")
Back to Journals » Drug Design, Development and Therapy » Volume 9
Pancreatic cancer: optimizing treatment options, new, and emerging targeted therapies
Authors Chiorean EG , Coveler A
Received 9 March 2015
Accepted for publication 28 April 2015
Published 7 July 2015 Volume 2015:9 Pages 3529—3545
DOI https://doi.org/10.2147/DDDT.S60328
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Shu-Feng Zhou
Elena Gabriela Chiorean, Andrew L Coveler
Department of Medicine, Division of Oncology, University of Washington, Seattle, WA, USA
Abstract: Pancreatic cancer is the fourth leading cause of cancer death in the US and is expected to become the second leading cause of cancer-related deaths in the next decade. Despite 5-fluorouracil/leucovorin with irinotecan and oxaliplatin (FOLFIRINOX) and gemcitabine/nab-paclitaxel significantly improving outcomes for metastatic cancer, refractory disease still poses significant challenges. Difficulties with early detection and the inherent chemo- and radio-resistant nature of this malignancy led to attempts to define the sequential biology of pancreatic cancer in order to improve survival outcomes. Pancreatic adenocarcinoma is characterized by several germline or acquired genetic mutations, the most common being KRAS (90%), CDK2NA (90%), TP53 (75%–90%), DPC4/SMAD4 (50%). In addition, the tumor microenvironment, chemoresistant cancer stem cells, and the desmoplastic stroma have been the target of some promising clinical investigations. Among the core pathways reproducibly shown to lead the development and progression of this disease, DNA repair, apoptosis, G1/S cell cycle transition, KRAS, Wnt, Notch, Hedgehog, TGF-beta, and other cell invasion pathways, have been the target of “precision therapeutics”. No single molecularly targeted therapeutic though has been uniformly successful, probably due to the tumor heterogeneity, but biomarker research is evolving and it hopes to select more patients likely to benefit. Recent reports note activity with immunotherapies such as CD40 agonists, CCR2 inhibitors, cancer vaccines, and novel combinations against the immunosuppressive tumor milieu are ongoing. While many obstacles still exist, clearly we are making progress in deciphering the heterogeneity within pancreatic cancers. Integrating conventional and immunological targeting will be the key to effective treatment of this deadly disease.
Keywords: pancreatic cancer, immunotherapies, signaling pathway inhibitors, targeted therapies
Introduction
Pancreatic cancer accounts for 277,000 new cases diagnosed each year in the world,1 among which approximately 49,000 each occur in the US2 and Europe.3 Despite modest improvements in detection, which may have contributed to its rise in incidence, the 5-year overall survival (OS) rate only increased from 5% to 6% in the past three decades.3,4 While currently pancreatic cancer represents the fourth leading cause of cancer death in the US (approximately 40,000 deaths annually), it is expected to become the second cause of cancer-related deaths in the US in the next decade.5 Pancreatic ductal adenocarcinoma is the most common histologic type of pancreatic cancer and will be reviewed in this paper.
Among the known risk factors for pancreatic cancer, smoking and overweight/obesity are major contributors. The significant decrease in tobacco smoking prevalence since the 1990s has been expected to reduce the incidence of pancreatic cancer, but it may have been overcome by the increased prevalence in overweight/obesity and type II diabetes with untoward effects on risk and mortality from pancreatic cancer.4,6 The high mortality rate of pancreatic cancer is attributed to the lack of reliable screening methodologies for early detection of precancerous neoplasia and early-stage invasive pancreatic cancers, as well as the relative poor efficacy of systemic chemotherapy treatments. More than 50% of pancreatic cancers are identified in metastatic stage where survival rates range from 7–11 months,7,8 and in 30%–40% of patients the disease is localized but not surgically resectable, with OS of 11–18 months.9–11 Patients with resected pancreatic cancer have poorer outcomes compared to other resected solid tumors, and the median survival after surgery and adjuvant therapy averages 24 months.12–16
While gemcitabine is still used as the adjuvant therapy of choice for resected pancreatic cancer, since 2011 two combination regimens for metastatic disease have become gold standard: 5-fluorouracil (5-FU)/leucovorin with irinotecan and oxaliplatin (FOLFIRINOX),7 and nab-paclitaxel with gemcitabine.8 With these approaches, response rates range between 23% and 31%, progression-free survival (PFS) rates are 5.5–6.6 months, and OS is between 8.5 and 11 months. Given the success with FOLFIRINOX and gemcitabine/nab-paclitaxel in metastatic disease, these regimens are being studied in several adjuvant and neoadjuvant trials for resectable and borderline resectable disease, as well as for the treatment of locally advanced but inoperable pancreatic cancers. Preliminary data with combination chemotherapy indicate superior responses and survival rates vs historical reports with gemcitabine alone.10,17 It is now evident that these more aggressive chemotherapy backbones can be used to build upon for combinations with novel targeted therapies.
Pancreatic adenocarcinoma is characterized by several germline or acquired genetic mutations, the most common being KRAS (90%), CDK2NA (90%), TP53 (75%–90%), SMAD4/DPC4 (50%), as well as genomic and epigenetic alterations, which can guide personalized cancer therapy. In addition, the tumor microenvironment, the chemoresistant cancer stem cells, and the desmoplastic stroma have been the target of recent clinical investigations.
Current treatment options and limitations
In the past three decades, the standard chemotherapies for pancreatic cancer consisted of fluoropyrimidines like 5-FU, and the antimetabolite drug gemcitabine, which were mostly equivalent in randomized clinical trials, contributing 0%–10% each to tumor response, and with PFS and OS rates of 4–6 months.18 Several combinations with other chemotherapies and/or biologically targeted agents were studied with almost invariably negative results (Table 1).19–23 In 2005, the first statistically significant survival improvement with a gemcitabine combination was noted with the epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor, erlotinib (6.2 vs 5.9 months, hazard ratio [HR] 0.82, P=0.004) in the NCIC Phase III study.24 Nevertheless, the benefit was small, and these results were not practice changing. Furthermore, no biomarkers have been identified to predict benefit from erlotinib.25 Due to grade ≥2 rash from erlotinib being associated with increased OS (10.5 months) in the NCIC study, Van Cutsem et al tested gemcitabine plus erlotinib dose escalated to rash, in the Phase II randomized RACHEL study.26 While more patients in the escalation group developed rash (41%) vs 9% in the standard group, the PFS and OS were not superior (3.5 vs 4.5 months, and 7 vs 8.4 months, respectively).
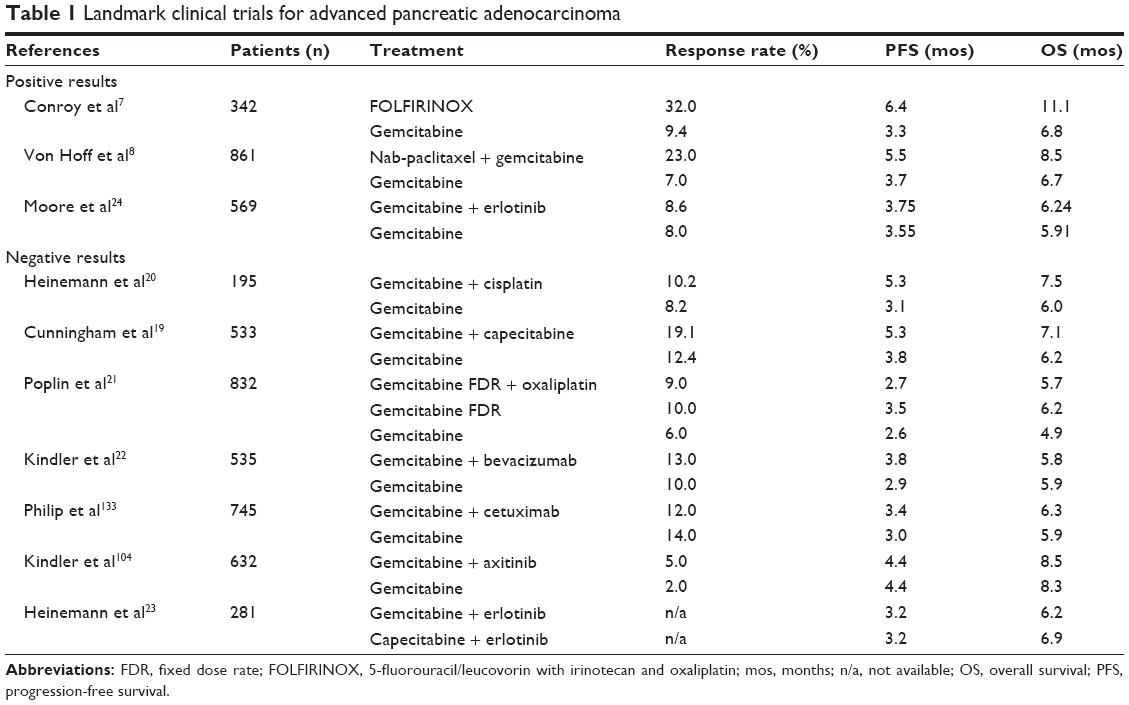 | Table 1 Landmark clinical trials for advanced pancreatic adenocarcinoma |
The excessive chemoresistance which characterizes pancreatic cancer has been studied in both xenograft and genetically engineered KrasLSL-G12D/+Trp53LSL-R172H/+Cre mouse models (KPC GEMM).27 These preclinical models, as well as several human clinical studies noted that the dense desmoplastic tumor stroma, mostly devoid of functional vasculature and infiltrated by an immunosuppressive environment, contributes to poor access by therapeutics, and confers chemo- and radiotherapy resistance.28,29 Targeting the tumor stroma has become an area of intense research.30–33
Nab-paclitaxel
Taxanes are important components in the systemic treatment of many cancers including breast, ovarian, lung, and gastro-esophageal. Paclitaxel and docetaxel have been studied in the treatment of pancreatic adenocarcinoma with few noticeable results, until recently with the new generation nanoparticle albumin-bound paclitaxel (nab-paclitaxel). Taxanes stabilize microtubules by increasing their polymerization, and induce cell cycle arrest at the G2/M phase, resulting in cell death.34 Nab-paclitaxel, a water soluble compound has enhanced distribution properties within the tumor microenvironment compared to paclitaxel, and increases intratumoral gemcitabine levels in mouse models.35 A putative mechanism for the intratumoral accumulation of nab-paclitaxel is the presence of albumin-binding proteins such as gp60 and secreted protein acidic and rich in cysteine (SPARC/osteonectin) in the tumor microenvironment.36,37 While SPARC expression in peritumoral stroma seems to correlate with worse outcomes in early stage pancreatic cancer,38,39 it is debatable whether it correlates to improved efficacy from nab-paclitaxel-based therapy.40–43
Targeting the tumor stroma has been one of the proposed mechanisms of action for nab-paclitaxel. The stromal disrupting effects of nab-paclitaxel have also been noted in a small neoadjuvant study for resectable pancreatic cancer patients. Among the ten patients who underwent surgical resection, treatment with two 4-week cycles of nab-paclitaxel and gemcitabine prior to surgery did not induce objective radiological responses, but caused increased “tumor softness” by endoscopic ultrasound-based elastography, and histopathologically resulted in one complete pathological response, and six major pathological responses (a few isolated malignant cells left).44 Compared to untreated controls, nab-paclitaxel with gemcitabine was associated with less abundant collagen matrix infiltration around tumor glands, and a decreased number of cancer-associated fibroblasts. A more recently described mechanism of nab-paclitaxel synergism with gemcitabine involves the inactivation by nab-paclitaxel of the gemcitabine catabolizing enzyme cytidine deaminase, via production of destabilizing reactive oxygen species.35
The most significant benefit to date with nab-paclitaxel has been reported in the setting of metastatic pancreatic cancer. The phase III international MPACT study among 861 randomized patients, showed a significant response (23% vs 7%, P<0.001), PFS (5.5 vs 3.7 months, P<0.001), and OS rate (8.5 vs 6.7 months, P<0.001) benefit with nab-paclitaxel plus gemcitabine compared to gemcitabine alone.8 Overall, the combination treatment was well tolerated, and the most significant grade ≥3 toxicities were neutropenia (38%), fatigue, and neuropathy (17% each), and thrombocytopenia (13%). These results led to the approval by the United States Food and Drug Administration (FDA) of nab-paclitaxel on September 6, 2013 for metastatic pancreatic cancer. In this trial, archival tumor biopsies have been collected to analyze the role of stromal and tumor SPARC expression, both as prognostic and predictive markers of benefit. While earlier studies proposed SPARC as a prognostic marker for worse outcomes in early stage pancreatic cancer,38 in MPACT high SPARC levels did not correlate with worse survival or with superior benefit from nab-paclitaxel therapy.43 This is consistent with recent preclinical data in pancreatic cancer mouse models indicating SPARC-independent desmoplasia, and intratumoral paclitaxel levels no different among SPARC+/+ (positive) and SPARC−/− (negative) tumors.45 The role of circulating SPARC is controversial, but it has been proposed that it may be linked to higher intravascular nab-paclitaxel levels.45
Given potential synergy seen in preclinical models, nab-paclitaxel/gemcitabine was combined with capecitabine, but likely due to lower administered doses (gemcitabine 750 mg/m2, nab-paclitaxel 100 mg/m2 both on day 4; and capecitabine 750 mg/m2 twice daily, days 1 to 7, every 14 days), results were modest: response rates 14% and median survival 7.5 months.46 Nab-paclitaxel has been tested as monotherapy or in combination with gemcitabine in the second-line treatment of advanced pancreatic cancer. In a Phase II trial, single agent nab-paclitaxel showed low response rates (5%) and PFS of 1.7 months, but the OS of 7.3 months suggests benefit from additional lines of therapy.47 Another retrospective analysis of nab-paclitaxel showed median PFS and OS of 2 and 3 months, respectively.48 Similarly, gemcitabine/nab-paclitaxel showed little benefit in the salvage setting, with median PFS of 3 months.49
Small clinical trials with nab-paclitaxel-based chemotherapy in the first or second line setting added biologically targeted agents such as the EGFR-tyrosine kinase inhibitor erlotinib,50 or the VEGFR-2 (vascular endothelial growth factor and receptor), EGFR, RET kinase inhibitor vandetanib,51 but the results are not clearly superior to chemotherapy alone (median PFS 5.3 months, and OS 8–9 months).
Given high response rates in advanced disease, several small clinical trials noted activity of nab-paclitaxel with gemcitabine in the preoperative setting. Radiological responses, likely an inadequate surrogate,52 with neoadjuvant therapy were only 0%–16%, but higher pathological response rates were noted (30%–70%); long-term survival data are not yet available.53,54 A Phase III trial with nab-paclitaxel and gemcitabine (APACT, NCT01964430) is ongoing in the United States for patients with resected pancreatic adenocarcinoma, and neoadjuvant studies integrating chemoradiotherapy with gemcitabine/nab-paclitaxel are planned or ongoing. In Europe, the NEONAX trial (NCT02047513) will test neoadjuvant plus adjuvant vs only adjuvant gemcitabine/nab-paclitaxel for resectable pancreatic cancer patients.
FOLFIRINOX
The combination of 5-FU, irinotecan, and oxaliplatin (FOLFIRINOX) first demonstrated activity in pancreatic cancer in a Phase I solid tumors trial published in 2003, which was based on the independent clinical activity previously noted with its constituent agents: 5-FU, irinotecan, and 5-FU/oxaliplatin.55 A subsequent Phase II study among 47 patients with pancreatic cancer demonstrated response rates of 26%, time to progression of 8.2 months, and the median OS was 10.2 months.56 These data led to the conduct of the Phase II–III PRODIGE4/ACCORD 11 study of 342 patients selected for excellent performance status (ECOG PS 0 or 1), randomized to FOLFIRINOX vs gemcitabine alone.7 The toxicity profile with FOLFIRINOX demonstrated an increased risk of grade 3/4 myelosuppression (46% neutropenia, 5.4% febrile neutropenia), fatigue (24%), vomiting (15%), diarrhea (13%), and sensory neuropathy (9%). The efficacy results showed remarkable responses of 31% (vs 9% with gemcitabine), PFS of 6.4 months (vs 3.3 months, HR =0.47, P<0.001), and median OS of 11.1 months (vs 6.8 months, HR 0.57, P<0.001), clearly setting FOLFIRINOX as one of the most effective first-line treatment options for good performance status patients younger than 76 years old. Nevertheless tolerability remains a big issue, and as many as a third of patients discontinue treatment early for toxicity reasons. FOLFIRINOX modifications have been implemented clinically in most practices, often with the omission of the 5-FU bolus and/or dose reductions for irinotecan, as well as growth factor support. Prospective studies with modified FOLFIRINOX will be able to define its efficacy, but interim reports do not seem to indicate detrimental effects compared to the standard regimen.57
The activity of FOLFIRINOX in advanced disease led to its use as a means of cancer downstaging to increase resectability rates for locally advanced pancreatic cancer (LAPC) and borderline resectable disease. Local institutional studies, mostly retrospective, report that FOLFIRINOX confers response rates of 27% or higher, PFS of 11–12 months, and resectability rates of approximately 10%–20% for patients with LAPC, but longer term survival data is not available.17,57–59
For patients with resected pancreatic cancer, the UNICANCER group in France will test adjuvant FOLFIRINOX vs gemcitabine, and the study is planned to complete a 490-patient accrual in 2018.
Radiotherapy
The role of chemoradiotherapy is still debated for both resectable and LAPC pancreatic adenocarcinomas. While it is clear that pancreatic cancer is an inherently systemic disease, up to 30% of patients die with local progression.60 In the past two decades, two randomized European studies (EORTC61 and ESPAC-112) concluded that radiation therapy offers no benefit after pancreatic cancer surgery. Currently the RTOG 0848 trial (NCT01013649) is studying the role of adjuvant radiotherapy in addition to gemcitabine vs gemcitabine alone after resection. For LAPC disease, similar controversy exists.9,62 The LAP-07 trial (gemcitabine with or without capecitabine-based chemoradiotherapy and with or without erlotinib), did not show a survival advantage when radiotherapy was added after a 4 months chemotherapy induction period.11 Nevertheless, contemporary studies are testing the addition of chemoradiotherapy to the more effective combination backbones of FOLFIRINOX (CONKO-7 trial in Germany due to complete 2018), and gemcitabine/nab-paclitaxel (LAPACT, NCT02301143). DPC4/SMAD4 has been identified as a putative genetic biomarker predictive of local progression when intact, or metastatic dissemination when mutated.60 The RTOG1201 Phase III randomized study (NCT01921751) will prospectively test whether the DPC4 gene status will identify LAPC patients more likely to benefit from chemoradiotherapy in addition to gemcitabine/nab-paclitaxel vs gemcitabine/nab-paclitaxel chemotherapy alone.
New and emerging treatments
Pancreatic cancer tumor behavior is influenced by both the malignant potential of the cancer cells as well as by the host microenvironment defined by fibroblasts, myofibroblasts, pancreatic stellate cells, vascular cells, immune cells, and the extracellular matrix with associated cytokines.63
Immunotherapies
One of the most frequently invoked reasons for the aggressiveness and chemoresistance associated with pancreatic cancer has been its immunosuppressive tumor microenvironment. Preclinical data suggest that the pancreatic cancer mass is represented roughly by 50% immune and inflammatory cells, with an abundance of regulatory T cells (CD4+FoxP3+Treg), myeloid-derived suppressor cells (MDSC), and macrophages, but a relative lack of cytotoxic T cells.64,65 Pancreatic cancer prognosis has been directly associated with the induction of a humoral response to the cancer antigens MUC-1 and mesothelin, as well as with the presence of intratumoral cytotoxic T lymphocytes and helper T cells.66–69 Furthermore, the tumor expression level of the negative immune checkpoint regulator programmed death-1 ligand,70 the intratumoral accumulation of Treg,71 and the ratio of Treg relative to CD4+ lymphocytes72 may inversely correlate with prognosis. On these premises, several approaches have been taken to augment antitumor immune recognition of pancreatic cancer (Table 2).
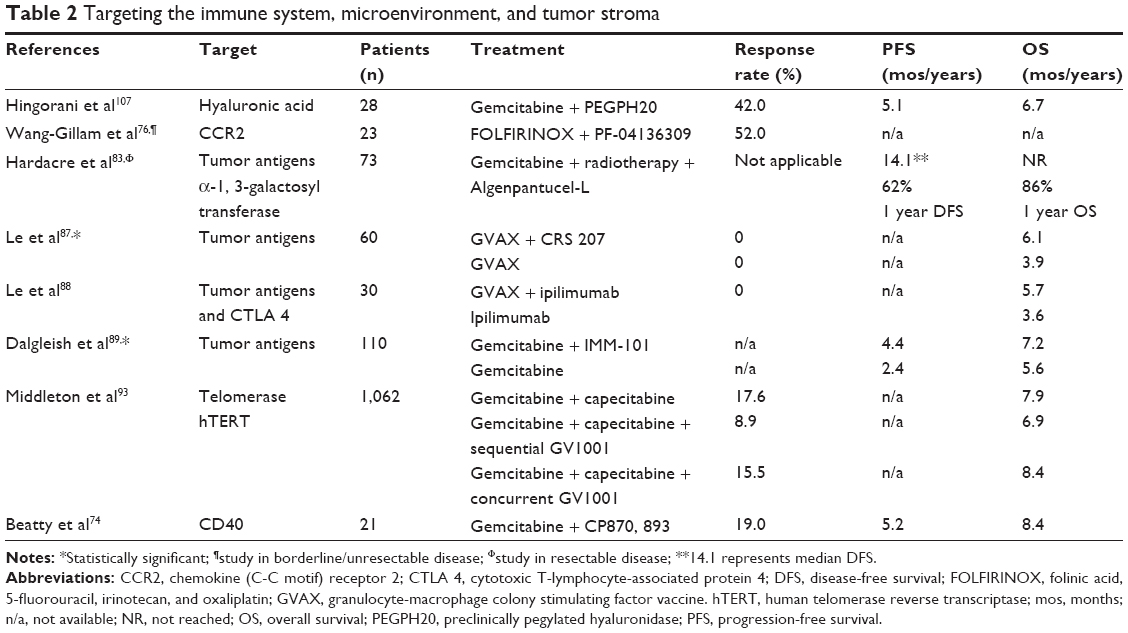 | Table 2 Targeting the immune system, microenvironment, and tumor stroma |
Macrophage-targeted therapies
CD40, a tumor necrosis factor receptor superfamily member regulates activation of T-cells and regulates cancer associated inflammation and fibrosis. CD40 activation has been shown preclinically to induce anti-tumor T cell responses. In a clinical trial in metastatic pancreatic cancer with anti-CD40 agonist antibody CP-870,893 with gemcitabine, responses were noted to be associated with tumor macrophage, but not lymphocyte, infiltration.73,74 Biologically, the activation of tumor-associated macrophages (TAM) with the induction of a cytotoxic macrophage phenotype was implicated in tumor stroma depletion.73 In other preclinical models, suppression of inhibitory TAM with chemokine (C-C motif) receptor 2 or colony-stimulating factor-1 receptor antagonists improved chemotherapy response and increased anti-tumor T cell responses.75 Given that TAM may manifest both pro- and anti-tumor effects, a productive macrophage-directed immunotherapy needs to induce anti-stroma and anticancer activity. Wang-Gillam et al reported on PF-04136309, a novel chemokine (C-C motif) receptor 2 inhibitor capable of depleting inflammatory immunosuppressive TAM, in combination with FOLFIRINOX in borderline and LAPCs.76 The combination therapy was safe and among 23 evaluable patients, response rates were 52%, but long-term outcomes were not available. These preliminary data about targeting the TAM component of the tumor microenvironment have promising results and should be further explored.
Cytotoxic T-lymphocytes-targeted therapies
Inducing an immunoreactive environment may be accomplished by stimulating endogenous CD8 T cells via adoptive T cell therapies, cancer vaccines, or interferons (IFNs), targeted depletion of inhibitory MDSC, checkpoint blockade inhibitors against the programmed cell death 1, the programmed cell death ligand-1, against indoleamine 2,3-dioxygenase, and against the cytotoxic T-lymphocyte-associated protein 4.
Immune checkpoint inhibitors
Possibly because of a reduced pancreatic cancer neo-antigenic potential,77 to date clinical trials with checkpoint blockade inhibitors alone (programmed cell death ligand-1 and cytotoxic T-lymphocyte-associated protein 4) have not demonstrated significant activity in pancreatic cancer.78,79 Nevertheless, combinatorial approaches of checkpoint blockade inhibitors plus vaccines (NCT02243371), with agents that target immunosuppressive cell populations (Treg, MDSC), and with chemotherapy (NCT02309177, NCT01313416) or radiotherapy (NCT02305186) are actively being pursued.
Adoptive T cell therapy
Adoptive immunotherapy utilizing patient’s own T cells expanded and stimulated ex vivo against various tumor antigens is currently being tested. In refractory pancreatic cancer patients, pilot data with cytokine-induced T cells showed safety and PFS rates of 11 weeks and OS of 27 weeks, comparable with second-line chemotherapy regimens.80 Genetic engineering of T cells to enhance the expression and affinity of tumor-antigen specific receptors or to render them refractory to inhibitory signals may bring promise to the immunotherapy field in pancreatic cancer.81
Cancer vaccines
Cancer vaccines can stimulate and mature dendritic cells for specific tumor antigen presentation with the ultimate goal for activating the adaptive immune response with effective cytotoxic T lymphocytes. Several types of cancer vaccines have been tested in pancreatic cancer: 1) whole-cell vaccines engineered to overexpress a certain epitope; 2) peptide vaccines; and 3) DNA vaccines, in which DNA coding a target antigen is inserted into a vector which is taken up by tumor cells.
Several whole cell vaccines have reported results in pancreatic cancer. Algenpantucel-L is an irradiated combination of two human allogeneic pancreatic cancer cell lines HAPa1 and HAPa2 genetically engineered to express the murine enzyme α-1,3-galactosyl transferase (αGT). Humans naturally do not express αGal epitopes but possess large amounts of anti-αGal antibodies; therefore, vaccination with algenpantucel-L results in “hyperacute rejection” based on complement-mediated lysis and antibody dependent cell mediated cytotoxicity (ADCC) against the allogeneic vaccine cells. Exposure to the entire vaccine cells antigenic repertoire results in the activation of the immune system, similar to that observed with transplant rejection.82 Hardacre et al reported on a Phase II study (NCT00569387) of adjuvant algenpantucel-L with gemcitabine and 5-FU based chemoradiotherapy after surgery for pancreatic cancer patients.83 The 1-year survival was 86%, which compared favorably with the adjuvant RTOG-9704 study (1-year survival 69%), the 1-year disease-free survival was 62%, and the treatment was well tolerated. Higher vaccine doses (300 million cells/dose) resulted in superior results (96% 1-year survival). Biomarker studies noted that patients who mounted an anti-membrane-bound recombinant mesothelin response (31% of patients) or anti-calreticulin antibody response (48% of patients) had improved OS (42 months vs 20 months, P=0.027, and 35.8 months vs 19.2 months, P=0.03, respectively.84,85 A Phase III study (NCT01072981) with algenpantucel-L and chemoradiotherapy for patients with resected pancreatic cancer completed accrual, and results are eagerly awaited. Currently, New Link Genetics is conducting a Phase III study of algenpantucel-L with FOLFIRINOX or gemcitabine/nab-paclitaxel plus chemoradiotherapy in borderline and LAPC (NCT01836432).
The GVAX pancreas vaccine is another allogeneic whole cell vaccine transfected with granulocyte-macrophage colony-stimulating factor which acts as a maturation factor for the antigen presenting cells/dendritic cells. GVAX vaccine has been studied with chemoradiotherapy after resection of pancreatic cancer, and showed modest median disease-free and OS rates of 17.3 and 24.8 months, respectively.86 For patients with metastatic pancreatic cancer, a randomized Phase II study tested the GVAX vaccine combined with a boost live-attenuated Listeria monocytogenes (Lm) vaccine modified to deliver the pancreatic tumor antigen mesothelin (CRS-207), vs GVAX alone.87 Among 90 patients, 50% previously treated with 2+ lines of chemotherapy, median OS was 6.1 vs 3.9 months for the GVAX +/− CRS-207 vaccine therapy, and toxicity was manageable. A larger Phase IIb randomized multicenter 3-arm trial of GVAX plus CRS-207, vs CRS-207 alone vs chemotherapy is ongoing in patients with refractory metastatic pancreatic adenocarcinoma (ECLIPSE, NCT02004262). GVAX pancreas has also been combined with ipilimumab (cytotoxic T-lymphocyte-associated protein 4 antibody) in a randomized study vs ipilimumab alone, for patients with previously treated locally advanced or metastatic pancreatic cancer.88 OS rates were 5.7 months with the combination, and 3.6 months with ipilimumab alone. In both of the GVAX pancreas vaccine studies referenced above, an immunological T cell response to mesothelin has been associated with increased OS.87,88 Building upon the results with ipilimumab combined with GVAX OS in refractory pancreatic cancer, the Johns Hopkins’ group is leading a randomized Phase II trial of FOLFIRINOX for 8–12 doses induction chemotherapy followed by GVAX vaccine plus ipilimumab every 3 weeks ×4, then every 8 weeks, vs continuing FOLFIRINOX until progression or toxicity (NCT01896869).
Dalgleish et al reported on IMM-101, a heat-killed whole cell vaccine of Mycobacterium obuense able to induce a systemic immune response, combined with gemcitabine among 110 untreated patients with advanced pancreatic cancer, and observed OS of 7.2 months vs 5.6 months with gemcitabine alone (P=0.022, HR =0.60), and a similar improvement of PFS 4.4 vs 2.3 months (P<0.001, HR =0.40).89 While statistically significant, these results need to be reproduced in combination with contemporary chemotherapy regimens in larger randomized studies.
Peptide vaccines are based on cancer-specific peptides capable of binding human leukocyte antigen class molecules and activating a CD4/CD8 immune response. Mutant KRAS peptide vaccines have been evaluated after surgical resection of pancreatic cancer and conferred average survival rates of 27 months, and 5-year survival rates of 20%, comparable with historical adjuvant chemotherapy data.90,91 Telomerase is a ribonucleotide enzyme which maintains telomeres and confers cancer cells immortality. The telomerase peptide vaccine GV1001 while promising in a Phase I/II study,92 failed to improve survival (median OS 7–8 months in all arms) when combined sequentially or concurrently with gemcitabine/capecitabine chemotherapy vs chemotherapy alone in the Phase III randomized TeloVac trial.93
To date, the overall benefit conferred by pancreatic cancer vaccines is still under investigation, as no large study has shown convincing results. Nevertheless, rational synergistic immuno-therapeutic approaches are ongoing, and results are eagerly awaited.
Interferon
IFNs have been studied since 1990s in various immunoresponsive malignancies such as malignant melanoma and renal cell carcinomas, but also in pancreatic adenocarcinomas based on preclinical evidence of immunostimulatory properties, and chemo- and radio-sensitization.94 Type I IFN, both α and β demonstrated inhibition of pancreatic cancer cells growth by induction of apoptosis and cell cycle arrest.95,96 Another mechanism invoked in IFN-mediated suppression of pancreatic tumor growth and metastasis has been the modulation of the host tumor response though increased nitric oxide production.97 Clinically, the role of IFNα has been debated, between reports of increased clinical activity in institutional series, and detrimental toxicity without statistical survival benefit in the larger Phase II and III studies. Picozzi et al initially reported on 43 patients treated with adjuvant 5-FU, cisplatin, and IFNα-2bplus radiotherapy followed by adjuvant 5-FU in patients with resected pancreatic adenocarcinoma, and 5-year OS rates were 55%.98 Updated data after a median follow up of 45 months showed median disease-free survival of 22 months, median OS of 42 months, with 5- and 10-year OS rates of 42% and 28%, respectively.99 Efficacy results with this regimen were not reproduced by the multicenter ACOSOG Z05031 study, which noted median disease-free survival and OS of 14.1 and 25.4 months, respectively, but all-cause grade ≥3 toxicity was 95% resulting in patient accrual stopping early.100 Furthermore, the CapRI randomized Phase III study comparing adjuvant chemoimmunoradiotherapy vs 5-FU/leucovorin noted no significant survival difference (median OS 32 vs 28 months, and 2-year OS 62% vs 52%, P=0.49), but significantly higher toxicities.101
Stromal disruption
Tumor stroma is an active component in the biology of pancreatic cancer. The quest of therapies capable of inducing stromal collapse and easing chemotherapy access inside fibrotic pancreatic tumors has shown mixed results to date. Targeting matrix metalloproteinases has been ineffective102 and inhibiting stromal related angiogenic factors such as the VEGF and receptor VEGFR or the platelet-derived growth factor/receptor demonstrated no activity when combined with gemcitabine.22,103–105 It is unclear whether in combination with other chemotherapy backbones like FOLFIRINOX or nab-paclitaxel anti-angiogenic agents may prove more successful.
One of the most exciting and anticipated stroma-directed approaches in pancreatic cancer has been the targeting of hyaluronic acid (HA). HA poses a physical barrier within the pancreatic tumor extracellular matrix by creating very high interstitial pressure, and therefore vascular compression and hypoperfusion.28–31,106 Preclinically pegylated hyaluronidase (PEGPH20), an enzyme capable of depleting HA, reduced the interstitial pressure, leading to increased activity of gemcitabine in the KPC GEMM model.32 Similar results were observed in a Phase I/Ib clinical trial when PEGPH20 was combined with gemcitabine as first-line treatment among 28 patients with metastatic pancreatic cancer.107 The toxicity profile included muscle spasms (54%), myalgias (39%), and arthralgias (29%), among the most common toxicities, but most were grade 1–2. Overall, while median PFS and OS were 154 and 200 days, in an exploratory analysis patients with high intratumoral HA content (HAhigh) showed median PFS of 219 days (95% CI: 159–276) and OS of 395 days (95% CI: 210–578). PEGPH20 is currently being tested in two national randomized Phase II clinical trials in combination with gemcitabine plus nab-paclitaxel (NCT01839487), and with FOLFIRINOX (SWOG1313, NCT01959139).
Other components of the tumor stroma, such as the connective tissue growth factor which is overexpressed in pancreatic cancer, are being targeted. FG-3019, a monoclonal antibody against connective tissue growth factor, has been studied with gemcitabine/erlotinib in 75 patients with advanced pancreatic cancer (localized or metastatic) and conferred OS rates of 9.4 months, with best outcomes among patients with high FG-3019 plasma levels at day 15, and low baseline connective tissue growth factor levels.108 This agent is currently being tested as neoadjuvant therapy with gemcitabine/nab-paclitaxel for LAPC in an attempt to reduce the fibrotic stroma and enhance the chemotherapy efficacy (NCT02210559).109 The tumor growth factor beta (TGF-β) activates pancreatic stellate cells to become activated myofibroblasts, responsible for the formation of the extracellular matrix, a component of the pancreatic cancer stroma, and also acts as an immunosuppressor. Targeting the TGF-β pathway is undergoing clinical trials in solid tumors including pancreatic cancer.
Targeting core pathways
Several critical signaling pathways have been identified spanning the pancreatic cancer genomic map. Jones et al, Biankin et al, and others characterized key genetic mutations and RNA transcripts which occur with most propensity in pancreatic tumors.110,111 Among the 12 key pathways encompassing an average of 63 genetic mutations per tumor, genetic alterations have been found in the DNA repair, apoptosis, G1/S cell cycle transition, KRAS, Wnt, Notch, and Hedgehog (Hh) signaling, TGF-beta, chromatin remodeling (ARID1A and MLL3 genes) and other cell invasion pathways, which are the target of “precision therapeutics”.
To date, despite several targeting attempts with farnesyl-transferase inhibitors112,113 no single agent has successfully targeted mutated KRAS, possibly because of its complex crystallographic nature. The KRAS effector pathways, PI3K (phosphatidylinositol 3-kinase)/AKT (alpha serine/threonine-protein kinase)/mTOR (mammalian target of rapamycin) and RAF (rapidly accelerated fibrosarcoma)/MEK/MAPK (mitogen-activated protein kinase) have been targeted in several clinical trials in patients with metastatic pancreatic cancer, and as single agents they do not have significant activity (PFS 2 months and OS 5 months in second line setting).114,115 Preclinical data suggest that MEK inhibitors may be active only in certain KRAS mutational subsets (KRAS V12 mutation and KRAS copy number variation-gains or loss, are resistant to MEK inhibitors).116 Others suggest that MEK inhibitors alone or in combination with EGFR pathway inhibition may be effective in epithelial but not mesenchymal subtypes of pancreatic cancer.117 Furthermore, MEK inhibitors alone result in feedback activation of the EGFR and PI3K pathways, thus combination strategies are expected to be more effective.118 Indeed, MEK inhibitors in combination with gemcitabine have not demonstrated superior results vs gemcitabine alone in clinical trials, with PFS of 4–6 months and OS rates of 6–9 months,119–121 but a subgroup of KRAS wild-type patients demonstrated higher than expected PFS and OS (9 and 18 months respectively).121 Dual MEK and EGFR inhibition showed median PFS and OS of 2.6 and 7.5 months, respectively in the second line treatment of metastatic pancreatic cancer.122 The combination of PI3K/mTOR and MEK inhibitors is synergistic preclinically,123 but clinical data to date show only modest activity when these pathways are concomitantly blocked (disease stabilization in 30%–50% of patients as best response, PFS and OS of 2 months and 5 months respectively).124,125 Results of the SWOG S1115 randomized Phase II trial (NCT01658943) with MEK plus AKT inhibition (MEK inhibitor selumetinib AZD6244, and AKT inhibitor MK2206) vs folinic acid, 5-FU, and oxaliplatin (FOLFOX) in second line treatment for metastatic pancreatic cancer will be presented at ASCO 2015. mTOR inhibitors alone126,127 or in combination with EGFR blockade128 similarly have not demonstrated notable activity in pancreatic cancer. Despite the general lack of meaningful activity even in combination with chemotherapy,129 there has been interest in PI3K/mTOR blockade for select patients who manifest an upregulation of this pathway, such as patients with TSC (tuberous sclerosis complex) gene mutations or with Peutz–Jeghers syndrome, where STK11/LKB1 gene mutations can lead to inactivation of the LKB1/AMPK/TSC signaling with subsequent mTORC1 activation.130–132 With several mechanisms involved in resistance to PI3K/AKT/mTOR and MEK inhibitors, including feedback loop activation of parallel pathways, and EGFR or HER3 upregulation,118 it seems that best targeted treatment strategies with these agents are yet to be identified for patients with epithelial vs mesenchymal subtypes, and for those with various types of KRAS mutations. While targeting the EGFR with erlotinib provided marginal benefit in combination with gemcitabine in the NCIC Phase III trial,24 cetuximab did not increase survival (6.3 vs 5.9 months, P=0.23) when added to gemcitabine in an unselected population.133 Several other trials reported on erlotinib in combination with other chemotherapy backbones and/or biologics such as insulin growth factor receptor 1 inhibitors or VEGFR inhibitors, and had modest results (Table 3).134–141 Another member of the EGFR pathway, the HER2 (neu/ERBB2) gene rarely exhibits amplification in pancreatic cancer (2%), but those cases seem to be associated with lung and brain metastases.142 Several trials tested trastuzumab or lapatinib with chemotherapy in HER2 overexpressing tumors, and none showed improved survival rates (OS 4–7 months).143–145 It is possible that better patient selection using genomic profiling such as the one used in the IMPaCT trial (Australasian Gastrointestinal Trials Group ACTRN12612000777897), may show improved results.
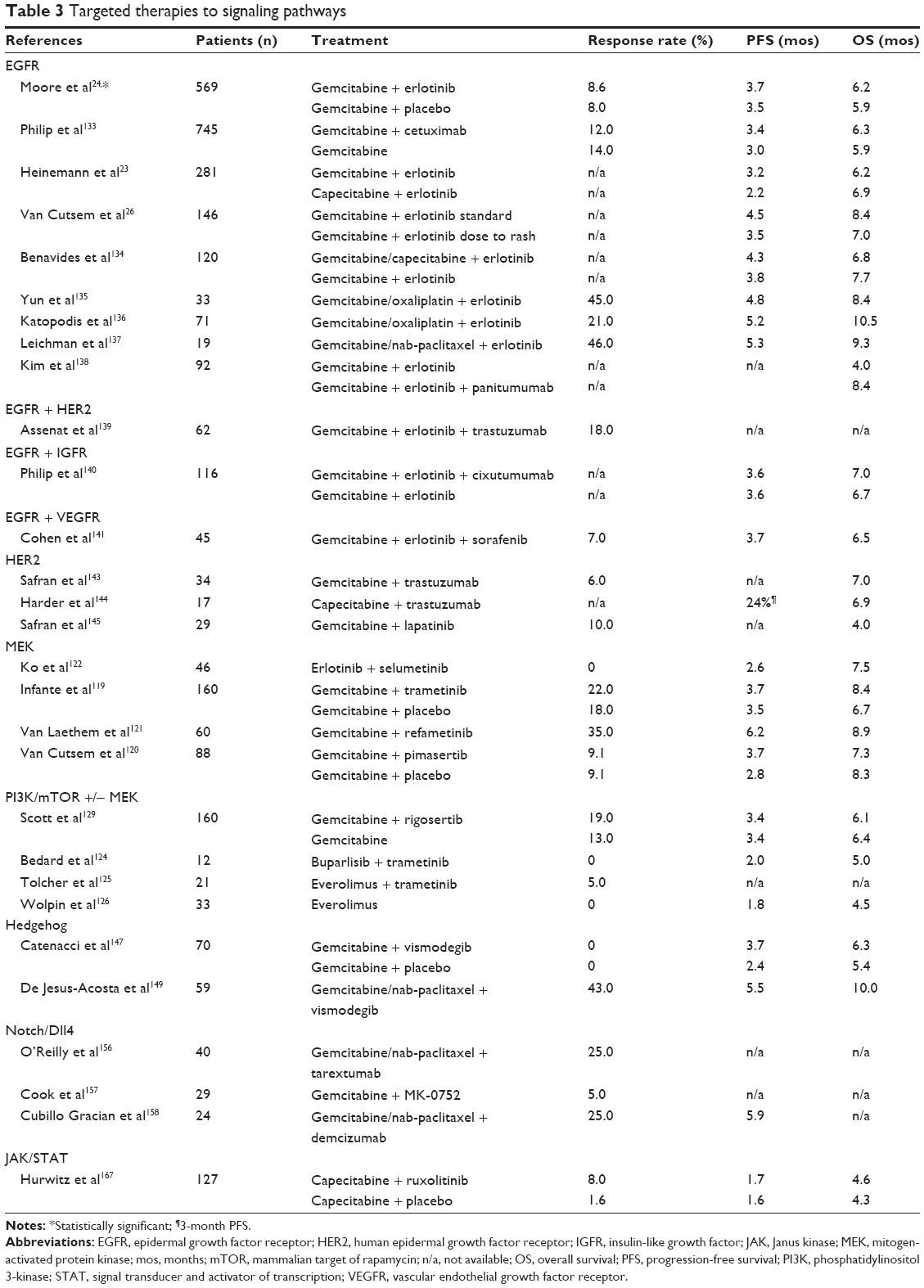 | Table 3 Targeted therapies to signaling pathways |
Another stumbling block showing discrepant results from tumor models occurred when Hh pathway was targeted with smoothened inhibitors. Despite promising evidence of Hh blockade causing depletion of cancer associated fibroblasts and improving perfusion in preclinical models,146 clinical trials with Hh inhibitors and chemotherapy did not demonstrate improved results compared to chemotherapy alone – vismodegib (GDC0449) plus gemcitabine showed PFS of 2.8–3.7 months and OS of 5.3–6.3 months (vs PFS 2.4 months and OS 5.4 months for gemcitabine alone),147,148 and in a recent trial in combination with gemcitabine/nab-paclitaxel PFS and OS rates were 5.5 and 10 months, respectively.149 The randomized Phase II trial of saridegib (IPI-926) with gemcitabine was terminated early due to lack of efficacy (OS >6 months in the gemcitabine plus placebo arm, and OS <6 months in gemcitabine plus IPI-926 arm) (NCT01130142), and a Phase I trial of saridegib with FOLFIRINOX (NCT01383538) is awaiting final results.150 One possible reason for the lack of activity of Hh inhibitors in pancreatic cancer could be the increased metastatic potential of cancer cells in the absence of a contained fibroblastic lattice, and possibly the increased perfusion within the tumor bed.151 The depletion of cancer associated fibroblasts has been recently associated with worse cancer aggressiveness, epithelial–mesenchymal transition (EMT), increased cancer stem cells, and reduced survival in pancreatic cancer mouse models.152 It is now apparent that cancer stroma can both restrain and enhance tumor growth, and future targeting needs to carefully elucidate its various components contribution to cancer progression.
The Delta–Notch pathway has been recognized as an important member of tumor angiogenesis and a contributor to cellular growth, differentiation, and stem cells self-renewal, including in pancreatic cancer.153,154 Preclinically, Notch/Dll4 inhibition has antitumor activity,155 and preliminary efficacy from Phase Ib studies with tarextumab (OMP-59R5)156 and MK-0752157 show early signs of activity when combined with chemotherapy. A Phase II study of single agent gamma-secretase inhibitor RO4929097 in previously treated advanced pancreatic cancer had poor results, with PFS and OS of 1.5 and 4.1 months, respectively, and the study accrual stopped early (NCT01232829). The novel Dll4 inhibitor demcizumab (OMP-21M18) in combination with gemcitabine and nab-paclitaxel showed encouraging PFS of 5.9 months in a Phase Ib study,158 and it is currently being tested in a larger randomized Phase II study (Onconova, NCT02289898). The emerging cardiovascular toxicity (such as pulmonary hypertension) seen with Dll4 inhibitors may be addressed by limiting treatment exposure to less than 100 days.
The Wnt-β-catenin developmental pathway has been implicated in pancreatic cancer progression, and cancer stem cells maintenance.159 PRI-724, a cAMP-response element-binding protein/β-catenin modulator which induces stem cells differentiation, is currently being tested in combination with gemcitabine in the second line treatment of metastatic pancreatic cancer (NCT01764477).
Chronic inflammation contributes to pancreatic cancer carcinogenesis. Deregulated cytokines like interleukin (IL)-6, and IL-1α with activation of downstream effectors signal transducer and activator of transcription (STAT)-3 and NF-kB lead to recruitment of proinflammatory cells from bone marrow to the pancreatic tumor stroma. Inflammatory signaling through STAT3, NFkB, and cyclooxygenase-2 (COX-2) also leads to pancreatic stellate cells proliferation, induces desmoplasia and promotes cancer stem cells.160–163 While COX-2 is frequently upregulated in pancreatic adenocarcinoma (50%–60%), clinically COX-2 inhibition did not provide additive benefits to chemotherapy.164,165 Another strategy targeting the inflammatory environment is by blocking the STAT and Janus kinase (JAK) pathways which have lead roles in cancer inflammation and immunity.160,161,166 Ruxolitinib, an oral inhibitor of JAK1 and JAK2 signaling, has been evaluated in combination with capecitabine in the randomized Phase II RECAP trial for second line treatment of pancreatic cancer.167 The addition of ruxolitinib conferred a slightly improved survival vs capecitabine/placebo (median OS 137 vs 130 days, HR =0.79, P=0.25), but this effect was limited to a subset of patients with higher C-reactive protein levels (>13 mg/L): median OS 83 vs 55 days, and 6-months OS 42% vs 11% (HR 0.47, P=0.01) (NCT01423604). The Phase III randomized Janus 1 study with this combination including only patients with CRP levels >10 mg/L is ongoing (NCT02117479).
A summary of targeted therapies against various signaling pathways is provided in Table 3.
Pancreatic cancer is characterized by genomic instability,110,111 and several DNA repair defects occur through mutations in DNA mismatch repair gene MLH1 (MutL homolog 1, 3%–15% incidence), tumor suppressor genes TP53 (50% incidence), and the Fanconi anemia pathway genes BRCA1/2 (breast cancer genes 1,2) and PALB2 (partner and localizer of BRCA2, 7% incidence in sporadic, and up to 17% in familial cases),168–171 as well as FANCC and FANCG (5%–10%).172–174 Other key factors and pathways in DNA repair including ATM/Chk2, ATR/Chk1, Rad51, ERCC1, and PTEN, can be mutated or inactivated in pancreatic cancer.175–177 Impaired DNA damage response pathways in pancreatic cancer create vulnerabilities that can be exploited therapeutically, and DNA cross-linking agents such as platinum and mitomycin C, topoisomerase inhibitors like irinotecan, and poly ADP-ribose polymerase (PARP) inhibitors are of particular interest.178 To date PARP inhibitors have demonstrated clinical activity as single agents in pancreatic cancers harboring BRCA1/2 or PALB2 mutations: olaparib demonstrated 22% response rates and median PFS and OS rates of 4.6 and 9.8 months, respectively in patients who received at least two prior lines of therapy, while veliparib showed 31% 4 months + stable disease rate, and a median PFS of approximately 2 months in refractory disease.179–181 Veliparib in combination with FOLFOX,182 gemcitabine/cisplatin183 or folinic acid, 5-FU, and irinotecan (FOLFIRI)184 chemotherapy in first or second line setting demonstrated sustained responses in BRCA mutated or non-mutated patients, and response rates were 14%–56%, and OS rates were up to 7.7 months. Clearly the DNA-repair pathway should be further explored and genomic characterization beyond BRCA1/2/PALB2 status should be determined for a “personalized” approach in a larger patient population.
Another hallmark of cancer is altered gene expression by variations that are independent from changes in DNA sequence.185–187 In pancreatic cancer, epigenetic dysregulation of tumor-associated genes, both tumor suppressors and oncogenes, can occur via DNA methylation, histone acetylation or microRNA (miRNA) expression. Epigenetic alterations have been implicated both in pancreatic cancer development and progression.188 Some the most common DNA methylation changes involve the promoter hypermethylation and silencing of the mismatch repair gene MLH1 (linked to the medullary subtype of pancreatic cancer),189 or of the CDKN2A/p16 gene,190 but several other methylation aberrations have been already reported.191 Histone acetylation can upregulate genes such as c-MYC,192 while deacetylation (with histone deacetylases – HDAC1 and HDAC2) results in transcriptional repression. HDAC1 can accelerate EMT and pancreatic cancer metastasis,193 and HDAC2 has been responsible for resistance to DNA damage.194 HDAC inhibitors were noted to have preclinical activity in pancreatic cancer mouse models by reinducing gene expression.195 Several miRNAs such as miRNA34a, a transcriptional target of p53, have been implicated in pancreatic tumor invasiveness, EMT, and stem cell maintenance.196,197 In pancreatic cancer cell lines re-expression of miRNA-34a after 5-aza-2′-deoxycytidine (Aza-dC) inhibited cell proliferation, cell cycle progression, EMT, and invasion.196 As many as 64 different miRNAs have been reported to be deregulated in pancreatic cancer, and many were associated with increased cancer risk or poor outcomes, thus could serve as future biomarkers.198 Epigenetically targeted therapeutics, including hypomethylating agents (eg, azacytidine, decitabine), and HDAC inhibitors (eg, vorinostat, belinostat, entinostat, panobinostat) have been explored in solid tumors clinical trials including pancreatic cancer patients, but miRNA-targeted therapeutics are not yet available.199,200 To date, despite promising preclinical data, HDAC inhibitors have not manifested meaningful efficacy in pancreatic cancer.201–204 CC-486 (oral azacytidine) is currently being tested in the adjuvant setting among high-risk-node-positive patients after pancreatic cancer resection and completion of adjuvant gemcitabine (NCT01845805).
Novel chemotherapies/formulations
MM-398 (Merrimack Pharmaceuticals) is a nanoparticle liposomal irinotecan formulation with improved biodistribution and pharmacokinetics which had been studied as monotherapy in second line treatment for pancreatic cancer, and showed moderate efficacy with PFS and OS rates of 2.4 and 5.2 months, respectively.205 The international Phase III NAPOLI 1 trial in second line metastatic pancreatic cancer tested MM-398 (80 mg/m2 iv over 90 minutes) with 5-FU every 2 weeks, vs 5-FU alone, vs MM-398 (120 mg/m2 iv over 90 minutes) every 3 weeks.206 MM-398 alone was not active, but in combination with 5-FU showed 16% response rates and it increased PFS and OS compared to 5-FU alone (PFS 3.1 vs 1.5 months, P=0.0001, and OS 6.1 vs 4.2 months, HR 0.57, P=0.0009).
Another novel chemotherapy, TH-302 is a hypoxia activated cytotoxic DNA cross-linking agent which has been tested with gemcitabine in a randomized Phase II study, and showed interesting activity at the 340 mg/m2 weekly dose ×3/4 weeks: response rates of 26%, median PFS 6 months and OS 9.2 months, compared to gemcitabine: 12% response, PFS and OS 3.6 and 6.9 months, respectively.207 Based on these encouraging results, the global Phase III MAESTRO trial (NCT01746979) of gemcitabine with and without TH-302 is currently ongoing, and due to preclinical synergy, a Phase Ib/II study of TH-302 with gemcitabine/nab-paclitaxel (NCT02047500) is also recruiting patients.
Conclusion
There currently are several promising therapies in pancreatic adenocarcinomas, between novel chemotherapeutics, stroma-and immune-targeted agents. While many obstacles still exist, such as defining biomarkers of benefit from signaling pathways inhibitors, and fostering an optimal immunological response in the context of an immunosuppressive tumor environment, clearly we are making progress in deciphering the heterogeneity within pancreatic cancers. Integrating conventional and immunological targeting will be the key to effective treatment of this deadly disease.
Disclosure
The authors have declared no conflict of interest.
References
Ferlay J, Soerjomataram I, Dikshit R, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136(5):E359–E386. | ||
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65(1):5–29. | ||
Bosetti C, Bertuccio P, Malvezzi M, et al. Cancer mortality in Europe, 2005–2009, and an overview of trends since 1980. Ann Oncol. 2013;24(10):2657–2671. | ||
Bosetti C, Bertuccio P, Negri E, La Vecchia C, Zeegers MP, Boffetta P. Pancreatic cancer: overview of descriptive epidemiology. Mol Carcinog. 2012;51(1):3–13. | ||
Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014;64(1):9–29. | ||
Finucane MM, Stevens GA, Cowan MJ, et al. National, regional, and global trends in body-mass index since 1980: systematic analysis of health examination surveys and epidemiological studies with 960 country-years and 9.1 million participants. Lancet. 2011;377(9765):557–567. | ||
Conroy T, Desseigne F, Ychou M, et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med. 2011;364(19):1817–1825. | ||
Von Hoff DD, Ervin T, Arena FP, et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med. 2013;369(18):1691–1703. | ||
Loehrer PJ Sr, Feng Y, Cardenes H, et al. Gemcitabine alone versus gemcitabine plus radiotherapy in patients with locally advanced pancreatic cancer: an Eastern Cooperative Oncology Group trial. J Clin Oncol. 2011;29(31):4105–4112. | ||
Moorcraft SY, Khan K, Peckitt C, et al. FOLFIRINOX for locally advanced or metastatic pancreatic ductal adenocarcinoma: The Royal Marsden Experience. Clin Colorectal Cancer. 2014;13(4):232–238. | ||
Hammel P, Huguet F, van Laethem JL, et al. Comparison of chemoradiotherapy and chemotherapy in patients with a locally advanced pancreatic cancer controlled after 4 months of gemcitabine with or without erlotinib: final results of the international phase III LAP-07 trial. J Clin Oncol. 2013;31 Suppl; abstr LBA4003. | ||
Neoptolemos JP, Stocken DD, Friess H, et al. A randomized trial of chemoradiotherapy and chemotherapy after resection of pancreatic cancer. N Engl J Med. 2004;350(12):1200–1210. | ||
Oettle H, Post S, Neuhaus P, et al. Adjuvant chemotherapy with gemcitabine vs observation in patients undergoing curative-intent resection of pancreatic cancer: a randomized controlled trial. JAMA. 2007;297(3):267–277. | ||
Oettle H, Neuhaus P, Hochhaus A, et al. Adjuvant chemotherapy with gemcitabine and long-term outcomes among patients with resected pancreatic cancer: the CONKO-001 randomized trial. JAMA. 2013;310(14):1473–1481. | ||
Neoptolemos JP, Stocken DD, Bassi C, et al. Adjuvant chemotherapy with fluorouracil plus folinic acid vs gemcitabine following pancreatic cancer resection: a randomized controlled trial. JAMA. 2010;304(10):1073–1081. | ||
Regine WF, Winter KA, Abrams R, et al. Fluorouracil-based chemoradiation with either gemcitabine or fluorouracil chemotherapy after resection of pancreatic adenocarcinoma: 5-year analysis of the U.S. Intergroup/RTOG 9704 phase III trial. Ann Surg Oncol. 2011;18(5):1319–1326. | ||
Faris JE, Blaszkowsky LS, McDermott S, et al. FOLFIRINOX in locally advanced pancreatic cancer: the Massachusetts General Hospital Cancer Center experience. Oncologist. 2013;18(5):543–548. | ||
Burris HA 3rd, Moore MJ, Andersen J, et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. J Clin Oncol. 1997;15(6):2403–2413. | ||
Cunningham D, Chau I, Stocken DD, et al. Phase III randomized comparison of gemcitabine versus gemcitabine plus capecitabine in patients with advanced pancreatic cancer. J Clin Oncol. 2009;27(33):5513–5518. | ||
Heinemann V, Quietzsch D, Gieseler F, et al. Randomized phase III trial of gemcitabine plus cisplatin compared with gemcitabine alone in advanced pancreatic cancer. J Clin Oncol. 2006;24(24):3946–3952. | ||
Poplin E, Feng Y, Berlin J, et al. Phase III, randomized study of gemcitabine and oxaliplatin versus gemcitabine (fixed-dose rate infusion) compared with gemcitabine (30-minute infusion) in patients with pancreatic carcinoma E6201: a trial of the Eastern Cooperative Oncology Group. J Clin Oncol. 2009;27(23):3778–3785. | ||
Kindler HL, Niedzwiecki D, Hollis D, et al. Gemcitabine plus bevacizumab compared with gemcitabine plus placebo in patients with advanced pancreatic cancer: phase III trial of the Cancer and Leukemia Group B (CALGB 80303). J Clin Oncol. 2010;28(22):3617–3622. | ||
Heinemann V, Vehling-Kaiser U, Waldschmidt D, et al. Gemcitabine plus erlotinib followed by capecitabine versus capecitabine plus erlotinib followed by gemcitabine in advanced pancreatic cancer: final results of a randomised phase 3 trial of the ‘Arbeitsgemeinschaft Internistische Onkologie’ (AIO-PK0104). Gut. 2013;62(5):751–759. | ||
Moore MJ, Goldstein D, Hamm J, et al. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol. 2007;25(15):1960–1966. | ||
da Cunha Santos G, Dhani N, Tu D, et al. Molecular predictors of outcome in a phase 3 study of gemcitabine and erlotinib therapy in patients with advanced pancreatic cancer: National Cancer Institute of Canada Clinical Trials Group Study PA.3. Cancer. 2010;116(24):5599–5607. | ||
Van Cutsem E, Li CP, Nowara E, et al. Dose escalation to rash for erlotinib plus gemcitabine for metastatic pancreatic cancer: the phase II RACHEL study. Br J Cancer. 2014;111(11):2067–2075. | ||
Hingorani SR, Wang L, Multani AS, et al. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell. 2005;7(5):469–483. | ||
Komar G, Kauhanen S, Liukko K, et al. Decreased blood flow with increased metabolic activity: a novel sign of pancreatic tumor aggressiveness. Clin Cancer Res. 2009;15(17):5511–5517. | ||
Mahadevan D, Von Hoff DD. Tumor-stroma interactions in pancreatic ductal adenocarcinoma. Mol Cancer Ther. 2007;6(4):1186–1197. | ||
Neesse A, Michl P, Frese KK, et al. Stromal biology and therapy in pancreatic cancer. Gut. 2011;60(6):861–868. | ||
Provenzano PP, Hingorani SR. Hyaluronan, fluid pressure, and stromal resistance in pancreas cancer. Br J Cancer. 2013;108(1):1–8. | ||
Provenzano PP, Cuevas C, Chang AE, Goel VK, Von Hoff DD, Hingorani SR. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell. 2012;21(3):418–429. | ||
Stromnes IM, DelGiorno KE, Greenberg PD, Hingorani SR. Stromal reengineering to treat pancreas cancer. Carcinogenesis. 2014;35(7):1451–1460. | ||
Fuchs DA, Johnson RK. Cytologic evidence that taxol, an antineoplastic agent from Taxus brevifolia, acts as a mitotic spindle poison. Cancer Treat Rep. 1978;62(8):1219–1222. | ||
Frese KK, Neesse A, Cook N, et al. nab-Paclitaxel potentiates gemcitabine activity by reducing cytidine deaminase levels in a mouse model of pancreatic cancer. Cancer Discov. 2012;2(3):260–269. | ||
Sato N, Fukushima N, Maehara N, et al. SPARC/osteonectin is a frequent target for aberrant methylation in pancreatic adenocarcinoma and a mediator of tumor-stromal interactions. Oncogene. 2003;22(32):5021–5030. | ||
Neuzillet C, Tijeras-Raballand A, Cros J, Faivre S, Hammel P, Raymond E. Stromal expression of SPARC in pancreatic adenocarcinoma. Cancer Metastasis Rev. 2013;32(3–4):585–602. | ||
Infante JR, Matsubayashi H, Sato N, et al. Peritumoral fibroblast SPARC expression and patient outcome with resectable pancreatic adenocarcinoma. J Clin Oncol. 2007;25(3):319–325. | ||
Sinn M, Sinn BV, Striefler JK, et al. SPARC expression in resected pancreatic cancer patients treated with gemcitabine: results from the CONKO-001 study. Ann Oncol. 2014;25(5):1025–1032. | ||
Yardley DA, Daniel BR, Inhorn RC, et al. SPARC microenvironment signature analysis of a phase II trial of neoadjuvant gemcitabine, epirubicin, and nab-paclitaxel in locally advanced breast cancer. J Clin Oncol. 2010;28 15 Suppl; abstr 10574. | ||
Markovic S, Suman V, Trieu VN, et al. Tumor SPARC microenvironment signature and plasma levels in a phase II trial of unresectable stage IV melanoma treated with nab-paclitaxel and carboplatin: a translational study of NCCTG trial N057E. J Clin Oncol. 2010;28 15 Suppl; abstr 8578. | ||
Von Hoff DD, Ramanathan RK, Borad MJ, et al. Gemcitabine plus nab-paclitaxel is an active regimen in patients with advanced pancreatic cancer: a phase I/II trial. J Clin Oncol. 2011;29(34):4548–4554. | ||
Hidalgo M, Plaza C, Illei P, et al. SPARC analysis in the phase III MPACT trial of nab-paclitaxel plus gemcitabine versus gemcitabine alone for patients with metastatic pancreatic cancer. Ann Oncol. 2014;25 Suppl 2; abstr O-0004. | ||
MacKenzie S, Zeh H, McCahill LE, et al. A pilot phase II multicenter study of nab-paclitaxel and gemcitabine as preoperative therapy forpotentially resectable pancreatic cancer. J Clin Oncol. 2013;31 Suppl; abstr 4038. | ||
Neesse A, Frese KK, Chan DS, et al. SPARC independent drug delivery and antitumour effects of nab-paclitaxel in genetically engineered mice. Gut. 2014;63(6):974–983. | ||
Ko AH, Truong TG, Kantoff E, et al. A phase I trial of nab-paclitaxel, gemcitabine, and capecitabine for metastatic pancreatic cancer. Cancer Chemother Pharmacol. 2012;70(6):875–881. | ||
Hosein PJ, de Lima Lopes G Jr, Pastorini VH, et al. A phase II trial of nab-Paclitaxel as second-line therapy in patients with advanced pancreatic cancer. Am J Clin Oncol. 2013;36(2):151–156. | ||
Ramfidis VS, Syrigos KN, Saif MW. New therapeutic strategies in the second line setting of advanced or metastatic pancreatic adenocarcinoma. JOP. 2013;14(4):344–346. | ||
Ernani V, Akunyili II, Hosein P, Macintyre J, Rocha Lima CMS. Gemcitabine and nab-paclitaxel in patients with refractory advanced pancreatic cancer. J Clin Oncol. 2012;30 Suppl 4; abstr 373. | ||
Leichman LP, O’Neil BH, Berlin J, et al. A phase IB study of erlotinib in combination with gemcitabine and nab-paclitaxel in patients with previously untreated advanced pancreatic cancer: an academic GI cancer consortium (AGICC). J Clin Oncol. 2012;30 Suppl; abstr 4052. | ||
El-Khoueiry AB, Iqbal S, Lenz H, et al. A phase I study of two different schedules of nab-paclitaxel with ascending doses of vandetanib with expansion in patients with pancreatic cancer. J Clin Oncol. 2011;29 Suppl; abstr 4124. | ||
Cassinotto C, Mouries A, Lafourcade JP, et al. Locally advanced pancreatic adenocarcinoma: reassessment of response with CT after neoadjuvant chemotherapy and radiation therapy. Radiology. 2014;273(1):108–116. | ||
Alvarez R, Musteanu M, Garcia-Garcia E, et al. Stromal disrupting effects of nab-paclitaxel in pancreatic cancer. Br J Cancer. 2013;109(4):926–933. | ||
MacKenzie S, Zeh H, McCahill LE, et al. A pilot phase II multicenter study of nab-paclitaxel and gemcitabine as preoperative therapy forpotentially resectable pancreatic cancer. J Clin Oncol. 2013;31 Suppl; abstr 4038. | ||
Ychou M, Conroy T, Seitz JF, et al. An open phase I study assessing the feasibility of the triple combination: oxaliplatin plus irinotecan plus leucovorin/5-fluorouracil every 2 weeks in patients with advanced solid tumors. Ann Oncol. 2003;14(3):481–489. | ||
Conroy T, Paillot B, Francois E, et al. Irinotecan plus oxaliplatin and leucovorin-modulated fluorouracil in advanced pancreatic cancer – a Groupe Tumeurs Digestives of the Federation Nationale des Centres de Lutte Contre le Cancer study. J Clin Oncol. 2005;23(6):1228–1236. | ||
Gunturu KS, Yao X, Cong X, et al. FOLFIRINOX for locally advanced and metastatic pancreatic cancer: single institution retrospective review of efficacy and toxicity. Med Oncol. 2013;30(1):361. | ||
Peddi PF, Lubner S, McWilliams R, et al. Multi-institutional experience with FOLFIRINOX in pancreatic adenocarcinoma. JOP. 2012;13(5):497–501. | ||
Boone BA, Steve J, Krasinskas AM, et al. Outcomes with FOLFIRINOX for borderline resectable and locally unresectable pancreatic cancer. J Surg Oncol. 2013;108(4):236–241. | ||
Iacobuzio-Donahue CA, Fu B, Yachida S, et al. DPC4 gene status of the primary carcinoma correlates with patterns of failure in patients with pancreatic cancer. J Clin Oncol. 2009;27(11):1806–1813. | ||
Klinkenbijl JH, Jeekel J, Sahmoud T, et al. Adjuvant radiotherapy and 5-fluorouracil after curative resection of cancer of the pancreas and periampullary region: phase III trial of the EORTC gastrointestinal tract cancer cooperative group. Ann Surg. 1999;230(6):776–782. | ||
Chauffert B, Mornex F, Bonnetain F, et al. Phase III trial comparing intensive induction chemoradiotherapy (60 Gy, infusional 5-FU and intermittent cisplatin) followed by maintenance gemcitabine with gemcitabine alone for locally advanced unresectable pancreatic cancer. Definitive results of the 2000–2001 FFCD/SFRO study. Ann Oncol. 2008;19(9):1592–1599. | ||
Feig C, Gopinathan A, Neesse A, Chan DS, Cook N, Tuveson DA. The pancreas cancer microenvironment. Clin Cancer Res. 2012;18(16):4266–4276. | ||
Clark CE, Hingorani SR, Mick R, Combs C, Tuveson DA, Vonderheide RH. Dynamics of the immune reaction to pancreatic cancer from inception to invasion. Cancer Res. 2007;67(19):9518–9527. | ||
Stromnes IM, Brockenbrough JS, Izeradjene K, et al. Targeted depletion of an MDSC subset unmasks pancreatic ductal adenocarcinoma to adaptive immunity. Gut. 2014;63(11):1769–1781. | ||
Dodson LF, Hawkins WG, Goedegebuure P. Potential targets for pancreatic cancer immunotherapeutics. Immunotherapy. 2011;3(4):517–537. | ||
Johnston FM, Tan MC, Tan BR Jr, et al. Circulating mesothelin protein and cellular antimesothelin immunity in patients with pancreatic cancer. Clin Cancer Res. 2009;15(21):6511–6518. | ||
Hamanaka Y, Suehiro Y, Fukui M, Shikichi K, Imai K, Hinoda Y. Circulating anti-MUC1 IgG antibodies as a favorable prognostic factor for pancreatic cancer. Int J Cancer. 2003;103(1):97–100. | ||
Pages F, Galon J, Dieu-Nosjean MC, Tartour E, Sautes-Fridman C, Fridman WH. Immune infiltration in human tumors: a prognostic factor that should not be ignored. Oncogene. 2010;29(8):1093–1102. | ||
Nomi T, Sho M, Akahori T, et al. Clinical significance and therapeutic potential of the programmed death-1 ligand/programmed death-1 pathway in human pancreatic cancer. Clin Cancer Res. 2007;13(7):2151–2157. | ||
Hiraoka N, Onozato K, Kosuge T, Hirohashi S. Prevalence of FOXP3+ regulatory T cells increases during the progression of pancreatic ductal adenocarcinoma and its premalignant lesions. Clin Cancer Res. 2006;12(18):5423–5434. | ||
Ino Y, Yamazaki-Itoh R, Shimada K, et al. Immune cell infiltration as an indicator of the immune microenvironment of pancreatic cancer. Br J Cancer. 2013;108(4):914–923. | ||
Beatty GL, Chiorean EG, Fishman MP, et al. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science. 2011;331(6024):1612–1616. | ||
Beatty GL, Torigian DA, Chiorean EG, et al. A phase I study of an agonist CD40 monoclonal antibody (CP-870,893) in combination with gemcitabine in patients with advanced pancreatic ductal adenocarcinoma. Clin Cancer Res. 2013;19(22):6286–6295. | ||
Mitchem JB, Brennan DJ, Knolhoff BL, et al. Targeting tumor-infiltrating macrophages decreases tumor-initiating cells, relieves immunosuppression, and improves chemotherapeutic responses. Cancer Res. 2013;73(3):1128–1141. | ||
Wang-Gillam A, Nyweing TM, Sanford DE, et al. Phase IB study of FOLFIRINOX plus PF-04136309 in patients with borderline resectable and locally advanced pancreatic adenocarcinoma. J Clin Oncol. 2015;33 3 Suppl; abstr 338. | ||
Lawrence MS, Stojanov P, Polak P, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013;499(7457):214–218. | ||
Brahmer JR, Tykodi SS, Chow LQ, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366(26):2455–2465. | ||
Royal RE, Levy C, Turner K, et al. Phase 2 trial of single agent Ipilimumab (anti-CTLA-4) for locally advanced or metastatic pancreatic adenocarcinoma. J Immunother. 2010;33(8):828–833. | ||
Chung MJ, Park JY, Bang S, Park SW, Song SY. Phase II clinical trial of ex vivo-expanded cytokine-induced killer cells therapy in advanced pancreatic cancer. Cancer Immunol Immunother. 2014;63(9):939–946. | ||
Stromnes IM, Schmitt TM, Chapuis AG, Hingorani SR, Greenberg PD. Re-adapting T cells for cancer therapy: from mouse models to clinical trials. Immunol Rev. 2014;257(1):145–164. | ||
Rossi GR, Mautino MR, Unfer RC, Seregina TM, Vahanian N, Link CJ. Effective treatment of preexisting melanoma with whole cell vaccines expressing alpha(1,3)-galactosyl epitopes. Cancer Res. 2005;65(22):10555–10561. | ||
Hardacre JM, Mulcahy M, Small W, et al. Addition of algenpantucel-L immunotherapy to standard adjuvant therapy for pancreatic cancer: a phase 2 study. J Gastrointest Surg. 2013;17(1):94–100. | ||
Rossi GR, Hardacre JM, Mulcahy MF, et al. Algenpantucel-L immunotherapy for pancreatic cancer induces anti-mesothelin antibody titers that positively correlate with improved overall survival. J Clin Oncol. 2013;31 Suppl; abstr 3007. | ||
Rossi GR, Rocha Lima CM, Hardacre JM, et al. Anti-calreticulin antibody titers correlate with improved overall survival in a phase 2 clinical trial of algenpantucel-l immunotherapy for patients with resected pancreatic cancer. J Clin Oncol. 2014;32 5s Suppl; abstr 3029. | ||
Lutz E, Yeo CJ, Lillemoe KD, et al. A lethally irradiated allogeneic granulocyte-macrophage colony stimulating factor-secreting tumor vaccine for pancreatic adenocarcinoma. A Phase II trial of safety, efficacy, and immune activation. Ann Surg. 2011;253(2):328–335. | ||
Le DT, Wang-Gillam A, Picozzi V, et al. Safety and survival with GVAX pancreas prime and listeria monocytogenes-expressing mesothelin (CRS-207) boost vaccines for metastatic pancreatic cancer. J Clin Oncol. Epub Jan 15, 2015. | ||
Le DT, Lutz E, Uram JN, et al. Evaluation of ipilimumab in combination with allogeneic pancreatic tumor cells transfected with a GM-CSF gene in previously treated pancreatic cancer. J Immunother. 2013;36(7):382–389. | ||
Dalgleish AG, The IMAGE I Trial Investigators. A multicenter randomized, open-label, proof-of-concept, phase II trial comparing gemcitabine with and without IMM-101 in advanced pancreatic cancer. J Clin Oncol. 2015;33 3 Suppl; abstr 336. | ||
Gjertsen MK, Buanes T, Rosseland AR, et al. Intradermal ras peptide vaccination with granulocyte-macrophage colony-stimulating factor as adjuvant: clinical and immunological responses in patients with pancreatic adenocarcinoma. Int J Cancer. 2001;92(3):441–450. | ||
Abou-Alfa GK, Chapman PB, Feilchenfeldt J, et al. Targeting mutated K-ras in pancreatic adenocarcinoma using an adjuvant vaccine. Am J Clin Oncol. 2011;34(3):321–325. | ||
Bernhardt SL, Gjertsen MK, Trachsel S, et al. Telomerase peptide vaccination of patients with non-resectable pancreatic cancer: a dose escalating phase I/II study. Br J Cancer. 2006;95(11):1474–1482. | ||
Middleton GW, Valle J, Wadsley J, et al. A phase III randomized trial of chemoimmunotherapy comprising gemcitabine and capecitabine with or without telomerase vaccine GV1001 in patients with locally advanced or metastatic pancreatic cancer. J Clin Oncol. 2013;31 Suppl; abstr LBA4004. | ||
Pfeffer LM, Dinarello CA, Herberman RB, et al. Biological properties of recombinant alpha-interferons: 40th anniversary of the discovery of interferons. Cancer Res. 1998;58(12):2489–2499. | ||
Vitale G, van Eijck CH, van Koetsveld Ing PM, et al. Type I interferons in the treatment of pancreatic cancer: mechanisms of action and role of related receptors. Ann Surg. 2007;246(2):259–268. | ||
Recchia F, Sica G, Casucci D, Rea S, Gulino A, Frati L. Advanced carcinoma of the pancreas: phase II study of combined chemotherapy, beta-interferon, and retinoids. Am J Clin Oncol. 1998;21(3):275–278. | ||
Wang B, Xiong Q, Shi Q, Le X, Abbruzzese JL, Xie K. Intact nitric oxide synthase II gene is required for interferon-beta-mediated suppression of growth and metastasis of pancreatic adenocarcinoma. Cancer Res. 2001;61(1):71–75. | ||
Picozzi VJ, Kozarek RA, Traverso LW. Interferon-based adjuvant chemoradiation therapy after pancreaticoduodenectomy for pancreatic adenocarcinoma. Am J Surg. 2003;185(5):476–480. | ||
Rocha FG, Hashimoto Y, Traverso LW, et al. Interferon-based adjuvant chemoradiation for resected pancreatic head cancer: long-term follow-up of the Virginia Mason Protocol. Ann Surg. Epub 2015 Mar 13. | ||
Picozzi VJ, Abrams RA, Decker PA, et al. Multicenter phase II trial of adjuvant therapy for resected pancreatic cancer using cisplatin, 5-fluorouracil, and interferon-alfa-2b-based chemoradiation: ACOSOG Trial Z05031. Ann Oncol. 2011;22(2):348–354. | ||
Marten A, Schmidt J, Debus J, et al. CapRI- final results of the open-label, multi-center, randomized phase III trial of adjuvant chemoradiation plus interferon-α2b (CRI) versus 5FU/FA for patients with resected pancreatic adenocarcinoma. J Clin Oncol. 2010;28:18s Suppl; abstr LBA4012. | ||
Bramhall SR, Rosemurgy A, Brown PD, Bowry C, Buckels JA, Marimastat Pancreatic Cancer Study G. Marimastat as first-line therapy for patients with unresectable pancreatic cancer: a randomized trial. J Clin Oncol. 2001;19(15):3447–3455. | ||
Cascinu S, Berardi R, Sobrero A, et al. Sorafenib does not improve efficacy of chemotherapy in advanced pancreatic cancer: a GISCAD randomized phase II study. Dig Liver Dis. 2014;46(2):182–186. | ||
Kindler HL, Ioka T, Richel DJ, et al. Axitinib plus gemcitabine versus placebo plus gemcitabine in patients with advanced pancreatic adenocarcinoma: a double-blind randomised phase 3 study. Lancet Oncol. 2011;12(3):256–262. | ||
Kindler HL, Wroblewski K, Wallace JA, et al. Gemcitabine plus sorafenib in patients with advanced pancreatic cancer: a phase II trial of the University of Chicago Phase II Consortium. Invest New Drugs. 2012;30(1):382–386. | ||
Jacobetz MA, Chan DS, Neesse A, et al. Hyaluronan impairs vascular function and drug delivery in a mouse model of pancreatic cancer. Gut. 2013;62(1):112–120. | ||
Hingorani SR, Harris WP, Beck JT, et al. Final results of a phase Ib study of gemcitabine plus PEGPH20 in patients with stage IV previously untreated pancreatic cancer. J Clin Oncol. 2015;33(3s) Suppl; abstr 359. | ||
Picozzi VJ, Pipas JM, Koong A, et al. FG-3019, a human monoclonal antibody to connective tissue growth factor, with gemcitabine/erlotinib in patients with locally advanced or metastatic pancreatic ductal adenocarcinoma. J Clin Oncol. 2014;32(5s) Suppl; abstr 4138. | ||
Rocha F, Helton S, Picozzi V. A randomized, open-label, phase I/II trial of gemcitabine plus nab-paclitaxel with or without FG-3019 as neoadjuvant chemotherapy in locally advanced, unresectable pancreatic cancer. J Clin Oncol. 2015;33(3s) Suppl; abstr TPS500. | ||
Jones S, Zhang X, Parsons DW, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321(5897):1801–1806. | ||
Biankin AV, Waddell N, Kassahn KS, et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature. 2012;491(7424):399–405. | ||
Macdonald JS, McCoy S, Whitehead RP, et al. A phase II study of farnesyl transferase inhibitor R115777 in pancreatic cancer: a Southwest oncology group (SWOG 9924) study. Invest New Drugs. 2005;23(5):485–487. | ||
Rich TA, Winter K, Safran H, et al. Weekly paclitaxel, gemcitabine, and external irradiation followed by randomized farnesyl transferase inhibitor R115777 for locally advanced pancreatic cancer. Onco Targets Ther. 2012;5:161–170. | ||
Rinehart J, Adjei AA, Lorusso PM, et al. Multicenter phase II study of the oral MEK inhibitor, CI-1040, in patients with advanced non-small-cell lung, breast, colon, and pancreatic cancer. J Clin Oncol. 2004;22(22):4456–4462. | ||
Bodoky G, Timcheva C, Spigel DR, et al. A phase II open-label randomized study to assess the efficacy and safety of selumetinib (AZD6244 [ARRY-142886]) versus capecitabine in patients with advanced or metastatic pancreatic cancer who have failed first-line gemcitabine therapy. Invest New Drugs. 2012;30(3):1216–1223. | ||
Hamidi H, Lu M, Chau K, et al. KRAS mutational subtype and copy number predict in vitro response of human pancreatic cancer cell lines to MEK inhibition. Br J Cancer. 2014;111(9):1788–1801. | ||
Collisson EA, Sadanandam A, Olson P, et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat Med. 2011;17(4):500–503. | ||
Mirzoeva OK, Collisson EA, Schaefer PM, et al. Subtype-specific MEK-PI3 kinase feedback as a therapeutic target in pancreatic adenocarcinoma. Mol Cancer Ther. 2013;12(10):2213–2225. | ||
Infante JR, Somer BG, Park JO, et al. A randomised, double-blind, placebo-controlled trial of trametinib, an oral MEK inhibitor, in combination with gemcitabine for patients with untreated metastatic adenocarcinoma of the pancreas. Eur J Cancer. 2014;50(12):2072–2081. | ||
Van Cutsem E, Hidalgo M, Bazin I, et al. Phase II randomized trial of MEK inhibitor pimasertib or placebo combined with gemcitabine in the first-line treatment of metastatic pancreatic cancer. J Clin Oncol. 2015;33(3s) Suppl; abstr 344. | ||
Van Laethem JL, Jassem J, Heinemann V, et al. Phase II study of refametinib (BAY 86-9766), an allosteric dual MEK 1/2 inhibitor, and gemcitabine in patients with unresectable, locally advanced, or metastatic pancreatic cancer. J Clin Oncol. 2014;32(5s) Suppl; abstr 4025. | ||
Ko AH, Tempero M, Bekaii-Saab TB, et al. Dual MEK/EGFR inhibition for advanced, chemotherapy-refractory pancreatic cancer: a multicenter phase II trial of selumetinib (AZD6244; ARRY-142886) plus erlotinib. J Clin Oncol. 2013;31(s) Suppl; abstr 4014. | ||
Alagesan B, Contino G, Guimaraes AR, et al. Combined MEK and PI3K inhibition in a mouse model of pancreatic cancer. Clin Cancer Res. 2015;21(2):396–404. | ||
Bedard PL, Tabernero J, Janku F, et al. A phase Ib dose-escalation study of the oral pan-PI3K inhibitor buparlisib (BKM120) in combination with the oral MEK1/2 inhibitor trametinib (GSK1120212) in patients with selected advanced solid tumors. Clin Cancer Res. 2015;21(4):730–738. | ||
Tolcher AW, Bendell JC, Papadopoulos KP, et al. A phase IB trial of the oral MEK inhibitor trametinib (GSK1120212) in combination with everolimus in patients with advanced solid tumors. Ann Oncol. 2015;26(1):58–64. | ||
Wolpin BM, Hezel AF, Abrams T, et al. Oral mTOR inhibitor everolimus in patients with gemcitabine-refractory metastatic pancreatic cancer. J Clin Oncol. 2009;27(2):193–198. | ||
Garrido-Laguna I, Tan AC, Uson M, et al. Integrated preclinical and clinical development of mTOR inhibitors in pancreatic cancer. Br J Cancer. 2010;103(5):649–655. | ||
Javle MM, Shroff RT, Xiong H, et al. Inhibition of the mammalian target of rapamycin (mTOR) in advanced pancreatic cancer: results of two phase II studies. BMC Cancer. 2010;10:368. | ||
Scott AJ, O’Neill B, Gomes C, et al. A phase II/III randomized study to compare the efficacy and safety of rigosertib plus gemcitabine versus gemcitabine alone in patients with previously untreated metastatic pancreatic cancer. J Clin Oncol. 2015;33(3s) Suppl; abstr 342. | ||
van Veelen W, Korsse SE, van de Laar L, Peppelenbosch MP. The long and winding road to rational treatment of cancer associated with LKB1/AMPK/TSC/mTORC1 signaling. Oncogene. 2011;30(20):2289–2303. | ||
Utomo WK, Narayanan V, Biermann K, et al. mTOR is a promising therapeutical target in a subpopulation of pancreatic adenocarcinoma. Cancer Lett. 2014;346(2):309–317. | ||
Klumpen HJ, Queiroz KC, Spek CA, et al. mTOR inhibitor treatment of pancreatic cancer in a patient with Peutz-Jeghers syndrome. J Clin Oncol. 2011;29(6):e150–e153. | ||
Philip PA, Benedetti J, Corless CL, et al. Phase III study comparing gemcitabine plus cetuximab versus gemcitabine in patients with advanced pancreatic adenocarcinoma: Southwest Oncology Group-directed intergroup trial S0205. J Clin Oncol. 2010;28(22):3605–3610. | ||
Benavides M, Gallego Plazas J, Guillen C, et al. Gemcitabine/erlotinib versus gemcitabine/erlotinib/capecitabine in the first-line treatment of patients with metastatic pancreatic cancer: efficacy and safety results of a phase IIb randomized study from the Spanish TTD Collaborative Group. J Clin Oncol. 2014;32(5s) Suppl; abstr 4122. | ||
Yun J, Kim HJ, Kim KH, et al. A phase II study of gemcitabine, oxaliplatin, and erlotinib (GEMOX-T) combination chemotherapy in previously untreated patients with locally advanced unresectable or metastatic pancreatic cancer. J Clin Oncol. 2014;32(s) Suppl; abstr e15260. | ||
Katopodis O, Souglakos J, Stathopoulos E, et al. Frontline treatment with gemcitabine, oxaliplatin and erlotinib for the treatment of advanced or metastatic pancreatic cancer: a multicenter phase II study of the Hellenic Oncology Research Group (HORG). Cancer Chemother Pharmacol. 2014;74(2):333–340. | ||
Leichman LP, O’Neill BH, Berlin J, et al. A phase IB study of erlotinib in combination with gemcitabine and nab-paclitaxel in patients with previously untreated advanced pancreatic cancer: an Academic GI Cancer Consortium (AGICC) study. J Clin Oncol. 2012;30 Suppl; abstr 4052. | ||
Kim GP, Foster NR, Salim M, et al. Randomized phase II trial of panitumumab, erlotinib, and gemcitabine versus erlotinib-gemcitabine in patients with untreated, metastatic pancreatic adenocarcinoma. J Clin Oncol. 2011;29(s) Suppl; abstr 4030. | ||
Assenat E, Mineur L, Mollevi C, et al. Phase II study evaluating the association of gemcitabine, trastuzumab, and erlotinib as first-line treatment in patients with metastatic pancreatic adenocarcinoma (GATE 1). J Clin Oncol. 2015;33(3s) Suppl; abstr 379. | ||
Philip PA, Goldman B, Ramanathan RK, et al. Dual blockade of epidermal growth factor receptor and insulin-like growth factor receptor-1 signaling in metastatic pancreatic cancer: phase Ib and randomized phase II trial of gemcitabine, erlotinib, and cixutumumab versus gemcitabine plus erlotinib (SWOG S0727). Cancer. 2014;120(19):2980–2985. | ||
Cohen DJ, Leichman LP, Love E, et al. Phase II study of sorafenib with gemcitabine and erlotinib in first-line advanced pancreatic cancer. J Clin Oncol. 2011;29 Suppl 4; abstr 266. | ||
Chou A, Waddell N, Cowley MJ, et al. Clinical and molecular characterization of HER2 amplified-pancreatic cancer. Genome Med. 2013;5(8):78. | ||
Safran H, Iannitti D, Ramanathan R, et al. Herceptin and gemcitabine for metastatic pancreatic cancers that overexpress HER-2/neu. Cancer Invest. 2004;22(5):706–712. | ||
Harder J, Ihorst G, Heinemann V, et al. Multicentre phase II trial of trastuzumab and capecitabine in patients with HER2 overexpressing metastatic pancreatic cancer. Br J Cancer. 2012;106(6):1033–1038. | ||
Safran H, Miner T, Bahary N, et al. Lapatinib and gemcitabine for metastatic pancreatic cancer. A phase II study. Am J Clin Oncol. 2011;34(1):50–52. | ||
Olive KP, Jacobetz MA, Davidson CJ, et al. Inhibition of hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science. 2009;324(5933):1457–1461. | ||
Catenacci DVT, Bahari N, Edelman MJ, et al. A phase IB/randomized phase II study of gemcitabine plus placebo or vismodegib, a hedgehog (Hh) pathway inhibitor, in patients with metastatic pancreatic cancer: interim analysis of a University of Chicago phase II consortium study. J Clin Oncol. 2012;30(s) Suppl; abstr 4022. | ||
Kim EJ, Sahai V, Abel EV, et al. Pilot clinical trial of hedgehog pathway inhibitor GDC-0449 (vismodegib) in combination with gemcitabine in patients with metastatic pancreatic adenocarcinoma. Clin Cancer Res. 2014;20(23):5937–5945. | ||
De Jesus-Acosta A, O’Dwyer PJ, Ramanathan RK, et al. A phase II study of vismodegib, a hedgehog pathway inhibitor, combined with gemcitabine and nab-paclitaxel in patients with untreated metastatic pancreatic ductal adenocarcinoma. J Clin Oncol. 2014;32(3s) Suppl; abstr 257. | ||
Ko AH, LoConte N, Kantoff E, et al. A phase Ib trial of FOLFIRINOX plus saridegib, an oral hedgehog inhibitor, in patients with advanced pancreatic cancer. J Clin Oncol. 2012;30(s) Suppl; abstr 3105. | ||
Rhim AD, Oberstein PE, Thomas DH, et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell. 2014;25(6):735–747. | ||
Ozdemir BC, Pentcheva-Hoang T, Carstens JL, et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell. 2014;25(6):719–734. | ||
Ranganathan P, Weaver KL, Capobianco AJ. Notch signalling in solid tumours: a little bit of everything but not all the time. Nat Rev Cancer. 2011;11(5):338–351. | ||
Avila JL, Kissil JL. Notch signaling in pancreatic cancer: oncogene or tumor suppressor? Trends Mol Med. 2013;19(5):320–327. | ||
Yen WC, Fischer MM, Hynes M, et al. Anti-DLL4 has broad spectrum activity in pancreatic cancer dependent on targeting DLL4-Notch signaling in both tumor and vasculature cells. Clin Cancer Res. 2012;18(19):5374–5386. | ||
O’Reilly EM, Smith L, Bendell JC, et al. Final results of phase Ib of anticancer stem cell antibody tarextumab (OMP-59R5, TRXT, anti-Notch 2/3) in combination with nab-paclitaxel and gemcitabine in patients with untreated metastatic pancreatic cancer. J Clin Oncol. 2015;33(3s) Suppl; abstr 278. | ||
Cook N, Basu B, Smith DM, et al. A phase I trial of the γ-secretase inhibitor MK-0752 in combination with gemcitabine in patients with pancreatic ductal adenocarcinoma. J Clin Oncol. 2014;32(5s) Suppl; abstr 4116. | ||
Cubillo Gracian A, Jameson M, Grande E, et al. A phase 1b study of the anticancer stem cell agent demcizumab and gemcitabine with or without paclitaxel protein bound particles (nab-paclitaxel) in patients with pancreatic cancer. J Clin Oncol. 2014;32(3s) Suppl; abstr 279. | ||
Morris JP, Wang SC, Hebrok M. KRAS, Hedgehog, Wnt and the twisted developmental biology of pancreatic ductal adenocarcinoma. Nat Rev Cancer. 2010;10(10):683–695. | ||
Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer. 2009;9(11):798–809. | ||
Steele CW, Jamieson NB, Evans TR, et al. Exploiting inflammation for therapeutic gain in pancreatic cancer. Br J Cancer. 2013;108(5):997–1003. | ||
Aoki H, Ohnishi H, Hama K, et al. Cyclooxygenase-2 is required for activated pancreatic stellate cells to respond to proinflammatory cytokines. Am J Physiol Cell Physiol. 2007;292(1):C259–C268. | ||
Panni RZ, Sanford DE, Belt BA, et al. Tumor-induced STAT3 activation in monocytic myeloid-derived suppressor cells enhances stemness and mesenchymal properties in human pancreatic cancer. Cancer Immunol Immunother. 2014;63(5):513–528. | ||
Pino MS, Milella M, Gelibter A, et al. Capecitabine and celecoxib as second-line treatment of advanced pancreatic and biliary tract cancers. Oncology. 2009;76(4):254–261. | ||
El-Rayes BF, Zalupski MM, Shields AF, et al. A phase II study of celecoxib, gemcitabine, and cisplatin in advanced pancreatic cancer. Invest New Drugs. 2005;23(6):583–590. | ||
Siveen KS, Sikka S, Surana R, et al.Targeting the STAT3 signaling pathway in cancer: role of synthetic and natural inhibitors. Biochim Biophys Acta. 2014;1845(2):136–154. | ||
Hurwitz H, Uppal N, Wagner SA, et al. A randomized double-blind phase 2 study of ruxolitinib or placebo with capecitabine as second-line therapy in patients with metastatic pancreatic cancer. J Clin Oncol. 2014;32(5s) Suppl; abstr 4000. | ||
Naderi A, Couch FJ. BRCA2 and pancreatic cancer. Int J Gastrointest Cancer. 2002;31(1–3):99–106. | ||
Wang SC, Shao R, Pao AY, Zhang S, Hung MC, Su LK. Inhibition of cancer cell growth by BRCA2. Cancer Res. 2002;62(5):1311–1314. | ||
Hruban RH, Maitra A, Schulick R, et al. Emerging molecular biology of pancreatic cancer. Gastrointest Cancer Res. 2008;2(4 Suppl):S10–S15. | ||
Goggins M, Schutte M, Lu J, et al. Germline BRCA2 gene mutations in patients with apparently sporadic pancreatic carcinomas. Cancer Res. 1996;56(23):5360–5364. | ||
van der Heijden MS, Brody JR, Gallmeier E, et al. Functional defects in the fanconi anemia pathway in pancreatic cancer cells. Am J Pathol. 2004;165(2):651–657. | ||
van der Heijden MS, Yeo CJ, Hruban RH, Kern SE. Fanconi anemia gene mutations in young-onset pancreatic cancer. Cancer Res. 2003;63(10):2585–2588. | ||
Couch FJ, Johnson MR, Rabe K, et al. Germ line Fanconi anemia complementation group C mutations and pancreatic cancer. Cancer Res. 2005;65(2):383–386. | ||
Smith J, Tho LM, Xu N, Gillespie DA. The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and cancer. Adv Cancer Res. 2010;108:73–112. | ||
Tong X, Liu B, Dong Y, Sun Z. Cleavage of ATM during radiation-induced apoptosis: caspase-3-like apoptotic protease as a candidate. Int J Rad Biol. 2000;76(10):1387–1395. | ||
McEllin B, Camacho CV, Mukherjee B, et al. PTEN loss compromises homologous recombination repair in astrocytes: implications for glioblastoma therapy with temozolomide or poly(ADP-ribose) polymerase inhibitors. Cancer Res. 2010;70(13):5457–5464. | ||
Gartner EM, Burger AM, Lorusso PM. Poly(adp-ribose) polymerase inhibitors: a novel drug class with a promising future. Cancer. 2010;16(2):83–90. | ||
Lowery MA, Kelsen DP, Stadler ZK, et al. An emerging entity: pancreatic adenocarcinoma associated with a known BRCA mutation: clinical descriptors, treatment implications, and future directions. Oncologist. 2011;16(10):1397–1402. | ||
Kaufman B, Shapira-Frommer R, Schmutzler RK, et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J Clin Oncol. 2015;33(3):244–250. | ||
Lowery MA, Kelsen DP, Smith SC, et al. Phase II trial of veliparib in patients with previously treated BRCA or PALB2 mutated pancreas adenocarcinoma. J Clin Oncol. 2015;33 Suppl 3:abstr 358. | ||
Pishvaian MJ, Wang H, Zhuang T, et al. A phase I/II study of ABT-888 in combination with 5-fluorouracil and oxaliplatin in patients with metastatic pancreatic cancer. J Clin Oncol. 2013;31(4s) Suppl; abstr 147. | ||
O’Reilly EM, Lowery MA, Segal MF, et al. Phase IB trial of cisplatin, gemcitabine, and veliparib in patients with known or potential BRCA or PALB2-mutated pancreas adenocarcinoma. J Clin Oncol. 2014;32(5s) Suppl; abstr 4023. | ||
Berlin J, Ramanathan RK, Strickler JH, et al. A phase 1 dose-escalation study of veliparib with bimonthly FOLFIRI in patients with advanced solid tumors. J Clin Oncol. 2014;32(5s) Suppl; abstr 2574. | ||
Holliday R. The inheritance of epigenetic defects. Science. 1987;238(4824):163–170. | ||
Ueki T, Toyota M, Sohn T, et al. Hypermethylation of multiple genes in pancreatic adenocarcinoma. Cancer Res. 2000;60(7):1835–1839. | ||
McCleary-Wheeler AL, Lomberk GA, Weiss FU, et al. Insights into the epigenetic mechanisms controlling pancreatic carcinogenesis. Cancer Lett. 2013;328(2):212–221. | ||
Hruban RH, Goggins M, Parsons J, Kern SE. Progression model for pancreatic cancer. Clin Ca Res. 2000;6(8):2969–2972. | ||
Wilentz RE, Goggins M, Redston M, et al. Genetic, immunohistochemical, and clinical features of medullary carcinoma of the pancreas: a newly described and characterized entity. Am J Pathol. 2000;156(5):1641–1651. | ||
Schutte M, Hruban RH, Geradts J, et al. Abrogation of the Rb/p16 tumor suppressive pathway in virtually all pancreatic carcinomas. Cancer Res. 1997;57(15):3126–3130. | ||
Vincent A, Omura N, Hong SM, Jaffe A, Eshleman J, Goggins M. Genome-wide analysis of promoter methylation associated with gene expression profile in pancreatic adenocarcinoma. Clin Ca Res. 2011;17(13):4341–4354. | ||
Koenig A, Linhart T, Schlengemann K, et al. NFAT-induced histone acetylation relay switch promotes c-Myc–dependent growth in pancreatic cancer cells. Gastroenterology. 2010;138(3):1189–1199. | ||
Ouaissi M, Cabral S, Tavares J, et al. Histone deacetylase (HDAC) encoding gene expression in pancreatic cancer cell lines and cell sensitivity to HDAC inhibitors. Cancer Biol Ther. 2008;7(4):523–531. | ||
Fritsche P, Seidler B, Schuler S, et al. HDAC2 mediates therapeutic resistance of pancreatic cancer cells via the BH3-only protein NOXA. Gut. 2009;58(10):1399–1409. | ||
Vincent A, Hong SM, Hu C, et al. Epigenetic silencing of EYA2 in pancreatic adenocarcinomas promotes tumor growth. Oncotarget. 2014;5(9):2575–2587. | ||
Bloomston M, Frankel WL, Petrocca F, et al. MicroRNA expression patterns to differentiate pancreatic adenocarcinoma from normal pancreas and chronic pancreatitis. JAMA. 2007;297(17):1901–1908. | ||
Nalls D, Tang SN, Rodova M, Srivastava RK, Shankar S. Targeting epigenetic regulation of miR-34a for treatment of pancreatic cancer by inhibition of pancreatic cancer stem cells. PLoS One. 2011;6:e24099. | ||
Lee EJ, Gusev Y, Jiang J, et al. Expression profiling identifies microRNA signature in pancreatic cancer. Int J Cancer. 2007;120(5):1046–1054. | ||
Koutsounas I, Giaginis C, Theocharis S. Histone deacetylase inhibitors and pancreatic cancer: are there any promising clinical trials? World J Gastroenterol. 2013;19(8):1173–1181. | ||
Neureiter D, Jager T, Ocker M, Kiesslich T. Epigenetics and pancreatic cancer: pathophysiology and novel treatment aspects. World J Gastroenterol. 2014;20(24):7830–7848. | ||
Millward M, Price T, Townsend A, et al. Phase 1 clinical trial of the novel proteasome inhibitor marizomib with the histone deacetylase inhibitor vorinostat in patients with melanoma, pancreatic and lung cancer based on in vitro assessments of the combination. Invest New Drugs. 2012;30(6):2303–2317. | ||
Pili R, Salumbides B, Zhao M, et al. Phase I study of the histone deacetylase inhibitor entinostat in combination with 13-cis retinoic acid in patients with solid tumours. Br J Cancer. 2012;106(1):77–84. | ||
Wang H, Cao Q, Dudek AZ. Phase II study of panobinostat and bortezomib in patients with pancreatic cancer progressing on gemcitabine-based therapy. Anticancer Res. 2012;32(3):1027–1031. | ||
Deming DA, Ninan J, Bailey HH, et al. A phase I study of intermittently dosed vorinostat in combination with bortezomib in patients with advanced solid tumors. Invest New Drugs. 2014;32(2):323–329. | ||
Ko AH, Tempero MA, Shan YS, et al. A multinational phase 2 study of nanoliposomal irinotecan sucrosofate (PEP02, MM-398) for patients with gemcitabine-refractory metastatic pancreatic cancer. Br J Cancer. 2013;109(4):920–925. | ||
Von Hoff D, Li CP, Wang-Gillam A, et al. Napoli-1: randomzied phase 3 study of MM-398 with or without 5-fluorouracil and leucovorin, versus 5-fluorouracil and leucovorin, in metastatic pancreatic cancer progressed on or following gemcitabine-based therapy. Ann Oncol. 2014;25 Suppl 2; abstract O-0003. | ||
Borad MJ, Reddy SG, Bahary N, et al. Randomized phase II trial of gemcitabine plus TH-302 versus gemcitabine in patients with advanced pancreatic cancer. J Clin Oncol. Epub Dec 17, 2014. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.