained a much slower growth rate. Furthermore, flow cytometry and histological analyses of the tumors revealed a significant reduction of MDSCs and macrophages in tumors and spleens in both single-agent pexidartinib and pexidartinib plus radiation.20 In a similar study done in a glioblastoma (GBM) mouse model pexidartinib was combined with ionizing radiation (IR).21 This study found that GBM xenografts treated with IR upregulated the CSF-1R ligand expression and increased the number of myeloid cells in the tumors. However, treatment with pexidartinib depleted myeloid cells and potentiated the response of the intracranial tumors to IR. Additionally, the median survival was significantly longer in mice that received pexidartinib in combination with IR compared to IR alone. Pexidartinib was also found to prevent IR-recruited monocyte cells from differentiating into immunosuppressive, pro-angiogenic tumor-associated macrophages.21 Moreover, DeNardo et al demonstrated a decrease in macrophage infiltration to the mammary tumors of MMTV-PyMT mice receiving paclitaxel in combination with pexidartinib, and a significant reduction in tumor growth and pulmonary metastases was observed compared to mice receiving paclitaxel alone.19
Other preclinical studies have found that pexidartinib can reduce cancer-related pain,22,23 maintain normal bone volumes in bone cancer,23 abrogate therapeutic resistance,24 and enhance anti-PD-L1 blockade.25 In a study using canine ACE-1 prostate cancer cells to induce osteoblastic bone metastases in athymic nude mice, sustained administration of pexidartinib had a significant effect on attenuating spontaneous bone metastases.26–28 The pain level was measured by monitoring nociceptive behaviors such as flinching and guarding of the tumor-bearing limb.23 Furthermore, Thompson et al found that pexidartinib attenuated skeletal pain, disease progression, and tumor-induced bone remodeling by nearly 50%.23 Resistance to therapy is often attributed to the infiltration of immunosuppressive myeloid cells in the tumor microenvironment. Interestingly, in a preclinical study by Yan et al they found that pexidartinib restored the sensitivity of glioma cells in a PDGF-B-driven proneural glioma mouse model to tyrosine kinase inhibitors by interfering with the tumor-mediated education of macrophages.24 Additionally, in a hepatocellular carcinoma (cell lines MHCC97H and HCC-LM) immunocompetent C57BL/6 mouse model, Zhu et al demonstrated that combining pexidartinib with a PD-L1 inhibitor prolonged survival in tumor-bearing mice, increased CD8+ T cell infiltration, and decreased TAM infiltration by histological and flow cytometric analysis.25
Clinical Trials with Pexidartinib
Phase I Trials
The use of pexidartinib first began in a phase I dose-escalation trial examining the safety, pharmacokinetics (PK), and pharmacodynamics (PD) of single-agent pexidartinib in patients with solid tumors (NCT01004861). A total of 41 patients were enrolled in the standard 3+3 study, each receiving single-agent pexidartinib administered by daily oral dosing in four-week cycles.11 The mean duration of treatment for patients in the study was 70.7 days. Pexidartinib was generally tolerated, and the number of adverse events (AEs) among the 41 patients enrolled in this study included three patients (7%) achieving a maximum of grade 1, eighteen (44%) with a maximum of grade 2, sixteen (39%) with a maximum of grade 3, and three (7%) with a maximum of grade 4 events.11 AEs of grade 3 or higher that occurred in more than one patient included anemia, increased aspartate aminotransferase level, and lymphopenia.11 A total of 35 patients completed at least one cycle of treatment, with eight patients (23%) experiencing stable disease and one patient (3%) experiencing a partial response per tumor volume score (TVS) efficacy assessment. Overall, pexidartinib was well tolerated and the maximum tolerated dose (MTD) was determined to be 1000 mg per day.11 This phase I dose-escalation study led to further trials utilizing single-agent pexidartinib, including studies examining Asian subjects with advanced solid tumors (NCT02734433),29 patients with GIST in the setting of moderate hepatic impairment (NCT04223635), unresectable or metastatic KIT-mutated melanoma (NCT02975700), and relapsed or refractory FLT-3-ITD-positive acute myeloid leukemia (NCT01349049). Additionally, a phase I trial in children with refractory leukemias and refractory solid tumors (NCT02390752) was established in order to determine the MTD and PK of pexidartinib in patients aged 3–31. Initially, 14 patients were analyzed and common AEs included fatigue, leukopenia, increase in creatinine kinase and serum amylase, headache, anorexia, vomiting, diarrhea, and hair hypopigmentation.30 It was concluded that in children, pexidartinib was tolerated at all dose levels and the recommended Phase 2 dose (RP2D) was found to be 800 mg/m2 once daily.
In addition to the first-in-human phase I dose-escalation trial examining safety, pharmacokinetics, and preliminary anti-tumor activity (NCT01004861), a subsequent extension study was performed in which 23 patients with recurrent, inoperable, or difficult to resect TGCT were treated with the maximum tolerated total daily dose (1000mg) of pexidartinib. The median age was 46 years and 18 (78%) of the patients had previous surgeries. An overall response rate of 52% was reported, with seven patients that experienced stable disease, conferring an 83% rate of disease control with pexidartinib.1 Eleven of the fourteen patients who had measurable tumor volume had at least a 50% reduction in tumor volume.1 Median progression-free survival was not reached. Four patients had previous treatment with imatinib or nilotinib (agents also used to treat TGCT although with modest efficacy) and showed responses to pexidartinib with tumor volume reductions of 40–55%.1 Common AEs observed included fatigue (78%), nausea (65%), arthralgia (39%) and periorbital edema (26%).1 Hair depigmentation (74%) was also observed in patients, indicating potential inhibition of the c-KIT signaling pathway.2,31 AEs of grade 3 or higher included increased aspartate aminotransferase or alanine aminotransferase levels or both (9%), hyponatremia (9%), fatigue (4%), diarrhea (4%), anemia (4%), and neutropenia (4%).1 Overall, 50% of patients had abnormal liver function tests with 13% grade 3 or greater. All cases resolved or decreased to grade 1 with cessation of pexidartinib.1 This study resulted in the FDA granting Breakthrough Therapy Designation for pexidartinib in the treatment of TGCT.
Phase I Combination Trials
Prior phase I trials using single-agent pexidartinib led to combination studies such as the phase Ib study analyzing the combination of pexidartinib and paclitaxel in patients with advanced solid tumors (NCT01525602).32 In part 1 of this study, 24 patients received escalating doses of pexidartinib (starting at 600 mg/day) with weekly paclitaxel (80 mg/m2). The MTD was not reached.20 The recommended Phase II dose of pexidartinib in combination with the standard dose and schedule of paclitaxel was established at 1600 mg daily based on pharmacodynamic and pharmacokinetic data. In part 2 of the study, 30 patients with metastatic solid tumors were enrolled to examine the safety, efficacy, and tolerability of the RP2D.20 A total of 51 patients recorded at least 1 AE, most commonly fatigue (65%), anemia (59%), diarrhea (39%), and nausea (39%). Additionally, 38 patients (70%) reported grade 3–4 AEs including anemia (26%), neutropenia (22%), lymphopenia (19%), fatigue (15%), and hypertension (11%).20 Grade 3 aspartate aminotransferase (AST) and alanine aminotransferase (ALT) elevations were relatively low at 7% and 2% of patients. Overall, 38 patients were evaluable for efficacy, including one patient (3%) with peritoneal carcinoma who had a complete response, five patients (13%) who experienced a partial response, thirteen patients (34%) who experienced stable disease, and seventeen patients (45%) who had progressive disease.20 Additionally, patient blood CD14dim/CD16+ monocyte levels decreased 57–100% after 2 weeks of pexidartinib treatment.20 This specific population of monocytes represents the non-classical sub-type of monocytes and is known to play a key role in pro-inflammatory processes by production of TNF-alpha.33 Furthermore, plasma CSF-1 levels increased 1.6- to 53-fold.20 These pharmacodynamic results indicated strong target inhibition of CSF-1R. The increase in CSF-1 levels could be due to the compensatory increase in production or decreased receptor-mediated endocytosis due to competitive binding by pexidartinib. Wesolowski et al concluded that the combination of pexidartinib and paclitaxel was well tolerated, additionally reporting the RP2D for pexidartinib as 1600 mg/day in combination with paclitaxel.32
A phase I trial of binimetinib, a MEK inhibitor, in combination with pexidartinib, was also conducted in patients with gastrointestinal stromal tumors (GIST) (NCT03158103). The MEK cascade is an intracellular signaling pathway that regulates cellular proliferation and the survival of tumor cells; the inhibition of this pathway in combination with other immunotherapeutic interventions has shown considerable promise.34 The purpose of the study was to test the safety and tolerability of the combination of pexidartinib and binimetinib examining different dosage concentrations for each drug.35 The investigators concluded that this combination was safe with only grade 3 creatinine phosphokinase reported. At the time of publication, one patient remained on treatment for at least 19 months and experienced a 27% decrease in tumor size. The other patient had 5 prior lines of treatment, had stable disease for 6.1 months, and survived for 14.6 months after starting treatment.35 Though small, this trial demonstrated that pexidartinib can be used in other disease states and that combination therapy with other kinase inhibitors may help overcome resistance mechanisms. Other combination studies using pexidartinib include a phase I trial examining the treatment of metastatic/advanced pancreatic or colorectal cancers with pexidartinib and the PD-L1 inhibitor durvalumab (NCT02777710),36 a phase I trial testing pexidartinib in combination with the BRAFV600E inhibitor vemurafenib in individuals with metastatic or unresectable melanoma (NCT01826448), and a phase I trial examining the effect of radiation and anti-hormone therapy in combination with pexidartinib on prostate adenocarcinoma (NCT2472275).
Phase I/II Trials
Additional single-agent pexidartinib studies include phase I trials with phase II extensions, such as the previously mentioned studies for unresectable or metastatic KIT-mutated melanoma (NCT02975700), refractory leukemias and solid tumors (NCT02390752), and relapsed/refractory FLT3-ITD-positive acute myeloid leukemia (NCT01349049). An additional phase I/II trial examined the safety and efficacy of three dose concentrations of pexidartinib in patients with relapsed/refractory acute myeloid leukemia (AML), which was recently completed (NCT01349049). This open-label, sequential dose escalation (part 1) was followed by a cohort expansion design (part 2) at the recommended phase II dose that was established in part 1. A total of 90 patients underwent treatment, receiving daily oral pexidartinib at either 800 mg, 1000 mg, 1200 mg, 1400 mg, 2000 mg, 3000 mg, 4000 mg, or 5000 mg. In part 2 of the study, patients receiving 3000 mg of pexidartinib daily (RP2D) had a median disease-free survival rate and overall survival rate of 289 days and 112 days, respectively. The most common AEs across all treatment groups included febrile neutropenia, anorexia, and fatigue.
Pexidartinib has also been used in several combination studies, including a phase I/II trial for advanced melanoma and other solid tumors in combination with the PD-1 inhibitor pembrolizumab (NCT02452424),37 a phase I/II trial treating newly diagnosed glioblastoma using pexidartinib in combination with radiation therapy and temozolomide (NCT01790503),38 a phase I/II study testing pexidartinib in combination with eribulin for metastatic breast carcinoma (NCT01596751), a phase I/II trial assessing the ability of pexidartinib and sirolimus to inhibit the growth of unresectable synovial sarcoma (NCT02584647), and a phase I/II trial studying the treatment of advanced gastrointestinal stromal tumors with pexidartinib and PLX9486, an inhibitor of the transmembrane protein and receptor tyrosine kinase c-KIT (NCT02401815).
Phase II Trials
There are many phase II trials testing the efficacy of pexidartinib that are ongoing or have been recently completed. One study examined KIT-mutated advanced acral and mucosal melanoma (NCT02071940), with results pending. Another study was recently completed for patients with relapsed or refractory Hodgkin’s lymphoma (NCT01217229).39 In this study, 20 patients received pexidartinib once or twice daily at 900 mg/day. However, the overall response rate (ORR) was only 5%. AEs included hair depigmentation (40%), fatigue (40%), anemia (30%), thrombocytopenia (30%), dyspnea (25%), rash (25%), and/or lung infection (5%).
Based on preclinical data showing that pexidartinib can delay the onset of castrate-resistance prostate cancer (CRPC) by reducing the number of infiltrating TAM, a phase II trial for bone-metastatic CRPC patients has been initiated but was terminated early after enrolling 6 patients. (NCT01499043).40,41 Serious grade 3 AEs included influenza (16.7%), pneumonia (16.7%), fall (16.7%), pain in extremity (16.7%), and/or urinary retention (16.7%). Furthermore, results from a Phase II, open-label study of 1000 mg daily oral dosing of pexidartinib in subjects with recurrent glioblastoma (NCT01349036) have been reported. A total of 37 patients were enrolled, 13 of whom were treated with surgical intervention in cohort 1 and 24 patients of whom were treated without surgery in cohort 2. Cohort 1 was treated with pexidartinib for 7 days prior to surgery, while cohort 2 was initiated only after the resected tumor tissue PK analysis from patients in cohort 1 demonstrated a pexidartinib concentration of ≥100 nM in at least 5 of 10 tumor tissue samples.42 This study showed no objective responses and a primary efficacy endpoint of 6-month progression-free survival (PFS6) of only 8.8%.43 It was therefore concluded that this immunotherapeutic regimen was well tolerated, but had limited clinical efficacy.32,33 Of note, the only phase II combination study currently evaluating the safety and efficacy of pexidartinib in combination with another agent is a trial with pexidartinib plus paclitaxel in breast carcinoma patients, which is currently recruiting (NCT01042379).
Phase III Trials
The ENLIVEN trial is a completed Phase III, double-blind, randomized, placebo-controlled, registration trial performed at 39 locations in 12 countries in patients with TGCT (NCT02371369).2 Patients included in the trial were adult men and women with advanced TGCT in which surgery was not an option or would result in severe morbidity. All patients had symptomatic disease with joint stiffness and measurable disease of at least 2 cm. Patients were randomly assigned (1:1) to the pexidartinib or placebo group. Patients in the pexidartinib group received 400 mg pexidartinib in the morning and 600 mg in the evening for 2 weeks followed by 400 mg twice daily for 22 weeks.2 Part 2 of the study was an open-label study in which cross over was allowed for those on placebo after 24 weeks.12 The primary endpoint was overall response rate at week 25 with secondary endpoints of mean change from baseline in range of motion, response via tumor volume score (TVS), duration of response, and mean change in patient-reported outcomes of functionality, stiffness, and pain.
Of the 174 patients who were screened, 120 met eligibility criteria and were randomized resulting in 61 patients assigned to the pexidartinib group and 59 patients assigned to placebo. The median age was 44 for the pexidartinib group and 45 for the placebo group.12 Between treatment groups, 61% of patients had TGCT located in the knee, 53% had at least one previous surgery, and only 9% had previous tyrosine kinase inhibitor therapy.12 Group demographics were well balanced, however, the pexidartinib group had a slightly higher proportion of patients with a history of prior surgeries and prior therapy with nilotinib and imatinib.
For the primary endpoint, overall response per RECIST version 1.1 at 25 weeks was 39% in the pexidartinib group vs 0% in the placebo group (p < 0.0001). Per RECIST criteria, 15% of patients had a complete response and 24% had a partial response. Overall response achieved by TVS was 56% vs 0% respectively (p < 0.0001). Per TVS, 5% of pexidartinib treated patients had a complete response and 51% had a partial response. Most patients who achieved a complete or partial response maintained the response during the open-label part of the treatment and at data cutoff. All patients who responded at 25 weeks were still responding at 6-month follow-up (longest 17 months), and no patients had progressed. Treatment with pexidartinib resulted in significantly increased relative range of motion and physical function with a greater improvement in stiffness. There was a trend towards less pain in the pexidartinib cohort, however, this was not statistically significant. Of note, improvements in other secondary endpoints correlated with tumor response seen above.
Although pexidartinib was studied with a load dose followed by a maintenance dose, the FDA-approved regimen is a flat dose of 400 mg twice daily.18 This is due to the second part of the ENLIVEN study where patients who were previously on placebo started pexidartinib 400 mg twice daily instead of the aforementioned loading dose of 1000 mg/day. There were no observations of liver function test (LFT) or bilirubin elevations in this treatment dose group and thus it is expected that the risk of hepatotoxicity with 400 mg twice daily may be lower. There was a similar overall response rate at 53% by RECIST and 67% by TVS. All prior or ongoing clinical trials of pexidartinib are summarized in Table 1.
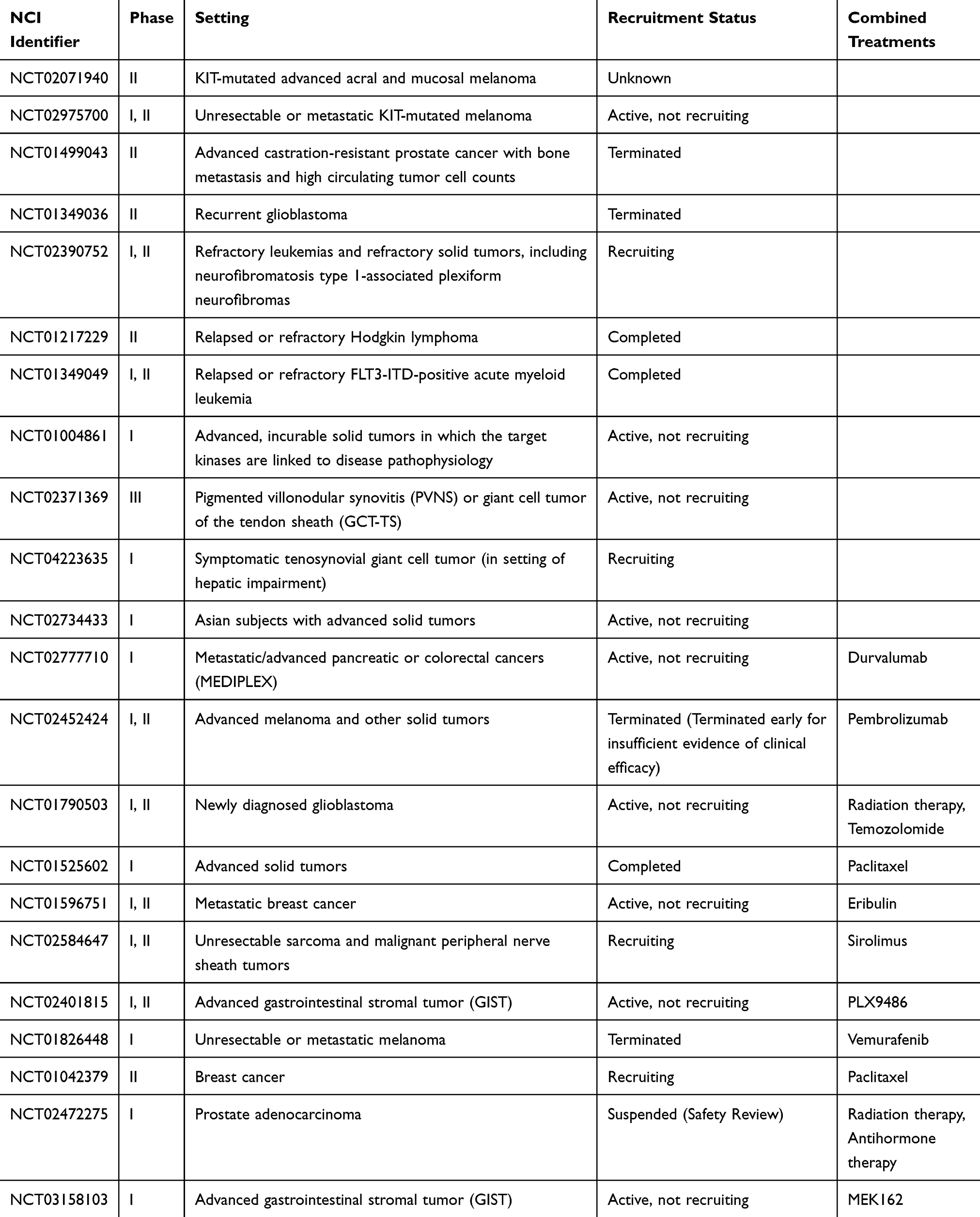 |
Table 1 Summary of Clinical Trials Using Pexidartinib |
Drug Toxicities
In the ENLIVEN trial, 23 of 61 patients (38%) in the pexidartinib group and 6 of 59 patients (10%) in the placebo group experienced a dose reduction or discontinued pexidartinib due to AEs.12 The most frequent pexidartinib-associated AEs included hair color changes (67%), fatigue (54%), increased aspartate aminotransferase (39%), nausea (38%), increased alanine aminotransferase (28%), and dysgeusia (25%).12 Other notable AEs included periorbital edema (18%) and peripheral edema (13%). The most common AEs seen in the placebo group were nausea (41%), fatigue (36%), diarrhea (25%), and arthralgia (25%).12 Side effects such as hair discoloration, periorbital edema, and peripheral edema have frequently been seen with other cKIT inhibitors and are most likely due to the effects of pexidartinib on cKIT.
The pexidartinib toxicity profile includes the potential for hepatic adverse events. The most common liver related AEs in the ENLIVEN trial included reversible AST and ALT elevations.2 These toxicities are likely the result of the inhibitory effects of pexidartinib on Kupffer cells, which are resident macrophages that are present in the sinusoids of the liver and express CSF-1R.32 Kupffer cells are involved in clearance of transaminases as well as other enzymes such as creatinine kinase and lactate dehydrogenase. Therefore, pexidartinib-mediated inhibition of Kupffer cells often results in increased levels of these enzymes. Of note, these elevations commonly return to baseline upon discontinuing pexidartinib.
In rare cases, pexidartinib can also cause more serious, potentially irreversible and/or fatal liver injury.18 Laboratory findings in affected patients typically show persistent elevations of total bilirubin and alkaline phosphatase (ALP) in the background of elevated transaminases. Liver biopsies performed in patients with pexidartinib-induced liver injury showed significant ductopenia and cholestasis.18 Overall, eight (13.11%) of the pexidartinib treated patients in the ENLIVEN trial experienced a serious AE that led to discontinuation, seven of which were liver-related.2 The seven liver-related serious AEs included four patients with increased liver enzymes (4.92%), one patient with hepatitis A and E infection (1.64%), one patient with an unspecified liver disorder (1.64%), and one patient due to hepatotoxicity (1.64%).2 Due to the risk of serious or potentially fatal liver injury, the FDA placed a black box warning for hepatotoxicity requiring frequent monitoring of liver function tests.18 AST, ALT, total bilirubin, direct bilirubin, ALP, and gamma-glutamyl transferase (GGT) should be monitored prior to initiation, weekly for 8 weeks, then every 2 weeks for a month, followed by every 3 months until discontinuation.18 Pexidartinib should be avoided in patients with preexisting liver or biliary tract diseases and in patients with elevated bilirubin or transaminases at baseline.18 Dose reductions for AEs and dose modifications for hepatotoxicity are summarized in Tables 2 and 3 respectively.
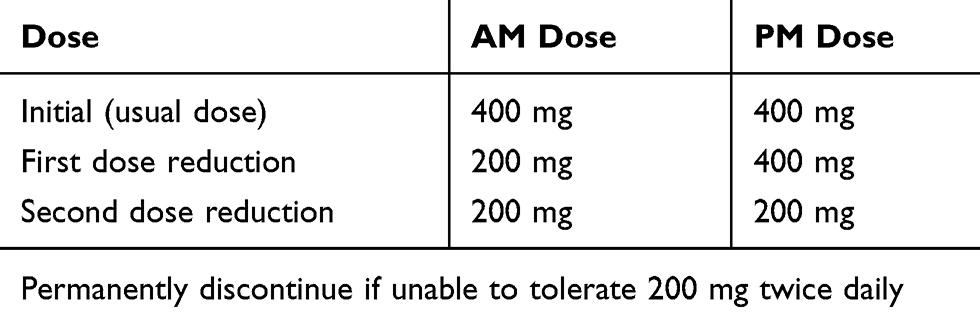 |
Table 2 Pexidartinib Dose Reduction for Adverse Events (AEs) |
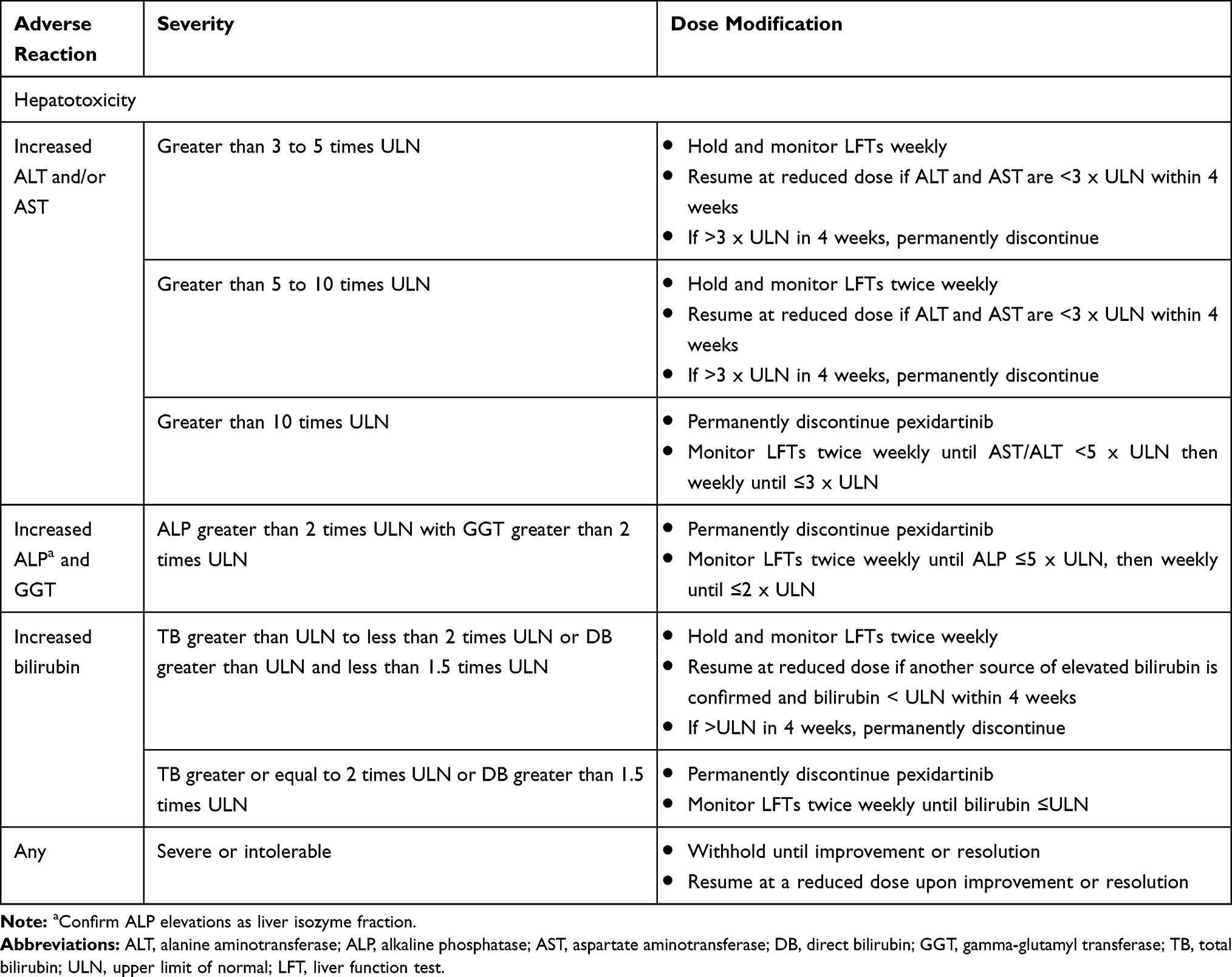 |
Table 3 Dose Modifications for Hepatotoxicity |
Conclusion and Future Perspective
Identifying novel targeted therapies to improve clinical outcomes are needed to provide new options to patients and improve their quality of life. Increased knowledge of CSF-1 overexpression and pathogenesis has led to a pipeline of multiple agents that inhibit CSF-1R in hopes of disease stabilization or tumor shrinkage. Pexidartinib is the first CSF-1R inhibitor to gain FDA approval, yet single-agent therapy has demonstrated very modest activity with the exception of its use in TGCT. Given the ability of pexidartinib to regulate the anti-tumor activity of M2 macrophages, combination studies with CSF-1R inhibitors have emerged as the next step to employ this agent to treat other solid tumors with preliminary data from preclinical studies guiding this development. To date, CSF-1R inhibition has been paired with chemotherapy, radiation therapy, and immunotherapy in various disease settings.10,20,42-44
In summary, pexidartinib is the first CSF-1R inhibitor to provide clinical benefit and offer an effective strategy to treat TGCT by improving patient symptoms and functional outcomes. Studies testing other inhibitors of CSF-1R are currently ongoing and hold the potential to treat a wide spectrum of other conditions.
Disclosure
Dr Robert Wesolowski reports non-financial support from Plexxikon, during the conduct of the study. The authors report no other conflicts of interest in this work.
References
1. Tap WD, Wainberg ZA, Anthony SP, et al. Structure-guided blockade of CSF1R kinase in tenosynovial giant-cell tumor. N Engl J Med. 2015;373:428–437. doi:10.1056/NEJMoa1411366
2. Tap WD, Gelderblom H, Palmerini E, et al. Pexidartinib versus placebo for advanced tenosynovial giant cell tumour (ENLIVEN): a randomized Phase 3 trial. Lancet. 2019;394:478–487. doi:10.1016/S0140-6736(19)30764-0
3. Giustini N, Bernthal NM, Bukata SV, Singh AS Tenosynovial giant cell tumor: case report of a patient effectively treated with pexidartinib (PLX3397) and review of the literature. Clin Sarcoma Res. 2018;8, 1–5.
4. Ehrenstein V, Andersen SL, Qazi I, et al. Tenosynovial giant cell tumor: incidence, prevalence, patient characteristics, and recurrence. A registry-based cohort study in Denmark. J Rheumatol. 2017;44(10):1476–1483. doi:10.3899/jrheum.160816
5. Granowitz S, D’Antonio J, Mankin H. The pathogenesis and long-term end results of pigmented villonodular synovitis. Clin Orthop Relat Res. 1976.
6. Griffin AM, Ferguson PC, Catton CN, et al. Long-term outcome of the treatment of high-risk tenosynovial giant cell tumor/pigmented villonodular synovitis with radiotherapy and surgery. Cancer. 2012;118:4901–4909. doi:10.1002/cncr.26529
7. West RB, et al. A Landscape Effect in Tenosynovial Giant-Cell Tumor from Activation of CSF1 Expression by a Translocation in a Minority of Tumor Cells. 2005.
8. Cupp J, Miller MA, Montgomery KD, et al. Translocation and expression of CSF1 in pigmented. Am J Surg Pathol. 2007;31:970–976.
9. Stanley ER, Chitu V. CSF-1 receptor signaling in myeloid cells. Cold Spring Harb Perspect Biol. 2014;6(6):a021857. doi:10.1101/cshperspect.a021857
10. Cannarile MA, Weisser M, Jacob W, et al. Colony-stimulating factor 1 receptor (CSF1R) inhibitors in cancer therapy. J Immunother Cancer. 2017;5:53. doi:10.1186/s40425-017-0257-y
11. Chang M, Hamilton JA, Scholz GM, et al. Phosphatidylinostitol-3 kinase and phospholipase C enhance CSF-1-dependent macrophage survival by controlling glucose uptake. Cell Signal. 2009;21(9):1361–1369. doi:10.1016/j.cellsig.2009.04.003
12. Stanley ER, Heard PM. Factors regulating macrophage production and growth. Purification and some properties of the colony stimulating factor from medium conditioned by mouse L cells. J Biol Chem. 1977;252(12):4305–4312.
13. Breton CS, Dhillon MK, Lim JY, et al. Molecular profiling reveals a tumor-promoting phenotype of monocytes and macrophages in human cancer progression. Immunity. 2014;41:33.
14. Ostuni R, Kratochvill F, Murray PJ, Natoli G. Macrophages and cancer: from mechanisms to therapeutic implications. Trends Immunol. 2015;36:229–239. doi:10.1016/j.it.2015.02.004
15. Pollard JW. Trophic macrophages in development and disease. Nat Rev Immunol. 2009;9(4):259–270. doi:10.1038/nri2528
16. Chitu V, Stanley ER. Colony-stimulating factor-1 in immunity and inflammation. Curr Opin Immunol. 2006;18(1):39–48. doi:10.1016/j.coi.2005.11.006
17. Chitu V, Nacu V, Charles JF, et al. PSTPIP2 deficiency in mice causes osteopenia and increased differentiation of multipotent myeloid precursors into osteoclasts. Blood. 2012;120:3126–3135. doi:10.1182/blood-2012-04-425595
18. Sankyo D. TURALIO (pexidartinib) prescribing information.
19. Denardo DG, Brennan DJ, Rexhepaj E, et al. Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer Discov. 2011;1:54–67. doi:10.1158/2159-8274.CD-10-0028.Leukocyte
20. Xu J, Escamilla J, Mok S, et al. CSF1R signaling blockade stanches tumor-infiltrating myeloid cells and improves the efficacy of radiotherapy. Cancer Res. 2013;73(9):2782–2794. doi:10.1158/0008-5472.CAN-12-3981
21. Stafford JH, Hirai T, Deng L, et al. Colony stimulating factor 1 receptor inhibition delays recurrence of glioblastoma after radiation by altering myeloid cell recruitment and polarization. Neuro Oncol. 2016;18(6):797–806. doi:10.1093/neuonc/nov272
22. Li M, Li Z, Ren H, et al. Colony stimulating factor 1 receptor inhibition eliminates microglia and attenuates brain injury after intracerebral hemorrhage. J Cereb Blood Flow Metab. 2017;37(7):2383–2395. doi:10.1177/0271678X16666551
23. Thompson M, Jimenez-Andrade JM, Chartier S, et al. Targeting cells of the myeloid lineage attenuates pain and disease progression in a prostate model of bone cancer. Pain. 2015;156(9):1692–1702. doi:10.1097/j.pain.0000000000000228
24. Yan D, Kowal J, Akkari L, et al. Inhibition of colony stimulating factor-1 receptor abrogates microenvironment-mediated therapeutic resistance in gliomas. Oncogene. 2017;36(43):6049–6058. doi:10.1038/onc.2017.261
25. Zhu Y, Yang J, Xu D, et al. Disruption of tumour-associated macrophage trafficking by the osteopontin-induced colony-stimulating factor-1 signalling sensitises hepatocellular carcinoma to anti-PD-L1 blockade. Gut. 2019;68(9):1653–1666. doi:10.1136/gutjnl-2019-318419
26. Thudi NK, Martin CK, Murahari S, et al. Dickkopf-1 (DKK-1) stimulated prostate cancer growth and metastasis and inhibited bone formation in osteoblastic bone metastases. Prostate. 2011;71(6):615–625. doi:10.1002/pros.21277
27. Thudi NK, Martin CK, Nadella MVP, et al. Zoledronic acid decreased osteolysis but not bone metastasis in a nude mouse model of canine prostate cancer with mixed bone lesions. Prostate. 2008;68(10):1116–1125. doi:10.1002/pros.20776
28. Halvorson KG, Kubota K, Sevcik MA, et al. A blocking antibody to nerve growth factor attenuates skeletal pain induced by prostate tumor cells growing in bone. Cancer Res. 2005;65(20):9426–9435. doi:10.1158/0008-5472.CAN-05-0826
29. Lee JH, Chen TW, Hsu CH, et al. A phase I study of pexidartinib, a colony-stimulating factor 1 receptor inhibitor, in Asian patients with advanced solid tumors. Invest New Drugs. 2019:99–110. doi:10.1007/s10637-019-00745-z.
30. Hittson L, Glod J, Amaya M, et al. Phase I study of pexidartinib (PLX3397) in children with refractory leukemias and solid tumors including neurofibromatosis type I (NF1) related plexiform neurofibromas (PN). J Clin Oncol. 2017;35(15_suppl):10546. doi:10.1200/JCO.2017.35.15_suppl.10546
31. Ricci F, De Simone C, Del Regno L, Peris K. Drug-induced hair colour changes. Eur J Dermatology. 2016;26(6):531–536. doi:10.1684/ejd.2016.2844
32. Wesolowski R, Sharma N, Reebel L, et al. Phase Ib study of the combination of pexidartinib (PLX3397), a CSF-1R inhibitor, and paclitaxel in patients with advanced solid tumors. Ther Adv Med Oncol. 2019;11:175883591985423. doi:10.1177/1758835919854238
33. Skrzeczyńska-Moncznik J, Bzowska M, Lo˝seke S, et al. Peripheral blood CD14 high CD16 + monocytes are main producers of IL-10. Scand J Immunol. 2008;67(2):152–159. doi:10.1111/j.1365-3083.2007.02051.x
34. Grimaldi A, Simeone E, Festino L, et al. MEK inhibitors in the treatment of metastatic melanoma and solid tumors. Am. J. Clin. Dermatol. 2017;18(6):745–754. doi:10.1007/s40257-017-0292-y
35. Rosenbaum E, Kelly C, D’Angelo SP, et al. A Phase I study of binimetinib (MEK162) combined with pexidartinib (PLX3397) in patients with advanced gastrointestinal stromal tumor. Oncologist. 2019;24(10):1309–e983. doi:10.1634/theoncologist.2019-0418
36. Cassier A, Garin G, Eberst L, et al. MEDIPLEX: A Phase 1 study of durvalumab (D) combined with pexidartinib (P) in patients (pts) with advanced pancreatic ductal adenocarcinoma (PDAC) and colorectal cancer (CRC). J Clin Oncol. 2019;37(15_suppl):2579. doi:10.1200/JCO.2019.37.15_suppl.2579
37. Wainberg Z, Eisenberg PD, Sachdev JC, et al. Phase 1/2a study of double immune suppression blockade by combining a CSF1R inhibitor (pexidartinib/PLX3397) with an anti PD-1 antibody (pembrolizumab) to treat advanced melanoma and other solid tumors. J Clin Oncol. 2017.
38. Colman H, Raizer JJ, Walbert T, et al. Phase 1b/2 study of pexidartinib (PEX) in combination with radiation therapy (XRT) and temozolomide (TMZ) in newly diagnosed glioblastoma. J Clin Oncol. 2018;36(15_suppl):2015. doi:10.1200/JCO.2018.36.15_suppl.2015
39. Moskowitz CH, Younes A, de Vos S, et al. CSF1R inhibition by PLX3397 in patients with relapsed or refractory hodgkin lymphoma: results from a Phase 2 single agent clinical trial. Blood. 2012;120(21):1638. doi:10.1182/blood.V120.21.1638.1638
40. Lo CH, Lynch CC. Multifaceted roles for macrophages in prostate cancer skeletal metastasis. Front Endocrinol (Lausanne). 2018;9:1–12. doi:10.3389/fendo.2018.00247
41. Buqué A, Bloy N, Aranda F, et al. Trial watch—small molecules targeting the immunological tumor microenvironment for cancer therapy. Oncoimmunology. 2016;5(6):e1149674. doi:10.1080/2162402X.2016.1149674
42. Butowski N, Colman H, De Groot JF, et al. Orally administered colony stimulating factor 1 receptor inhibitor PLX3397 in recurrent glioblastoma: an Ivy Foundation Early Phase Clinical Trials Consortium phase II study. Neuro Oncol. 2016;18:557–564. doi:10.1093/neuonc/nov245
43. Zhu Y, Knolhoff BL, Meyer MA, et al. CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res. 2014;74:5057–5069. doi:10.1158/0008-5472.CAN-13-3723
44. Bryant C, Chang-Strachan D, Chan V, et al. Macrophage IL-10 blocks CD8+ T cell-dependent responses to chemotherapy by suppressing IL-12 expression in intratumoral dendritic cells. Cancer Cell. 2009;19:6218–6221.
45. Plexxikkon. Pexidartinib; 2019.
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.