")
Back to Journals » Drug Design, Development and Therapy » Volume 9
Novel levocetirizine HCl tablets with enhanced palatability: synergistic effect of combining taste modifiers and effervescence technique
Authors Labib G
Received 11 July 2015
Accepted for publication 6 August 2015
Published 7 September 2015 Volume 2015:9 Pages 5135—5146
DOI https://doi.org/10.2147/DDDT.S92245
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Shu-Feng Zhou
Gihan S Labib1,2
1Department of Pharmaceutics and Industrial Pharmacy, Faculty of Pharmacy, King Abdulaziz University, Jeddah, Saudi Arabia; 2Department of Pharmaceutics, Faculty of Pharmacy, Alexandria University, Alexandria, Egypt
Objectives: Levocetirizine HCl, a second-generation piperazine derivative and H1-selective antihistaminic agent, possesses few side effects. The first objective of the study was to compare and evaluate the taste-masking effect of different ratios of 2-hydroxypropyl-β-cyclodextrin and mannitol on levocetirizine HCl using an inclusion complex and solid dispersion, respectively. The second objective was to study the possibility of preparing and evaluating effervescent tablets from the best-chosen taste-masked blends for the purpose of their use either as orodispersible tablets or as water-soluble effervescent tablets, according to patients’ will.
Materials and methods: Prepared taste-masked blends were prepared and subjected to palatability, Fourier-transform infrared spectroscopy, and differential scanning calorimetry studies. Tablets containing different percentages of effervescent mixtures were prepared by direct compression on the selected taste-modified blends. Evaluation tests were conducted, including flowability and compressibility on the precompressed blends and hardness, friability, wetting time, effervescent time, in vitro, in vivo disintegration time, and in vitro dissolution study on the compressed tablets. Formulated tablets were evaluated and compared to marketed orodispersible tablets for mouth feel and palatability.
Results: All prepared tablets showed convenient physical and palatability properties compared to the selected brand. The in vitro drug-release study revealed fast release of levocetirizine HCl within 5 minutes from all prepared tablets.
Conclusion: Levocetirizine HCl effervescent tablets are likely to increase patient compliance with drug administration. Moreover, the use of these effervescent tablets in an orodispersible dosage form can improve oral drug bioavailability and act as an attractive pediatric dosage form.
Keywords: antihistaminic, effervescent tablets, 2-hydroxypropyl-β-cyclodextrin, mannitol, taste masking
Introduction
Oral drug-delivery systems are the most common and preferred route for systemic administration of drugs.1 Compared to other oral drug-dosage forms, tablets are considered the most economic form concerning their manufacturing equipment, packaging, and transportation.2 However, geriatric, pediatric, and bedridden patients find difficulty in swallowing tablets and capsules.3 Alternatives for such patients include liquid-dosage preparations, effervescent and dispersible tablets, and buccal orodispersible films or tablets. Direct compression for tablet manufacturing using conventional equipment and commonly available excipients with a limited number of processing steps is considered the easiest way for tablet preparation. Compressed tablet disintegration and solubilization depends mainly on the presence of either single or combined action of disintegrants, water-soluble excipients, and effervescent agents that hasten the process of disintegration.4 However, patient compliance with buccal or liquid-drug administration is related to the efficient taste-masking effect of sour, bitter, and salty drugs.5–7
Drugs possessing an unacceptable, unpleasant taste were classified into bitter, sour, or salty. An intimate relationship between the chemical structure and the taste of any drug was postulated, taking into consideration the different characteristics of the drug, including its solubility, degree of ionization, and types of ion produced that have a direct effect on the taste-transmission signals to the brain to interpret the taste.8 The presence of a hydrogen ion in a compound was explained as the main cause for sour-tasting drugs. On the other hand, bitter-tasting drugs were related to the presence of free bases, such as alkaloids and amides, polyhydroxyl compounds with molecular weights greater than 300 g/mol, and halogenated or thiolated compounds. Moreover, salty drugs were supposed to possess both anions and cations in their structure, as all salts do.9
Allergic rhinitis is one of the most common diseases characterized by the expression of inflammatory symptoms in the upper respiratory tract. These symptoms comprise nasal congestion, sneezing, pruritus, and rhinorrhea in most cases. Traditional antihistaminic drugs in combination with intranasal corticosteroids are used as first-line treatment for such cases. The addition of corticosteroid drugs in the treatment strategy was based on the fact that most previously used antihistaminic drugs had little or no effect in the treatment of nasal congestion.10 However, relatively recent generations of antihistaminic drugs proved to possess a decongestant effect in addition to their known antihistaminic properties. Levocetirizine is one of the newest generation of antihistaminics that suppress the cutaneous allergic responses induced by chronic idiopathic urticaria. It is effective in the treatment of nasal congestion caused by allergic rhinitis. It acts as a selective inhibitor of H1 receptors.11 However, levocetirizine HCl is characterized by its bitter and sour taste due to the presence of chlorine and hydrogen ions in its structure.12 It also suffers from low oral bioavailability, due to its high first-pass metabolism.4
The first objective of the current work was to compare the effect of two types of taste modifiers in different ratios on the taste masking of levocetirizine HCl. The second objective was to formulate, prepare, and evaluate directly compressed effervescent tablets using different percentages of effervescent mixtures. The effervescent mixture will probably increase the release rate of the drug while permitting an additional synergistic immediate taste-masking effect. The formulated tablets can be used as orodispersible tablets with the aim of increasing oral bioavailability or can be dissolved before use, according to the patient’s convenience.
Several methods to achieve taste masking adopted in the literature have comprised the use of physical barriers, chemical and solubility modification of drugs, and the use of taste modifiers applying either the solid dispersion (SD) or inclusion complex (IC) techniques.13–17 Among the methods adopted have been the microencapsulation of the drug,13 the use of cyclodextrin (CD) ICs,12 SD,18 chemical modification,16 sublimation,3 lipoproteins, ion-exchange resin, or multiemulsion techniques.9,15 Several studies have investigated different techniques in the preparation of fast-dissolving, orodispersible, and effervescent tablets, including direct compression, disintegrant addition,4 wet granulation,19 and lyophilization.20
Materials and methods
Levocetirizine HCl was obtained as a gift sample (Global Napi Pharmaceuticals, Cairo, Egypt). Anhydrous 2-hydroxypropyl-β-CD (HPβ-CD; molecular weight 1,454 g/mol), mannitol (MN), Avicel (Sigma-Aldrich Co, St Louis, MO, USA), menthol (SD Fine-Chem Ltd, Mumbai, India), and polyethylene glycol (PEG) 6000 (BDH Laboratory Supplies, Poole, UK) were purchased. All other excipients and reagents were of pharmaceutical and analytical grade.
Preparation of taste-masked levocetirizine HCl blends
Levocetirizine HCl (Levocet), IC using HPβ-CD, and SD using MN were prepared by solvent evaporation.21 Accurately weighed amounts of the Levocet and HPβ-CD or MN at different molar and weight ratios, respectively, were thoroughly mixed (Table 1). A suitable amount of solvent (ethanol:water 1:2) was added portion-wise while mixing until complete solubility or dispersion was achieved. The different blends were dried at 40°C in a thermostatically controlled oven. The dried powder was ground, sieved through 80#, and stored in well-closed amber-glass containers.
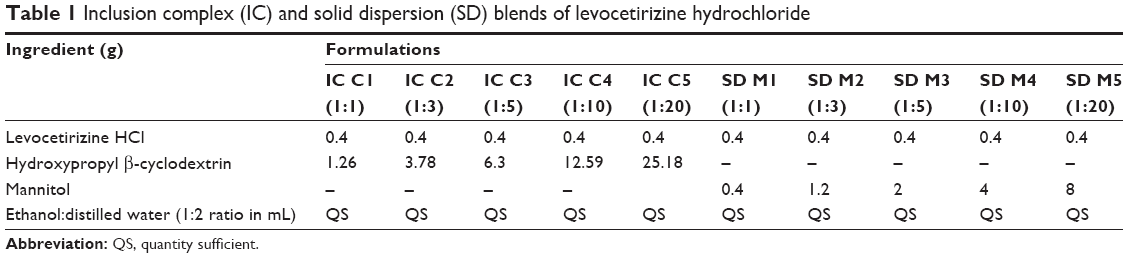 | Table 1 Inclusion complex (IC) and solid dispersion (SD) blends of levocetirizine hydrochloride |
Physical characterization of the prepared blends
Determination of degree of bitterness
Six healthy female volunteers aged 35–48 years were selected to assess the degree of bitterness of the drug in its raw form and assign a score from 0 to 5: 0 indicated the absence of bitterness, while 5 indicated very bitter taste. The volunteers were asked to retain a small amount of the drug (5 mg) in their mouths at the posterior lobe of the tongue for 10 seconds and register their scores. After thorough rinsing of the mouth, the volunteers were asked to repeat the experiment on each prepared formulation using amounts of the prepared blends equivalent to 5 mg drug. The experiments were done with 20-minute spacing to ensure accuracy. Mean scores were calculated ± standard deviation. The Biomedical Ethics Research Committee at King Abdulaziz University under reference 256-14 gave ethical approval for the conduction of all tests related to the participating volunteers in this study. All volunteers had to give formal signed consent before participating in the experiments.
FT-IR analysis
Levocet, HPβ-CD, MN, physical mixtures, and the prepared blend were subjected to Fourier-transform infrared (FT-IR) spectroscopy (PerkinElmer Inc, Waltham, MA, USA). Samples were prepared using KBr pellets according to a previously reported method.22 The spectra were recorded over a range of 4,000 cm−1–400 cm−1.
Differential scanning calorimetry analysis
Differential scanning calorimetry (DSC) thermograms of Levocet, HPβ-CD, MN, physical mixtures, and the prepared blend using a differential scanning calorimeter (PerkinElmer) were recorded. The adopted method has been previously described in detail.23
Preparation of effervescent tablets by direct compression
Based on the taste-masking effect of the prepared blends, the best taste-masked combination from each taste modifier was chosen to prepare the tablets. Effervescent content in the different formulations represented 10%, 20%, and 30% of the total weight of the prepared tablets. Ingredients used in the preparation of the different formulations of effervescent tablets are listed in Table 2. For each formulation, all accurately weighed ingredients were sieved through 80 times and mixed together in a mortar applying geometric addition. The blends were compressed using a single-punch tablet machine (Erweka GmbH, Heusenstamm, Germany) to get suitable hardness using a round, concave, 13 mm-diameter punch and die set (Shanghai Tianxiang & Chentai Pharmaceutical Machinery Co Ltd, Shanghai, People’s Republic of China). Tablets were dried in a thermostated oven at 40°C for 2 hours and stored in well-closed glass bottles in a desiccator pending evaluation.
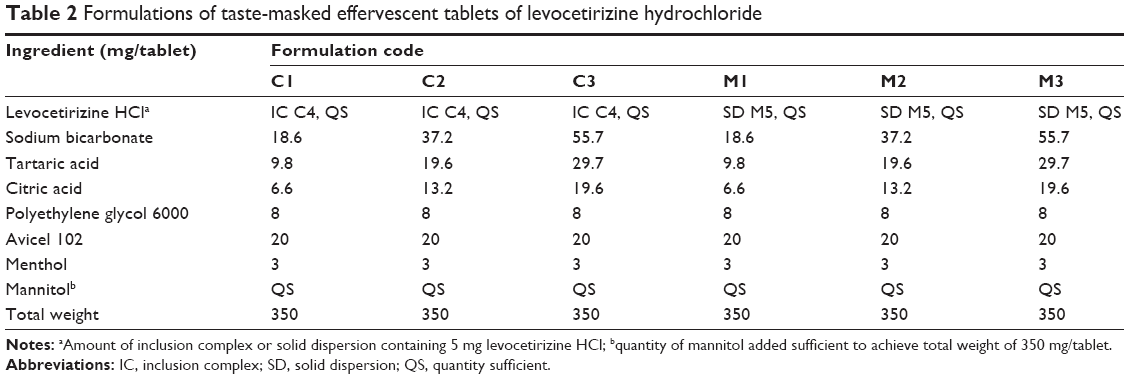 | Table 2 Formulations of taste-masked effervescent tablets of levocetirizine hydrochloride |
Precompression evaluation of the effervescent blends
Angle of repose
The fixed funnel method for the determination of the angle of repose was adopted according to a previously reported method.24 The following equation was used to calculate the angle of repose of the tested tablet blends:
θ = tan−1 (h/r) | (1) |
where θ is the angle of repose, h the height of the formed pile, and r the radius of the pile.
Compressibility of blends
Compressibility was determined from the bulk and tapped densities of the blends. Definite weighed quantity of the tablet blend was poured into a suitable-size cylinder, and the height of the mixture was recorded. The cylinders were tapped until a constant decrease in height was measured. Bulk and tapped densities were calculated. Carr’s index ([1− bulk density/tapped density] ×100) and the Hausner ratio (tapped density/bulk density), representing the compressibility of the blends, were calculated for each blend.
Evaluation of tablets
Drug content
Drug content was assessed according to a previously reported method with slight modification.25 Twenty tablets were ground, and an accurate weight equivalent to 5 mg of the drug was suitably diluted and spectrophotometrically measured at 232 nm. The test was done in triplicate.
Weight variation, thickness, and diameter
Twenty tablets of each formulation were weighed individually, and their average weight was calculated. The thickness and diameter of these tablets were measured using a digital caliper (Thermo Fisher Scientific, Waltham, MA, USA) to ensure uniformity of dimensions of the compressed tablets. All results are presented as means ± standard deviation.
Hardness
The resistance of tablets to breaking under different conditions of transportation and storage was measured by hardness values. This study was done on six tablets of each formulation using a hardness tester (TBH210; Erweka).
Friability
Tablet strength was measured by the friability test. The test was performed using a friability tester (PTFE D-63512; Pharma Test GmbH, Hainburg, Germany). Ten tablets from each formulation were weighed. The tester was rotated at a fixed speed of 25 rpm for 4 minutes. The tablets were dedusted and reweighed. The percentage of weight loss was calculated using the formula:
% friability = (W0 − W1)/W0 ×100 | (1) |
where W0 is the initial weight and W1 the final weight.
Moisture content
The Karl Fischer method was used to measure the moisture content according to a previously reported method.1 Accurately weighed amounts of powdered tablets were quickly transferred to the titration vessel containing 50 mL methanol (previously titrated to end point with Karl Fischer reagent), stirred to dissolve, and titrated with Karl Fischer reagent to end point:
|
|
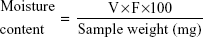
where F is the factor of Karl Fischer reagent and V the volume in milliliters of Karl Fischer reagent consumed for sample titration.
Wetting and in vitro disintegration time
Six circular tissue papers were placed in a petri dish of 15 cm diameter. Water (20 mL) containing 0.5% amaranth, a water-soluble dye, was added to the petri dish. One tablet was carefully placed on the surface of each of the tissue papers. The time required for the upper surface of the tablets to be colored was noted as the wetting time. Wetting time was recorded using a stopwatch.24 Mean values were calculated ± standard deviation.
In vitro disintegration time of the effervescent tablets was determined on six tablets of each formulation under study using a disintegration apparatus (automatic disintegration tester; Logan Instruments, Somerset, NJ, USA). Each tablet was placed in a 1,000 mL beaker containing distilled water and maintained at 37°C±0.5°C. The apparatus was started and the disintegration time in seconds was determined.
Carbon dioxide content, effervescence time, and pH of effervescent solution
Each weighed tablet was immersed in a preweighed flask containing 100 mL of 10% sulfuric acid solution and the total weight was calculated. After complete effervescence, the flask was reweighed and the difference in weight determined. The result represented the percentage CO2 released with respect to the total weight/tablet.1 For each tablet formulation, three tablets were placed in three beakers of water and the effervescence time was recorded using a stopwatch.26 The pH of the solution was determined immediately after completion of the effervescence time using a pH meter (Metrohm 632; Metrohm AG, Herisau, Switzerland). Experiments were done in triplicate for each formulation. Mean values were calculated ± standard deviation.
Mouth feel, palatability, and in vivo disintegration time
Mouth-feel and palatability testing was carried out on six healthy female volunteers for each tablet formulation under study. Scores of 0–5 were given, which were interpreted as excellent, very good, good, bad, very bad, and worst, respectively. They were asked to determine the time required in seconds for the total disintegration of the tablets. Perceptions of taste and grittiness of the disintegrated tablet were then recorded according to the aforementioned scale. The volunteers were asked to thoroughly rinse out total tablet residue after complete disintegration between each trial. All results were evaluated in comparison to the taste and palatability of a marketed orodispersible tablet of the same drug under study using the same volunteers.
In vitro dissolution
The in vitro dissolution study was carried using a USP type II dissolution apparatus (Erweka). The study was carried out in 500 mL pH 6.8 phosphate-buffered solution or pH 1.2 0.1 N HCl as dissolution media maintained at 37°C±0.5°C and 50 rpm. Samples (5 mL) were collected at 2-, 5-, 10-, 15-, and 30-minute intervals. Samples were recompensed using fresh preheated media. Samples were analyzed using an ultraviolet spectrophotometer (PD-303UV; Apel Co Ltd, Kawaguchi, Japan) at 232 nm. Each test was performed in triplicate.
Moisture absorption and stability
Preweighed tablets were placed for 1 month under conditions of 40°C/relative humidity 75%. Tablets were reweighed after 2, 14, and 30 days. The mean percentage of absorbed moisture was calculated. Tablets were subjected to evaluation after 1 month for physical appearance, disintegration time, effervescence time, and in vitro dissolution.
Results
Levocet IC and SDs using HPβ-CD and MN, respectively, were prepared using solvent evaporation method.
Physical characterization of the prepared taste-masked blends
Determination of degree of bitterness
Results revealed that an increase in either of the taste modifiers used caused a remarkable decrease in unacceptable taste, as shown in Figure 1A.
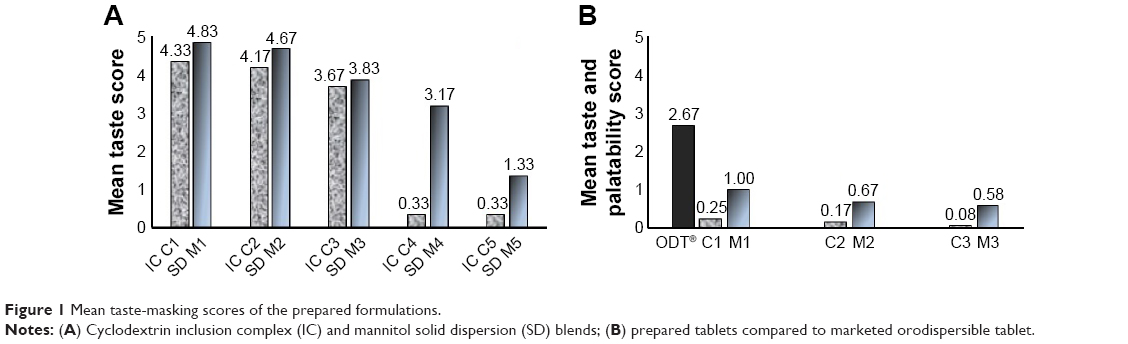 | Figure 1 Mean taste-masking scores of the prepared formulations. |
FT-IR analysis
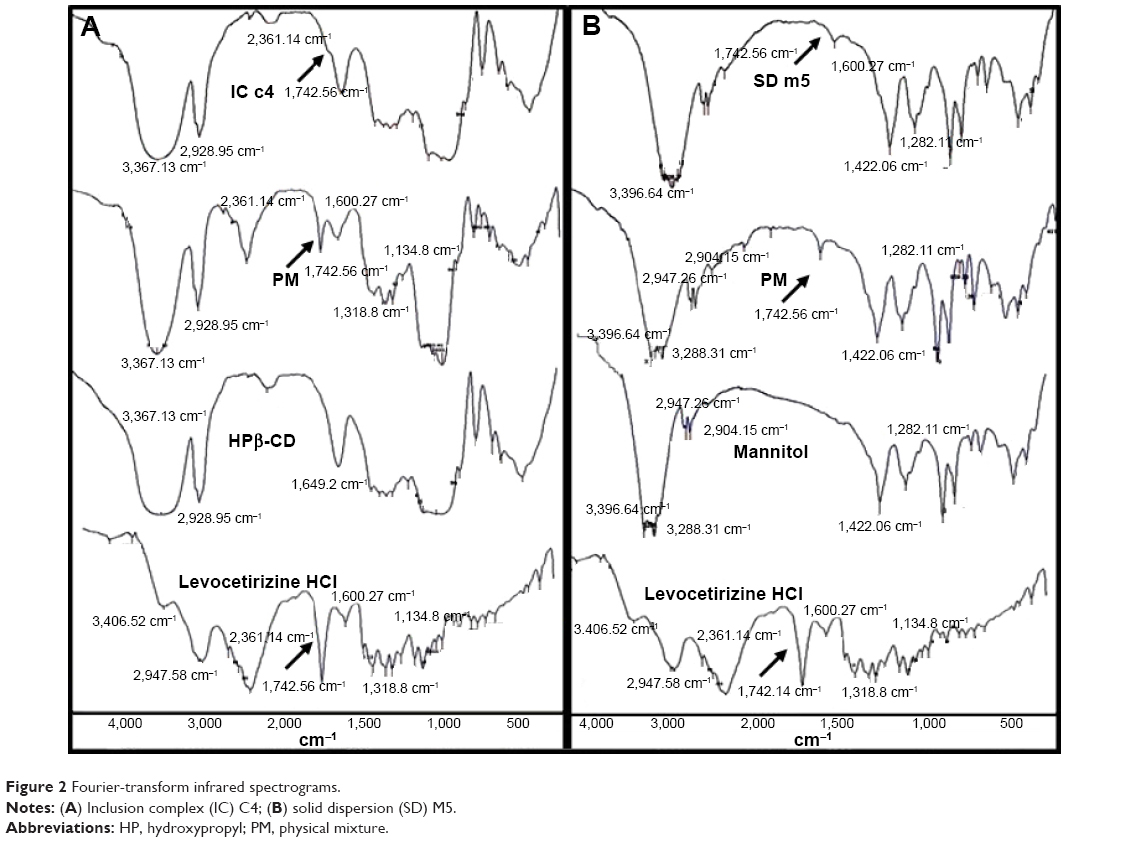
FT-IR spectra of Levocet, HPβ-CD, MN, physical mixtures, IC C4 and SD M5 are presented in Figure 2.
 | Figure 2 Fourier-transform infrared spectrograms. |
Differential scanning calorimetry analysis
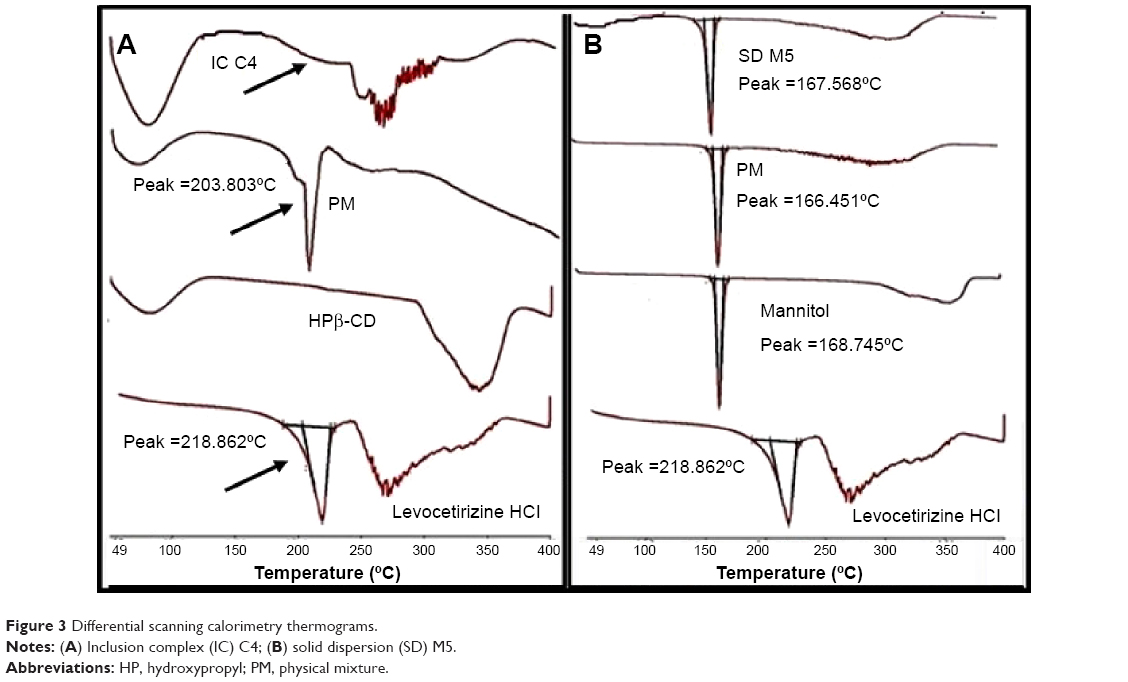
The DSC thermograms of Levocet, HPβ-CD, MN, physical mixtures, IC C4, and SD M5 are presented in Figure 3.
 | Figure 3 Differential scanning calorimetry thermograms. |
Precompression evaluation of the effervescent blends
Angle of repose
Calculated angles of repose for the prepared effervescent blends ranged from 19.65° to 22.2° (Table 3).
 | Table 3 Precompression evaluation parameters of the effervescent blends |
Compressibility of blends
Calculated Carr’s index values for the prepared effervescent blends were in the range of 12.24–18.86. The Hausner ratio was in the range of 1.13–1.23 (Table 3).
Preparation and evaluation of effervescent tablets
Effervescent tablets were prepared by direct compression using different percentages of the effervescent blends ranging from 10% to 30%.
Drug content
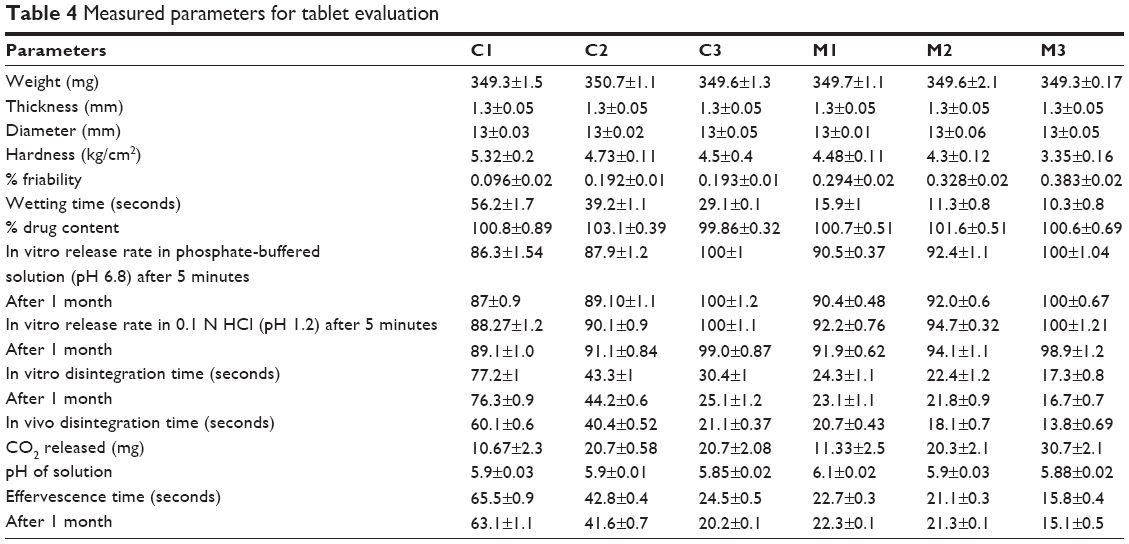
Determined % drug content from all formulations ranged from 99.86±0.32 to 103.1±0.39 (Table 4).
 | Table 4 Measured parameters for tablet evaluation |
Weight variation, thickness, and diameter
All formulated tablets ranged in weight from 349.3±0.17 to 350.7±1.1 mg. The thickness of all tablets was 1.3±0.05 mm, and their diameter ranged from 13±0.01 to 13±0.06 mm (Table 4).
Hardness
All C tablets showed increased values of harness compared to M tablets. The maximum tablet hardness was observed for C1 (5.32±0.2 kg/cm2), while the minimum was observed for M3 formulations (3.35±0.16 kg/cm2) (Table 4).
Friability
All tablets under study showed percentage-friability values within acceptable limits (less than 1%) (Table 4).
Moisture content
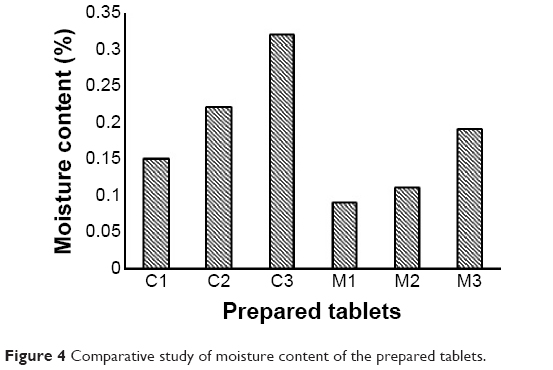
Moisture content determined by Karl Fischer test showed that tablets containing higher ratios of effervescent mixture had the maximum moisture content (Figure 4).
 | Figure 4 Comparative study of moisture content of the prepared tablets. |
Wetting, effervescence, and in vitro disintegration time
It was observed that all tablets started effervescence once subjected to the media. Wetting, effervescence, and in vitro disintegration time in general were superior for C tablets compared to those of M tablets (Table 4).
Carbon dioxide content and pH of effervescent solution
Table 4 shows the results of CO2 content released from all formulations under study. C1, C2, and C3 released 10.67±2.31, 20.7±0.58, and 20.7±2.1 mg CO2, respectively, while M1, M2, and M3 released 11.33±2.5, 20.3±2.1, and 30.7±2.1 mg CO2, respectively. The measured pH of all formulations ranged from 5.88±0.02 to 6.1±0.02 (Table 4).
Mouth feel, palatability, and in vivo disintegration time
Figure 1B shows the recorded mouth-feel and palatability scores for the prepared tablet formulations compared to the marketed orodispersible tablet. It could be observed that tablets prepared from the chosen IC of HPβ-CD (IC C4) and SD of MN (SD M5) had a significantly lower score for taste masking by the use of effervescent mixtures compared to the original taste-masking scores of the IC C4 and SD M5. This effect was more pronounced with IC tablets. It is worth stating that the in vivo disintegration time was slightly less than the in vitro disintegration time for all prepared tablets (Table 4).
In vitro dissolution
All M-tablet formulations released drug at a comparatively faster rate than C tablets after 5 minutes in both media under study (Table 4).
Moisture absorption and stability
Figure 5 shows the different amounts of water absorbed by the prepared tablets when subjected to 75% relative humidity at 40°C after 2, 14, and 30 days.
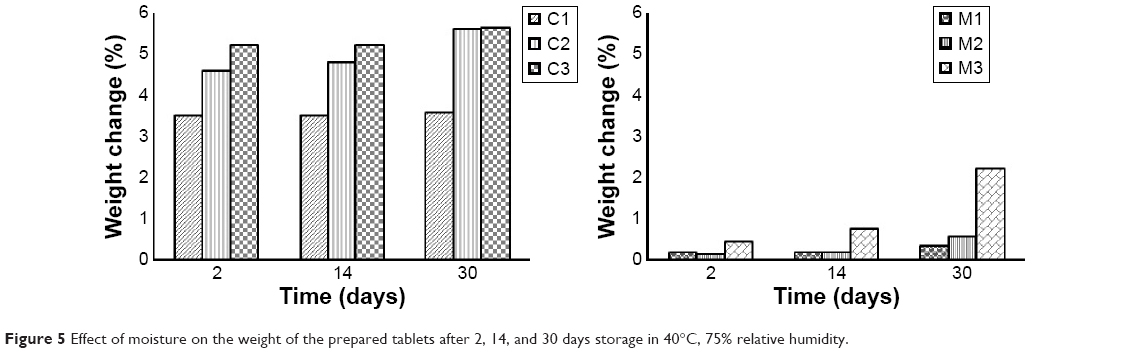 | Figure 5 Effect of moisture on the weight of the prepared tablets after 2, 14, and 30 days storage in 40°C, 75% relative humidity. |
Discussion
Physical characterization of the prepared taste-masked blends
Determination of the degree of bitterness
The dried powders were evaluated for the effect of different concentrations of the excipient used on the taste masking of the unacceptable taste of the drug. HPβ-CD had a more prominent effect than MN on taste masking. Drug molecules assumed to be incorporated in the HPβ-CD cavity or within its crystal lattice led to a remarkable taste-masking effect achieved, as previously reported.27 A hexahydric alcohol related to mannose, MN, was previously used as a taste-masking agent.28 The degree of taste masking however in the latter case depended mainly on its ratio, as shown in the current study.
FT-IR analysis
FT-IR spectroscopic analysis was conducted to determine the possible drug–carrier interactions. Prominent peaks of Levocet were observed at 3,406.52 cm−1 due to N–H stretching, at 2,947.58 cm−1 due to C–H stretching, at 2,361.14 cm−1 due to tertiary amine salt, and at 1,742.56 cm−1 and 1,600.27 cm−1 due to the carbonyl group and phenyl nucleus skeletal stretching, respectively. At lower frequencies, 1,318.8 cm−1 for C–N stretching and 1,134.8 cm−1 for CO stretching were observed.11,12 With regard to the most prominent bands found in the IR spectra of HPβ-CD, an intense broadband at 3,367.13 cm−1 corresponded to the free –OH stretching vibration. The vibration of C–H and CH2 was expressed as a sharper peak detected at 2,928.95 cm−1. At 1,649.2 cm−1, a sharp peak was detected, which was explained in previous literature to correspond to the hydrated bonds between the HPβ-CD molecules.29,30 Upon correlating the FT-IR spectra of the physical mixture with those of HPβ-CD and the pure drug, the author observed that the broadband peaks of HPβ-CD masked the peaks of the Levocet peak lying in the region of 3,406.52 cm−1 and 2,947.58 cm−1. However, other characteristic peaks of Levocet remained unchanged in the physical mixture. On the other hand, the FT-IR spectra of the prepared complex in the chosen formulation (molar ratio 1:10) showed complete disappearance of the main peak of Levocet at 1,742.56 cm−1 and a remarkable decrease in the intensity of the other characteristic peak of the drug at 2,361.14 cm−1. The disappearance of the most important peaks of the drug and the masking of others could be explained based on complete inclusion of Levocet within the HPβ-CD cavity. Moreover, the presence of one band at the OH region (3,398.64 cm−1) could further be explained as a possible intermolecular hydrogen bonding of Levocet with HPβ-CD.27
The FT-IR spectra of MN (Figure 2B) shows two main peaks corresponding to OH– stretching and hydrogen bonding at 3,396.64 cm−1 and 3,288.31 cm−1, respectively. Another two peaks were detected at 2,947.26 cm−1 and 2,904.15 cm−1 corresponding to C–H stretching. Correlating the FT-IR spectra of the physical mixture and the selected SD of Levocet with MN (1:20 weight ratio) with the main peaks of Levocet and MN, it could be observed that in both cases, Levocet retained overall symmetry of the molecule, with a decrease in most peak intensities. Although previous FT-IR study revealed that there were no interactions taking place between Levocet and some used excipients,11 the remarkable decrease in the intensity of the most characterizing Levocet peaks in the physical mixture and the prepared SD may indicate possible intermolecular hydrogen bonding of Levocet with MN.
Differential scanning calorimetry analysis
DSC studied thermal behavior of HPβ-CD IC in order to confirm the formation of total IC suggested by the FT-IR study. DSC thermograms of Levocet showed one sharp characteristic endothermic peak at 218.862°C, which corresponds to its melting temperature. In the case of HPβ-CD, a broad endothermic peak was observed at approximately 60°C, owing to its amorphous nature.27 The physical mixture of Levocet with HPβ-CD showed an endothermic peak at 203.803°C, while the solid IC formed by solvent evaporation showed a complete disappearance of the endothermic peak of Levocet. This could be attributed to the formation of a total true IC through the incorporation of the drug molecule into the crystal lattice of HPβ-CD, as previously suggested.27,31 On the other hand, Figure 3B shows the DSC thermograms of MN, Levocet, physical mixture, and SD M5. It was clear that the thermal peak of the drug completely disappeared in the physical mixture and the prepared SD. This could be attributed to the large ratio of MN used compared to the drug amount, with a possible transformation of the drug to the amorphous form.30
Precompression evaluation of the effervescent blends
Angle of repose
All prepared blends were evaluated for their flow properties. From the results of precompression studies of effervescent blends C1, C2, C3, M1, M2, and M3, it could be concluded that all powder blends had excellent-to-good flow property with regard to their determined angle of repose ranging from 19.65° to 22.2°. However, it is worth mentioning that C blends showed relatively better flowability compared to M blends.
Compressibility of blends
Compressibility of a blend was defined as the ability of the powder to decrease in volume when subjected to pressure. Determined values of both Carr’s index and Hausner ratio suggested good compressibility properties, with a more pronounced effect for blends containing HPβ-CD/Levocet ICs.
Preparation and evaluation of effervescent tablets
Effervescent tablets were prepared using different percentages of the effervescent blends in combination with MN as the major water-soluble diluent with the aim of decreasing its disintegration time with faster dissolution of the drug. Avicel 102 was used in smaller ratios as a binder for better compression. PEG 6000 was chosen as lubricant in all formulations, as it is water-soluble and would aid in the fast tablet-disintegration process.32
Drug content
All prepared tablets showed an acceptable range of drug content, indicating uniformity of the drug throughout the prepared compressed blends.
Weight variation, thickness, and diameter
Physical parameters were measured to ensure the uniformity of the prepared tablets. All tablets had acceptable range limits of weight variation, thickness, and diameter.33
Hardness
The resistance of tablets for breakage and shipping under different conditions of storage depends on their hardness. It was previously shown that tablets prepared by direct compression usually possessed lower hardness values, in accordance with the current study.34 However, most prepared tablets possessed suitable hardness values to resist possible breakage during different steps of packaging and transportation.19,35
Friability study
Tablet strength can be measured by its friability values. Maximum friability values were determined for M2 and M3, probably due to their decreased hardness values compared to other prepared tablets. A remarkable observation was that all C tablets had lower friability values compared to M tablets. This fact is in accordance with the hardness values determined.
Moisture content
Results obtained are in accordance with a previous study explaining that the presence of higher ratios of sodium bicarbonate allows for more moisture absorbance.35 However, no tablets under study exceeded 0.32% in their moisture content, which can be considered acceptable.1 It is worth mentioning that tablets containing HPβ-CD had higher values of moisture content compared to those containing MN, which was previously reported to have high resistance to moisture adsorption.36
Wetting, effervescence, and in vitro disintegration time
Although wetting time is not required for the evaluation of effervescent tablets, the test was done to determine the time required for the tablets to get wet if suggested to be used as an orodispersible tablet. Maximum wetting, in vitro disintegration, and effervescence time was observed with C1 tablets, while the lowest values were observed for M3 tablets. This could be explained on the basis of lower effervescent mixture (10%) present in C1 compared to the presence of 30% effervescent mixture in M3. Although C3 contained 30% effervescent mixture, it showed higher determined values for all measured parameters compared to the similar formulation of M3. This could be attributed to the lower recorded hardness value and the highly soluble nature of MN, which could have added additional factors. The effervescence time of tablets must be less than 3 minutes, which was in accordance for all prepared tablets in the current study.37 Same pattern of results was observed for in vitro disintegration time.
Carbon dioxide content and pH of effervescent solution
It was previously reported that each gram of an effervescent tablet mixture containing citric acid and sodium bicarbonate contains 292 mg CO2, which is comparable to our study in the cases of C1, C2, M1, M2, and M3 on the basis of the weight of effervescence-mixture calculation.37 In the current study, the tablets contained smaller portions of effervescent mixture used as taste synergistic agent compared to the total weight of the tablet. However, C3 showed a lower degree of CO2 content. This could be explained based on the increased affinity of C3 tablets for water absorption with the possible beginning of an effervescent reaction on a small scale, as previously explained by other researchers.32 The measured pH of all formulations were comparable to previously reported pH values of similarly prepared effervescent tablets.35 The low values of the calculated standard deviation for the pH measurement in different samples of one formulation indicated that all prepared mixtures were uniform.
Mouth feel, palatability, and in vivo disintegration time
A remarkable observation was that all prepared tablets showed superiority in taste masking compared to the taste of the marketed orodispersible tablet. Moreover, all volunteers confirmed the acceptability of the taste and mouth feel of all prepared tablets and the absence of any grittiness after the in vivo disintegration test. However, all agreed that although increasing the percentage of effervescent content aided in the complete masking of the taste of the drug, it was not much favored, due to its higher oral effervescence. The slight decrease in the in vivo disintegration time compared to the in vitro disintegration time for all prepared tablets may be attributed to the presence of enzymatic factor in the saliva that aided in the disintegration process of the sugar-based tablets.
In vitro dissolution
The higher release rate of M tablets compared to C tablets could be explained on the basis of the higher solubility property of MN compared to HPβ-CD and the faster disintegration time of M tablets. C3 and M3 formulations showed 100% dissolution rates after 5 minutes. This is further explained by the presence of 30% effervescent mixture, which aided in increasing water penetration and drug release compared to other formulations. However, all other formulations showed fast release rate of the drug after 5 minutes, ranging from 88.27%±1.02% to 94.7%±0.32%. These results are in accordance with a previously reported study on Levocet orodispersible tablets that compared prepared orodispersible tablets to a marketed conventional tablet.11
Moisture absorption and stability
The presence of effervescent compounds in the prepared tablets allowed for the absorption of moisture, as explained previously.35 It could be seen that the liability of C formulations to absorb water was much higher than M tablets. This is explained based on the limited ability of MN for moisture adsorption compared to HPβ-CD. C1 tablets showed an increase in weight relative to the moisture uptake – 3.5%±0.208% of the total tablet weight – after 2 days, which was nearly unchanged throughout the period of study to reach a maximum of 3.57%±0.19% by the end of the month. As for C2, the increase in weight was observed to be 4.58%±0.12%, and increased by the end of the study to be 5.59%±1.55%. Moreover, C3 showed the maximum water uptake: 5.21%±0.54% after 2 days, increasing to 5.62%±0.16% by the end of the study with a sticky texture. This could be explained by the decrease in MN content used as filler in the latter compared to C2 and C1, with an increase in the liability to water uptake. The same thing was observed with M tablets with much less effect, as moisture adsorption did not exceed 2.2% after 1 month for the M3 formulation. On the other hand, slight decreases in disintegration time and effervescent time were observed for all formulations, to a slightly greater extent in C3 tablets, accompanied by an insignificant increase in the in vitro release of drug after 30 days’ exposure to 40°C and 75% relative humidity (P<0.05). Similar results have been previously reported.1
Conclusion
The present study investigated the possibility of preparing taste-masked blends of levocetirizine HCl using HPβ-CD by IC technique or MN by SD technique. Based on the taste-masking test results, the chosen blends from each technique were incorporated into different formulations of effervescent tablets containing different percentages of effervescent mixtures. The use of HPβ-CD at a molar ratio of 1:10 showed a considerable positive effect on the taste masking of the drug through the complete inclusion of the drug within the crystal lattice of the used CD. This mechanism was confirmed through FT-IR and DSC studies. On the other hand, the SD of levocetirizine HCl:MN prepared at a weight ratio of 1:20 showed an obvious increase in the acceptability of the taste of the drug, devoid of any possible drug–carrier chemical interactions. The effervescent tablet blends prepared were subjected to several in vitro pre- and postcompression evaluation tests, including flow properties, compressibility on prepared blends, hardness, friability, wetting time, effervescent time, in vitro and in vivo disintegration time, and in vitro dissolution study of the prepared tablets. Formulated tablets were further evaluated and compared to marketed orodispersible tablets for mouth feel and palatability. Results revealed that all prepared effervescent blends possessed acceptable flowability and compressibility properties. Prepared compressed tablets showed an immediate high in vitro drug release of within 5 minutes. All prepared tablets disintegrated within 14–60 seconds in saliva with pleasant taste and smooth mouth feel, and within 17–77 seconds when allowed to dissolve in water. Based on the palatability and stability studies done on the formulated tablets, it could be concluded that the best effervescent tablet formulations are those with 20% effervescent mixture for both C and M tablets.
Acknowledgments
This work was supported by the Deanship of Scientific Research (DSR), King Abdulaziz University, Jeddah, under grant 166-665-D1435. The author, therefore gratefully acknowledges the DSR technical and financial support.
Disclosure
The author reports no conflicts of interest in this work.
References
Rajalakshmi G, Vamsi CH, Balachandar R, Damodharan N. Formulation and evaluation of diclofenac potassium effervescent tablets. Int J Pharm Biomed Res. 2011;2:237–243. | ||
Kothapally NK, Devareddy S. Formulating taste-masked orally disintegrating tablets of a bitter drug ibuprofen. Int Res J Pharm. 2013;4:71–78. | ||
Bagul U, Gujar K, Patel N, Aphale S, Dhat S. Formulation and evaluation of sublimed fast melt tablets of levocetirizine dihydrochloride. Int J Pharm Sci. 2010;2:76–80. | ||
Voleti VK, Gunasekharan V, Bolla SP, Kumar JM, Rayaprolu M. Formulation and evaluation of levo citerizine dihydro chloride fast dissolving tablets using superdisintegrants. Int J Pharm Pharm Sci. 2010;5:324–328. | ||
Koizumi IK. New method of preparing highly porous rapidly saliva soluble tablets by sublimation technique. Int J Pharm. 1997;152:127–131. | ||
Arun A, Amrish C. Fast drug delivery systems: a review. Pharm Lett. 2010;2:350–361. | ||
Bi YX, Sunada H, Yonezawa Y, Danjoe K. Evaluation of rapidly disintegrating tablets prepared by a direct compression method. Drug Dev Ind Pharm. 1999;25:571–581. | ||
University of the Sciences in Philadelphia. Remington: The Science and Practice of Pharmacy. 21st ed. Philadelphia: Lippincott Williams & Wilkins; 2005. | ||
Shet N, Vaidya I. Taste masking: a pathfinder for bitter drugs. Int J Pharm Sci Rev Res. 2013;18:1–12. | ||
Nettis E, Calogiuri GF, Di Leo E, et al. Once daily levocetirizine for treatment of allergic rhinitis and chronic idiopathic urticaria. J Asthma Allergy. 2009;2:17–23. | ||
Gandhi GS, Mundhada DR, Bhaskaran S. Levocetirizine orodispersible tablet by direct compression method. J Appl Pharm Sci. 2011;1:145–150. | ||
Renuka M, Avani A. Optimization and characterization of rapidly dissolving films of cetirizine hydrochloride using cyclodextrins for taste masking. Int J Pharmtech Res. 2013;5:536–552. | ||
Al Omran M, Al-Suwayeh S A, El-Helw AM, Saleh SI. Taste masking of diclofenac using microencapsulation. J Microencapsul. 2002;19:45–52. | ||
Kurasumi T, Imamori K, Iwasa A, inventors; SS Pharmaceutical Co, assignee. Carbetapentane citrate-containing composition. Japanese patent JPH03236316. 1991 Oct 22. | ||
Kasturagi Y, Sugiura Y, Lee C, Otsuji K, Kurihara K. Selective inhibition of bitter taste of various drugs by lipoprotein. Pharm Res. 1995;12:658–662. | ||
Hussain M, Aungst B, Koval C, Shefter E. Improved buccal delivery of opioid analgesics and antagonists with bitterless prodrugs. Pharm Res. 1988;5:615–618. | ||
Lewis S. Taste masking technologies: a review. Int J Pharm Pharm Sci. 2010;2:6–13. | ||
Rahman Z. Risperidone solid dispersion for orally disintegrating tablet: its formulation design and nondestructive methods of evaluation. Int J Pharm. 2010;400:49–58. | ||
Khemariya P, Gajbhiye KR, Vaidya VD, Jadon RS, Mishra S, Shukla A. Preparation and evaluation of mouth dissolving tablets of meloxicam. Int J Drug Deliv. 2010;2:76–80. | ||
Corveleyn S, Remon JP. Formulation and production of rapidly disintegrating tablets by lyophilization using hydrochlorothiazide as a model drug. Int J Pharm. 1997;152:215–225. | ||
Rao YS, Vijaya L, Varalakshmi TS, Chandana R, Chowdary KP. Formulation and evaluation of carvedilol solid dispersions for dissolution rate enhancement. Int J Adv Pharm Biol Chem. 2012;1:489–495. | ||
Shenoy V, Pandey S. Meloxicam-PEG6000 solid dispersions in rapidly disintegrating tablets: preparation, in vitro and in vivo characterization. Asian J Pharm Sci. 2008;3:142–150. | ||
Guleria R, Kaith NS, Singh R. PEG based solid dispersions of gliclazide: a comparative study. Int J Pharm Pharm Sci. 2012;4:507–511. | ||
Garg A, Gupta MM. Taste masking and formulation development and evaluation of mouth dissolving tablets of levocetirizine dihydrochloride. J Drug Deliv Ther. 2013;3(3):123–130. | ||
Shende P, Shah V, Ghodke D, Shah R, Patil S, Chougule D. Validation of UV spectrophotometric method for estimation of levocetirizine dihydrochloride in bulk and pharmaceutical formulation. J Pharm Res. 2010;3:2386–2387. | ||
Masareddy R, Yellanki SK, Patil BR, Manvi FV. Development and evaluation of floating matrix tablets of riboflavin. Int J Pharmtech Res. 2010;2:1439–1445. | ||
Talegaonkar S, Khan AY, Khar RK, Ahmad FJ, Khan ZI. Development and characterization of paracetamol complexes with hydroxypropyl-β-cyclodextrin. Iran J Pharm Res. 2007;6:95–99. | ||
Kulkarni MS, Zeeshan A, Bhise KS, Somwanshi SV. Formulation and evaluation of orodispersible tablet of ornidazole. Int J Pharm Stud Res. 2010;1:39–47. | ||
Jun SW, Kim MS, Kim JS, et al. Preparation and characterization of simvastatin/hydroxypropyl-β-cyclodextrin inclusion complex using supercritical antisolvent (SAS) process. Eur J Pharm Biopharm. 2007;66:413–421. | ||
Badr-Eldin SM1, Elkheshen SA, Ghorab MM. Inclusion complexes of tadalafil with natural and chemically modified β-cyclodextrins. I: Preparation and in-vitro evaluation. Eur J Pharm Biopharm. 2008;70:819–827. | ||
Cappello B, Carmingnani C, Iervolino M, La Rotonda MI, Saettone MF. Solubilization of tropicamide by hydroxypropyl-β-cyclodextrin and water-soluble polymers: in vitro/in vivo studies. Int J Pharm. 2001;213:75–81. | ||
Aslani A, Fattahi F. Formulation, characterization and physicochemical evaluation of potassium citrate effervescent tablets. Adv Pharm Bull. 2013;3:217–225. | ||
Gosai AR, Patil SB, Sawant KK. Formulation and evaluation of oro dispersible tablets of ondansetron hydrochloride by direct compression using superdisintegrants. Int J Pharm Sci Nanotechnol. 2008;26:106–111. | ||
Bhardwaj V, Bansal M, Sharma PK. Formulation and evaluation of fast dissolving tablets of amlodipine besylate using different super disintegrants and camphor as sublimating agent. Am Euras J Sci Res. 2010;5:264–269. | ||
Aslani A, Jahangiri H. Formulation, characterization and physicochemical evaluation of ranitidine effervescent tablets. Adv Pharm Bull. 2013;3:315–322. | ||
Bauer H, Herkert T, Bartels M, Kovar KA, Schwarz E, Schmidt PC. Investigations on polymorphism of mannitol/sorbitol mixtures after spray drying using differential scanning calorimetry, X-ray diffraction and near infrared spectroscopy. Pharm Ind. 2000;62:231–235. | ||
Yanze FM, Duru C, Jacob M. A process to produce effervescent tablets: fluidized bed dryer melt granulation. Drug Dev Ind Pharm. 2000;26:1167–1176. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.