")
Back to Journals » International Journal of Nanomedicine » Volume 13
Overcoming tumor hypoxia as a barrier to radiotherapy, chemotherapy and immunotherapy in cancer treatment
Received 13 April 2018
Accepted for publication 11 July 2018
Published 4 October 2018 Volume 2018:13 Pages 6049—6058
DOI https://doi.org/10.2147/IJN.S140462
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Thomas Webster
Video abstract presented by Kaitlin Graham.
Views: 2535
Kaitlin Graham, Evan Unger
NuvOx Pharma, Tucson, AZ, USA
Abstract: Hypoxia exists to some degree in most solid tumors due to inadequate oxygen delivery of the abnormal vasculature which cannot meet the demands of the rapidly proliferating cancer cells. The levels of oxygenation within the same tumor are highly variable from one area to another and can change over time. Tumor hypoxia is an important impediment to effective cancer therapy. In radiotherapy, the primary mechanism is the creation of reactive oxygen species; hypoxic tumors are therefore radiation resistant. A number of chemotherapeutic drugs have been shown to be less effective when exposed to a hypoxic environment which can lead to further disease progression. Hypoxia is also a potent barrier to effective immunotherapy in cancer treatment. Because of the recognition of hypoxia as an important barrier to cancer treatment, a variety of approaches have been undertaken to overcome or reverse tumor hypoxia. Such approaches have included breathing hyperbaric oxygen, artificial hemoglobins, allosteric hemoglobin modifiers, hypoxia activated prodrugs and fluorocarbons (FCs). These approaches have largely failed due to limited efficacy and/or adverse side effects. Oxygen therapeutics, based on liquid FCs, can potentially increase the oxygen-carrying capacity of the blood to reverse tumor hypoxia. Currently, at least two drugs are in clinical trials to reverse tumor hypoxia; one of these is designed to improve permeability of oxygen into the tumor tissue and the other is based upon a low boiling point FC that transports higher amounts of oxygen per gram than previously tested FCs.
Keywords: fluorocarbon, solid tumor, oxygen therapeutics, dodecafluoropentane emulsion
Introduction
The biology of tumor hypoxia
There are several mechanisms involved in the development of hypoxia in tumors, including perfusion-limited O2 delivery, diffusion-limited O2 delivery and anemic hypoxia. The varying mechanisms cause substantial heterogeneity in the tissue oxygenation levels of the tumor.1 Tumor blood vessels are chaotic and lack fundamental architecture of blood vessels in normal tissues. Perfusion-limited O2 delivery is caused by the severe structural and functional abnormality of the tumor vasculature. The abnormal vessel shapes cause geometric resistance, which disrupts blood flow.2 Furthermore, the vessel walls in tumor vasculature are more permeable as they are deficient in smooth muscle cells and often have an irregular endothelial cell lining and basement membrane.3 These structural abnormalities of the tumor vasculature lead to ischemic hypoxia. This type of hypoxia is also referred to as “acute” hypoxia. Diffusion-limited O2 delivery is due to the deterioration of the diffusion geometry of the blood vessels. In normal tissues, blood vessels are arranged in a controlled and systematic manner such that the distance from the cells to the capillaries is maintained to establish a constant and uniform oxygen gradient. In neoplastic tissues, blood vessels can form further from the cells, which deprive the cells of oxygen. Figure 1 shows a tumor supplied by a principal feeding artery. The cells in the periphery are further from the vascular supply and are thus, hypoxic. This diffusion-limited hypoxia is also referred to as “chronic” hypoxia.1 Anemic hypoxia can be either tumor-associated or therapy-induced. Tumors that have low perfusion rates are especially susceptible to anemic hypoxia.4 Normal tissues are able to compensate for ischemia by increasing the amount of oxygen drawn from the blood and can counteract anemia by accelerating the rate of local blood flow. However, tumors are not able to regulate the diminishing oxygen levels, leading to the development of hypoxia.
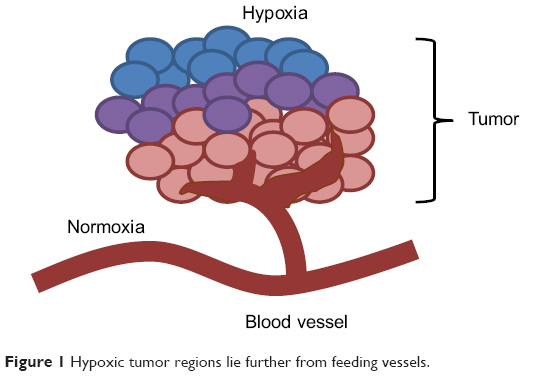 | Figure 1 Hypoxic tumor regions lie further from feeding vessels. |
Hypoxia is a characteristic hallmark of solid tumors that directly contribute to the malignant properties of cancers.4–6 As tumors develop regions of hypoxia, they must adjust their metabolism to adapt to this oxygen-depleted microenvironment. Tumors acclimate through the activation of hypoxia inducible factors (HIFs), which play an essential role in shifting to an anaerobic energy production.7 HIFs in turn upregulate the expression of multiple genes associated with angiogenesis, metabolic regulation, pH balance and cell apoptosis, which promote tumor survival. The essential role of HIFs in vascular protection, recovery of tumor blood and nutrient supply, makes solid tumors difficult to treat, leading to resistance in radiotherapy, chemotherapy (CT) and immunotherapy.7
Methods of detecting tumor hypoxia
As cancer therapy is significantly affected by hypoxia, a number of methods have been developed to measure and assess tumor hypoxia.1,8 These include invasive measurements such as oxygen analysis with polarographic electrodes (see Figure 2)4 and fiber-optic probes, immunohistochemical detection of exogenously administered drugs (eg, pimonidazole, EF5 and 2-nitroimidazole drugs) that label hypoxic cells, and noninvasive imaging methods such as positron emission tomography (PET) using hypoxia tracers (eg, 2-nitroimidazole,18 F-labeled tracers (MISO, FMISO, EF5, FAZA and HX4), 11C-acetate and others) and MRI, specifically blood oxygenation level dependent (BOLD) or tissue oxygenation level dependent (TOLD) MRI.8,9
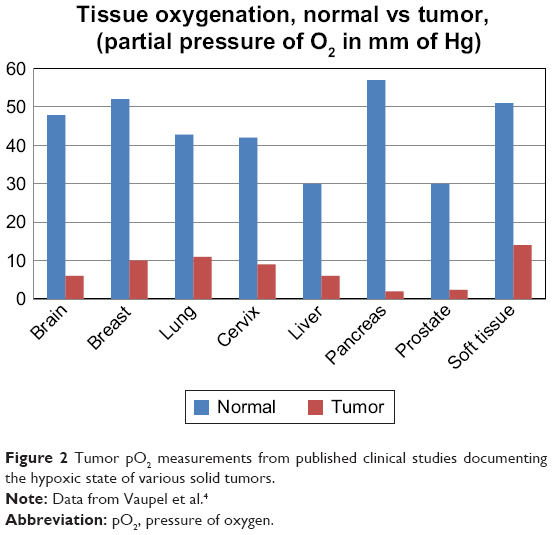 | Figure 2 Tumor pO2 measurements from published clinical studies documenting the hypoxic state of various solid tumors. |
Oxygen electrodes have been used extensively to provide the oxygenation status of solid tumors.4 Figure 2 demonstrates the collective results of 125 clinical studies that show the hypoxic state of varying types of solid tumors.4 Although the use of polarographic measurement of partial pressure of oxygen (pO2) is a common approach to identify hypoxia in solid tumors, it has the disadvantage of being an invasive technique that is prone to variability and measurement bias depending on the location of the sensor.1,10
Hypoxyprobe™ (pimonidazole) is a non-toxic exogenous 2-nitroimidazole that forms adducts with thiol groups in proteins in hypoxic cells. Detection of pimonidazole adducts can then be detected through immunohistochemistry (IHC) for qualitative assessment of tumor hypoxia. Pimonidazole can be administered orally or IV in humans and can also be administered intra-peritoneally for animal studies. As shown in Figure 3, pimonidazole staining provides evidence of deep tissue hypoxia. IHC staining of ovarian tumor samples detected activation of Hypoxyprobe™-1 (adduct formation) in six different tumors.
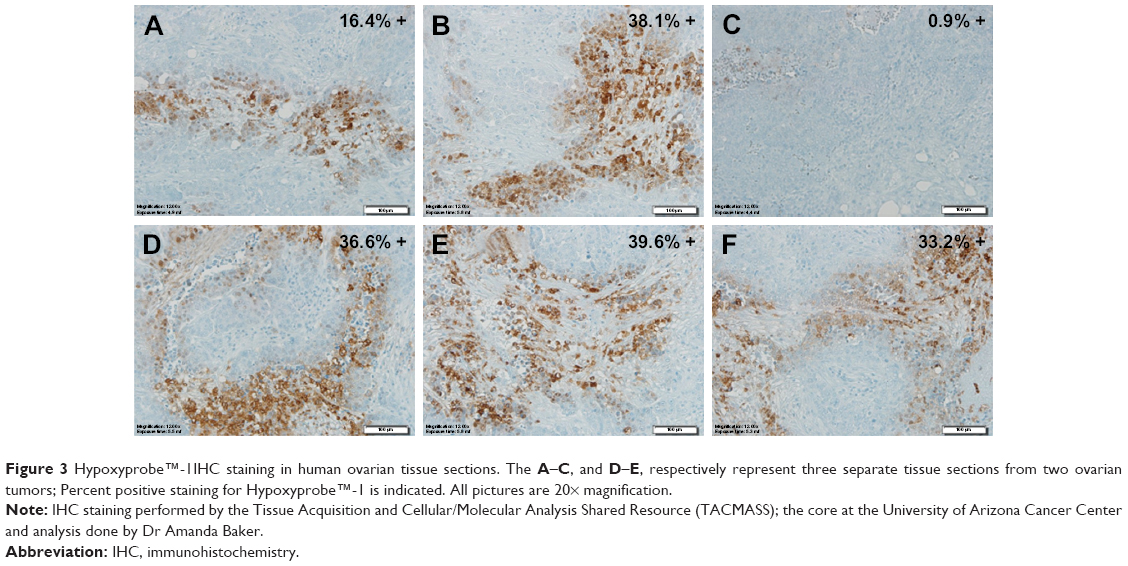 | Figure 3 Hypoxyprobe™-1IHC staining in human ovarian tissue sections. The A–C, and D–E, respectively represent three separate tissue sections from two ovarian tumors; Percent positive staining for Hypoxyprobe™-1 is indicated. All pictures are 20× magnification. |
Five of the six tumor samples showed greater than 15% staining of the tumor regions represented (see percent positive staining in upper right corner of each picture in Figure 3). The data also suggests that at least one of the ovarian tumors (comparing A, B and C) had significant intra-tumor heterogeneity. Due to the spatial and temporal heterogeneity of hypoxia, sampling bias is problematic and the clinical utility of endogenous biomarkers depends on the number, location and timing of samples biopsied.8
Precision medicine for cancer treatment will most likely require multiple biomarker assessments over time in a single patient, with individualized treatment based on the changing tumor and patient characteristics.8 For this type of assessment, noninvasive imaging methods offer an important benefit of providing the location of the hypoxic microregions throughout the tumor. PET imaging using 2-nitroimidazole 18F-labeled tracers can provide spatial mapping of hypoxia over time. However, the success of the imaging is dependent upon successful delivery of the tracer to the neoplastic tissue.10 11C-acetate is a PET radiotracer that has recently gained popularity due to its role in tumor metabolism, specifically lipid synthesis. Not only is 11C-acetate overexpressed in certain types of cancers such as prostate cancer, renal cell carcinoma and hepatocellular carcinoma, but it may also be useful in the detection of tumor recurrence.11,12 Due to the novelty of 11C-acetate, limited information is available on its prognostic value, but it is predicted to be an emerging imaging tool in the field of oncology.11
Oxygen-enhanced MRI is an alternative approach in noninvasive imaging. BOLD MRI is a well-known imaging modality to monitor oxygen levels in the blood, as transverse relaxation rate (R2*) is influenced by the conversion of deoxy- to oxyhemoglobin.13 However, BOLD MRI is largely dependent on blood flow and is not directly interpretable as tissue pO2 or O2 concentration.1,8,13 TOLD MRI is a novel approach in imaging hypoxia that uses the spin lattice relaxation rate (R1) to measure the concentration of free oxygen molecules in the plasma or interstitial tissue fluid and thus, pO2.13,14 TOLD MRI is a more robust biomarker for tumor hypoxia than BOLD MRI and is more cost-effective than PET imaging, as it does not require the use of a radiotracer.
Tumor hypoxia as an impediment to radiotherapy, CT and immunotherapy
Table 1 below shows the critical oxygen tensions (millimeters of mercury, mmHg) at which typical cellular functions in solid tumors progressively cease or anticancer treatments are impaired as a result of inadequate O2 availability in tumor tissue.15 Profound hypoxia promotes changes in the proteome and genome.1 Genomic instability, in turn, promotes dedifferentiation and progression of the aggressive cancer geno and phenotype.
 | Table 1 Effect of oxygen tension on cancer therapya |
The primary mechanism of radiation is creation of reactive oxygen species, which in turn irreversibly damage tumor cell DNA resulting in apoptosis and cell death.16 In the 1950s, Gray et al performed experiments showing that a three-fold higher dose of radiation was required to kill hypoxic as opposed to normoxic cells.17 Due to the limited tolerance of normal tissues to radiation, it is generally not possible to increase the dose of radiation to compensate for tumor hypoxia. After radiation, hypoxic tumor cells may persist and then divide, resulting in tumor persistence and development of an even more aggressive tumor phenotype.
The generation of reactive oxygen species can also be achieved through photodynamic therapy (PDT). PDT requires the administration of a photosensitizer, which accumulates in tumor tissues, and is activated by local irradiation of a specific wavelength of light. Once activated, the photosensitizer transfers its energy to the oxygen available in the surrounding tissues, causing incomplete reduction of oxygen and thus, the generation of reactive oxygen species.18,19 The antitumor effects of PDT are hindered by hypoxic tumor microenvironments and would benefit from normalizing the tumor oxygen levels.
Hypoxia has been associated with treatment resistance to a number of anticancer agents.20,21 Since the tumor has limited vasculature due to its rapid growth, there exists a diffusion barrier between the drug-supplying blood vessels and the tumor cells.21 Table 2 lists chemotherapeutic drugs that have been demonstrated to be less effective in a hypoxic as opposed to a normoxic or hyperoxic environment.20–25 The drugs listed in Table 2, were tested against human cancer cell lines in vitro under varying oxygen concentrations. The results vary depending upon the cell line. The drugs that generally had poor performance in hypoxic rather than normoxic conditions are listed.
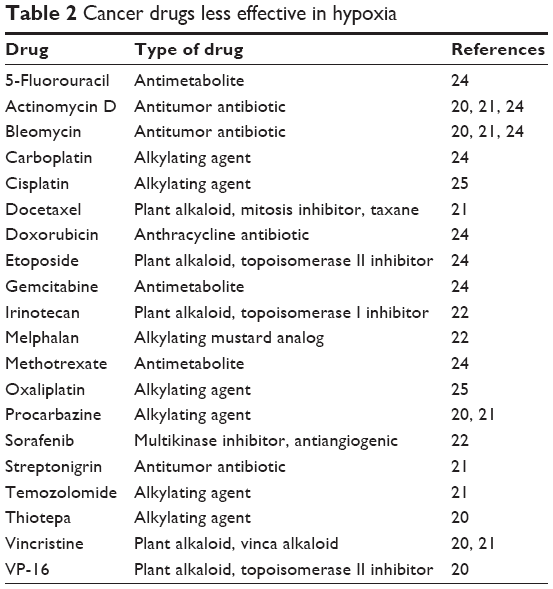 | Table 2 Cancer drugs less effective in hypoxia |
Immunotherapy has emerged as arguably the most exciting and potentially effective means of cancer treatment. Four therapeutic approaches in immuno-oncology have entered the clinic (approved by the US Food and Drug Administration [FDA]). The first of these is a prostate cancer vaccine, PROVENGE(R) (sipuleucel-T), designed to stimulate proliferation and infiltration of prostate cancer-specific T cells with tumoricidal properties.26 The second approach to gain FDA approval is an immune checkpoint inhibitor using antibody CTLA-4 (ipilimumab) and the third approach targets PD-1 (pemrolizumab, inolumab and nivolumab). Both of these receptors are expressed on T cells after activation and their expression blocks T cell proliferation and differentiation.27,28 To date, the most effective immunotherapies have been against CTLA-4, PD-1 and the PD-1 ligand (atezolizumab, durvalumab and avelumab).28–32 Still, only a minority of cancer patients currently respond to immunotherapy.
The most recent therapy to be approved is chimeric antigen receptor (CAR)-T cell immunotherapy, which is the first form of gene transfer therapy to receive commercial approval from the FDA.33 This therapy is broadly categorized as adoptive T cell transfer, referring to a transfusion medicine that infuses lymphocytes into a patient to mediate antitumor effects. CAR-T cell immunotherapy consists of autologous T cells that are engineered to express a CAR specific to CD19 B lymphocytes. This treatment is transforming the outcomes for hematologic malignancies, specifically refractory pre-B cell acute lymphoblastic leukemia and diffuse large B cell lymphoma. Although this is a strong advancement in the field of immunotherapy, there are still challenges to overcome as CAR-T cells received contingent approval due to the potential adverse effects of severe cytokine release syndrome and neurologic toxicities.33 Further progress will have to be made before CAR-T cells can be applied more broadly to solid tumors.
Hypoxia is an important factor preventing effective immunotherapy of cancer. Anaerobic glycolysis by tumor cells result in over-production of adenosine, which is excreted by the tumor cells into the extracellular tumor matrix.34 Extracellular intratumoral adenosine is a potent suppressor of T-cells.35,36 Thus, tumor hypoxia results in infiltration and accumulation of suppressor T-cells in the tumor matrix.37,38 Furthermore, HIF-1α is known to be inhibitory to cells of the adaptive immune system.34,35 Increasing tumor oxygenation to improve the efficacy of radiation, CT and immunotherapy would be of significant clinical benefit to cancer patients.
Strategies to overcome tumor hypoxia
Given the importance of hypoxia as a barrier to effective cancer treatment, several approaches have been undertaken to overcome tumor hypoxia. One of these approaches has been to develop hypoxia-activated prodrugs (HAPs), also known as bioreductive alkylating agents. HAPs are generally inactive prodrugs, which can be enzymatically reduced under hypoxic conditions, usually by one or two electron oxidoreductases to generate cytotoxic species.39,40 Early work on HAPs focused on developing derivatives of Mitomycin-C, as the drug was proven more effective in a hypoxic environment.41 However, despite more than 40 years of work, there are no HAPs approved by the FDA. Nevertheless, prior work on HAPs has advanced the field and more promising HAPs are currently under preclinical and clinical development.40 HAPs currently making clinical progress include Evofosfamide (TH-302), PR-104 and Apaziquone (EO9).42 A detailed review of HAPs is beyond the scope of this work.
Under room-air breathing, arterial hemoglobin is nearly saturated and breathing supplemental oxygen results in minimal increase in arterial pO2. Under normobaric conditions, the oxygen carrying capacity of the plasma is very limited. The oxygen carrying capacity of the plasma can be increased by breathing oxygen under hyperbaric conditions.43,44 In one experiment, the effect of normo- and hyperbaric oxygen (HBO) breathing on hypoxic regions and spatial pO2 distribution was tested in a tumor model in rats. At ambient pressure (one atm), hyperoxia only slightly enhanced tumor oxygenation. In comparison, rats tested under elevated ambient pressure (two atm) while breathing supplemental oxygen had a median pO2 five-fold higher than the normobaric group.45 A study was performed on 320 patients with cervical cancer treated with radiotherapy and HBO. Both local control and patient survival were significantly improved in the HBO group.46 Similar results were obtained in another study that enrolled 505 patients with various types of cancers in a clinical trial investigating the impact of HBO with radiotherapy.47 Studies have also been performed with HBO in humans with malignant brain tumors. HBO has been shown to restore normoxia in hypoxic regions in brain tumor tissue in patients.48 Clinical studies of HBO in brain cancer patients with glioma, treated with radiotherapy suggest that HBO improves survival.49 Other studies, however, suggest a high rate of complications from HBO + radiotherapy, including radiation necrosis and seizures.50 Overall, the results from HBO + radiotherapy are encouraging, but HBO has not emerged as the mainstream therapy and further study is warranted and necessary to establish the efficacy of HBO in cancer treatment.51
Recently, the use of 60% supplemental oxygen has been explored preclinically to increase the effects of the immune system in the tumor microenvironment.38,52 In these studies, the reversal of hypoxia decreased the accumulation of extracellular adenosine in the tumor, thereby decreasing the intensity of the A2A adenosine receptor (A2AR) mediated immunosuppression. This led to an increase in the activity of T and NK cells, promoting tumor regression and survival.38 The results from these studies demonstrate the potential value that supplemental oxygen could have in the clinic when administered concomitantly with existing cancer immunotherapies. A challenge associated with this immunological coadjuvant is that preclinical studies suggest that patients would require 24 hour/day respiratory hyperoxia to show optimal results. Further studies are required to determine the best method to move this practice from bench to bedside.
Another approach, to potentially reverse tumor hypoxia, has been to use allosteric hemoglobin modifiers. RSR13 (Efaproxiral) is a small molecule that binds non-covalently in the central water cavity of the hemoglobin tetramer and reduces hemoglobin oxygen-binding affinity. By facilitating the release of oxygen from hemoglobin, RSR13 causes an increase in whole blood p50 (pO2 for 50% hemoglobin saturation) and an increase in tissue pO2.53,54 Research in Efaproxiral was discontinued when the results of the Phase III ENRICH trial in patients with breast cancer and hypoxic brain metastases treated with whole brain radiation therapy (RT) and Efaproxiral failed to demonstrate significant improvement in overall survival.55
Molecules to improve the diffusion of oxygen are another strategy being tested to reverse tumor hypoxia. Trans sodium crocetinate (TSC) is a kosmotrope that causes physical changes in blood plasma allowing oxygen to diffuse more quickly from red blood cells to the vascular wall.56 TSC is under clinical development as a radiosensitizer for glioblastoma multiforme (GBM), pancreatic cancer and brain metastases. A Phase Ib/II clinical trial in GBM patients treated with chemoradiation and TSC has been completed.56 Diffusion Pharmaceuticals, the company developing TSC, plans to perform a subsequent randomized, placebo-controlled Phase II/III clinical trial of TSC in association with chemoradiation treatment of GBM.
Oxygen transport agents have also been developed to reverse tumor hypoxia. The two basic types include those using hemoglobin and those using fluorocarbons (FCs).16 Hemoglobin-based agents can be prepared using several different methods such as cross-linked human and animal hemoglobins, polymerized hemoglobins, polyethylene glycol (PEG) conjugated hemoglobins and genetically-engineered hemoglobins. Preclinical studies of both PEG-hemoglobin and liposome-encapsulated hemoglobin demonstrated reversal of tumor hypoxia.57,58 However, questions remain about the nonlinear dose response of both hemoglobin-based agents. More recently, a company called Omniox has developed an oxygen carrier, OMX, based on the heme nitric oxide/oxygen-binding protein that has been shown, preclinically in FL261 tumor-bearing mice, to decrease hypoxia levels and thereby reverse the immunosuppressive tumor microenvironment.59 In the future, OMX may work synergistically with radio and immunotherapy to improve GBM patient outcomes.
FC-based agents consist of fluorochemicals stabilized in emulsions. FCs can dissolve considerable amounts of oxygen based on a diffusion process from higher to lower concentration areas and thus, are able to deliver oxygen through passive diffusion in regions of hypoxia.60 Unlike hemoglobin’s localized chemical binding to oxygen molecules, a FCs’ oxygen solubility is directly proportional to the gas’ partial pressure.61 Therefore, oxygen can be largely extracted from FCs at a rapid pace when needed. This dramatically differs from the highly regulated chemical binding and release of oxygen from hemoglobin, resulting in significant differences in oxygen transport and delivery.16 Furthermore, contrary to supplemental oxygen, FCs are not limited by the carrying capacity of hemoglobin or plasma, leading to maximal oxygen delivery.
Recently, FCs, specifically perfluorohexane, have been tested preclinically to enhance PDT by loading a photosensitizer directly into the FC nanodroplets.62 While still under development, this novel technology could have a wide impact on the use of PDT in the clinic. A number of different FCs have also been studied as potential radiosensitizers, including Oxygent™ (perfluorooctylbromide (PFOBe), perflubron) and Fluosol(R)-DA 20% (perfluorodecalin (PFDe) with perfluorotrypropylamine).63–66 However, they required high doses of the FC, which negatively affected the safety profile of the drug. Up until recently, Fluosol was the only FC to be studied clinically as a radiosensitizer specifically for the treatment of GBM. Like other drug products from this class of therapeutics, the high doses of Fluosol required to demonstrate efficacy led to significant adverse events in patients.
A more recent FC to be tested as a radiosensitizer is (DDFPe, NVX-108). NVX-108 is an emulsion of 2% DDFP in stabilizers (sucrose, PEG-Telomer-B [PTB]) and phosphate buffered saline (pH 7.2). Stoichiometrically, DDFP has 200 times the oxygen carrying capacity compared to human hemoglobin. Compared to Oxygent and Fluosol, DDFPe carries far more oxygen per gram of FC, as shown below in Figure 4.67 To generate the data summarized below in Figure 4, 2% w/vol emulsions of each FC, DDFPe, PFDe and PFOBe were prepared, and the oxygen carrying capacity for each emulsion was compared at 37°C. As shown below, DDFPe carries far more oxygen per gram of FC. Compared to PFOBe and PFDe (boiling points=142°C), the boiling point of DDFPe is much lower (29°C). As the DDFPe nanodroplets are exposed to oxygen, particle sizing studies show an increase in particle diameter from about 200 nm to about 400 nm. The low boiling of DDFPe and ability of the particles to increase in diameter as they imbibe oxygen may at least partly explain the enhanced oxygen carrying capacity compared to the higher boiling point FCs. Additionally, DDFP has a higher proportion of methyl groups than the higher molecular weight FCs, which may also account for some of the improved oxygen carrying capacity per gram of FC.60
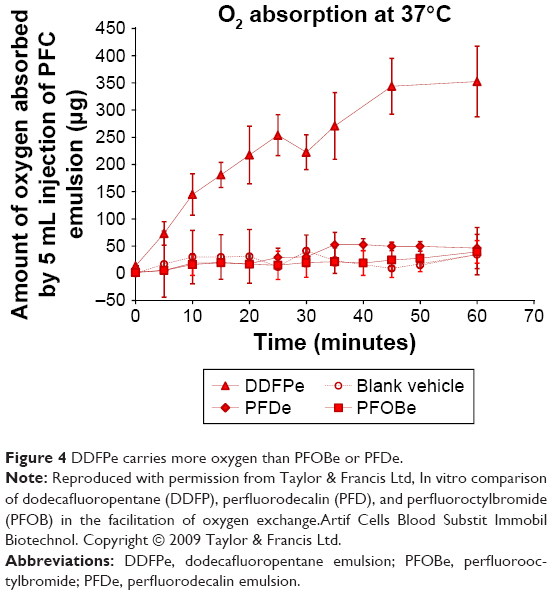 | Figure 4 DDFPe carries more oxygen than PFOBe or PFDe. |
NVX-108 as a radiosensitizer
Initial preclinical studies for NVX-108 demonstrated its promise as a radiosensitizer. In female SCID mice (n=29) implanted in the flank with human pancreatic tumor, xenografts (Hs-766T), survival of the NVX-108 treated group (NVX-108 + RT + carbogen) was twice that of mice treated with RT alone (RT + carbogen). Tumor pO2 was measured in the pancreatic xenografts with an oxygen fiber-optic probe (OxyLab). Compared to carbogen alone, NVX-108 + carbogen resulted in a 400% increase in tumor oxygen levels.68 Note that results from a preclinical study in a different tumor model analyzing the effects of TSC only saw an increased tumor pO2 of 40%.69 In another study of DDFPe (identical material to NVX-108 but without a physiological buffer), administration with carbogen completely reversed radiation resistance in rats with Morris hepatoma.70
NVX-108 is being tested clinically in patients with primary GBM. GBM is a primary malignant tumor of the brain with average survival of only about 14–15 months after standard treatment71 that consists of surgery (if operable) followed by RT with concomitant radiosensitizing CT (temozolomide [TMZ]). RT is administered in fractions; usually 30 fractions of 2 Gray each over 6 weeks.71 GBM is known to be hypoxic with pO2 measurements about 1/10th the level of pO2 in normal brain tissue.4,72–74 The cause of the hypoxia is uncertain, but it might arise from thrombosis of small blood vessels due to production of pro-coagulant factors by tumor cells.75,76 Hypoxia leads to necrosis of parts of the GBM and also the switching on of expression of angiogenic factors, most notably vascular endothelial growth factor.77–80 As mentioned previously, an important consequence of tumor hypoxia is relative resistance to therapeutic radiation. This occurs because radiation-induced killing of malignant cells depends on the production of oxygen free radicals by the ionizing radiation.17 Similarly, hypoxic cells can become chemoresistant due to decrease in drug action in the absence of O2, limited drug diffusion and inability to deliver to cells distant from functional vasculature.81 NVX-108 is being developed to deliver oxygen to hypoxic GBM tissue, which will boost the effectiveness of frontline treatment with both RT and CT.
Specific to the CT used in GBM, a recent in vitro study demonstrated the effects of hyperoxia on human glioblastoma cells resistant to TMZ.23 TMZ-sensitive GBM cells were repetitively exposed to TMZ to develop subclones of TMZ-resistant GBM cells. These TMZ-resistant cells were then exposed to varying oxygen levels, with or without TMZ treatment. The results showed that the TMZ sensitivity of both chemo-sensitive and resistant cells was significantly increased under hyperoxia. The specific mechanism of hyperoxia resulting in enhanced TMZ toxicity was hypothesized to be due to the induction of apoptosis, specifically via MAPK-related pathways.23 This study suggests that increased oxygen delivery may potentially result in an improved clinical outcome in both TMZ responders and non-responders.
NVX-108 was tested in an Australian Phase Ib/II dose finding, pharmacodynamic study of NVX-108 combined with radiation and TMZ in patients with newly-diagnosed GBM (NCT02189109).82 All patients received standard chemoradiation consisting of 30 fractions of focal brain radiation (total 60 Gray, given as 2 Gray fractions, 5 days per week for 6 weeks) with concurrent oral TMZ at a dose of 75 mg/m2 day, 7 days per week for 6 weeks. NVX-108 was administered by i.v. infusion over 30 minutes in combination with each fraction of radiation. Patients continuously breathed either 100% oxygen or carbogen (a mixture of oxygen 98% and carbon dioxide 2%) from the start of each NVX-108 infusion. Preliminary studies at the University of Arizona have demonstrated that there is no difference between carbogen and 100% oxygen with respect to oxygenation of tumors in mice.10 Reversal of tumor hypoxia was monitored using TOLD MRI on some of the patients, pre and post-administration of NVX-108. TOLD MRI showed significant decrease in T1 in tumor tissue (P=0.015) with no significant change in normal brain tissue.82
The Phase Ib/II study consisted of an initial dose exploration phase with a starting dose of 0.05 mL/kg and a dose expansion phase at the MTD of 0.1 mL/kg. An accelerated dose-escalation scheme was employed, using NVX-108 dose levels of 0.05, 0.1, 0.17, 0.25 and 0.35 mL/kg. The MTD was determined to be 0.1 mL/kg, with dose limiting toxicity in the 0.17 mL/kg group being delayed radiation necrosis. Presently, nine patients have been treated at the MTD.
Human exposure to NVX-108 prior to the Phase Ib/II clinical study had been performed when the material was tested as an ultrasound contrast agent with the hypobarically-activated compound (activated by pulling back on the filled syringe to apply negative pressure). In clinical pharmacokinetic studies, human volunteers received two doses of DDFPe (24 hours apart). The maximum administered dose (0.35 mL/kg via rapid i.v. bolus) resulted in no significant adverse effects. Review of the earlier PK data indicates a terminal half-life for DDFP of about 90 minutes in humans with clearance via exhalation.
As mentioned previously, Fluosol is the only FC emulsion besides NVX-108 to be tested in clinical studies as a radiosensitizer. However, the doses of Fluosol required in the clinical trials were approximately 800–1600-fold higher than for NVX-108, on a gram basis of FC. Table 3 summarizes the results of clinical studies of NVX-108 and Fluosol as radiosensitizers.
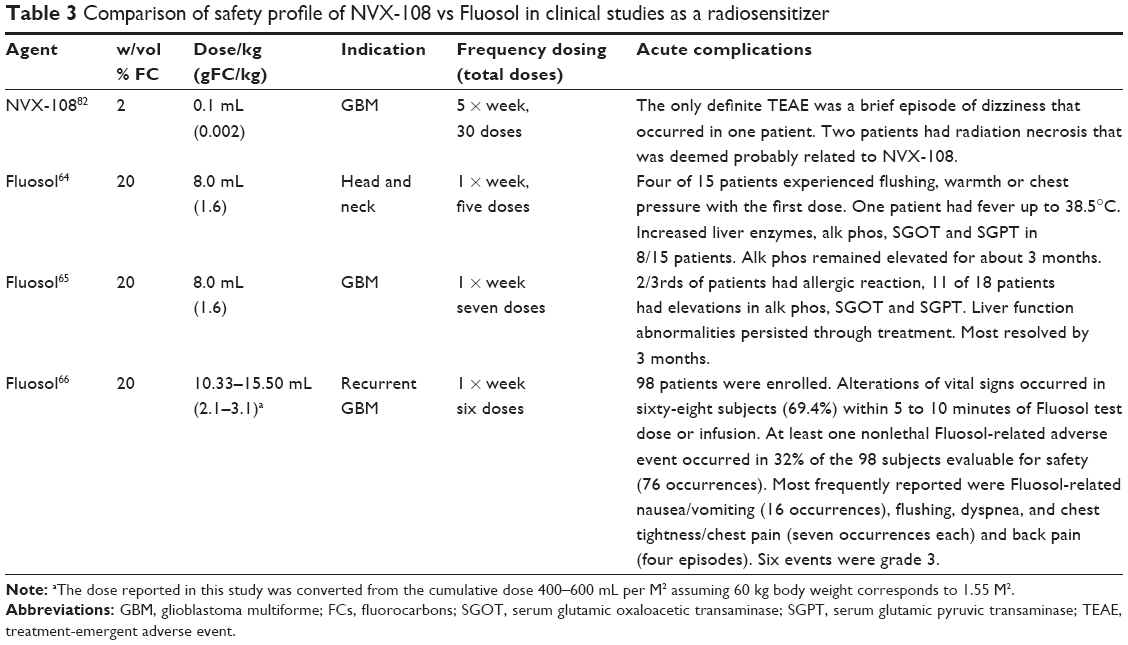 | Table 3 Comparison of safety profile of NVX-108 vs Fluosol in clinical studies as a radiosensitizer |
Not only were the doses of Fluosol in these trials much higher than those of NVX-108, but Fluosol could only be administered a maximum of one time per week (unlike NVX-108 which can be administered during each fraction of radiation – five times per week). While there have been minimal and no serious acute adverse events with administration of NVX-108, Fluosol administration was associated with significant adverse events. In the largest study of 98 patients with recurrent GBM, 68% of patients had alterations of vital signs. Overall, allergic reactions including altered liver enzymes, nausea and vomiting as well as other adverse reactions were common. NVX-108 is the first oxygen-therapeutic capable of administration during each fraction of RT without causing significant adverse reactions. Current results from the Phase Ib study of NVX-108 in GBM patients indicate safety and a trend of improvement in the overall survival of patients.
Conclusion
Tumor hypoxia is a prevalent and major obstacle to effective cancer treatment with radiotherapy, CT and immunotherapy. A variety of different methods have been tried to reverse tumor hypoxia and to date there are no FDA approved treatments to reverse tumor hypoxia. At least two different products designed to reverse tumor hypoxia are currently under clinical trials. One of these is designed to improve diffusion of oxygen. The other is a low boiling point FC that carries more oxygen per gram than other FCs that had been previously tested. Randomized, prospective, placebo-controlled clinical trials will be necessary to show efficacy of the new drugs in treating tumor hypoxia.
Acknowledgment
We would like to thank Dr Amanda Baker, PharmD, PhD for her edits to this document and for providing the material for Figure 3.
Disclosure
Ms Kaitlin Graham is an employee of NuvOx Pharma and owns stock in the company. Dr Evan Unger is President and CEO of NuvOx Pharma, serves on the Board of Directors, and owns stock in the company. Dr Unger is also a patent holder of the NuvOx Pharma technology. The authors report no other conflicts of interest in this work.
References
Vaupel P, Mayer A, Höckel M. Tumor hypoxia and malignant progression. Methods Enzymol. 2004;381:335–354. | ||
Less JR, Posner MC, Skalak TC, Wolmark N, Jain RK. Geometric resistance and microvascular network architecture of human colorectal carcinoma. Microcirculation. 1997;4(1):25–33. | ||
Siemann DW. The unique characteristics of tumor vasculature and preclinical evidence for its selective disruption by Tumor-Vascular Disrupting Agents. Cancer Treat Rev. 2011;37(1):63–74. | ||
Vaupel P, Höckel M, Mayer A. Detection and characterization of tumor hypoxia using pO2 histography. Antioxid Redox Signal. 2007;9(8):1221–1235. | ||
Zhu W, Dong Z, Fu T, et al. Modulation of Hypoxia in Solid Tumor Microenvironment with MnO2 Nanoparticles to Enhance Photodynamic Therapy. Adv Funct Mater. 2016;26(30):5490–5498. | ||
Moulder JE, Rockwell S. Hypoxic fractions of solid tumors: experimental techniques, methods of analysis, and a survey of existing data. Int J Radiat Oncol Biol Phys. 1984;10(5):695–712. | ||
Zeng W, Liu P, Pan W, Singh SR, Wei Y. Hypoxia and hypoxia inducible factors in tumor metabolism. Cancer Lett. 2015;356(2 Pt A):263–267. | ||
Dhani N, Fyles A, Hedley D, Milosevic M. The clinical significance of hypoxia in human cancers. Semin Nucl Med. 2015;45(2):110–121. | ||
Kizaka-Kondoh S, Konse-Nagasawa H. Significance of nitroimidazole compounds and hypoxia-inducible factor-1 for imaging tumor hypoxia. Cancer Sci. 2009;100(8):1366–1373. | ||
Schafer R, Gmitro AF. Dynamic oxygenation measurements using a phosphorescent coating within a mammary window chamber mouse model. Biomed Opt Express. 2015;6(2):639–650. | ||
Kitajima K, Murphy RC, Nathan MA, Sugimura K. Update on positron emission tomography for imaging of prostate cancer. Int J Urol. 2014;21(1):12–23. | ||
Spick C, Herrmann K, Czernin J. Evaluation of Prostate Cancer with 11C-Acetate PET/CT. J Nucl Med. 2016;57(Suppl 3):30S–37S. | ||
White DA, Zhang Z, Li L, et al. Developing oxygen-enhanced magnetic resonance imaging as a prognostic biomarker of radiation response. Cancer Lett. 2016;380(1):69–77. | ||
O’Connor JP, Boult JK, Jamin Y, et al. Oxygen-Enhanced MRI Accurately Identifies, Quantifies, and Maps Tumor Hypoxia in Preclinical Cancer Models. Cancer Res. 2016;76(4):787–795. | ||
Höckel M, Vaupel P. Tumor hypoxia: definitions and current clinical, biologic, and Molecular Aspects. J Natl Cancer Inst. 2001;93(4):266–276. | ||
Rockwell S, Dobrucki IT, Kim EY, Marrison ST, Vu VT, Vt V. Hypoxia and radiation therapy: past history, ongoing research, and future promise. Curr Mol Med. 2009;9(4):442–458. | ||
Gray LH, Conger AD, Ebert M, Hornsey S, Scott OC. The concentration of oxygen dissolved in tissues at the time of irradiation as a factor in radiotherapy. Br J Radiol. 1953;26(312):638–648. | ||
Dang J, He H, Chen D, Yin L. Manipulating tumor hypoxia toward enhanced photodynamic therapy (PDT). Biomater Sci. 2017;5(8):1500–1511. | ||
Moghissi K, Dixon K, Gibbins S. A Surgical View of Photodynamic Therapy in Oncology: A Review. Surg J (N Y). 2015;1(1):e1–e15. | ||
Teicher BA, Holden SA, Al-Achi A, Herman TS. Classification of antineoplastic treatments by their differential toxicity toward putative oxygenated and hypoxic tumor subpopulations in vivo in the FSaIIC murine fibrosarcoma. Cancer Res. 1990;50(11):3339–3344. | ||
Teicher BA, Lazo JS, Sartorelli AC. Classification of antineoplastic agents by their selective toxicities toward oxygenated and hypoxic tumor cells. Cancer Res. 1981;41(1):73–81. | ||
Strese S, Fryknäs M, Larsson R, Gullbo J. Effects of hypoxia on human cancer cell line chemosensitivity. BMC Cancer. 2013;13:331. | ||
Sun S, Lee D, Lee NP, et al. Hyperoxia resensitizes chemoresistant human glioblastoma cells to temozolomide. J Neurooncol. 2012;109(3):467–475. | ||
Wouters A, Pauwels B, Lardon F, Vermorken JB. Review: implications of in vitro research on the effect of radiotherapy and chemotherapy under hypoxic conditions. Oncologist. 2007;12(6):690–712. | ||
Baker AF, Koh MY, Williams RR, et al. Identification of thioredoxin-interacting protein 1 as a hypoxia-inducible factor 1 alpha-induced gene in pancreatic cancer. Pancreas. 2008;36(2):178–186. | ||
Cheever MA, Higano CS. PROVENGE (Sipuleucel-T) in prostate cancer: the first FDA-approved therapeutic cancer vaccine. Clin Cancer Res. 2011;17(11):3520–3526. | ||
Ascierto PA, Addeo R, Cartenì G, et al. The role of immunotherapy in solid tumors: report from the Campania Society of Oncology Immunotherapy (SCITO) meeting, Naples 2014. J Transl Med. 2014;12:291. | ||
Markovic SN, Kumar AB. Therapeutic Targets of FDA-Approved Immunotherapies in Oncology. The Basics of Cancer Immunotherapy. Dong H, Markovic SN, editors. Cham: Springer International Publishing; 2018:21–37. | ||
Hodi FS, O’Day SJ, Mcdermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363(8):711–723. | ||
Hamid O, Robert C, Daud A, et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med. 2013;369(2):134–144. | ||
Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366(26):2443–2454. | ||
Herbst RS, Soria JC, Kowanetz M, et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature. 2014;515(7528):563–567. | ||
June CH, O’Connor RS, Kawalekar OU, Ghassemi S, Milone MC. CAR T cell immunotherapy for human cancer. Science. 2018;359(6382):1361–1365. | ||
Leone RD, Horton MR, Powell JD. Something in the air: hyperoxic conditioning of the tumor microenvironment for enhanced immunotherapy. Cancer Cell. 2015;27(4):435–436. | ||
Hatfield SM, Sitkovsky M. A2A adenosine receptor antagonists to weaken the hypoxia-HIF-1α driven immunosuppression and improve immunotherapies of cancer. Curr Opin Pharmacol. 2016;29:90–96. | ||
Sitkovsky MV, Hatfield S, Abbott R, Belikoff B, Lukashev D, Ohta A. Hostile, hypoxia-A2-adenosinergic tumor biology as the next barrier to overcome for tumor immunologists. Cancer Immunol Res. 2014;2(7):598–605. | ||
Facciabene A, Peng X, Hagemann IS, et al. Tumour hypoxia promotes tolerance and angiogenesis via CCL28 and T(reg) cells. Nature. 2011;475(7355):226–230. | ||
Hatfield SM, Kjaergaard J, Lukashev D, et al. Immunological mechanisms of the antitumor effects of supplemental oxygenation. Sci Transl Med. 2015;7(277):277ra230. | ||
Brown JM. Tumor hypoxia in cancer therapy. Methods Enzymol. 2007;435:297–321. | ||
Yeh JJ, Kim WY. Targeting tumor hypoxia with hypoxia-activated prodrugs. J Clin Oncol. 2015;33(13):1505–1508. | ||
Lin AJ, Cosby LA, Shansky CW, Sartorelli AC. Potential bioreductive alkylating agents. 1. Benzoquinone derivatives. J Med Chem. 1972;15(12):1247–1252. | ||
Guise CP, Mowday AM, Ashoorzadeh A, et al. Bioreductive prodrugs as cancer therapeutics: targeting tumor hypoxia. Chin J Cancer. 2014;33(2):80–86. | ||
Brizel DM, Lin S, Johnson JL, Brooks J, Dewhirst MW, Piantadosi CA. The mechanisms by which hyperbaric oxygen and carbogen improve tumour oxygenation. Br J Cancer. 1995;72(5):1120–1124. | ||
Jain KK, editor. Physical, physiological, and biochemical aspects of hyperbaric oxygenation. In: Jain KK, editor. Textbook of Hyperbaric Medicine. Cham: Springer International Publishing; 2017:11–22. | ||
Thews O, Vaupel P. Spatial oxygenation profiles in tumors during normo- and hyperbaric hyperoxia. Strahlenther Onkol. 2015;191(11):875–882. | ||
Watson ER, Halnan KE, Dische S, et al. Hyperbaric oxygen and radiotherapy: a Medical Research Council trial in carcinoma of the cervix. Br J Radiol. 1978;51(611):879–887. | ||
Cade IS, McEwen JB. Clinical trials of radiotherapy in hyperbaric oxygen at Portsmouth, 1964–1976. Clin Radiol. 1978;29(3):333–338. | ||
Beppu T, Kamada K, Yoshida Y, Arai H, Ogasawara K, Ogawa A. Change of oxygen pressure in glioblastoma tissue under various conditions. J Neurooncol. 2002;58(1):47–52. | ||
Chang CH. Hyperbaric oxygen and radiation therapy in the management of glioblastoma. Natl Cancer Inst Monogr. 1977;46:163–169. | ||
Kohshi K, Kinoshita Y, Terashima H, Konda N, Yokota A, Soejima T. Radiotherapy after hyperbaric oxygenation for malignant gliomas: a pilot study. J Cancer Res Clin Oncol. 1996;122(11):676–678. | ||
Stępień K, Ostrowski RP, Matyja E. Hyperbaric oxygen as an adjunctive therapy in treatment of malignancies, including brain tumours. Med Oncol. 2016;33(9):101. | ||
Hatfield SM, Kjaergaard J, Lukashev D, et al. Systemic oxygenation weakens the hypoxia and hypoxia inducible factor 1α-dependent and extracellular adenosine-mediated tumor protection. J Mol Med (Berl). 2014;92(12):1283–1292. | ||
Khandelwal SR, Kavanagh BD, Lin PS, et al. RSR13, an allosteric effector of haemoglobin, and carbogen radiosensitize FSAII and SCCVII tumours in C3H mice. Br J Cancer. 1999;79(5–6):814–820. | ||
Suh JH, Stea B, Nabid A, et al. Phase III study of efaproxiral as an adjunct to whole-brain radiation therapy for brain metastases. J Clin Oncol. 2006;24(1):106–114. | ||
Suh JH, Stea B, Tankel K, et al. Results of the Phase III ENRICH (RT-016) Study of Efaproxiral Administered Concurrent with Whole Brain Radiation Therapy (WBRT) in Women with Brain Metastases from Breast Cancer. Int J Radiat Oncol Biol Phys. 2008;72(1):S50–S51. | ||
Gainer JL, Sheehan JP, Larner JM, Jones DR. Trans sodium crocetinate with temozolomide and radiation therapy for glioblastoma multiforme. J Neurosurg. 2017;126(2):460–466. | ||
Murayama C, Kawaguchi AT, Ishikawa K, et al. Liposome-encapsulated hemoglobin ameliorates tumor hypoxia and enhances radiation therapy to suppress tumor growth in mice. Artif Organs. 2012;36(2):170–177. | ||
Teicher BA, Ara G, Herbst R, Takeuchi H, Keyes S, Northey D. PEG-hemoglobin: effects on tumor oxygenation and response to chemotherapy. In Vivo. 1997;11(4):301–311. | ||
Moan NL, Leung P, Ng S, et al. Abstract 4686: Omx a hypoxia modulator reverses the immunosuppressive glioblastoma microenvironment by stimulating T cell infiltration and activation that results in increased number of long-term survivors. Cancer Res. 2017;77(13 Supplement):4686–4686. | ||
Johnson JLH. Oxygen carriers: are they enough for cellular support? In: Lapchak PA, Zhang JH, editors. Neuroprotective Therapy for Stroke and Ischemic Disease. Cham: Springer International Publishing; 2017:621–640. | ||
Riess JG. Understanding the fundamentals of perfluorocarbons and perfluorocarbon emulsions relevant to in vivo oxygen delivery. Artif Cells Blood Substit Immobil Biotechnol. 2005;33(1):47–63. | ||
Cheng Y, Cheng H, Jiang C, et al. Perfluorocarbon nanoparticles enhance reactive oxygen levels and tumour growth inhibition in photodynamic therapy. Nat Commun. 2015;6:8785. | ||
Rockwell S, Irvin CG, Kelley M, et al. Effects of hyperbaric oxygen and a perfluorooctylbromide emulsion on the radiation responses of tumors and normal tissues in rodents. Int J Radiat Oncol Biol Phys. 1992;22(1):87–93. | ||
Rose C, Lustig R, Mcintosh N, Teicher B. A clinical trial of Fluosol DA 20% in advanced squamous cell carcinoma of the head and neck. Int J Radiat Oncol Biol Phys. 1986;12(8):1325–1327. | ||
Evans RG, Kimler BF, Morantz RA, et al. A phase I/II study of the use of Fluosol as an adjuvant to radiation therapy in the treatment of primary high-grade brain tumors. Int J Radiat Oncol Biol Phys. 1990;19(2):415–420. | ||
Hochberg F, Prados M, Russell C, et al. Treatment of recurrent malignant glioma with BCNU-fluosol and oxygen inhalation. A phase I–II study. J Neurooncol. 1997;32(1):45–55. | ||
Johnson JL, Dolezal MC, Kerschen A, Matsunaga TO, Unger EC. In vitro comparison of dodecafluoropentane (DDFP), perfluorodecalin (PFD), and perfluoroctylbromide (PFOB) in the facilitation of oxygen exchange. Artif Cells Blood Substit Immobil Biotechnol. 2009;37(4):156–162. | ||
Johnson JL, Leos RA, Baker AF, Unger EC. Radiosensitization of Hs-766T Pancreatic Tumor Xenografts in Mice Dosed with Dodecafluoropentane Nano-Emulsion-Preliminary Findings. J Biomed Nanotechnol. 2015;11(2):274–281. | ||
Sheehan J, Sherman J, Cifarelli C, et al. Effect of trans sodium crocetinate on brain tumor oxygenation. Laboratory investigation. J Neurosurg. 2009;111(2):226–229. | ||
Koch CJ, Oprysko PR, Shuman AL, Jenkins WT, Brandt G, Evans SM. Radiosensitization of hypoxic tumor cells by dodecafluoropentane: a gas-phase perfluorochemical emulsion. Cancer Res. 2002;62(13):3626–3629. | ||
Stupp R, Mason WP, van den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352(10):987–996. | ||
Collingridge DR, Piepmeier JM, Rockwell S, Knisely JP. Polarographic measurements of oxygen tension in human glioma and surrounding peritumoural brain tissue. Radiother Oncol. 1999;53(2):127–131. | ||
Meixensberger J, Dings J, Kuhnigk H, Roosen K. Studies of tissue PO2 in normal and pathological human brain cortex. Acta Neurochir Suppl. 1993;59:58–63. | ||
Rampling R, Cruickshank G, Lewis AD, Fitzsimmons SA, Workman P. Direct measurement of pO2 distribution and bioreductive enzymes in human malignant brain tumors. Int J Radiat Oncol Biol Phys. 1994;29(3):427–431. | ||
Bluff JE, Brown NJ, Reed MW, Staton CA. Tissue factor, angiogenesis and tumour progression. Breast Cancer Res. 2008;10(2):204. | ||
Ogiichi T, Hirashima Y, Nakamura S, Endo S, Kurimoto M, Takaku A. Tissue factor and cancer procoagulant expressed by glioma cells participate in their thrombin-mediated proliferation. J Neurooncol. 2000;46(1):1–9. | ||
Maity A, Pore N, Lee J, Solomon D, O’Rourke DM. Epidermal growth factor receptor transcriptionally up-regulates vascular endothelial growth factor expression in human glioblastoma cells via a pathway involving phosphatidylinositol 3′-kinase and distinct from that induced by hypoxia. Cancer Res. 2000;60(20):5879–5886. | ||
Peles E, Lidar Z, Simon AJ, Grossman R, Nass D, Ram Z. Angiogenic factors in the cerebrospinal fluid of patients with astrocytic brain tumors. Neurosurgery. 2004;55(3):562–567; discussion 567–568. | ||
Wachsberger P, Burd R, Dicker AP. Improving tumor response to radiotherapy by targeting angiogenesis signaling pathways. Hematol Oncol Clin North Am. 2004;18(5):1039–1057. | ||
Yao XH, Ping YF, Chen JH, et al. Production of angiogenic factors by human glioblastoma cells following activation of the G-protein coupled formylpeptide receptor FPR. J Neurooncol. 2008;86(1):47–53. | ||
Bristow RG, Hill RP. Hypoxia and metabolism. Hypoxia, DNA repair and genetic instability. Nat Rev Cancer. 2008;8(3):180–192. | ||
Unger EC. A phase Ib/II clinical trial of a novel oxygen therapeutic in chemoradiation of glioblastoma. 2017 ASCO Annual Meeting; June 2 – June 6, 2017 Chicago, IL, USA. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.