")
Back to Journals » Drug Design, Development and Therapy » Volume 9
Paget’s disease of bone: an osteoimmunological disorder?
Authors Numan M , Amiable N, Brown JP, Michou L
Received 19 May 2015
Accepted for publication 13 June 2015
Published 14 August 2015 Volume 2015:9 Pages 4695—4707
DOI https://doi.org/10.2147/DDDT.S88845
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Shu-Feng Zhou
Mohamed S Numan,1,2 Nathalie Amiable,1 Jacques P Brown,1–3 Laëtitia Michou1–3
1CHU de Québec Research Centre, 2Division of Rheumatology, Department of Medicine, 3Department of Rheumatology, CHU de Québec-Université Laval, Quebec City, QC, Canada
Abstract: Osteoimmunology represents a large area of research resulting from the cross talk between bone and immune systems. Many cytokines and signaling cascades are involved in the field of osteoimmunology, originating from various cell types. The RANK/receptor activator of nuclear factor Kappa-B ligand (RANKL)/osteoprotegerin (OPG) signaling has a pivotal role in osteoimmunology, in addition to proinflammatory cytokines such as tumor necrosis factor-α, interleukin (IL)-1, IL-6, and IL-17. Clinically, osteoimmunological disorders, such as rheumatoid arthritis, osteoporosis, and periodontitis, should be classified according to their pattern of osteoimmunological serum biomarkers. Paget’s disease of bone is a common metabolic bone disorder, resulting from an excessively increased bone resorption coupled with aberrant bone formation. With the exception of the cellular responses to measles virus nucleocapsid protein and the interferon-gamma signature, the exact role of the immune system in Paget’s disease of bone is not well understood. The cytokine profiles, such as the increased levels of IL-6 and the interferon-gamma signature observed in this disease, are also very similar to those observed in other osteoimmunological disorders. As a potential osteoimmunological disorder, the treatment of Paget’s disease of bone may also benefit from progress made in targeted therapies, in particular for receptor activator of nuclear factor Kappa-B ligand and IL-6 signaling inhibition.
Keywords: Paget’s disease of bone, SQSTM1/p62, osteoimmunology, osteoclast, RANKL
Introduction
Osteoimmunology at a glance
This narrative review of the literature presents first, data on osteoimmunology and osteoimmunological disorders, and second, discusses why Paget’s disease of bone should be considered as a potential osteoimmunological disease. Osteoimmunology is an emerging research area that is the result of the cross talk regarding the relationship between bones and the immune systems. Various cell types are involved in osteoimmunology processes, but most of them originate from the hematopoietic tissue. At the third week of human embryonic development, stem cell lineages are formed in the yolk sac.1,2 These lineages multiply asymmetrically, maintaining their original population and differentiating to other types of more specialized blood cells.3 The immunological basic steps begin with the primary formation of hematopoietic stem cells lineage, which first appears in the yolk sac, followed by mesoderms of aorta, gonads, and nephrons (mesonephrons), and from there will migrate later to liver, spleen, and lymph nodes. Around the fourth month of fetal life, these hematopoietic stem cells migrate to bone marrow, and at the time of birth, bone marrow is responsible for all hematopoietic function.4,5 Likewise, bone marrow pluripotent stem cells – which are stimulated by growth factors – are divided into two types of multipotent progenitor stem cells: common lymphoid cells and common myeloid cells (Figure 1).
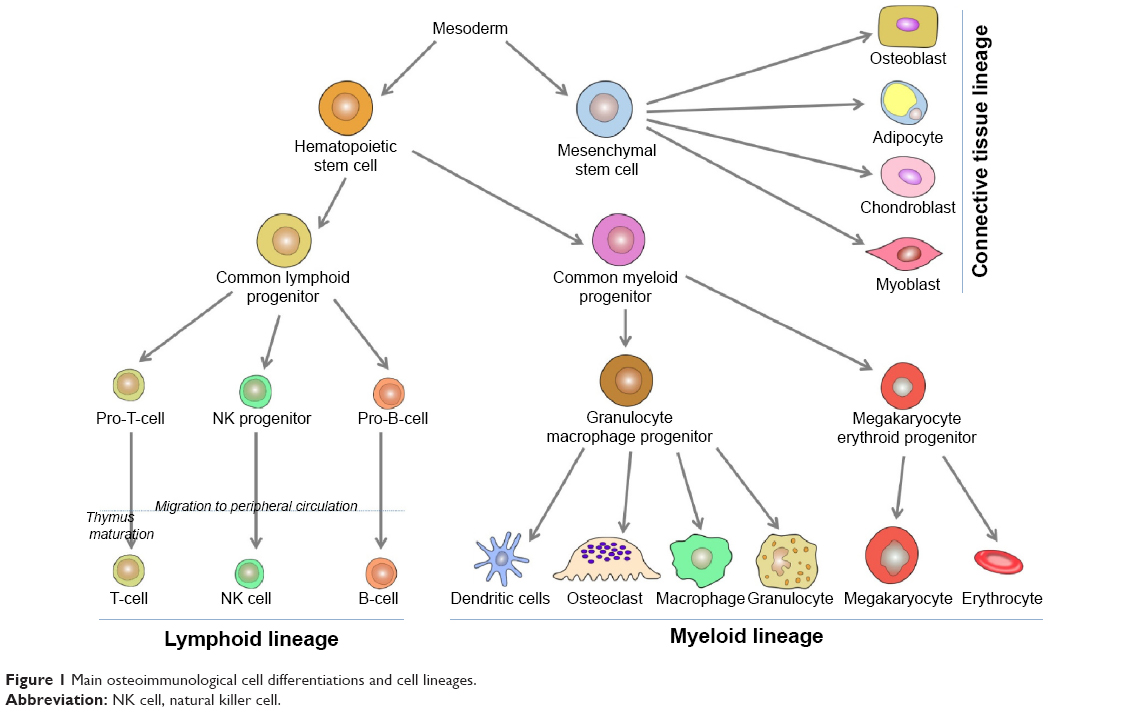 | Figure 1 Main osteoimmunological cell differentiations and cell lineages. |
Lymphoid cells
Common lymphoid cells are further divided into three cell types: committed pro-natural killer stem cells, pro-T stem cells, and pro-B stem cells. Pro-natural killer stem cells migrate to peripheral circulation, where they become natural killer cells. Natural killer cells differ from B-cells and T-cells by not having clusters of differentiation 3 (CD3) like T-cells, nor CD19 and CD20 like B-cells. Pro-T stem cells (naïve T-cells) migrate from bone marrow to peripheral circulation, mature in thymus gland, and then some return to peripheral circulation where they become mature T-cells. Some of these mature cells will become T helper (TH) naïve (containing markers CD3, CD4) lymphocytes or T cytotoxic (containing markers CD3, CD8) lymphocytes. TH naïve cells differentiate into TH1 cells and TH2 cells. TH1 cells when activated by interleukin (IL)-4 and IL-5 contribute to convert B-cells to plasma cells called active B-cells producing immunological antibodies. TH2, when activated by interferon-gamma (IFN-γ) and tumor necrosis factor-α (TNF-α), contribute to activate monocytes to be highly active macrophages, epithelioid cells (modified monocytes), and giant cells. IFN-γ receptor knockout mice showed exaggerated bone destruction in inflammatory arthritis in comparison with normal mice. IFN-γ induces TNF receptor-associated factor 6 (TRAF6) ubiquitination and degrades proteolytic TRAF6, ultimately leading to the inhibition of receptor activator of nuclear factor Kappa-B ligand (RANKL)-mediated osteoclastogenesis.6,7 Activities of TH2 cells constitute the humoral immunity, while activities of TH1 cells and T cytotoxic cells create the cellular immunity. Pro-B stem cells differentiate in turn to be mature B-cells in peripheral blood circulation.
Myeloid cells
Common myeloid progenitor cells differentiate into granulocyte/macrophage progenitor and megakaryocyte erythroid progenitor (MKEP) cells (Figure 1). Granulocyte/macrophage progenitor cells are divided into monocytes and granulocytes. Later, monocytes migrate to some tissues, reside there, and change their name depending on the tissue, such as monocytes that migrate to inflammation sites are called macrophages, monocytes that migrate to skin are called Langerhans cells, and monocytes that migrate and reside in bone tissue, which will be differentiated into bone resorbing cells are called osteoclasts.8,9 Osteoclasts work mainly at the bone resorption activity controlling the bone turnover cycle.10 Osteoclasts are large multinucleated cells of hematopoietic origin. They have the capability of removing organic and mineral components of bone. The macrophage lineages and the myeloid dendritic cell originate hematopoietically, and they are affected by the cytokines and produce many of them. On the other hand, osteoblasts originate from mesenchymal stems cells and play important roles in bone formation, osteoclasts differentiation, and hematopoietic cell growth and differentiation.11,12 The interaction or cross talk between osteoblast and osteoclasts play a central role in the osteoimmunological processes. Osteoblasts can control the osteoclastogenesis by two important cytokines; first the RANKL, which is a TNF member superfamily of proteins (Table 1). RANKL is a protein produced by Tumor Necrosis Factor ligand Super-Family member 11 (TNFSF11) gene. It has also been called TNF-related activation-induced cytokine or osteoprotegerin (OPG) ligand because it can be a ligand for osteoprotegerin decoy receptor. RANKL binds to RANK receptors, which normally are present at the pre-osteoclasts’ cell membrane. Its crucial role as a transmembrane protein synthesized by osteoblasts is to perform maturation, differentiation, and activation of osteoclasts.13 Second, OPG is also a member of the TNF superfamily and plays a role of a decoy receptor of RANKL leading to inhibition of osteoclasts maturation, differentiation, and activation and then leading to osteoclast apoptosis (Table 1). So, the balance between RANKL and OPG can modulate the level of bone resorption.14–16 OPG works like a brake against the excessive bone resorption activity. A new inhibitory mechanism against OPG via autoantibodies has been revealed by studies of Riches et al. Indeed, they discovered autoantibodies against OPG in a man with celiac disease, severe osteoporosis, and high bone turnover.17
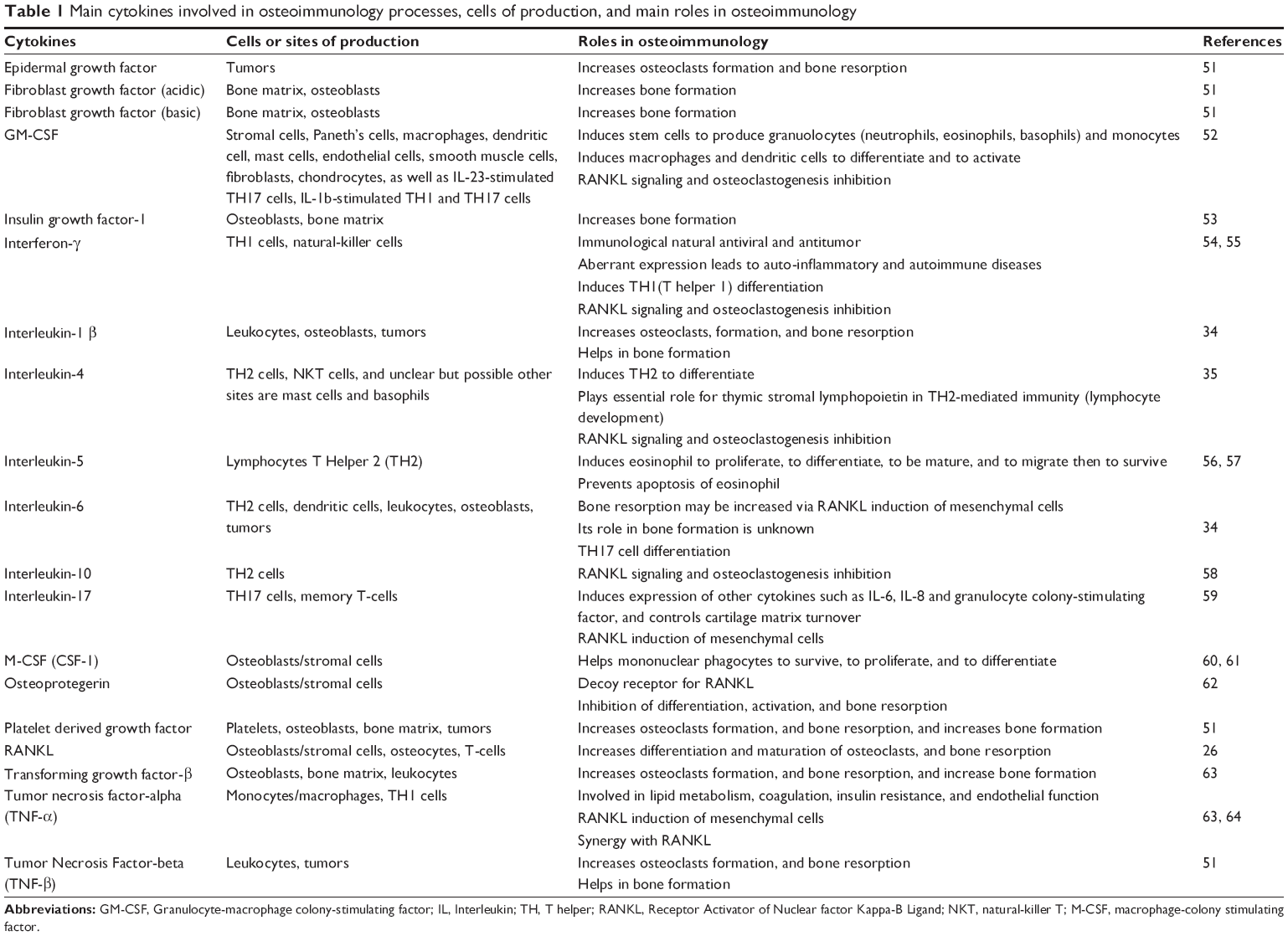 | Table 1 Main cytokines involved in osteoimmunology processes, cells of production, and main roles in osteoimmunology |
Osteoimmunological cytokines
IL-1 is a very essential cytokine in osteoimmunological processes. The analysis of supernatants from phytohemagglutinin-stimulated peripheral blood monocytes in healthy humans suggested that IL-1 acts as the main stimulus of osteoclast-activating factor, which has a central role in osteoclastogenic activity. Subsequently, the same bone resorbing stimulating activity was found in TNF-α and IL-6. Indeed, IL-1, IL-6, and TNF-α increase the osteoclasts response to RANKL and consequently osteolysis (Table 1). Estrogen withdrawal after menopause has the same stimulating effect, increasing osteoclastic activity through IL-1, IL-6, and TNF-α effects.18 Proinflammatory cytokines such as TNF- α, IL-1, IL-6, and IL-17 (Table 1) are also elevated in patients with rheumatoid arthritis, contributing to increased RANKL expression and subsequent osteolysis.19 Schett et al have reviewed the important relation between autoimmunity and joint erosion in rheumatoid arthritis, revealing the presence of anti-citrullinated protein antibodies and anti-carbamylated protein antibodies in serum of the patients with rheumatoid arthritis. Molecular interaction between anti-citrullinated protein antibodies and the surface of osteoclast precursor cells via citrullinated vimentin induces differentiation and production of bone-resorbing osteoclasts, resulting in excessive bone resorption.20 Vitamin D3, prostaglandin E2, parathyroid hormone, in addition to IL-1, IL-6, IL-11, and TNF-α, can also induce RANKL expression, leading to excessive osteoclastogenesis (Figure 2).21 Activated T-cells were also reported to regulate bone loss and activation of osteoclastogenesis in vitro through RANKL.22 Contrariwise, TNF-stimulated gene 6 protein is an inflammation-induced protein that can inhibit osteoblastogenesis and osteoclast activation.23 In addition, immunoreceptor tyrosine-based activation motif (ITAM) pathway may contribute to the relationship between immune system and bone as a co-stimulatory pathway in osteoclasts. ITAM-dependent receptors regulate myeloid-derived cells functions. Furthermore, ITAM-containing adapter proteins such as DNAX activation protein-12 and the Fc epsilon receptor I gamma chain (FCER1G) play an essential role in osteoclast differentiation. Suppression of calcineurin–nuclear factor of activated T-cells signaling can reduce the activity of ITAM pathway in the late stage of osteoclast differentiation, leading to the reduction of osteoclast differentiation and activity.24,25 Calcium signaling induces the calmodulin-dependent kinase pathway role in osteoclast formation and plays a crucial role in the autoamplification of the transcription factor nuclear factor of activated T-cells cytoplasmic-1. Further, activation of TRAF6 and c-Fos pathways by RANKL leads to autoamplification of nuclear factor of activated T-cells cytoplasmic-1 and enhances osteoclastogenesis.21
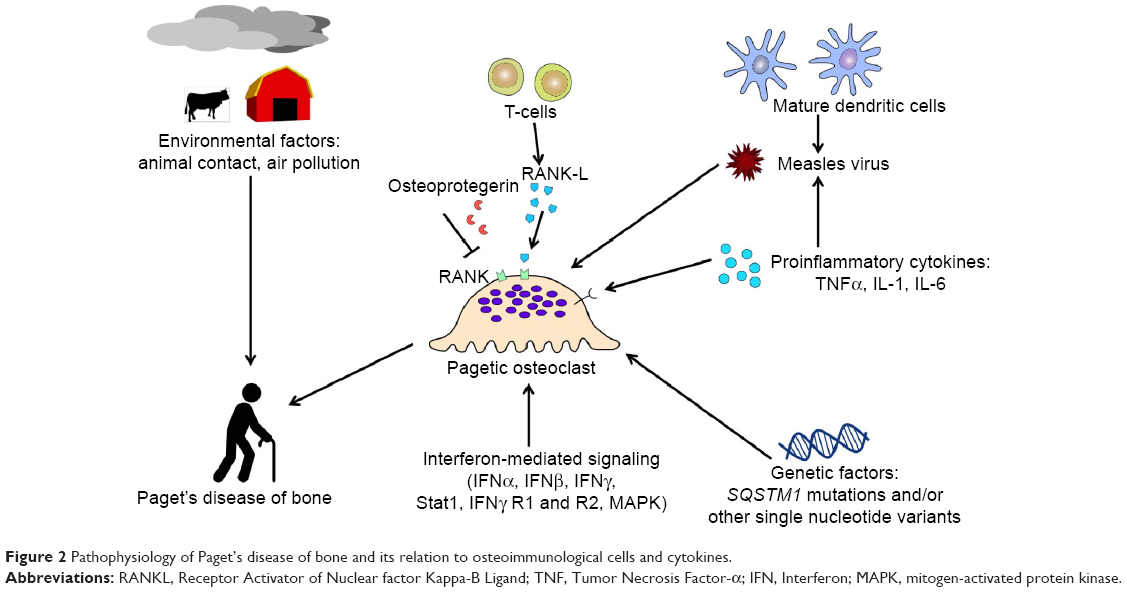 | Figure 2 Pathophysiology of Paget’s disease of bone and its relation to osteoimmunological cells and cytokines. |
Most frequent rheumatic osteoimmunological disorders and their related serum biomarkers
The most frequent rheumatic osteoimmunological disorders regroup bone metabolic diseases, such as osteoporosis and Paget’s disease of bone, systemic autoimmune diseases such as rheumatoid arthritis, systemic lupus erythematosus and systemic sclerosis, and other rheumatic diseases including osteoarthritis and spondyloarthropathies, whereas periodontitis is frequently associated with systemic rheumatic conditions (Table 2). In almost all these disorders, serum levels of osteoimmunological biomarkers have been characterized in the literature (Table 2), and they can be combined to define a specific osteoimmunological pattern associated with a given rheumatic disease. For example, bone formation markers are usually increased in all these diseases, except in osteoporosis and rheumatoid arthritis (decreased level) and in systemic lupus erythematosus and systemic sclerosis (normal level). Bone resorption markers are also usually increased except for osteoarthritis and systemic lupus erythematosus (normal level). In addition, some proinflammatory cytokines may be of paramount importance at differentiating different rheumatic disorders. IL-17 and IL-6 levels are usually simultaneously increased in the same disorders, except for Paget’s disease of bone and systemic sclerosis; in both diseases, IL-6 is elevated but not IL-17. Finally, the pattern of IFN-γ serum levels in rheumatic diseases is very interesting: it is increased in almost all diseases, with the exception of osteoporosis and psoriatic arthritis (decreased level), and rheumatoid arthritis and ankylosing spondylitis (normal level). Overall, a combination of one serum biomarker of bone formation, one bone resorption biomarker, two proinflammatory cytokines such as IL-17 and IL-6 in addition to IFN-γ serum levels would be able to classify with a good sensitivity the most frequent rheumatic osteoimmunological disorders. Adding other already available biomarkers in clinical practice, such as autoantibodies, would increase the specificity of such a combination, and its clinical utility (ie, combination of markers for outcome/prognosis prediction and/or a pharmacogenomic test to guide the choice of any targeted biotherapy) may further be validated in prospective cohorts.
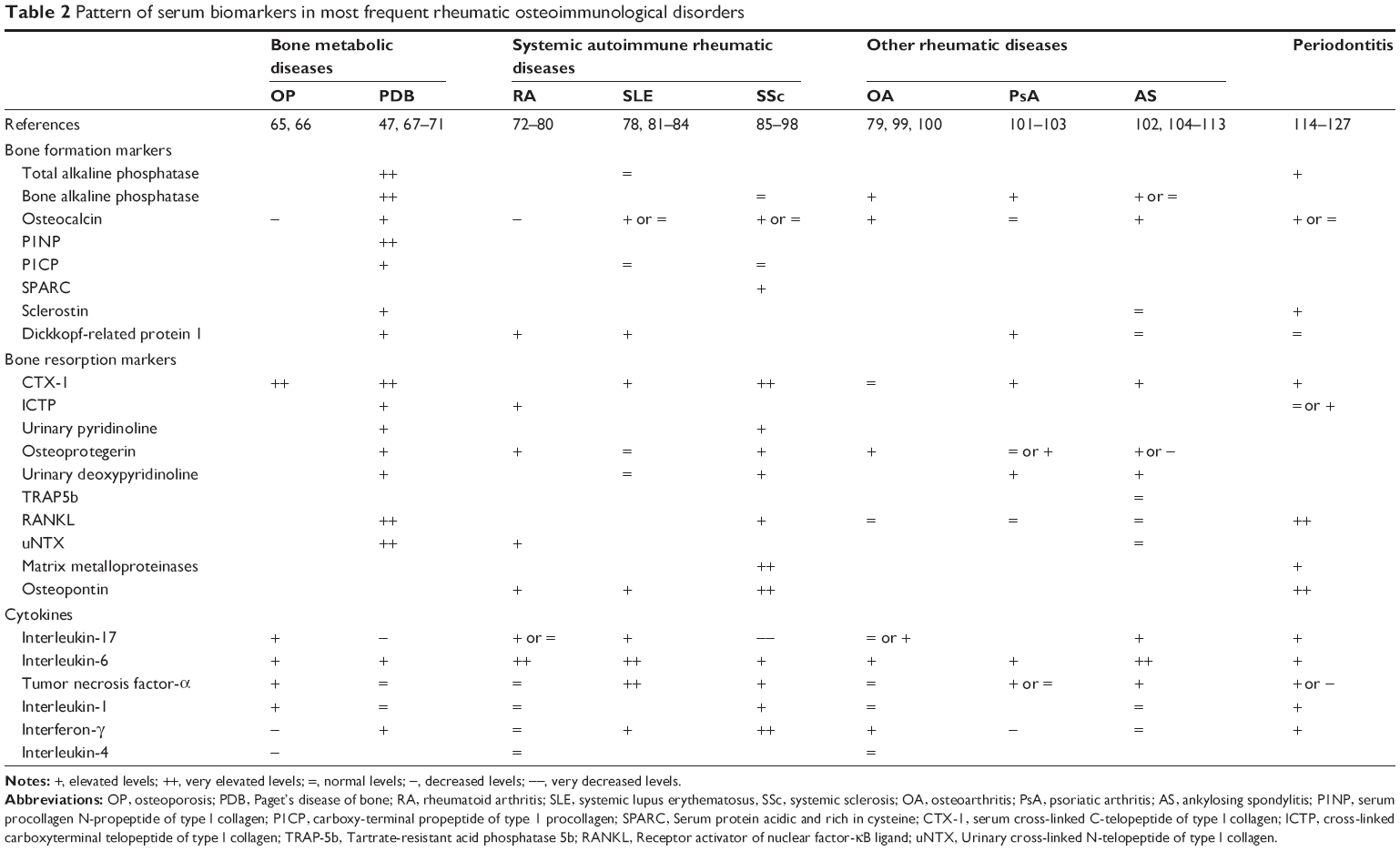 | Table 2 Pattern of serum biomarkers in most frequent rheumatic osteoimmunological disorders |
Paget’s disease of bone as a potential osteoimmunological disorder
Paget’s disease of bone
Paget’s disease of bone is the second most frequent metabolic bone disorder after osteoporosis,26 where more than 3% of Caucasians older than 55 years are affected. This disorder is characterized by an excessive increased bone resorption by osteoclasts accompanied by aberrant osteoblastic bone formation. This aberrant bone remodeling causes fragile and weaker bones. To date, about 30 mutations in SQSTM1/p62 gene have been reported in familial forms and unrelated patients with Paget’s disease of bone. Furthermore, several common single nucleotide polymorphisms have been associated with Paget’s disease of bone, in genome-wide association study, in particular in CSF1, OPTN, TNFRSF11A, PML, RIN3, and NUP205 genes.26–28 The consequences of these polymorphisms on osteoclast phenotype and activity are yet unknown.
SQSTM1/p62 role and importance of osteoclastogenesis in Paget’s disease
The SQSTM1/p62 protein anatomical structure has some important domains that regulate their essential functions such as Phox and Bem1p (PB1), ZZ, TRAF6 binding domain, LIR, KIR, and ubiquitin-associated (UBA) domains.29 PB1 plays a role in adipogenesis by inhibiting ERK1, and it also activates NF-κB pathway through interaction with PKCζ. ZZ domain activates NF-κB through the interaction with receptor interacting protein. Interaction of TRAF6 with the TRAF6 binding domain can activate NF-κB pathway. SQSTM1/p62 can activate autophagy by interaction of LC3 with LIR domain, and autophagy can be inhibited by mTOR. The UBA domain of SQSTM1/p62 has a very important role for this protein function; it interacts non-covalently with ubiquitin protein to perform post-transcriptional modifications and degradation by 26S multisubunit protease or by autophagy. The UBA domain also has an important role in induction and activation of some transcription factors such as NF-kB. In osteoclasts, NF-kB–RANK signaling pathway is very important for osteoclastogenesis. With impairment of UBA functions, ubiquitin protein cannot interact with its domain in SQSTM1/p62 disrupting the autophagy and NF-kB signaling pathways, and consequently, osteoclastogenesis.26,30,31 The KIR domain of SQSTM1/p62 plays a role in oxidative stress with Keap1 (cysteine-rich protein) that has antioxidant elements such as antioxidant response elements/electrophile response element in Nrf2 pathway. Keap1-Nrf2 has a cytoprotective action against oxidative stress, and Keap1 can be downregulated by SQSTM1/p62.32 Autophagy dysfunction may result as a consequence of SQSTM1 gene mutations and proteasomal pathway impairment. Ubiquitinated proteins are usually directed by SQSTM1/p62 protein to autophagosome. After binding to Atg8/LC3 at the surface of the autophagosome, they aggregate into polyubiquitinated non-functional proteins within the autophagosome in the autophagosome. Mutation in SQSTM1/p62 can induce abnormal autophagy process, leading to the accumulation of aggregated ubiquitinated proteins, which stimulate osteoclastogenesis by triggering of NF-kB pathway, and may contribute to Paget’s disease of bone.33 Athanasiou’s group reported very interesting data on pathogenic excessive osteoclastogenesis in Paget’s disease of bone.34 Neale et al found that macrophage-colony stimulating factor and IL-6 induce osteoclast formation and bone resorption in Paget’s disease. Furthermore, elevated macrophage-colony stimulating factor in the serum may correlate with disease activity in patients with Paget’s disease.34 Kudo et al have also shown that IL-6 and IL-11 can enhance osteoclast formation and bone resorption through RANKL-independent mechanism. Then, the role of dexamethasone in osteoclastogenesis was suggested.35 Indeed, dexamethasone can enhance proliferation and differentiation of human osteoclast precursors and suppress the bone resorption by mature osteoclasts.36
Role of Optineurin in Paget’s disease of bone
Mutations of Optineurin (OPTN) gene have been reported in glaucoma and amyotrophic lateral sclerosis, as well as common genetic variants; in particular, the intronic variant rs1561570 was found to be associated with Paget’s disease of bone.37 The OPTN gene encodes a 67 kDa cytosolic protein that consists of 577 amino acids. The importance of OPTN was first raised after discovering disease-causing mutations in primary open-angle glaucoma.38 In addition, OPTN may be involved in neurofibrillary tangles and dystrophic neuritis that leads to Alzheimer’s disease, amyotrophic lateral sclerosis, Parkinson’s disease, Creutzfeldt–Jakob disease, and glial cytoplasmic inclusions that lead to a rare neurological disorder called multiple system atropy.39 Although the exact role of OPTN in Paget’s disease of bone pathogenesis is unknown yet, interaction between OPTN and TANK-binding kinase 1 may suggest a connection with the immune system. Indeed, TANK-binding kinase 1 is a TNF-α-activated protein kinase, which may be activated by viral double-stranded DNA or by lipopolysaccharide. On the other hand, induction of IFN-β due to RNA virus infection can be suppressed by OPTN.39–41
Environmental factors
Environmental factors may also contribute to Paget’s disease of bone.26 Although controversial in the literature, several studies have found a relationship between viral infections and excessive enhanced osteoclasts activity.42 Inclusion bodies contained in osteoclasts were reported to be similar to Paramyxoviral nucleocapsids. Measles virus, respiratory syncytial virus, and canine distemper virus may play a role in Paget’s disease of bone.43 The expression of measles virus nucleocapsid protein (MVNP) in osteoclasts was reported to lead to the formation of pagetic-like osteoclasts. MVNP is known to increase the production of IL-6 that in turn leads to increase the production of TAFII-17 and increase the sensitivity of osteoclasts to 1,25-(OH)2D3. The pagetic phenotype of osteoclast is characterized by hypermultinucleation and hypersensitivity to 1,25-(OH)2D3. NF-kB signaling can be increased in cells by increasing the production of IL-6 and IL-1.44
Paget’s disease as a potential osteoimmunological disorder
Paget’s disease of bone should be considered as a potential osteoimmunological disorder for several reasons. First, the RANKL-NF-κB signaling has a major role in pagetic osteoclast differentiation and activation, and the cytokine profile observed in this disease is very similar to those observed in other osteoimmunological diseases (Table 1). However, the exact role of the immune system in Paget’s disease of bone is not very well understood, except for cellular responses to MNVP. Second, dendritic cells may also play a role in the pathogenesis of Paget’s disease of bone.45 Immature myeloid dendritic cells express CDw150, a signaling lymphocyte activation molecule acting as a receptor for measles virus. Dendritic cells matured by stimulation of Toll-like receptors 2 and 4 will overexpress CDw150 up to fivefold. Then, human dendritic cells may increase the expression of measles virus, the latter contributing to Paget’s disease of bone (Figure 2).46 In addition, Nagy et al were the first authors to show in the literature a remarkable increase in IFN-mediated signaling in Paget’s disease of bone. Increasing expression of IFN-α, IFN-β, and IFN-γ messenger (m)RNA, STAT-1, IFN-γ receptors 1 and 2, and mitogen-activated protein kinase were found in monocytes and lymphocytes from patients with Paget’s disease in comparison with healthy controls, suggesting a possible post-viral reaction in Paget’s disease of bone.47
Main osteoimmunological cytokines as therapeutic targets
A large majority of the already known osteoimmunological cytokines or their receptors have been targeted by monoclonal antibodies, humanized or not, and they are now indicated in several rheumatic disorders, mostly in rheumatoid arthritis (Table 3). The treatment of Paget’s disease of bone mostly relies on bisphosphonates to control disease activity, and no monoclonal antibodies have been investigated in this indication yet. But as a potential osteoimmunological disorder, the treatment of Paget’s disease of bone may also benefit from progresses in targeted therapies. For instance, denosumab, a monoclonal antibody inhibiting RANKL, could be relevant to treat Paget’s disease of bone as it prevents the osteoclastogenesis and promotes the apoptosis of mature osteoclasts. A case report has now been published on using denosumab in a patient with Paget’s disease of bone and impaired renal function, which contraindicated the use of bisphosphonates. In this case, the authors reported that denosumab 60 mg subcutaneously administrated at 0 month, 6 months, 9 months, 12 months, and 15 months has rapidly decreased the activity of Paget’s disease of bone as measured by biochemical markers and bone scan.48 It is worth mentioning that IL-6 was found to be elevated in pagetic osteoclasts in bone marrow and in peripheral blood of patients with Paget’s disease of bone.32 However, some other studies by Neale et al found low serum levels of IL-1 beta, IL-6, and TNF-α in 13 patients with Paget’s disease of bone in comparison with eight healthy controls.34 As the IL-6 signaling is induced by MVNP, inhibition of IL-6 signaling should inhibit the development of pageitc osteoclasts.49 Then, considering the high levels of IL-6 that usually characterize Paget’s disease of the bone, inhibiting the IL-6 signaling by drugs should also be considered as a future therapeutic avenue that is as yet unexplored.50
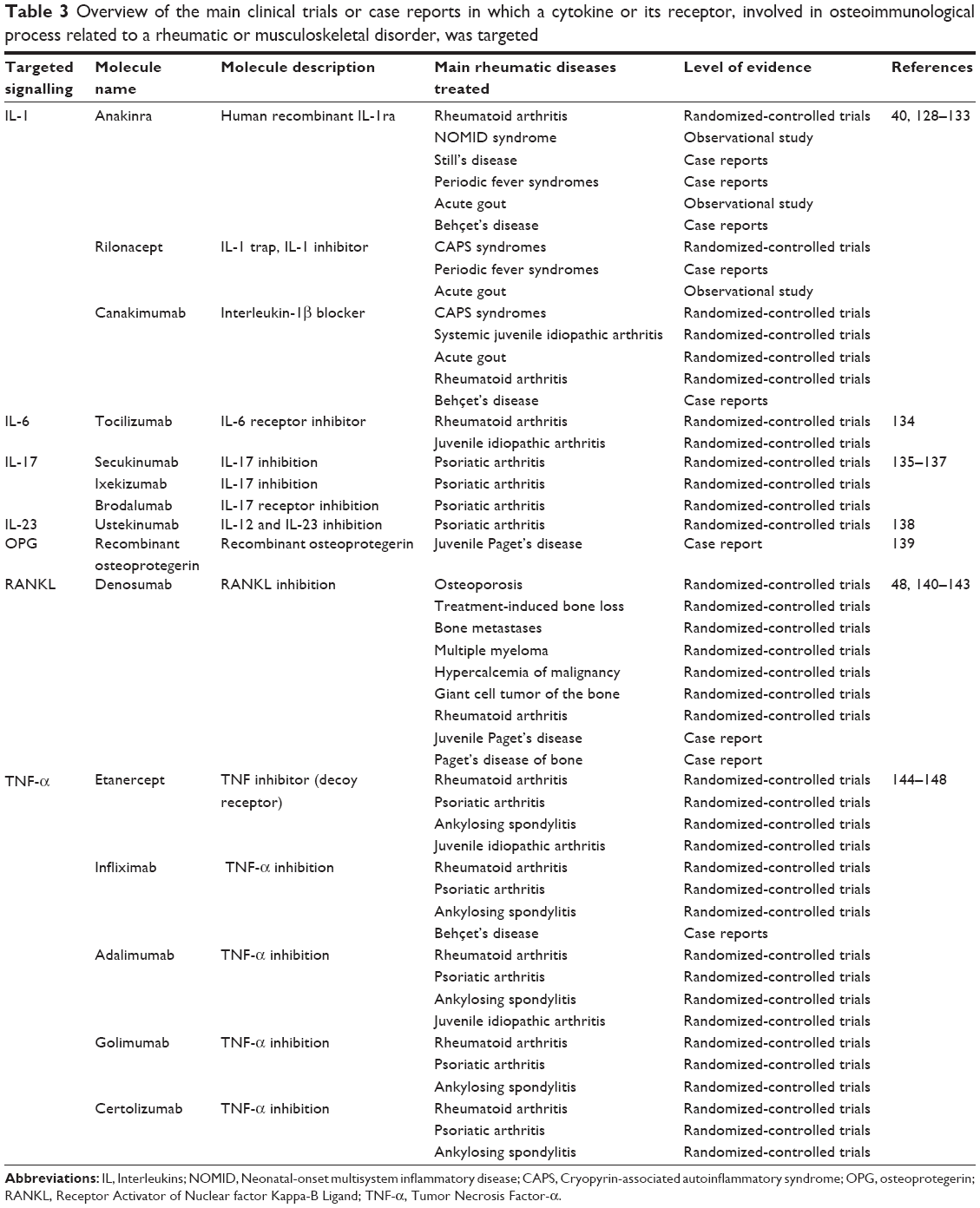 | Table 3 Overview of the main clinical trials or case reports in which a cytokine or its receptor, involved in osteoimmunological process related to a rheumatic or musculoskeletal disorder, was targeted |
Conclusion
In conclusion, Paget’s disease of bone should be considered as a new addition to the large family of osteoimmunological disorders. The cytokine profiles observed in this disease are also very similar to those observed in other osteoimmunological disorders that should probably be classified accordingly. The treatment of Paget’s disease of bone may also benefit from progresses in osteoimmunology-targeted therapies, in particular, RANKL and IL-6 signaling inhibition.
Acknowledgments
MSN was supported in part by an MSc student award from the Department of Medicine (Université Laval). NA is supported by a postdoctoral award from The Arthritis Society. LM is supported by a career award from the FRQ-S. The authors thank Mr Thomas Pornin for technical help with Figures 1 and 2. We acknowledge fundings from the Canadian Institute of Health Research (Catalyst grant: environments, genes and chronic diseases), the Fondation du CHU de Québec, the Canadian Foundation for Innovation, the FRQ-S, the Laval University, and the CHU de Québec Research Centre.
LM declares one grant per year for a congress attendance from Amgen Canada and in kind contribution for bone biochemical markers’ detection kits by Roche Diagnostics Canada. JPB declares research grants and/or consulting or speaking fees from Amgen Inc., Eli Lilly, Novartis, Radius.
Disclosure
The authors report no conflicts of interest in this work.
References
Tavian M, Peault B. Embryonic development of the human hematopoietic system. Int J Dev Biol. 2005;49(2–3):243–250. | ||
Regenerative Medicine. In Stem Cell Information [World Wide Web Site]. Chapter 5. Bethesda, MD: National Institutes of Health, U.S. Department of Health and Human Services; 2011 [cited Monday, May 18, 2015]. Available from: http://stemcells.nih.gov/info/scireport/Pages/2006report.aspx. Accessed February 11, 2015. | ||
Jones DL, Wagers AJ. No place like home: anatomy and function of the stem cell niche. Nat Rev Mol Cell Biol. 2008;9(1):11–21. | ||
Orkin SH. Development of the hematopoietic system. Curr Opin Genet Dev. 1996;6(5):597–602. | ||
Orkin SH, Zon LI. Hematopoiesis and stem cells: plasticity versus developmental heterogeneity. Nat Immunol. 2002;3(4):323–328. | ||
Jones DH, Kong YY, Penninger JM. Role of RANKL and RANK in bone loss and arthritis. Ann Rheum Dis. 2002;61(suppl 2):ii32–ii39. | ||
Zhao B, Ivashkiv LB. Negative regulation of osteoclastogenesis and bone resorption by cytokines and transcriptional repressors. Arthritis Res Ther. 2011;13(4):234. | ||
Yin T, Li L. The stem cell niches in bone. J Clin Invest. 2006;116(5):1195–1201. | ||
Imai Y, Youn MY, Inoue K, Takada I, Kouzmenko A, Kato S. Nuclear receptors in bone physiology and diseases. Physiol Rev. 2013;93(2):481–523. | ||
Hadjidakis DJ, Androulakis II. Bone remodeling. Ann N Y Acad Sci. 2006;1092:385–396. | ||
Lorenzo J, Choi Y. Osteoimmunology. Immunol Rev. 2005;208:5–6. | ||
Bar-Shavit Z. The osteoclast: a multinucleated, hematopoietic-origin, bone-resorbing osteoimmune cell. J Cell Biochem. 2007;102(5):1130–1139. | ||
Wong BR, Rho J, Arron J, et al. TRANCE is a novel ligand of the tumor necrosis factor receptor family that activates c-Jun N-terminal kinase in T cells. J Biol Chem. 1997;272(40):25190–25194. | ||
Wuyts W, Van Wesenbeeck L, Morales-Piga A, et al. Evaluation of the role of RANK and OPG genes in Paget’s disease of bone. Bone. 2001;28(1):104–107. | ||
Roodman GD, Galson DL. The Origins of Bone Cells: Osteoclasts Chapter 2, Osteoimmunology. Vol Osteoimmunology. 1st ed. 2011. | ||
Lorenzo J, Horowitz M, Choi Y, Schett G, Takayanagi H. Osteoimmunology: Interactions of the Immune and Skeletal Systems, Chapter 2. Academic Press Elsevier, London; 2010. | ||
Riches PL, McRorie E, Fraser WD, Determann C, van’t Hof R, Ralston SH. Osteoporosis associated with neutralizing autoantibodies against osteoprotegerin. N Engl J Med. 2009;361(15):1459–1465. | ||
Anderson DM, Maraskovsky E, Billingsley WL, et al. A homologue of the TNF receptor and its ligand enhance T-cell growth and dendritic-cell function. Nature. 1997;390(6656):175–179. | ||
Jung SM, Kim KW, Yang CW, Park SH, Ju JH. Cytokine-mediated bone destruction in rheumatoid arthritis. J Immunol Res. 2014;2014:263625. | ||
Schett G, Gravallese E. Bone erosion in rheumatoid arthritis: mechanisms, diagnosis and treatment. Nat Rev Rheumatol. 2012;8(11):656–664. | ||
Danks L, Takayanagi H. Immunology and bone. J Biochem. 2013;154(1):29–39. | ||
Kong YY, Feige U, Sarosi I, et al. Activated T cells regulate bone loss and joint destruction in adjuvant arthritis through osteoprotegerin ligand. Nature. 1999;402(6759):304–309. | ||
Mahoney DJ, Mikecz K, Ali T, et al. TSG-6 regulates bone remodeling through inhibition of osteoblastogenesis and osteoclast activation. J Biol Chem. 2008;283(38):25952–25962. | ||
Zawawi MS, Dharmapatni AA, Cantley MD, McHugh KP, Haynes DR, Crotti TN. Regulation of ITAM adaptor molecules and their receptors by inhibition of calcineurin-NFAT signalling during late stage osteoclast differentiation. Biochem Biophys Res Commun. 2012;427(2):404–409. | ||
Humphrey MB, Lanier LL, Nakamura MC. Role of ITAM-containing adapter proteins and their receptors in the immune system and bone. Immunol Rev. 2005;208:50–65. | ||
Numan MS, Brown JP, Michou L. Impact of air pollutants on oxidative stress in common autophagy-mediated aging diseases. Int J Environ Res Public Health. 2015;12(2):2289–2305. | ||
Michou L, Collet C, Laplanche JL, Orcel P, Cornelis F. Genetics of Paget’s disease of bone. Joint Bone Spine. 2006;73(3):243–248. | ||
Albagha OM, Wani SE, Visconti MR, et al; Genetic Determinants of Paget’s Disease (GDPD) Consortium. Genome-wide association identifies three new susceptibility loci for Paget’s disease of bone. Nat Genet. 2011;43(7):685–689. | ||
Rea SL, Walsh JP, Layfield R, Ratajczak T, Xu J. New insights into the role of sequestosome 1/p62 mutant proteins in the pathogenesis of Paget’s disease of bone. Endocr Rev. 2013;34(4):501–524. | ||
Layfield R, Ciani B, Ralston SH, et al. Structural and functional studies of mutations affecting the UBA domain of SQSTM1 (p62) which cause Paget’s disease of bone. Biochem Soc Trans. 2004;32(pt 5):728–730. | ||
Goode A, Layfield R. Recent advances in understanding the molecular basis of Paget disease of bone. J Clin Pathol. 2010;63(3):199–203. | ||
Copple IM, Lister A, Obeng AD, et al. Physical and functional interaction of sequestosome 1 with Keap1 regulates the Keap1-Nrf2 cell defense pathway. J Biol Chem. 2010;285(22):16782–16788. | ||
Mathew R, Karp CM, Beaudoin B, et al. Autophagy suppresses tumorigenesis through elimination of p62. Cell. 2009;137(6):1062–1075. | ||
Neale SD, Schulze E, Smith R, Athanasou NA. The influence of serum cytokines and growth factors on osteoclast formation in Paget’s disease. QJM. 2002;95(4):233–240. | ||
Kudo O, Sabokbar A, Pocock A, Itonaga I, Fujikawa Y, Athanasou NA. Interleukin-6 and interleukin-11 support human osteoclast formation by a RANKL-independent mechanism. Bone. 2003;32(1):1–7. | ||
Hirayama T, Sabokbar A, Athanasou NA. Effect of corticosteroids on human osteoclast formation and activity. J Endocrinol. 2002;175(1):155–163. | ||
Albagha OM, Visconti MR, Alonso N, et al. Genome-wide association study identifies variants at CSF1, OPTN and TNFRSF11A as genetic risk factors for Paget’s disease of bone. Nat Genet. 2010;42(6):520–524. | ||
Rezaie T, Child A, Hitchings R, et al. Adult-onset primary open-angle glaucoma caused by mutations in optineurin. Science. 2002;295(5557):1077–1079. | ||
Ying H, Yue BY. Cellular and molecular biology of optineurin. Int Rev Cell Mol Biol. 2012;294:223. | ||
Mankouri J, Fragkoudis R, Richards KH, et al. Optineurin negatively regulates the induction of IFNβ in response to RNA virus infection. PLoS Pathog. 2010;6(2):e1000778. | ||
Morton S, Hesson L, Peggie M, Cohen P. Enhanced binding of TBK1 by an optineurin mutant that causes a familial form of primary open angle glaucoma. FEBS Lett. 2008;582(6):997–1002. | ||
Singer FR. The etiology of Paget’s disease of bone: viral and genetic interactions. Cell Metab. 2011;13(1):5–6. | ||
Ralston SH, Layfield R. Pathogenesis of Paget disease of bone. Calcif Tissue Int. 2012;91(2):97–113. | ||
Ehrlich LA, Roodman GD. The role of immune cells and inflammatory cytokines in Paget’s disease and multiple myeloma. Immunol Rev. 2005;208:252–266. | ||
Takayanagi H. Osteoimmunology: shared mechanisms and crosstalk between the immune and bone systems. Nat Rev Immunol. 2007;7(4):292–304. | ||
Murabayashi N, Kurita-Taniguchi M, Ayata M, Matsumoto M, Ogura H, Seya T. Susceptibility of human dendritic cells (DCs) to measles virus (MV) depends on their activation stages in conjunction with the level of CDw150: role of toll stimulators in DC maturation and MV amplification. Microbes Infect. 2002;4(8):785–794. | ||
Nagy ZB, Gergely P, Donath J, Borgulya G, Csanad M, Poor G. Gene expression profiling in Paget’s disease of bone: upregulation of interferon signaling pathways in pagetic monocytes and lymphocytes. J Bone Miner Res. 2008;23(2):253–259. | ||
Schwarz P, Rasmussen AQ, Kvist TM, Andersen UB, Jorgensen NR. Paget’s disease of the bone after treatment with Denosumab: a case report. Bone. 2012;50(5):1023–1025. | ||
Galson DL, Roodman GD. Pathobiology of Paget’s disease of bone. J Bone Metab. 2014;21(2):85–98. | ||
Michou L, Brown JP. Emerging strategies and therapies for treatment of Paget’s disease of bone. Drug Des Devel Ther. 2011;5:225–239. | ||
Mills BG, Frausto A. Cytokines expressed in multinucleated cells: Paget’s disease and giant cell tumors versus normal bone. Calcif Tissue Int. 1997;61(1):16–21. | ||
Francisco-Cruz A, Aguilar-Santelises M, Ramos-Espinosa O, et al. Granulocyte-macrophage colony-stimulating factor: not just another haematopoietic growth factor. Med Oncol. 2014;31(1):774. | ||
Wang Y, Nishida S, Elalieh HZ, Long RK, Halloran BP, Bikle DD. Role of IGF-I signaling in regulating osteoclastogenesis. J Bone Miner Res. 2006;21(9):1350–1358. | ||
Schoenborn JR, Wilson CB. Regulation of interferon-gamma during innate and adaptive immune responses. Adv Immunol. 2007;96:41–101. | ||
Sokol CL, Barton GM, Farr AG, Medzhitov R. A mechanism for the initiation of allergen-induced T helper type 2 responses. Nat Immunol. 2008;9(3):310–318. | ||
Garlisi CG, Falcone A, Billah MM, Egan RW, Umland SP. T cells are the predominant source of interleukin-5 but not interleukin-4 mRNA expression in the lungs of antigen-challenged allergic mice. Am J Respir Cell Mol Biol. 1996;15(3):420–428. | ||
Garcia G, Taille C, Laveneziana P, Bourdin A, Chanez P, Humbert M. Anti-interleukin-5 therapy in severe asthma. Eur Respir Rev. 2013;22(129):251–257. | ||
Zhang Q, Chen B, Yan F, et al. Interleukin-10 inhibits bone resorption: a potential therapeutic strategy in periodontitis and other bone loss diseases. Biomed Res Int. 2014;2014:284836. | ||
Hymowitz SG, Filvaroff EH, Yin JP, et al. IL-17s adopt a cystine knot fold: structure and activity of a novel cytokine, IL-17F, and implications for receptor binding. EMBO J. 2001;20(19):5332–5341. | ||
Stanley ER, Berg KL, Einstein DB, et al. Biology and action of colony – stimulating factor-1. Mol Reprod Dev. 1997;46(1):4–10. | ||
Udagawa N, Takahashi N, Jimi E, et al. Osteoblasts/stromal cells stimulate osteoclast activation through expression of osteoclast differentiation factor/RANKL but not macrophage colony-stimulating factor: receptor activator of NF-kappa B ligand. Bone. 1999;25(5):517–523. | ||
Chamoux E, Houde N, L’Eriger K, Roux S. Osteoprotegerin decreases human osteoclast apoptosis by inhibiting the TRAIL pathway. J Cell Physiol. 2008;216(2):536–542. | ||
Itonaga I, Sabokbar A, Sun SG, et al. Transforming growth factor-beta induces osteoclast formation in the absence of RANKL. Bone. 2004;34(1):57–64. | ||
Online Mendelian Inheritance in Man® (OMIM®). Tumor Necrosis Factor (TNF), OMIM 191160; 2015. Available from: http://www.omim.org/entry/191160. Accessed March 3, 2015. | ||
Zhang J, Fu Q, Ren Z, et al. Changes of serum cytokines-related Th1/Th2/Th17 concentration in patients with postmenopausal osteoporosis. Gynecol Endocrinol. 2015;31(3):183–90. Epub 2014 Nov 11. | ||
Al-Zahrani MK, Elnasieh AM, Alenezi FM, et al. A 3-month oral vitamin D supplementation marginally improves diastolic blood pressure in Saudi patients with type 2 diabetes mellitus. Int J Clin Exp Med. 2014;7(12):5421–5428. | ||
Alvarez L, Peris P, Guañabens N, et al. Long-term biochemical response after bisphosphonate therapy in Paget’s disease of bone. Proposed intervals for monitoring treatment. Rheumatology. 2004;43(7):869–874. | ||
Marshall MJ, Evans SF, Sharp CA, Powell DE, McCarthy HS, Davie MW. Increased circulating dickkopf-1 in Paget’s disease of bone. Clin Biochem. 2009;42(10–11):965–969. | ||
Polyzos SA, Anastasilakis AD, Efstathiadou Z, et al. The effect of zoledronic acid on serum dickkopf-1, osteoprotegerin, and RANKL in patients with Paget’s disease of bone. Horm Metab Res. 2009;41(11):846–850. | ||
Werner de Castro GR, Buss Z, Da Rosa JS, Frode TS. Inflammatory cytokines in Paget’s disease of bone. Int Immunopharmacol. 2014;18(2):277–281. | ||
Yavropoulou MP, van Lierop AH, Hamdy NA, Rizzoli R, Papapoulos SE. Serum sclerostin levels in Paget’s disease and prostate cancer with bone metastases with a wide range of bone turnover. Bone. 2012;51(1):153–157. | ||
Rossini M, Viapiana O, Adami S, et al. In patients with rheumatoid arthritis, dickkopf-1 serum levels are correlated with parathyroid hormone, bone erosions and bone mineral density. Clin Exp Rheumatol. 2015;33(1):77–83. | ||
Hong Q, Xu J, Xu S, Lian L, Zhang M, Ding C. Associations between serum 25-hydroxyvitamin D and disease activity, inflammatory cytokines and bone loss in patients with rheumatoid arthritis. Rheumatology. 2014;53(11):1994–2001. | ||
Cortet B, Cotten A, Boutry N, et al. Percutaneous vertebroplasty in patients with osteolytic metastases or multiple myeloma. Rev Rhum. 1997;64(3):177–183. | ||
Al-Awadhi A, Olusi S, Al-Zaid N, Prabha K. Serum concentrations of interleukin 6, osteocalcin, intact parathyroid hormone, and markers of bone resorption in patients with rheumatoid arthritis. J Rheumatol. 1999;26(6):1250–1256. | ||
Seriolo B, Ferretti V, Sulli A, Caratto E, Fasciolo D, Cutolo M. Serum osteocalcin levels in premenopausal rheumatoid arthritis patients. Ann N Y Acad Sci. 2002;966:502–507. | ||
Wong PK, Young L, Vaile JH, et al. Telopeptides as markers of bone turnover in rheumatoid arthritis and osteoarthritis. Intern Med J. 2004;34(9–10):539–544. | ||
Long L, Liu Y, Wang S, et al. Dickkopf-1 as potential biomarker to evaluate bone erosion in systemic lupus erythematosus. J Clin Immunol. 2010;30(5):669–675. | ||
Heard BJ, Rosvold JM, Fritzler MJ, El-Gabalawy H, Wiley JP, Krawetz RJ. A computational method to differentiate normal individuals, osteoarthritis and rheumatoid arthritis patients using serum biomarkers. J R Soc Interface. 2014;11(97):20140428. | ||
Ji HI, Lee SH, Song R, et al. Serum level of osteopontin as an inflammatory marker does not indicate disease activity or responsiveness to therapeutic treatments in patients with rheumatoid arthritis. Clin Rheumatol. 2014;33(3):397–402. | ||
Korczowska I, Olewicz-Gawlik A, Hrycaj P, Lacki J. The effect of long-term glucocorticoids on bone metabolism in systemic lupus erythematosus patients: the prevalence of its anti-inflammatory action upon bone resorption. Yale J Biol Med. 2003;76(2):45–54. | ||
Rullo OJ, Woo JM, Parsa MF, et al. Plasma levels of osteopontin identify patients at risk for organ damage in systemic lupus erythematosus. Arthritis Res Ther. 2013;15(1):R18. | ||
Lyn-Cook BD, Xie C, Oates J, et al. Increased expression of toll-like receptors (TLRs) 7 and 9 and other cytokines in systemic lupus erythematosus (SLE) patients: ethnic differences and potential new targets for therapeutic drugs. Mol Immunol. 2014;61(1):38–43. | ||
Vincent FB, Northcott M, Hoi A, Mackay F, Morand EF. Clinical associations of serum interleukin-17 in systemic lupus erythematosus. Arthritis Res Ther. 2013;15:R97. | ||
La Montagna G, Baruffo A, Abbadessa S, Maja L, Tirri R. Evidence for bone resorption in systemic sclerosis. J Rheumatol. 1995;22(4):797–799. | ||
Ištok R, Czirják L, Lukáč J, Stančíková M, Rovenský J. Increased urinary pyridinoline cross-link compounds of collagen in patients with systemic sclerosis and Raynaud’s phenomenon. Rheumatology. 2001;40(2):140–146. | ||
Atteritano M, Sorbara S, Bagnato G, et al. Bone mineral density, bone turnover markers and fractures in patients with systemic sclerosis: a case control study. PLoS One. 2013;8(6):e66991. | ||
Allanore Y, Borderie D, Lemaréchal H, Cherruau B, Ekindjian OG, Kahan A. Correlation of serum collagen I carboxyterminal telopeptide concentrations with cutaneous and pulmonary involvement in systemic sclerosis. J Rheumatol. 2003;30(1):68–73. | ||
Macko RF, Gelber AC, Young BA, et al. Increased circulating concentrations of the counteradhesive proteins SPARC and thrombospondin-1 in systemic sclerosis (scleroderma). Relationship to platelet and endothelial cell activation. J Rheumatol. 2002;29(12):2565–2570. | ||
Castellino G, Corallini F, Bortoluzzi A, et al. The tumour necrosis factor-related apoptosis-inducing ligand-osteoprotegerin system in limited systemic sclerosis: a new disease marker? Rheumatology. 2010;49(6):1173–1176. | ||
Dovio A, Data V, Carignola R, et al. Circulating osteoprotegerin and soluble RANK ligand in systemic sclerosis. J Rheumatol. 2008;35(11):2206–2213. | ||
Maekawa T, Komine M, Murata S, Ohtsuki M. Peritoneal loose body: a case report and comparison with encapsulated fat necrosis. J Dermatol. 2013;40(12):1058–1059. | ||
Andersen GN, Nilsson K, Nagaeva O, Rantapaa-Dahlqvist S, Sandstrom T, Mincheva-Nilsson L. Cytokine mRNA profile of alveolar T lymphocytes and macrophages in patients with systemic sclerosis suggests a local Tr1 response. Scand J Immunol. 2011;74(3):272–281. | ||
Wu M, Schneider DJ, Mayes MD, et al. Osteopontin in systemic sclerosis and its role in dermal fibrosis. J Invest Dermatol. 2012;132(6):1605–1614. | ||
Olewicz-Gawlik A, Danczak-Pazdrowska A, Kuznar-Kaminska B, et al. Interleukin-17 and interleukin-23: importance in the pathogenesis of lung impairment in patients with systemic sclerosis. Int J Rheum Dis. 2014;17(6):664–670. | ||
Pehlivan Y, Onat AM, Ceylan N, et al. Serum leptin, resistin and TNF-alpha levels in patients with systemic sclerosis: the role of adipokines in scleroderma. Int J Rheum Dis. 2012;15(4):374–379. | ||
Hasegawa M, Sato S, Fujimoto M, Ihn H, Kikuchi K, Takehara K. Serum levels of interleukin 6 (IL-6), oncostatin M, soluble IL-6 receptor, and soluble gp130 in patients with systemic sclerosis. J Rheumatol. 1998;25(2):308–313. | ||
Kim WU, Min SY, Cho ML, et al. Elevated matrix metalloproteinase-9 in patients with systemic sclerosis. Arthritis Res Ther. 2005;7(1):R71–R79. | ||
Pantsulaia I, Kalichman L, Kobyliansky E. Association between radiographic hand osteoarthritis and RANKL, OPG and inflammatory markers. Osteoarthr Cartil. 2010;18(11):1448–1453. | ||
Kumm J, Tamm A, Lintrop M. Diagnostic and prognostic value of bone biomarkers in progressive knee osteoarthritis: a 6-year follow-up study in middle-aged subjects. Osteoarthr Cartil. 2013;21(6):815–822. | ||
Dalbeth N, Pool B, Smith T, et al. Circulating mediators of bone remodeling in psoriatic arthritis: implications for disordered osteoclastogenesis and bone erosion. Arthritis Res Ther. 2010;12(4):R164. | ||
Grisar J, Bernecker PM, Aringer M, et al. Ankylosing spondylitis, psoriatic arthritis, and reactive arthritis show increased bone resorption, but differ with regard to bone formation. J Rheumatol. 2002;29(7):1430–1436. | ||
Amital H, Barak V, Winkler RE, Rubinow A. Impact of treatment with infliximab on serum cytokine profile of patients with rheumatoid and psoriatic arthritis. Ann N Y Acad Sci. 2007;1110(1):649–660. | ||
Taylan A, Sari I, Akinci B, et al. Biomarkers and cytokines of bone turnover: extensive evaluation in a cohort of patients with ankylosing spondylitis. BMC Musculoskelet Disord. 2012;13:191. | ||
Marhoffer W, Stracke H, Masoud I, et al. Evidence of impaired cartilage/bone turnover in patients with active ankylosing spondylitis. Ann Rheum Dis. 1995;54(7):556–559. | ||
Toussirot E, Ricard-Blum S, Dumoulin G, Cedoz J, Wendling D. Relationship between urinary pyridinium cross-links, disease activity and disease subsets of ankylosing spondylitis. Rheumatology. 1999;38(1):21–27. | ||
Mitra D, Elvins D, Collins A. Biochemical markers of bone metabolism in mild ankylosing spondylitis and their relationship with bone mineral density and vertebral fractures. J Rheumatol. 1999;26(10):2201–2204. | ||
Yilmaz N, Ozaslan J. Biochemical bone turnover markers in patients with ankylosing spondylitis. Clin Rheumatol. 2000;19(2):92–98. | ||
Park MC, Chung SJ, Park YB, Lee SK. Bone and cartilage turnover markers, bone mineral density, and radiographic damage in men with ankylosing spondylitis. Yonsei Med J. 2008;49(2):288–294. | ||
Vosse D, Landewe R, Garnero P, van der Heijde D, van der Linden S, Geusens P. Association of markers of bone-and cartilage-degradation with radiological changes at baseline and after 2 years follow-up in patients with ankylosing spondylitis. Rheumatology. 2008;47(8):1219–1222. | ||
Mei Y, Pan F, Gao J, et al. Increased serum IL-17 and IL-23 in the patient with ankylosing spondylitis. Clin Rheumatol. 2011;30(2):269–273. | ||
Bal A, Unlu E, Bahar G, Aydog E, Eksioglu E, Yorgancioglu R. Comparison of serum IL-1 beta, sIL-2R, IL-6, and TNF-alpha levels with disease activity parameters in ankylosing spondylitis. Clin Rheumatol. 2007;26(2):211–215. | ||
Franck H, Meurer T, Hofbauer LC. Evaluation of bone mineral density, hormones, biochemical markers of bone metabolism, and osteoprotegerin serum levels in patients with ankylosing spondylitis. J Rheumatol. 2004;31(11):2236–2241. | ||
Hou LT, Liu CM, Liu BY, Lin SJ, Liao CS, Rossomando EF. Interleukin-1beta, clinical parameters and matched cellular-histopathologic changes of biopsied gingival tissue from periodontitis patients. J Periodontal Res. 2003;38(3):247–254. | ||
Takeichi O, Haber J, Kawai T, Smith DJ, Moro I, Taubman MA. Cytokine profiles of T-lymphocytes from gingival tissues with pathological pocketing. J Dent Res. 2000;79(8):1548–1555. | ||
Kawai T, Matsuyama T, Hosokawa Y, et al. B and T lymphocytes are the primary sources of RANKL in the bone resorptive lesion of periodontal disease. Am J Pathol. 2006;169(3):987–998. | ||
Ohyama H, Kato-Kogoe N, Kuhara A, et al. The involvement of IL-23 and the Th17 pathway in periodontitis. J Dent Res. 2009;88(7):633–638. | ||
Malhotra R, Grover V, Kapoor A, Kapur R. Alkaline phosphatase as a periodontal disease marker. Indian J Dent Res. 2010;21(4):531. | ||
Marcaccini AM, Novaes AB Jr, Meschiari CA, et al. Circulating matrix metalloproteinase-8 (MMP-8) and MMP-9 are increased in chronic periodontal disease and decrease after non-surgical periodontal therapy. Clin Chim Acta. 2009;409(1–2):117–122. | ||
Nakajima T, Honda T, Domon H, et al. Periodontitis-associated up-regulation of systemic inflammatory mediator level may increase the risk of coronary heart disease. J Periodontal Res. 2010;45(1):116–122. | ||
Duarte PM, da Rocha M, Sampaio E, et al. Serum levels of cytokines in subjects with generalized chronic and aggressive periodontitis before and after non-surgical periodontal therapy: a pilot study. J Periodontol. 2010;81(7):1056–1063. | ||
Liu K, Meng H, Tang X, et al. Elevated plasma calcifediol is associated with aggressive periodontitis. J Periodontol. 2009;80(7): 1114–1120. | ||
Zhang X, Meng H, Sun X, et al. Elevation of vitamin D-binding protein levels in the plasma of patients with generalized aggressive periodontitis. J Periodontal Res. 2013;48(1):74–79. | ||
Miricescu D, Totan A, Calenic B, et al. Salivary biomarkers: relationship between oxidative stress and alveolar bone loss in chronic periodontitis. Acta Odontol Scand. 2014;72(1):42–47. | ||
Ozcaka O, Nalbantsoy A, Bicakci N, Kose T, Buduneli N. Plasma levels of C-telopeptide pyridinoline cross-links of type I collagen and osteocalcin in chronic periodontitis. Inflammation. 2011;34(3):203–208. | ||
Gursoy UK, Könönen E, Pradhan-Palikhe P, et al. Salivary MMP-8, TIMP-1, and ICTP as markers of advanced periodontitis. J Clin Periodontol. 2010;37(6):487–493. | ||
Hans S, Mali AM. Estimation and comparison of osteopontin levels in plasma in subjects with healthy periodontium and generalized chronic periodontitis and its assessment after scaling and root planing. J Indian Soc Periodontol. 2012;16(3):354–357. | ||
Kone-Paut I, Galeotti C. Anakinra for cryopyrin-associated periodic syndrome. Expert Rev Clin Immunol. 2014;10(1):7–18. | ||
Cavalli G, Franchini S, Aiello P, et al. Efficacy and safety of biological agents in adult-onset Still’s disease. Scand J Rheumatol. 2015:1–6. | ||
Saygin C, Uzunaslan D, Hatemi G. Currently used biologic agents in the management of Behcet’s syndrome. Curr Med Chem. 2015;22(16):1976–1985. | ||
Highlights of prescribing information, Arcalyst [rilonacept]. Regeneron Pharmaceuticals, Inc. Available from: http://www.accessdata.fda.gov/drugsatfda_docs/label/2008/125249lbl.pdf. Accessed October 20, 2014. | ||
European Medicines Agency Docs; 2015. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_–_Product_Information/human/001109/WC500031680.pdf. Accessed February 28, 2015. | ||
Cantarini L, Lopalco G, Caso F, et al. Effectiveness and tuberculosis-related safety profile of interleukin-1 blocking agents in the management of Behcet’s disease. Autoimmun Rev. 2015;14(1):1–9. | ||
PRODUCT MONOGRAPH, ACTEMRA® [tocilizumab]; 2015. Available from: http://www.rochecanada.com/fmfiles/re7234008/Research/ClinicalTrialsForms/Products/ConsumerInformation/MonographsandPublicAdvisories/Actemra/Actemra_PM_E.pdf. Accessed February 28, 2015. | ||
MPR. Ixekizumab vs Etanercept for Plaque Psoriasis: Phase 3 Results Announced; 2015. Available from: http://www.empr.com/ixekizumab-vs-etanercept-for-plaque-psoriasis-phase-3-results-announced/article/367354/. Accessed February 28, 2015. | ||
Novartis Pharmaceuticals Canada Inc. Secukinumab [AIN457]; 2015. Available from: http://www.novartis.ca/cs/www.novartis.ca-v2/downloads/en/News/Novartis_PR_Nov172014_EN.pdf. Accessed February 28, 2015. | ||
MPR. Brodalumab Demonstrates Efficacy in Plaque Psoriasis Study; 2015. Available from: http://www.empr.com/ixekizumab-vs-etanercept-for-plaque-psoriasis-phase-3-results-announced/article/367354/. Accessed February 28, 2015. | ||
JANSSEN, STELARA®; 2015. Available from: http://www.janssen.ca/product/190. Accessed February 28, 2015. | ||
Cundy T, Davidson J, Rutland MD, Stewart C, DePaoli AM. Recombinant osteoprotegerin for juvenile Paget’s disease. N Engl J Med. 2005;353(9):918–923. | ||
PRODUCT MONOGRAPH, PROLIA [denosumab]; 2015. Available from: https://www.amgen.ca/Prolia_PM.pdf. Accessed February 28, 2015. | ||
PRODUCT MONOGRAPH, XGEVA [denosumab]; 2015. Available from: https://www.amgen.ca/Xgeva_PM.pdf. Accessed February 28, 2015. | ||
Grasemann C, Schündeln MM, Hövel M, et al. Effects of RANK-ligand antibody (denosumab) treatment on bone turnover markers in a girl with juvenile Paget’s disease. J Clin Endocrinol Metab. 2013;98(8):3121–3126. | ||
Polyzos SA, Singhellakis PN, Naot D, et al. Denosumab treatment for juvenile Paget’s disease: results from two adult patients with osteoprotegerin deficiency (“Balkan” mutation in the TNFRSF11B gene). J Clin Endocrinol Metab. 2014;99(3):703–707. | ||
PRODUCT MONOGRAPH, ENBREL [etanercept]; 2015. Available from: https://www.amgen.ca/Enbrel_PM.pdf. Accessed February 28, 2015. | ||
PRODUCT MONOGRAPH, REMICADE [infliximab]; 2015. Available from: http://www.mun.ca/pharmacy/aboutpharmacy/REMICADEPME.pdf. Accessed February 28, 2015. | ||
PRODUCT MONOGRAPH, HUMIRA® [adalimumab]; 2015. Available from: http://www.abbvie.ca/content/dam/abbviecorp/ca/english/docs/HUMIRA_PM_EN.pdf. Accessed February 28, 2015. | ||
PRODUCT MONOGRAPH, SIMPONI [golimumab]; 2015. Available from: http://www.mun.ca/pharmacy/aboutpharmacy/SIM11182011CPM.SNDS.pdf. Accessed February 28, 2015. | ||
PRODUCT MONOGRAPH, CIMZIA® [certolizumab pegol]; 2015. Available from: http://www.ucb-canada.ca/_up/ucbpharma_ca_en/documents/cimzia_pm_en_15jan2014.pdf. Accessed February 28, 2015. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.