")
Back to Journals » International Journal of Nanomedicine » Volume 14
Ocular anti-inflammatory activity of prednisolone acetate loaded chitosan-deoxycholate self-assembled nanoparticles
Authors Hanafy AF , Abdalla AM , Guda TK , Gabr KE , Royall PG , Alqurshi A
Received 24 November 2018
Accepted for publication 11 March 2019
Published 29 May 2019 Volume 2019:14 Pages 3679—3689
DOI https://doi.org/10.2147/IJN.S195892
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Anderson Oliveira Lobo
Ahmed F Hanafy,1,2 Ahmed M Abdalla,1 Tawheda K Guda,1 Khairy E Gabr,1,3 Paul G Royall,4 Abdulmalik Alqurshi1
1Department of Pharmaceutics and Pharmaceutical Technology, College of Pharmacy, Taibah University, Medina, KSA; 2Research and Development Department, Al Andalous for Pharmaceutical Industries, Giza, Egypt; 3Department of Pharmaceutics, Faculty of Pharmacy, Mansoura University, Mansoura, Egypt; 4Institute of Pharmaceutical Science, King’s College London, London, SE1 9NH, UK
Background and purpose: Conventional topical ophthalmic aqueous solutions and suspensions are often associated with low bioavailability and high administration frequency, pulsatile dose and poor exposure to certain ocular parts. The aim of this study was to develop an ophthalmic nanoparticles loaded gel, for delivering prednisolone acetate (PA), to increase dosing accuracy, bioavailability, and accordingly, efficiency of PA in treating inflammatory ocular diseases.
Methods: A novel formulation of self-assembled nanoparticles was prepared by the complexation of chitosan (CS) and, the counter-ion, sodium deoxycholate (SD), loaded with the poorly-water-soluble PA. Particle size, zeta potential, encapsulation efficiency (EE) and drug loading content (LC) of prepared nanoparticles were assessed. Moreover, the nanoparticles were characterized using differential scanning calorimetry (DSC) and Fourier transform infrared spectroscopy (FTIR). Drug release and eye anti-inflammatory potential of the prepared novel formulation was investigated.
Results: Mean particle size of the nanoparticles have dropped from 976 nm ±43 (PDI 1.285) to 480 nm ±28 (PDI 1.396) when the ratio of CS-SD was decreased. The incorporation of 0.1-0.3% of polyvinyl alcohol (PVA), in the preparation stages, resulted in smaller nanoparticles: 462 nm ±19 (PDI 0.942) and 321 nm ±22 (PDI 0.454) respectively. DSC and FTIR results demonstrated the interaction between CS and SD, however, no interactions were detected between PA and CS or SD. Drug release of PA as received, in simulated tears fluid (pH 7.4), showed a twofold increase (reaching an average of 98.6% in 24 hours) when incorporated into an optimized nanoparticle gel formulation (1:5 CS-SD).
Conclusion: The anti-inflammatory effect of PA nanoparticles loaded gel on female guinea pig eyes was significantly superior to that of the micronized drug loaded gel (P < 0.05).
Keywords: nanoparticles, ocular drug delivery, chitosan, ionic gelation, prednisolone acetate
Introduction
Inflammation of the eye is a common disease occurring to all ages and both genders. The symptoms associated with this disease include periorbital pain, proptosis, eyelid ptosis or edema, and reduced ocular motility.1 Ocular inflammation is most commonly treated using topical corticosteroids. Treatment with conventional topical prednisolone and dexamethasone is usually started with hourly administration of drops for the first 4 days to stop the aggressive inflammation, this is followed by gradual decrease of medication and cessation of therapy. The major problems faced with all topical conventional ophthalmic aqueous solution and suspensions are low bioavailability and high administration frequency, pulsatile dose and poor exposure to certain ocular parts.2 Prednisolone acetate (PA) nanoparticles (NPs) incorporated into ophthalmic gel might be a good solution of the aforementioned problems in treating eye inflammation.
Chitosan (CS) is a natural hydrophilic cationic polysaccharide obtained from chitin by alkaline N-deacetylation. It is one of the most broadly employed polysaccharides with a sugar backbone composed of β-(1–4)-linked D-glucosamine and N-cetyl-D-glucosamine.3,4 CS has the advantage of being non-toxic, biocompatible, and biodegradable. These features make CS an excellent candidate for various pharmaceutical and biomedical fields.5,6
CS has mucoadhesive characteristics which can prolong the residence time of drug delivery systems at the site of drug absorption.7 In addition, CS is capable of transiently opening and penetrating the tight junctions between epithelial cells, which makes it an ideal material for drug delivery.3 Several studies have applied CS, and its derivatives, as drug carriers8-11 or as a support material in gene delivery.12,13 A variety of CS-based drug delivery formulations, in the forms of gels, tablets, and films, have been developed and investigated.14,15
NPs of CS were studied as a carrier for various drugs. CS NPs loaded with insulin demonstrated a significant lowering of the blood glucose level after nasal administration to rabbits compared to insulin applied in a CS solution.16 Prego et al17 reported that calcitonin-coated CS NPs displayed a significantly higher hypocalcemic effect in rats than that achieved from the administration of an oral nano-emulsion formulation of the same drug. CS NPs and CS coated NPs were also shown to deliver tetanus toxoid to the immune system, thereby leading to a humoral and mucosal immune response.18 Moreover, CS NPs were used as a carrier for Gancyclovir and nitric oxide.10,19
Sodium deoxycholate (SD) is one of the bile salts classed as an endogenous surfactant. SD has been employed as an absorption enhancer to increase drug transport across various biological barriers. Yamamoto et al20 indicated that SD enhances the rectal penetration of insulin in the albino rabbit. Gandhi21 stated that the transbuccal delivery of salicylic acid was increased by SD. They related this improvement to the penetration enhancement effect on the protein domain that involves uncoiling and extending of the protein helix, thereby opening the polar pathway. Senyigit et al22 prepared a stable gel containing betamethasone-17-valerate for topical skin application. The produced gel had no irritant effect on the skin and has achieved an improvement of the drug in vitro in flux and in vivo anti-inflammatory activity compared to a commercially available cream.
Deoxycholic acid, the acid moiety of SD, forms self-aggregated NPs with CS and modified CS. These NPs are formed by covalent attachment of deoxycholic acid to CS. The self-aggregated NPs have been used as a carrier for drugs, such as Adriamycin,23 paclitaxel,24 doxorubicin,25 DNA12 and gene.26 The NPs prepared by self-assembly of the amphiphilic polymer in aqueous solution can encapsulate hydrophobic drugs into its hydrophobic core, leaving its hydrophilic parts on the surface. NPs CS–SD produced by mild ionic gelation procedure using different weight ratios of CS and SD were used to encapsulate plasmid.27
The aim of this study was to develop a PA self-assembled NPs, formulated into hydrogels, using the ionic gelation properties of CS and SD, as counter-ions, to increase dosing accuracy, bioavailability, and accordingly, efficiency of PA in treating inflammatory ocular diseases. In this work, physicochemical properties and the loading capacity (LC) of the particles were assessed. Moreover, the produced NPs were formulated as an ophthalmic gel for ocular delivery. Drug release was investigated from the loaded particles in simulated tears fluid pH 7.4. Finally, the potential ocular anti-inflammatory effect of the formulated gel was estimated using female guinea pigs.
Materials and methods
Materials
PA, purity ≥98%, was kindly donated from Jamjoom Pharma co. (Jeddah, Saudi Arabia); CS, MW 1250 (Poly (D-glucosamine) deacetylated), was purchased from Sigma Aldrich (Missouri, USA); SD was purchased from Alfa Aesar (Karlsruhe, Germany); polyvinyl alcohol (PVA) (RIA international LLC, K44534850) was kindly donated from Jamjoom Pharma company (Jeddah, Saudi Arabia). Sodium bicarbonate, calcium chloride, and sodium chloride used in this study were of analytical grade.
Preparation of CS–SD NPs loaded with PA
The self-assembled CS–SD NPs were prepared using an ionic gelation technique.27 The composition of the different formulations are shown in Table 1. CS was dissolved in 5% w/v acetic acid solution to produce 1% solution which was diluted by deionized water to 1 mg/mL. PVA 2% w/v solution was prepared by dissolving PVA in preheated deionized water at 80°C then cooled to room temperature. The required amount of PVA solution was added to CS solution. SD was separately dissolved in deionized water to obtain a 1 mg/mL solution. PA was dissolved in ethanol to produce a solution of 5 mg/mL and the required volume was added to SD solution. PA was added in an amount equivalent to 40% w/v of the total solid of both CS and SD. NPs were formed when the specified amounts of SD-PA solution were added dropwise to a CS-PVA solution and magnetically stirred at 300 rpm for 15 mins at room temperature. NPs were collected by centrifugation at 24,000 rpm for 1 hr at 25°C (Centrifuge, Centurion Scientific K3 series, UK). NPs were then washed thoroughly with deionized water, for three times, and then left to dry in a desiccator under vacuum for 48 hrs.
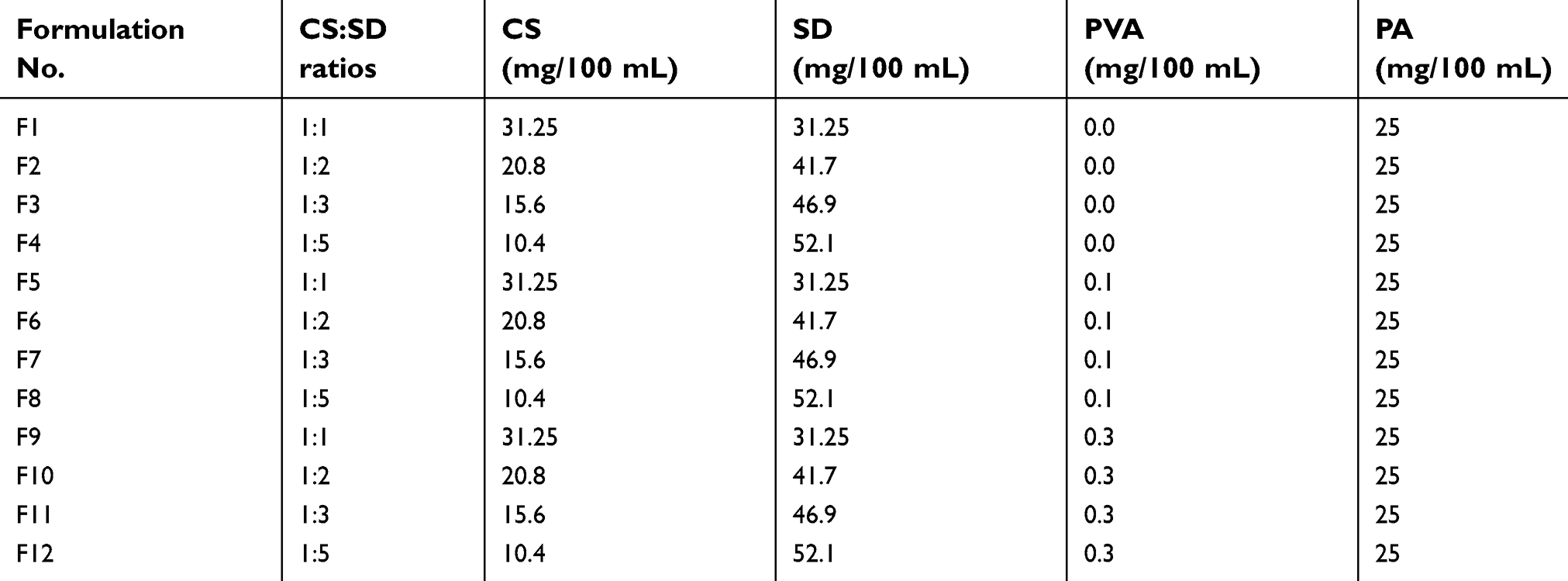 | Table 1 CS–SD self-assembled PA loaded formulations |
Characterization of CS–SD loaded PA NPs
Mean particle size and poly dispersibility index (PDI) of all the preparations were determined by a dynamic light scattering analyzer (Nanotrac Wave II, Microtrack, FL, USA) equipped with appropriate analysis software (Nanotrac Flex Version 11.1.0.1). Size measurements were performed by measuring each sample three times for 120 s at 25°C. To prepare the samples for analysis, approximately 5 mg of dried particles were suspended in 10 mL of deionized water and vortexed for 5 mins followed by sonication for 30 s.
Morphology of the F 12 formulation was studied using scanning electron microscope (SEM, SSX-550 superscan, Shimadzu, Japan) after gold coating of NPs using gold sputtering coater (GSL-1100X-SPC-12 compact plasma sputtering, MTI corporation, People's Republic of China).
To measure encapsulation efficiency (EE)% and loading content (LC)%, 20 mg of dried NPs were added to 25 mL of ethanol, and the mixture was stirred for 30 mins followed by sonication for 5 mins. The solution was then filtered through a 0.22 µm filter (Millipore, Ireland) to separate the polymer precipitates from the sample. The filtered sample was tested for drug content spectrophotometrically at 248 nm. The EE% and LC% were calculated for triplicate samples and slandered deviation (STD) calculated using the following equations (1) and (2), respectively:28
(1) (2)
where WA is the amount of drug (mg) found in the drug-loaded NPs and WD is the amount of drug (mg) initially used in the preparation of NPs. Finally, WNP is the amount of drug-loaded NPs (mg).
Thermal analysis for CS–SD loaded PA NPs
Thermal analysis was performed using differential scanning calorimeter (DSC-200 F3 Maia, NETZSCH, Germany). The instrument was calibrated, following manufacturer’s instructions, using indium. Thermal analysis was carried out for pure powder of PA, CS, SD, physical mixture of the F12 formula ingredients and the F12 formulation (dried NPs). Samples of about 5 mg were sealed in pin holed aluminum pans and the experiment was carried out under nitrogen atmosphere at a flow rate of 40 mL/min. Samples were heated at a rate of 10°C/min in the range of 0–300°C.
FTIR spectroscopy for CS–SD loaded PA NPs
FTIR spectral analysis of PA powder, CS, SD, physical mixture of the F12 formulation ingredients as well as F12 formulation NPs were performed using Fourier transform infrared spectroscopy (FTIR) (IR Affinity-1 FTIR spectrophotometer, Shimadzu, Japan) in 4,000–750 cm−1 wave number range, using a total reflectance method.
Formulation of micronized PA and PA loaded NPs ophthalmic gel
Micronized PA and selected formulations F11 and F12 NPs were formulated as ophthalmic gel. Methyl and propylparaben were included as preservatives, in concentrations of 0.15% w/v and 0.05% w/v, respectively, by dissolving them in water at approximately 75°C followed by cooling to room temperature using magnetic stirrer (Scilogex MS-H Pro, North America Inc., USA). The gel was prepared by slowly dispersing hydroxypropyl methylcellulose K4M in the preservative solution at a concentration of 2.5% w/v using Ultra-Turrax T25 digital homogenizer (IKA-Werke, Germany). Drug in concentration of 0.125% w/w or their equivalent of the NPs29 was incorporated in the gel by mixing using propeller stirrer (IKA-Werke, Germany). Preparation of these formulations was done in an aseptic environment (Biohazard safety cabinet, JSR, JSCB-900SB, Republic of Korea).30
Drug release study
A weighed amount (4 g) of the PA gel, or the F11 and or F12 NPs gel formulation, equivalent to 0.125%w/w of drug, was added into a cellophane membrane tube (Dialysis tubing cellulose membrane, 33 mm width, D9654, Sigma-Aldrich) whereby one end was sealed with closures (Universal closures, Spectrum laboratories Inc., USA). Once loaded with the gel, the cellophane membrane tube was filled with 10 mL of simulated tears fluid (STF), pH 7.4, and sealed from the second end.31 To characterize drug release profiles, the cellophane membrane tube was suspended into a USP dissolution tester vessel, apparatus 2 (paddle) (Erweka dissolution tester apparatus, Germany). Dissolution volume was kept at 500 mL, to maintain sink conditions, and 34±0.5°C to simulate the ocular surface temperature.32 Samples of 5 mL were collected and filtered through 0.22 μm filters (Millipore, Ireland) at predetermined time intervals, while necessary substitutions were made using fresh STF medium. Concentrations of PA released were quantified spectrophotometrically (Thermo Scientific Evolution 201 UV/Vis. Spectrophotometer, USA) at λmax of 248 nm after appropriate dilution.33 A mean cumulative percentage of PA released from triplicate samples for each formulation was determined and plotted against time with STD indicated as an error bar. A control dissolution test was also performed on a drug-free NPs gel formulation.
In vivo assessment of ocular anti-inflammatory effect
Procedures were approved by the Ethics Committee of Taibah University. In vivo tests were performed in the Medical Research Institute at Alexandria University in agreement with the guidelines for care and use of laboratory animals published by the United States National Institutes of Health. The in vivo study was performed on female guinea-pigs weighing (400–500 g). Benzalkonium chloride (BKC) was applied to guinea pig eyes as an inflammation induction agent to assess the anti-inflammatory effectiveness of the tested formulations.34 One milliliter of 0.2% w/v BKC was applied on cotton swab and instilled into each eye for 15 s to induce eye inflammation. This process was repeated every 1 hr for a period of up to 3 hrs till maximum ocular inflammation scoring was achieved. Animals were randomized into 4 groups each consisting of 5 guinea pigs. Groups classification was based on inflammation and drug administration; group 1 the healthy control group (normal control, saline was used instead of 0.2% BKC), group 2 the inflammation control group (non-treated eye inflammation-induced guinea pigs), group 3 the eye inflammation-induced guinea pigs group received conventional formulation containing PA dispersed in HPMC eye gel (PA-MG treated group), group 4 the eye inflammation-induced guinea pigs group received formulation F12 (PA NPs complex) loaded HPMC eye gel formulation (PA-NG treated group). After 5 hrs of inducing eye inflammation, group 3 received PA-MG gel containing 0.125% PA and group 4 received the selected PA-NG gel formulations containing 0.125% of the drug once daily.29 The inflammation control group 2 received the drug-free gel formulation at the same time. Clinical examination for ocular inflammation including conjunctiva, cornea and inflammatory changes were graded after 1 and 24 hrs as described by Monnickendam et al35 while the maximum assigned score for each eye was 69, constituted of a maximum score of 3 for each sign observed in female guinea pigs' inflamed eye (e.g. discharge & lacrimation). Scoring process for eye inflammation was performed by a consultant ophthalmologist.
All values in the in vivo study were expressed as means±STD. Statistical analysis was performed using SPSS® software (SPSS Inc., Chicago, IL, USA). Results were processed using Student’s t-test. Differences were considered significant when the associated P-value was less than 0.05.
Ethics statement
The in vivo study procedures on animals were performed in the Medical Research Institute, Alexandria University after approval by ethical committee and were consistent with the guidelines and regulations for the appropriate use of animals in biomedical studies. Female guinea-pigs of weight (400–500 g) were used in the study.
Results and discussion
The aim of this work was the production of self-assembled NPs of a poorly soluble drug by changing the weight ratio of CS and SD. The formation of CS–SD NPs is a process based on the complexation of the oppositely charged CS, as a polycation, and SD, as a negatively charged molecule. Since the critical micelle concentration (CMC) of SD is ranged from 2 to 6 mM, the SD concentration was kept below CMC. Singh et al36 stated that SD at higher concentrations could lead to the formation of micellar aggregates instead of NPs, thus the highest concentration of SD used, in all solutions of this study, did not exceed 1.258 mM.
Formulations F1–F4 showed a mean particle size ranging from 976 to 480 nm, which is considered relatively large on the nanoscale. Under the assumption that this was caused by particle aggregation, PA loaded NPs, with the same ratios of CS and SD, were prepared in the presence of a stabilizing agent, PVA, at a concentration of 0.1% w/v. Subsequently, formulations F5–F8 have shown a decrease in the mean particle size. This was further repeated with a higher concentration of PVA (0.3% w/v), resulting in a more acceptable mean particle size, ranging from 786 to 321 nm (shown by formulations F9–F12, respectively). Increasing the concentration of PVA has also shown to decrease PDI and zeta potential, this may be a result of PVA chains adsorbing to the surface of the complexed NPs, thus preventing further growth of particles, by aggregation, and charges. To avoid the interference of PVA in further characterizations, a thorough purification step, whereby NPs were washed three times with deionized water, was performed on formulations F5–F12.
Increasing the weight fraction of SD has also shown to reduce particle size and zeta potential as shown by the difference between the formulations F9 and F12 (most opalescent formulation presented in Figure 1), where increasing the weight fraction of SD by five times has reduced mean particle size by more than half, while the zeta potential was reduced from +33.7 to +28.8 mV (Table 2).
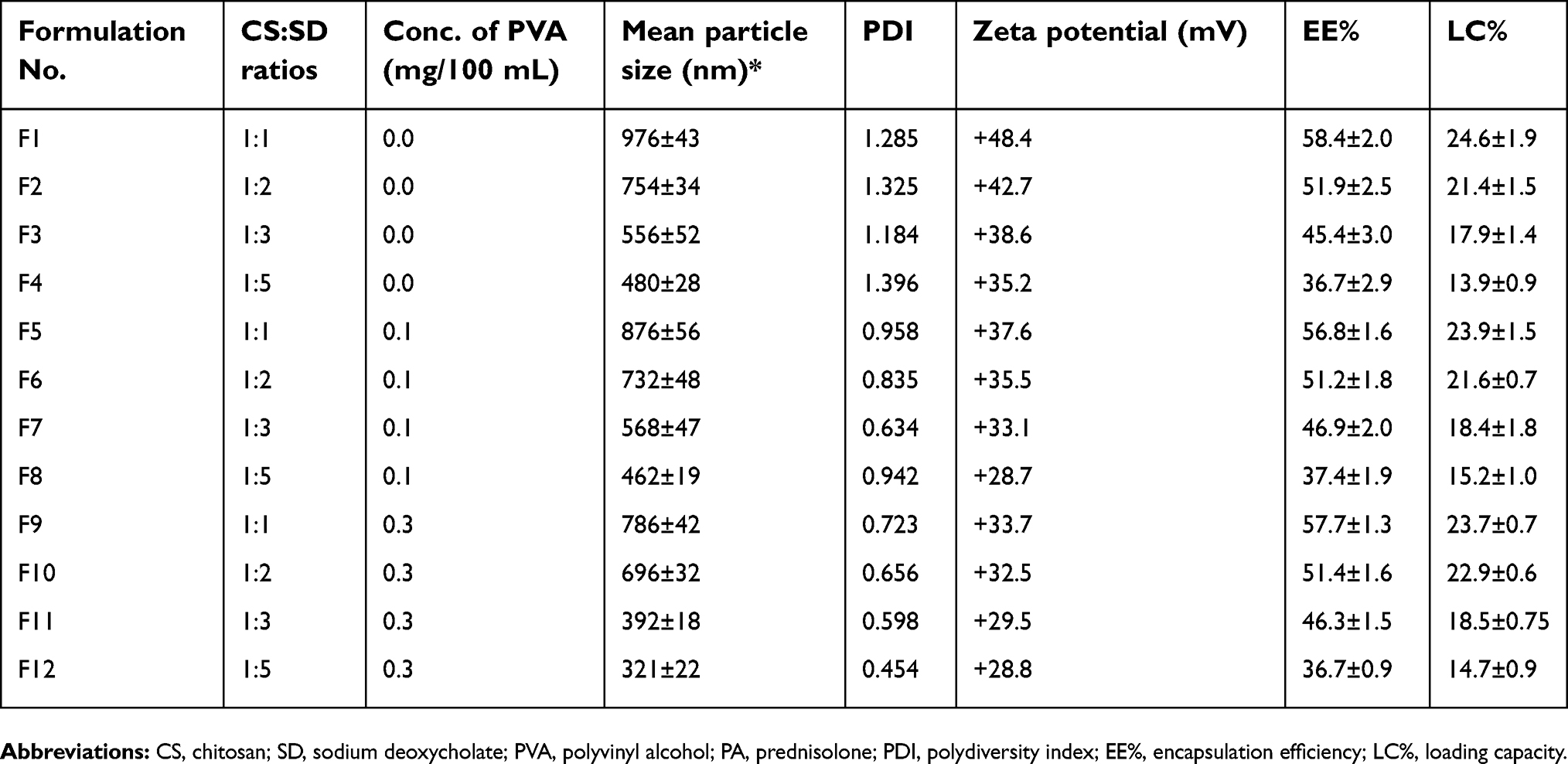 | Table 2 Characterization of PA loaded CS–SD NPs including mean particle size, PDI, zeta potential, EE% and drug LC%. The ratio of CS to SD for each formulation is also presented in addition to the concentration of the stabilizing agent (PVA) in preparation solutions |
 | Figure 1 Photograph showing the macroscopic appearance of F12 formulation NPs dispersion (1:5 CS–SD complexes prepared using 0.3% polyvinyl alcohol).Abbreviations: NPs, nanoparticles; CS, chitosan; SD, sodium deoxycholate. |
The weight fraction of SD has also shown to have an inverse effect on the value of PDI, whereby formulation F 12, with the highest weight fraction of SD, expressed a PDI value of 0.45 (Table 2). The SEM micrograph presented in Figure 2 shows NPs of formulation F 12.
 | Figure 2 SEM pictures of the F12 formulation NPs (×15,000). |
Due to their significantly lower mean particle size values, positive surface charge and PDI values, attributed to their high weight fraction of SD (Table 2), formulations F11 and F12 were chosen for further in vitro and in vivo studies. However, unlike the mean particle size, the PA EE% and the drug LC% were shown to be inversely affected by the weight fraction of SD, giving F12 the lowest EE and LC percentages (Table 2). This inverse relation may be attributed to SD positive effect on the aqueous solubility of the drug, thus increasing the drug’s affinity to the liquid phase.27
DSC thermograms of CS, SD and PA powders as well as the F12 physical mixture and the F12 NPs are presented in Figure 3. CS showed a glass transition (Tg) at 25°C followed by a broad endothermic peak starting from 100°C to 170°C which might be due to the loss of water. Polymer degradation started at 205°C and greater degradation at 280°C. This was in conformance with Madeleine-Perdrillat et al37 and Liu et al38 findings. PA showed a melting endothermic peak onset at 247°C in accordance with that stated by Halim and Salah.33 SD showed a sharp endothermic peak at 132°C with broad tailing till 170°C.39
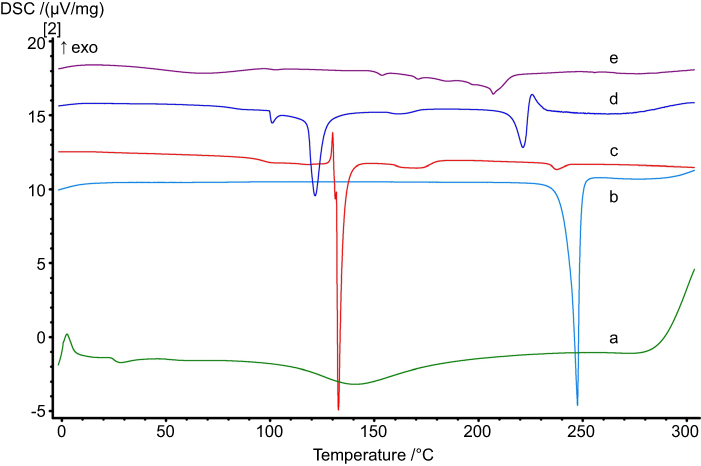 | Figure 3 DSC thermograms of (a) CS, (b) PA, (c) SD, (d) F12 ingredients physical mixture, and (e) F12 NPs formulation. Abbreviations: SEM, scanning electron microscope; NPs, nanoparticles. |
The F12 physical mixture showed broad endothermic peaks at 121.6°C and 221.3°C for SD and PA, respectively. These results indicate a lowering of the endothermic peak of both SD and PA. This might be due to the effect of moisture evaporated during dehydration of CS which was reported to cause partial solubilization or affect the outer environment thus causing a decrease of about 20°C in the melting points.40
The F12 NPs thermogram showed an expansive blunted endothermic peak at 207°C ±2 (n=3) while the SD endothermic peak disappeared. This may be due to the complex formation between CS and SD. The presence of a PA melting peak is evidence that a great proportion of the drug is still in its crystalline state, while the decrease in melting peak and the drop in the melting point onset may be attributed to the increased thermal sensitivity observed when nano-sizing a material, as the surface area is significantly increased. Furthermore, the reduction in peak size may also be attributed to the reduction in particle size as less drug–drug intermolecular forces are present.41
The FTIR of SD (Figure 4) shows very strong characteristic absorption bands of the conjugated carboxyl group at 1,640 cm−1 assigned to the (C=O) stretching and other characteristic bands 1,558 and 1,400 cm−1.39 CS IR spectrum (Figure 4) shows distinctive absorption bands, indicative of the (N-H) bond, at 1,540 cm−1, and the (C=O) bond at 1,640 cm−1.25 Drug-free complexes of CS and SD show a diminish of the N-H and C=O IR absorption peaks while a new peak appears at 1,700 cm−1 indicating a complex formation of CS–SD. Similar findings were reported by Shi et al25.
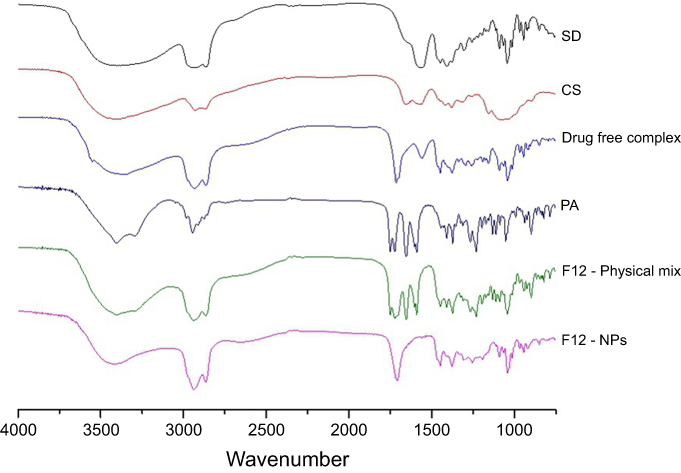 | Figure 4 FTIR spectra of, from the top, sodium deoxycholate as purchased powder (SD), chitosan as purchased powder (CS), drug-free complex containing dry NPs of CS–SD at a ratio of 1:3, as purchased prednisolone acetate (PA), F12 formulation components as a physical mix and finally the F12 NPs formulation.Abbreviations: SD, sodium deoxycholate; CS, chitosan; PA, prednisolone acetate; NPs, nanoparticles. |
The FTIR spectra of PA (Figure 4) show the principal peaks at 1,700 cm−1 corresponding to C=O stretching vibration, 1,660 cm−1 corresponding to cyclohexadiene, and other characteristic peaks at 1,610 and 1,246 cm−1.42 FTIR spectra of F12 physical mixture showed the same characteristic bands of the CS, SD, and PA in the same regions with decreasing intensity (Figure 4).
The FTIR spectra of the drug in both F12 physical mixture and NPs (Figure 4) show the same characteristic bands at the same regions and range. However, the intensity of the bands was decreased which may be due to the dilution factor in both physical mixture and the NPs. There were no new bands observed for the drug indicating no chemical interactions between the drug and components of the preparation.
The cumulative percent of PA released as a function of time from different formulations are shown in Figure 5. The drug release from F11 and F12 NPs gel exhibited a significantly improved release effect compared to the release profile of the gel loaded with micronized PA. This enhanced drug release might be attributed to the presence of drug in the nanosize range in the CS–SD complex as well as the wetting and solubilizing effect of SD.27 The percent of drug released from F11 and F12 NPs gel were similar during the first 8 hrs, giving a release of 72%. However, after 8 hrs of release the F12 NPs gel showed higher release than that of the F11 NPs. This could be attributed to the higher weight fraction of SD in the 1:5 CS–SD ratio resulting in a substantial improvement of the wetting and solubilization effect.21 The cumulative percent drug released for 24 hrs was 48.5%, 85.3%, and 98.6% from gel containing PA alone, F11 NPs and F12 NPs, respectively.
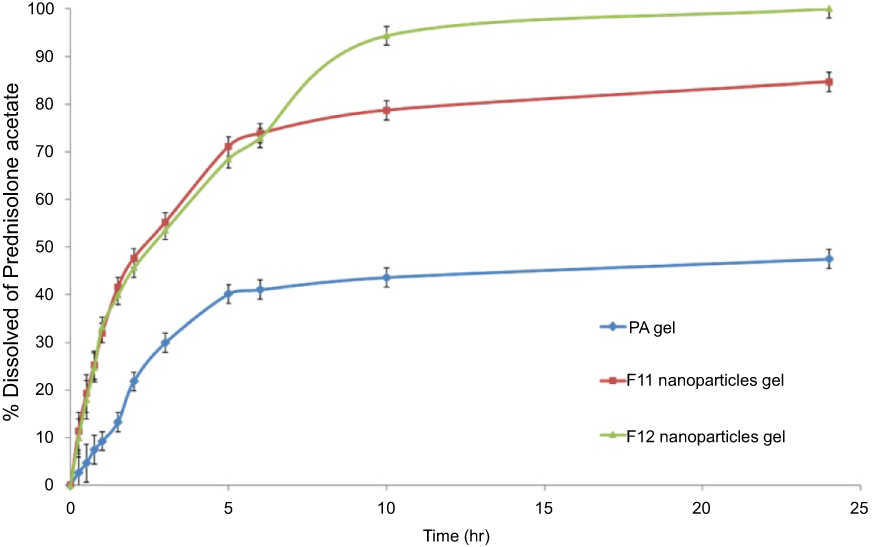 | Figure 5 Mean±STD of (n=3) percentage PA dissolved as a function of time (hr) from PA loaded gel (♦), 1:3 CS–SD complex (■) and 1:5 CS–SD complex (▲) in simulated tears fluid (STF) pH 7.4.Abbreviations: SD sodium deoxycholate; CS, chitosan, PA, prednisolone acetate; NPs nanoparticles. |
F12 (PA loaded 1:5 CS–SD) NPs gel was selected for the in vivo study because of the relatively smaller particle size and the better release characteristics. Clinical examination scorings for eye inflammation in female guinea pigs in different groups after induction of eye inflammation are presented in Table 3. Average score for eye inflammation for the control group (group 1) was zero before and after treatment with saline solution throughout the study (Figure 6A). All other groups showed an average score for eye inflammation slightly higher than 40 after induction of eye inflammation (Figure 6B).
 | Table 3 Clinical examination scoring for eye inflammation in female guinea pigs in different groups after induction of eye inflammation |
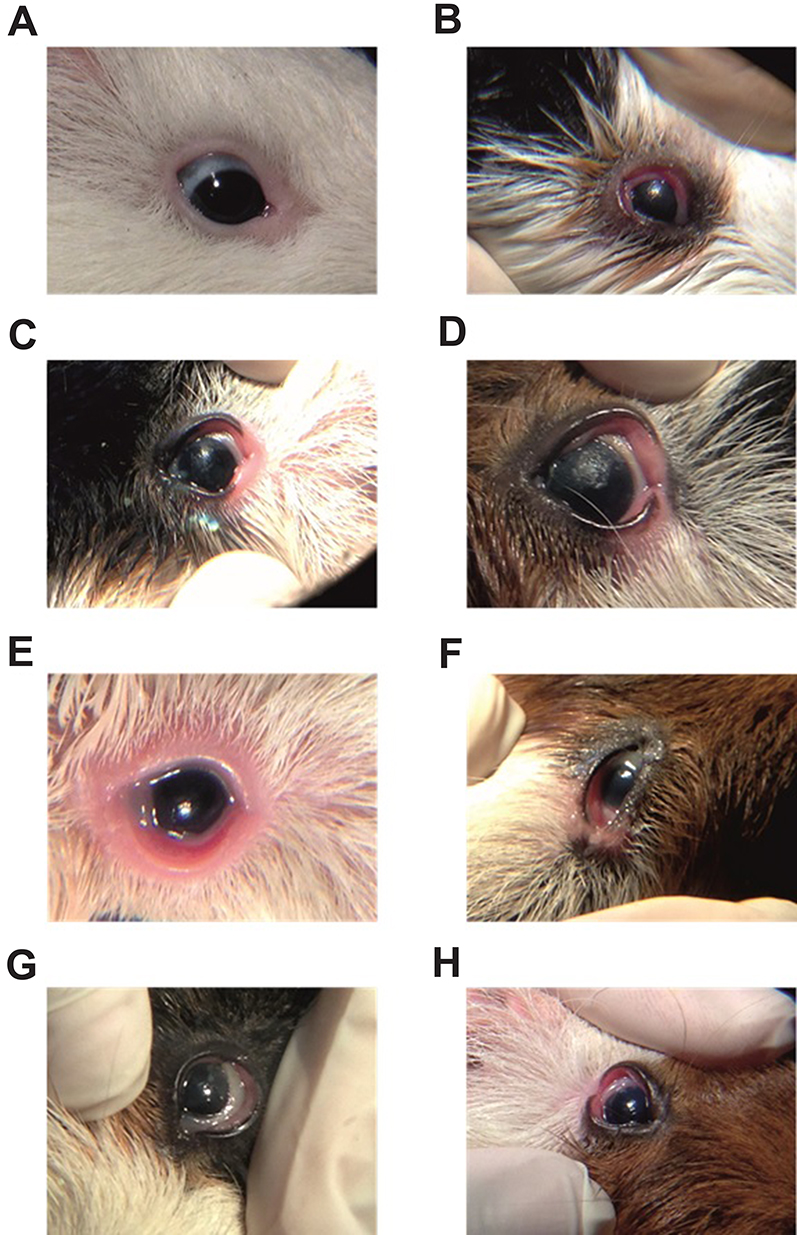 | Figure 6 Photographic picture showing eye of a female guinea pig of (A) control group, (B) zero time after induction of eye inflammation, (C) inflammation control group (group 2) after 1 hr of application of drug-free gel, (D) inflammation control group (group 2) after 24 hrs of application of drug-free gel, (E) group 3 after 1 hr of therapy with a conventional PA gel, (F) group 3 after 24 hrs of therapy with a conventional PA gel, (G) group 4 after 1 hr of therapy with a PA nanogel and (H) group 4 after 24 hrs of therapy with a PA nanogel.Abbreviation: PA, prednisolone acetate. |
The results shown in Table 3 and Figure 6C D illustrate that the inflammation control group (group 2) showed no decrease in inflammation scoring after 1 (Figure 6C) and 24 hrs (Figure 6D) of application of a drug-free gel. On the other hand, significant decrease in inflammation (p<0.05) was observed in group 3 after receiving a conventional treatment with a gel loaded PA after 1 hr (Figure 6E) and 24 hrs (Figure 6F). The average inflammation scores of this group ranged from 30±4 and 28±3, respectively. Group 4 treated with PA loaded F12 NPs gel showed significantly the highest anti-inflammatory effect (p<0.05) when compared with other groups resulting in the lowest eye inflammation score after 1 (Figure 6G) and 24 hrs (Figure 6H). The average score of this group is 18±3 and 19±2, respectively. The higher anti-inflammatory effect of F12 PA NPs gel in comparison to treatment using conventional gel treatment may be attributed to the nano-nature of drug in the formulation43 and the penetration enhancing the effect of CS and SD. Moreover, F12 PA NPs gel seem to offer anti-inflammatory effect throughout the treatment period (24 hrs) which might be attributed to CS mucoadhesive properties.27
Conclusions
PA loaded NPs prepared in PVA containing solutions, as a stabilizer, in concentration of 0.3% w/v (F12) showed successful complex formation as well as superior particle size characteristics. It achieved smallest mean particle size of 321 nm, with the lowest PDI, while expressing a relatively good encapsulation efficiency and loading capacity when compared to the other prepared formulations. The F12 self-assembled CS–SD NPs complex were also successfully included in gel formulation. NPs gel formulations have significantly improved PA release patterns compared to that of micronized drug loaded gel. Furthermore, the anti-inflammatory effect of PA loaded in self-assembled CS–SD NPs gel, on female guinea pigs, was shown to be significantly superior to that of gel loaded with micronized PA.
Acknowledgments
This work was supported by the Deanship of Scientific Research at Taibah University (grant number 1320/1434). Authors also would like to acknowledge Jamjoom Pharmaceutical Company for kindly supplying prednisolone acetate. The authors would also like to acknowledge Dr Sufian Abdulgadir Aboush for his help in performing IR analysis for this study.
Author contributions
All authors contributed to data analysis, drafting or revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
Ahmed F Hanafy is employeed by Al Andalous for Pharmaceutical Industries, (Giza, Egypt). The authors report no other conflicts of interest in this work.
References
1. Glass LRD, Freitag SK. Orbital inflammation: corticosteroids first. Surv Ophthalmol. 2016;61(5):670–673. doi:10.1016/j.survophthal.2016.01.005
2. Thadani SM, Foster CS. Treatment of ocular inflammation in children. Pediatr Drugs. 2004;6(5):289–301. doi:10.2165/00148581-200406050-00003
3. Ilium L. Chitosan and its use as a pharmaceutical excipient. Pharm Res. 1998;15(9):1326–1331.
4. Du J, Zhang S, Sun R, Zhang LF, Xiong CD, Peng YX. Novel polyelectrolyte carboxymethyl konjac glucomannan-chitosan nanoparticles for drug delivery. II. Release of albumin in vitro. J Biomed Mater Res B Appl Biomater. 2005;72(2):299–304.
5. Khor E, Lim LY. Implantable applications of chitin and chitosan. Biomaterials. 2003;24(13):2339–2349.
6. Cai G, Jiang H. pH-sensitive nanoparticles self-assembled from a novel class of biodegradable amphiphilic copolymers based on chitosan. J Mater Sci Mater Med. 2009;20(6):1315–1320.
7. Soane RJ, Frier M, Perkins AC, Jones NS, Davis SS, Illum L. Evaluation of the clearance characteristics of bioadhesive systems in humans. Int J Pharm. 1999;178(1):55–65.
8. Ruel-Gariepy E, Leclair G, Hildgen P, Gupta A, Leroux JC. Thermosensitive chitosan-based hydrogel containing liposomes for the delivery of hydrophilic molecules. J Control Release. 2002;82(2–3):373–383.
9. Wang Q, Zhang N, Hu X, Yang J, Du Y. Chitosan/polyethylene glycol blend fibers and their properties for drug controlled release. J Biomed Mater Res A. 2008;85(4):881–887.
10. Patel R, Gajra B, Parikh R, Patel G. Preparation, characterization and in-vitro drug release evaluation of Ganciclovir loaded Chitosan nanoparticles. Appl Nanomed. 2016;1(1):20–27.
11. Pourramezan Z, Kasra Kermanshahi R, Katbab A. Investigation of chitosan nanoparticles durability in combination with antioxidant-antibacterial fraction extracted from Lactobacillus casei and possible increase of antibacterial activity of the fraction in hybrid nanoparticle. Nanomed Res J. 2017;2(2):123–130.
12. Kim TH, Park IK, Nah JW, Choi YJ, Cho CS. Galactosylated chitosan/DNA nanoparticles prepared using water-soluble chitosan as a gene carrier. Biomaterials. 2004;25(17):3783–3792. doi:10.1016/j.biomaterials.2003.10.063
13. Williams SJ, Wang Q, Macgregor RR, Siahaan TJ, Stehno-Bittel L, Berkland C. Adhesion of pancreatic beta cells to biopolymer films. Biopolymers. 2009;91(8):676–685. doi:10.1002/bip.21196
14. Hu Y, Jiang X, Ding Y, Ge H, Yuan Y, Yang C. Synthesis and characterization of chitosan-poly(acrylic acid) nanoparticles. Biomaterials. 2002;23(15):3193–3201.
15. Yang Y, Wang S, Wang Y, Wang X, Wang Q, Chen M. Advances in self-assembled chitosan nanomaterials for drug delivery. Biotechnol Adv. 2014;32(7):1301–1316. doi:10.1016/j.biotechadv.2014.07.007
16. Fernandez-Urrusuno R, Calvo P, Remunan-Lopez C, Vila-Jato JL, Alonso MJ. Enhancement of nasal absorption of insulin using chitosan nanoparticles. Pharm Res. 1999;16(10):1576–1581.
17. Prego C, Garcia M, Torres D, Alonso MJ. Transmucosal macromolecular drug delivery. J Control Release. 2005;101(1–3):151–162. doi:10.1016/j.jconrel.2004.07.030
18. Vila A, Sanchez A, Janes K, et al. Low molecular weight chitosan nanoparticles as new carriers for nasal vaccine delivery in mice. Eur J Pharm Biopharm. 2004;57(1):123–131.
19. Pelegrino M, Weller R, Chen X, Bernardes J, Seabra A. Chitosan nanoparticles for nitric oxide delivery in human skin. MedChemComm. 2017;8(4):713–719. doi:10.1039/c6md00502k
20. Yamamoto A, Hayakawa E, Kato Y, Nishiura A, Lee VH. A mechanistic study on enhancement of rectal permeability to insulin in the albino rabbit. J Pharmacol Exp Ther. 1992;263(1):25–31.
21. Gandhi R, Robinson J. Mechanisms of penetration enhancement for transbuccal delivery of salicylic acid. Int J Pharm. 1992;85:129–140. doi:10.1016/0378-5173(92)90142-O
22. Senyigit T, Tekmen I, Sonmez U, Santi P, Ozer O. Deoxycholate hydrogels of betamethasone-17-valerate intended for topical use: in vitro and in vivo evaluation. Int J Pharm. 2011;403(1–2):123–129. doi:10.1016/j.ijpharm.2010.10.036
23. Lee K, Kim J-H, Kwon I, Jeong S. Self-aggregates of deoxycholic acid-modified chitosan as a novel carrier of adriamycin. Colloid Polym Sci. 2000;278(12):1216–1219. doi:10.1007/s003960000389
24. Park J, Kim T-H, Nam J-P, et al. Bile acid conjugated chitosan oligosaccharide nanoparticles for paclitaxel carrier. Macromol Res. 2014;22:310–317. doi:10.1007/s13233-014-2034-9
25. Shi Z, Guo R, Li W, et al. Nanoparticles of deoxycholic acid, polyethylene glycol and folic acid-modified chitosan for targeted delivery of doxorubicin. J Mater Sci Mater Med. 2014;25(3):723–731. doi:10.1007/s10856-013-5113-0
26. Chae SY, Son S, Lee M, Jang MK, Nah JW. Deoxycholic acid-conjugated chitosan oligosaccharide nanoparticles for efficient gene carrier. J Control Release. 2005;109(1–3):330–344. doi:10.1016/j.jconrel.2005.09.040
27. Cadete A, Figueiredo L, Lopes R, Calado CC, Almeida AJ, Goncalves LM. Development and characterization of a new plasmid delivery system based on chitosan-sodium deoxycholate nanoparticles. Eur J Pharm Sci. 2012;45(4):451–458. doi:10.1016/j.ejps.2011.09.018
28. Wei WH, Dong XM, Liu CG. In vitro investigation of self-assembled nanoparticles based on hyaluronic acid-deoxycholic acid conjugates for controlled release doxorubicin: effect of degree of substitution of deoxycholic acid. Int J Mol Sci. 2015;16(4):7195–7209. doi:10.3390/ijms16047195
29. Sendrowski DP, Jaanus SD, Semes LP, Stern ME. Chapter 12 – anti-inflammatory drugs. In: Clinical Ocular Pharmacology.
30. Alldredge BK, Corelli RL, Ernst ME, et al. Koda-Kimble and Young’s Applied Therapeutics: The Clinical Use of Drugs. Wolters Kluwer Health Adis (ESP). Philadelphia, PA: Lippincott Williams & Wilkins; 2013.
31. Wu W, Li J, Wu L, et al. Ophthalmic delivery of brinzolamide by liquid crystalline nanoparticles: in vitro and in vivo evaluation. AAPS PharmSciTech. 2013;14(3):1063–1071. doi:10.1208/s12249-013-9997-2
32. Morsi N, Ibrahim M, Refai H, El Sorogy H. Nanoemulsion-based electrolyte triggered in situ gel for ocular delivery of acetazolamide. Eur J Pharm Sci. 2017;104:302–314. doi:10.1016/j.ejps.2017.04.013
33. Halim SA, Salah S. Development of nanoparticulate formulations for ocular delivery of prednisolone acetate: preparation and characterization. J Drug Deliv Sci Technol. 2014;24(2):159–165.
34. Droy-Lefaix MT, Bueno L, Caron P, Belot E, Roche O. Ocular inflammation and corneal permeability alteration by benzalkonium chloride in rats: a protective effect of a myosin light chain kinase inhibitor. Invest Ophthalmol Vis Sci. 2013;54(4):2705–2710.
35. Monnickendam MA, Darougar S, Treharne JD, Tilbury AM. Guinea-pig inclusion conjunctivitis as a model for the study of trachoma: clinical, microbiological, serological, and cytological studies of primary infection. Br J Ophthalmol. 1980;64(4):279–283.
36. Singh J, Unlu Z, Ranganathan R, Griffiths P. Aggregate properties of sodium deoxycholate and dimyristoylphosphatidylcholine mixed micelles. J Phys Chem B. 2008;112(13):3997–4008.
37. Madeleine-Perdrillat C, Karbowiak T, Debeaufort F, Delmotte L, Vaulot C, Champion D. Effect of hydration on molecular dynamics and structure in chitosan films. Food Hydrocoll. 2016;61:57–65.
38. Liu C, Zhang H, Xiao R, Wu S. Value-added organonitrogen chemicals evolution from the pyrolysis of chitin and chitosan. Carbohydr Polym. 2017;156:118–124.
39. Hao F, He Y, Sun Y, et al. Improvement of oral availability of ginseng fruit saponins by a proliposome delivery system containing sodium deoxycholate. Saudi J Biol Sci. 2016;23(1, Supplement):S113–S125.
40. Kumar S, Randhawa JK. High melting lipid based approach for drug delivery: solid lipid nanoparticles. Mater Sci Eng C. 2013;4(33):1842–1852.
41. Fu Q, Ma M, Li M, et al. Improvement of oral bioavailability for nisoldipine using nanocrystals. Powder Technol. 2017;305:757–763.
42. Huanbutta K, Sangnim T, Limmatvapirat S, Nunthanid J, Sriamornsak P. Design and characterization of prednisolone-loaded nanoparticles fabricated by electrohydrodynamic atomization technique. Chem Eng Res Des. 2016;109:816–823.
43. Sabzevari A, Adibkia K, Hashemi H, et al. Polymeric triamcinolone acetonide nanoparticles as a new alternative in the treatment of uveitis: in vitro and in vivo studies. Eur J Pharm Biopharm. 2013;84(1):63–71.
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.