")
Back to Journals » Drug Design, Development and Therapy » Volume 11
Novel 5-oxo-hexahydroquinoline derivatives: design, synthesis, in vitro P-glycoprotein-mediated multidrug resistance reversal profile and molecular dynamics simulation study
Authors Shahraki O, Edraki N, Khoshneviszadeh M, Zargari F, Ranjbar S, Saso L , Firuzi O, Miri R
Received 17 August 2016
Accepted for publication 16 November 2016
Published 14 February 2017 Volume 2017:11 Pages 407—418
DOI https://doi.org/10.2147/DDDT.S119995
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Jianbo Sun
Omolbanin Shahraki,1,2 Najmeh Edraki,1 Mehdi Khoshneviszadeh,1,2 Farshid Zargari,1 Sara Ranjbar,1,2 Luciano Saso,3 Omidreza Firuzi,1 Ramin Miri1
1Medicinal and Natural Products Chemistry Research Center, 2Department of Medicinal Chemistry, School of Pharmacy, Shiraz University of Medical Sciences, Shiraz, Iran; 3Department of Physiology and Pharmacology “Vittorio Ersparmer”, Sapienza University of Rome, Rome, Italy
Abstract: Overexpression of the efflux pump P-glycoprotein (P-gp) is one of the important mechanisms of multidrug resistance (MDR) in many tumor cells. In this study, 26 novel 5-oxo-hexahydroquinoline derivatives containing different nitrophenyl moieties at C4 and various carboxamide substituents at C3 were designed, synthesized and evaluated for their ability to inhibit P-gp by measuring the amount of rhodamine 123 (Rh123) accumulation in uterine sarcoma cells that overexpress P-gp (MES-SA/Dx5) using flow cytometry. The effect of compounds with highest MDR reversal activities was further evaluated by measuring the alterations of MES-SA/Dx5 cells’ sensitivity to doxorubicin (DXR) using MTT assay. The results of both biological assays indicated that compounds bearing 2-nitrophenyl at C4 position and compounds with 4-chlorophenyl carboxamide at C3 demonstrated the highest activities in resistant cells, while they were devoid of any effect in parental nonresistant MES-SA cells. One of the active derivatives, 5c, significantly increased intracellular Rh123 at 100 µM, and it also significantly reduced the IC50 of DXR by 70.1% and 88.7% at 10 and 25 µM, respectively, in MES-SA/Dx5 cells. The toxicity of synthesized compounds against HEK293 as a noncancer cell line was also investigated. All tested derivatives except for 2c compound showed no cytotoxicity. A molecular dynamics simulation study was also performed to investigate the possible binding site of 5c in complex with human P-gp, which showed that this compound formed 11 average H-bonds with Ser909, Thr911, Arg547, Arg543 and Ser474 residues of P-gp. A good agreement was found between the results of the computational and experimental studies. The findings of this study show that some 5-oxo-hexahydroquinoline derivatives could serve as promising candidates for the discovery of new agents for P-gp-mediated MDR reversal.
Keywords: cancer, P-glycoprotein, multidrug resistance, 1,4-dihydropyridine, molecular dynamics simulation
Introduction
Despite recent progress in the elucidation of cancer biology and development of novel strategies in cancer diagnosis and treatment, this disease is still among the leading causes of death around the world.1 The resistance of malignant cells to structurally and mechanistically unrelated classes of anticancer agents is recognized as MDR.2 Different mechanisms are involved in drug resistance; a very important one is overexpression of P-gp (ABCB1, MDR1), which has been one of the first members of ABC transporters to be studied.3 P-gp extrudes a wide variety of endogenous molecules and xenobiotics and plays an important physiological role in detoxifying cells from exogenous toxic agents.4 However, overexpression of P-gp in cancer cells leads to reduced accumulation of chemotherapeutic drugs and results in resistance against these agents.5 In this context, inhibition of P-gp by small-molecule inhibitors seems to be a promising approach for overcoming MDR in cancer cells.6
Three different generations of P-gp inhibitors have been discovered. The first-generation inhibitors such as verapamil, cyclosporine A and quinidine7 were developed for other applications and then tested for P-gp inhibition. The second- (such as valspodar8) and third-generation inhibitors (such as elacridar,9 tariquidar,10 laniquidar11 and zosuquidar12) were specifically designed for MDR reversal and did not display other pharmacological effects. However, clinical results of these agents were unsatisfactory due to insufficient therapeutic benefit and unacceptable systemic toxicity.
Initial studies on calcium channel blockers demonstrated that verapamil and diltiazem were able to reverse MDR by increasing intracellular levels of chemotherapeutic agents.13 Afterward, Safa et al confirmed P-gp-inhibitory effect of calcium channel blocker azidopine belonging to the family of 1,4-DHP derivatives.14 Other authors have also reported the MDR reversal effect of a set of synthesized DHPs.15
SAR studies of DHPs show that replacement of carboxylate esters at C3 and C5 positions with aryl carboxamide groups dramatically diminishes their cardiovascular effects. We have previously studied the synthesis and biological evaluation of symmetric and asymmetric 1,4-DHPs containing different carboxamide substitutions at mentioned positions and observed that these compounds had poor calcium-channel-blocking activity compared to nifedipine.16–19
The compounds bearing DHP core have been shown to display a broad range of biological and pharmacological effects.16 Besides their calcium-channel-blocking activity,20–22 they have shown therapeutic effects against Alzheimer’s disease and atherosclerosis23 and have also shown MDR reversal effect.20,24,25
Three decades after the discovery of P-gp, lots of efforts have been made on the development of novel P-gp inhibitors, but no small-molecule inhibitor has been identified for clinical use. In an attempt to respond to this crucial need for novel therapeutic agents and as part of our research program toward the discovery of useful MDR reversal agents, herein we report synthesis, biological evaluation and MD simulations study of a set of 26 novel 5-oxo-hexahydroquinoline derivatives. The experimental results showed that these compounds can indeed inhibit P-gp, and these findings were in good agreement with the computational findings.
Results and discussion
Fragment-based design
Several efforts have been made to clarify the basic features that are responsible for the MDR reversal effect of DHPs. In our previous studies, we described the design, synthesis and in vitro data of a series of new inhibitors of P-gp with 1,4-DHP structure carrying 2- or 3-pyridyl methyl carboxylate at C3 with different alkyl carboxylate moieties at C5 position as well as nitrophenyl or heteroaromatic rings at C4 of central DHP core.26,27 Herein, we report a fragment-based design for a new set of 5-oxo-hexahydroquinoline derivatives based on a previously established SAR for reported DHPs (Scheme 1).
- The 5-oxo-hexahydroquinoline scaffold was chosen based on the observation that DHP pharmacophore exhibits MDR reversal effect.15
- The presence of nitroaryl substituents at C4 position has previously proved to enhance MDR reversal effect compared to heteroaryl rings.
- Substitution of different moieties bearing nitrogen at C3 such as carboxamide group resulted in increasing MDR reversal effect of designed compounds. Accordingly, different pyridyl methyl carboxylates at C3 position26,27 were bioisosterically replaced with alkyl or aryl carboxamides.
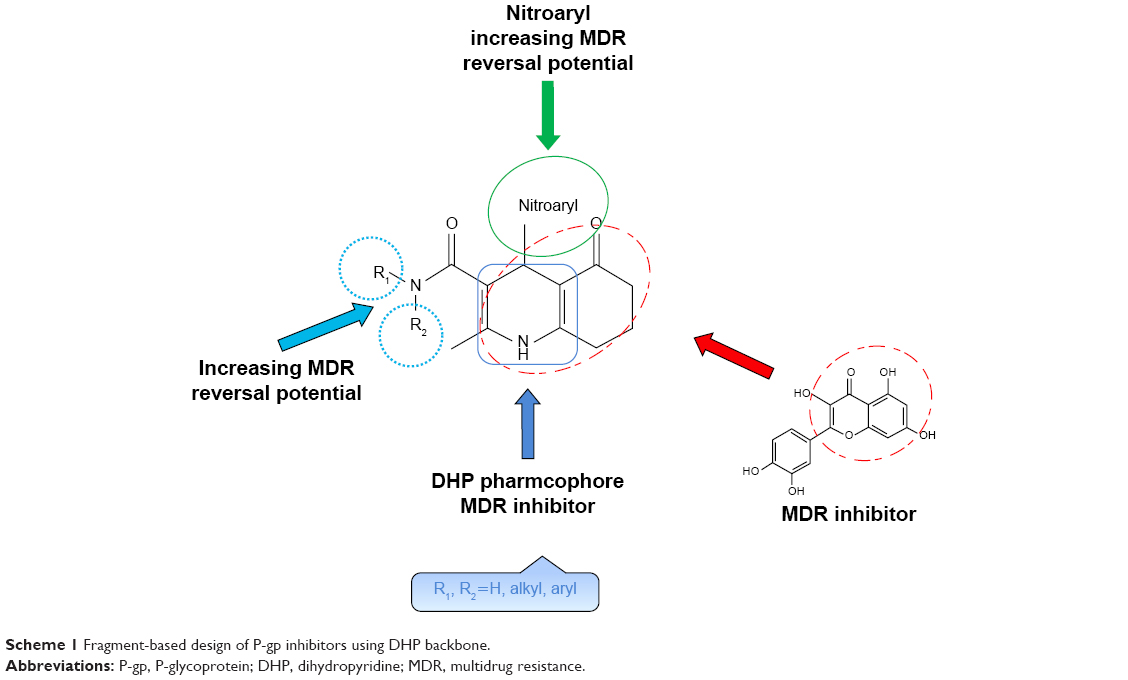 | Scheme 1 Fragment-based design of P-gp inhibitors using DHP backbone. |
Many studies have revealed that natural products with fused rings such as quercetin are MDR reversal agents.28 Based on these findings, bioisosteric replacement and hybridization of 4H-chromen-4-one ring of quercetin with 4,6,7,8-tetrahydroquinolin-5(1H)-one in DHP-based designed compounds would be expected to improve MDR reversal activity.29,30
Thus, in this study, a library of 26 compounds with 5-oxo-hexahydroquinolone skeleton bearing different nitrophenyl moieties at C4 and different aryl or alkyl carboxamides at C3 was designed.
Chemistry
The applied route for synthesis of designed compounds is outlined in Scheme 2, and the structures are presented in Table 1. By the reaction of the commercially available different primary and secondary amines (1) with 2,2,6-trimethyl-4H-1,3-dioxin-4-one (2), corresponding 3-oxobutanamides (1a–6a) were obtained (Table 2). The final products were synthesized by the reaction of obtained intermediates (1a–6a) with different aryl aldehydes and 1,3-cyclohexadione in the presence of excess amounts of ammonium acetate (Table 1). All compounds were completely purified and characterized using 1H NMR, 13C NMR and mass and IR spectroscopy (Supplementary materials).
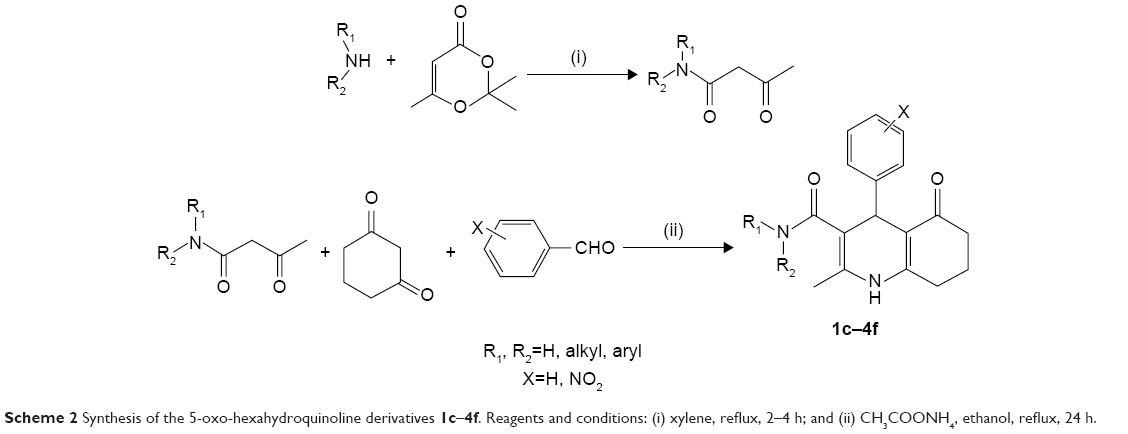 | Scheme 2 Synthesis of the 5-oxo-hexahydroquinoline derivatives 1c–4f. Reagents and conditions: (i) xylene, reflux, 2–4 h; and (ii) CH3COONH4, ethanol, reflux, 24 h. |
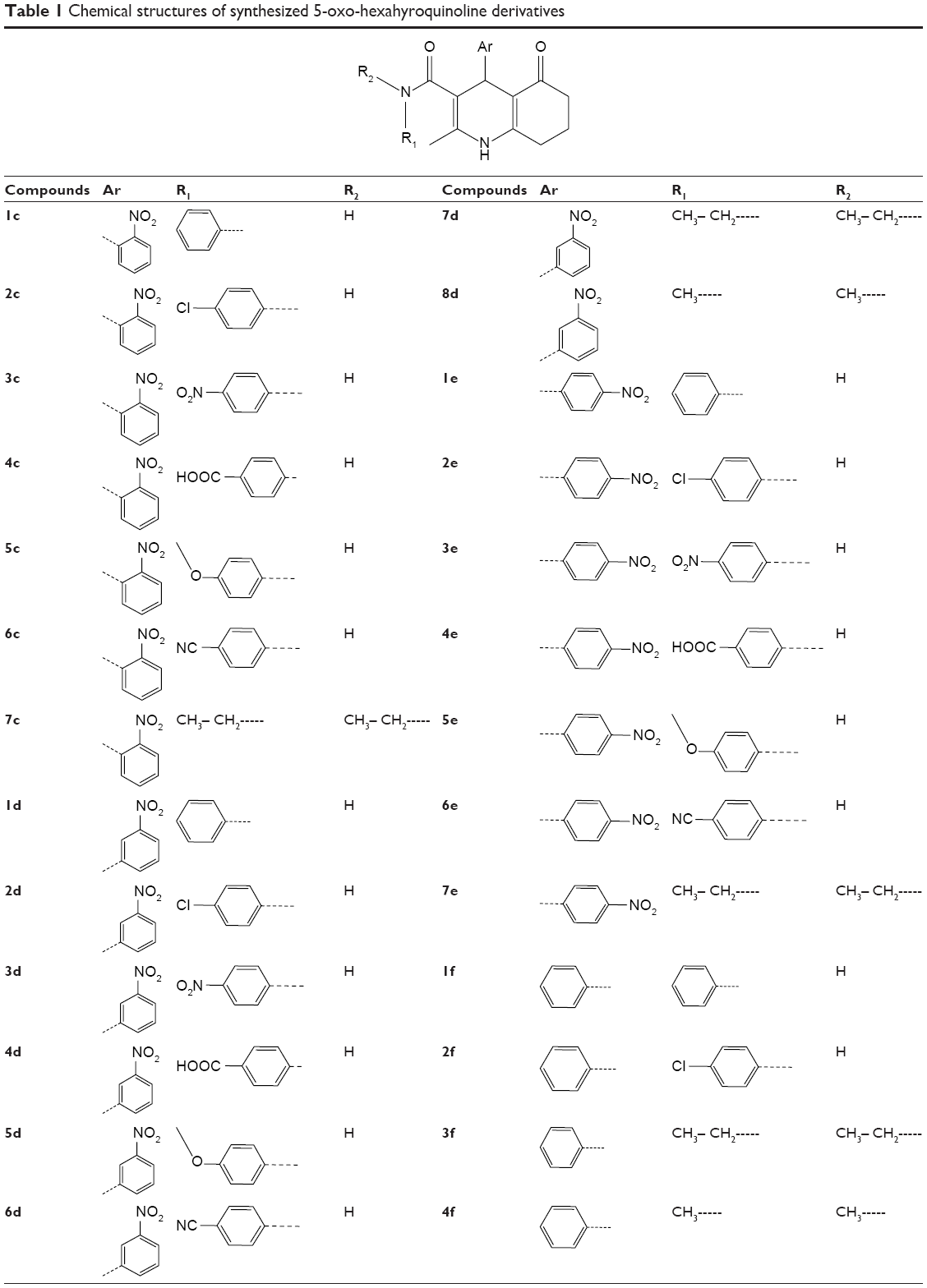 | Table 1 Chemical structure of synthesized 5-oxo-hexahyroquinoline derivatives |
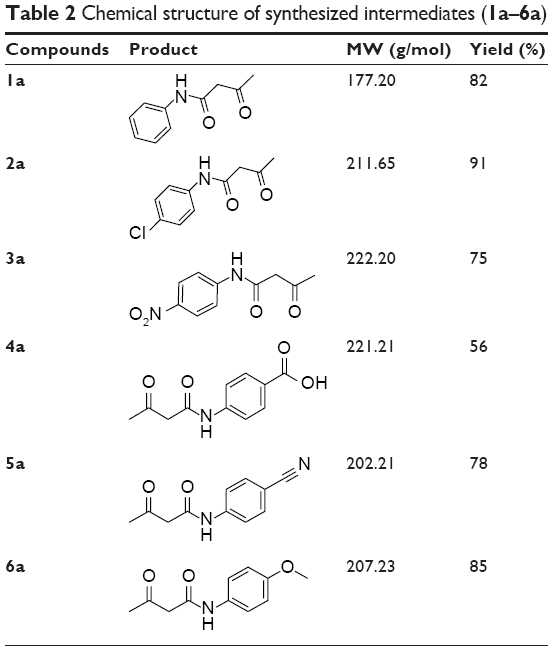 | Table 2 Chemical structure of synthesized intermediates (1a–6a) |
Biological evaluation
Rh123 accumulation assay
The P-gp-mediated Rh123 efflux was determined by flow cytometry on the DXR-resistant uterine sarcoma MES-SA/Dx5 cell line and its parental nonresistant MES-SA cell line.31 Verapamil was used as a positive control. MES-SA/Dx5 is a resistant cell line, which overexpresses P-gp as a result of continuous exposure to DXR. Alterations in the amount of the fluorescent Rh123 retained inside the MES-SA-Dx5 cells can be logically related to the inhibition of the activity of P-gp efflux pump in the cells. Figure 1 shows a representative histogram of the effect of different doses of compound 2f on the intracellular accumulation of Rh123 in resistant cells. A dose-dependent response can be clearly observed at 5, 25 and 100 μM on Rh123 accumulation as the histogram shifts to the right. All the active compounds except 2c showed a dose-dependent effect. The results are expressed as Rh123 accumulation fold increase, representing the ratio of Rh123 fluorescence in the presence and absence of synthesized compounds (Figure 2). Compound 2f bearing phenyl substitute at C4 and N-(4-chlorophenyl) carboxamide at C3 position of the DHP ring showed the highest levels of Rh123 accumulation and P-gp inhibition at 100 μM. Compounds 2c and 5c also exhibited a considerable MDR reversal activity at 25 and 100 μM, respectively.
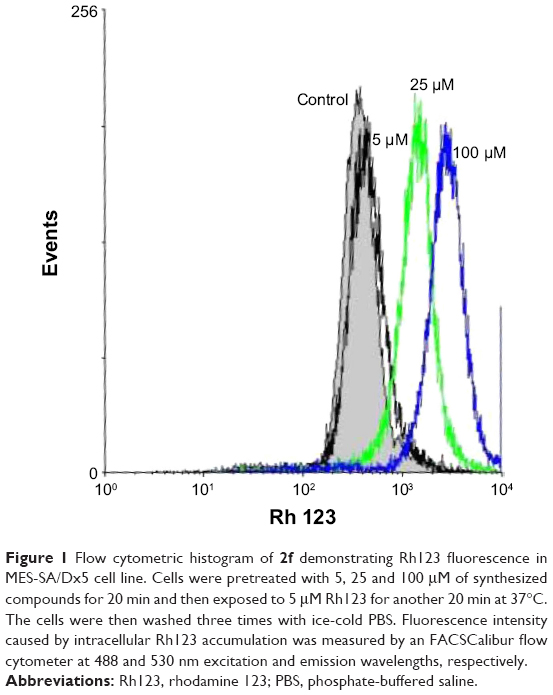 | Figure 1 Flow cytometric histogram of 2f demonstrating Rh123 fluorescence in MES-SA/Dx5 cell line. Cells were pretreated with 5, 25 and 100 μM of synthesized compounds for 20 min and then exposed to 5 μM Rh123 for another 20 min at 37°C. The cells were then washed three times with ice-cold PBS. Fluorescence intensity caused by intracellular Rh123 accumulation was measured by an FACSCalibur flow cytometer at 488 and 530 nm excitation and emission wavelengths, respectively. |
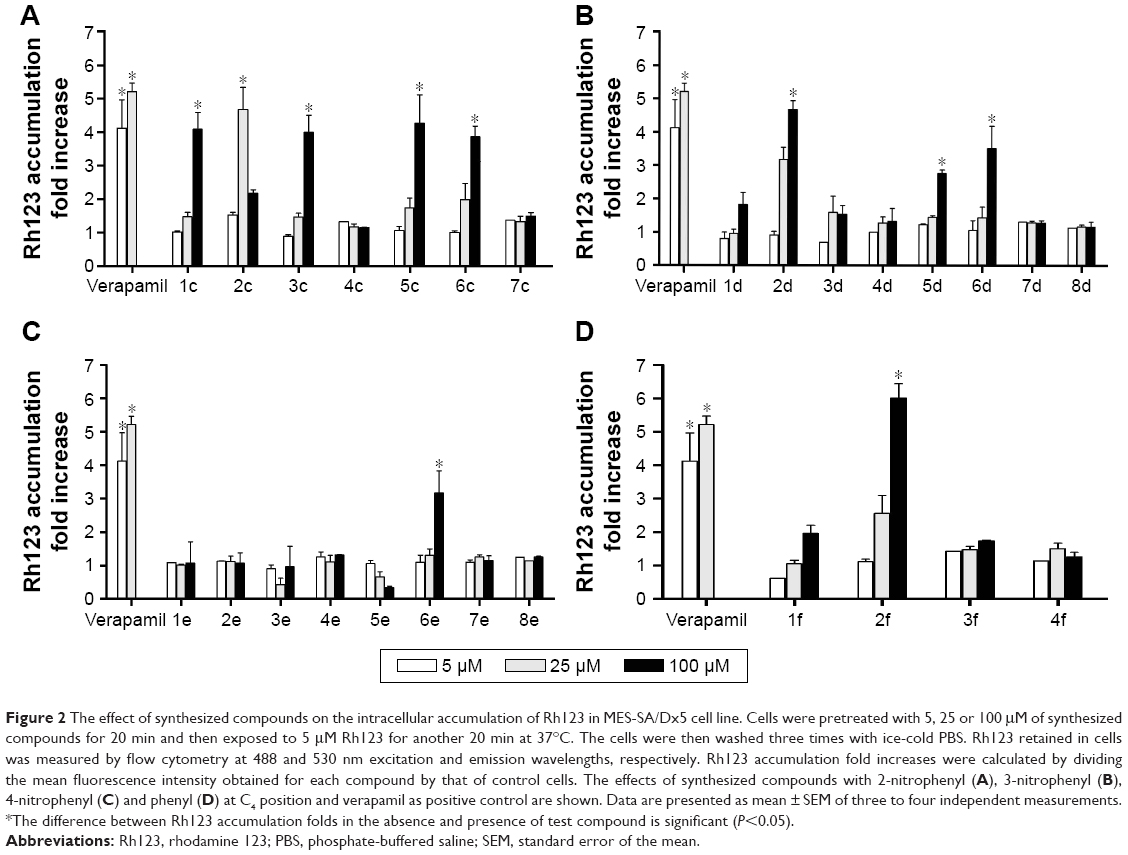 | Figure 2 The effect of synthesized compounds on the intracellular accumulation of Rh123 in MES-SA/Dx5 cell line. Cells were pretreated with 5, 25 or 100 μM of synthesized compounds for 20 min and then exposed to 5 μM Rh123 for another 20 min at 37°C. The cells were then washed three times with ice-cold PBS. Rh123 retained in cells was measured by flow cytometry at 488 and 530 nm excitation and emission wavelengths, respectively. Rh123 accumulation fold increases were calculated by dividing the mean fluorescence intensity obtained for each compound by that of control cells. The effects of synthesized compounds with 2-nitrophenyl (A), 3-nitrophenyl (B), 4-nitrophenyl (C) and phenyl (D) at C4 position and verapamil as positive control are shown. Data are presented as mean ± SEM of three to four independent measurements. *The difference between Rh123 accumulation folds in the absence and presence of test compound is significant (P<0.05). |
Considering the data presented in Figure 2, a brief SAR can be deduced as shown in Figure 3.
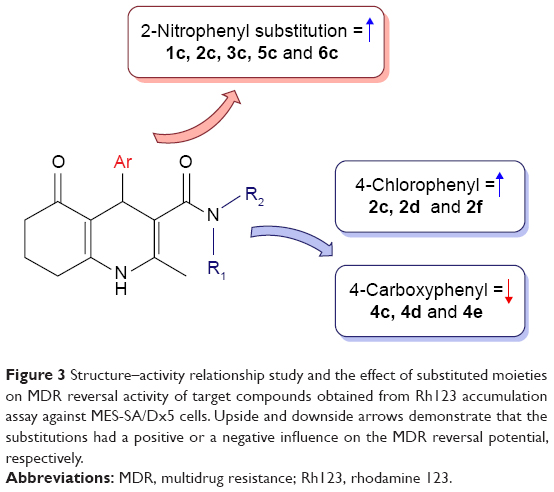 | Figure 3 Structure–activity relationship study and the effect of substituted moieties on MDR reversal activity of target compounds obtained from Rh123 accumulation assay against MES-SA/Dx5 cells. Upside and downside arrows demonstrate that the substitutions had a positive or a negative influence on the MDR reversal potential, respectively. |
In different series of synthesized compounds, those bearing 2-nitrophenyl substitution at C4 such as 1c, 2c, 3c, 5c and 6c were the most potent derivatives. This reveals that the presence of 2-nitrophenyl moiety is crucial in enhancing the potency.
- Compounds containing N-(4-chlorophenyl) carboxamide at C3 (2c, 2d and 2f) exhibited good MDR reversal effect. For example, compound 2f showed 6.0-fold Rh123 accumulation relative to the negative control.
- None of the compounds bearing 4-carboxyphenyl moiety at C3 (4c, 4d and 4e) were effective MDR reversal agents. 4c and 4d exhibited only 1.32-fold Rh123 accumulation.
As shown in Figure 4, the effects of synthesized compounds were specific against resistant MES-SA/Dx5 cell line, because Rh123 accumulation was not observed in the parental MES-SA cells.
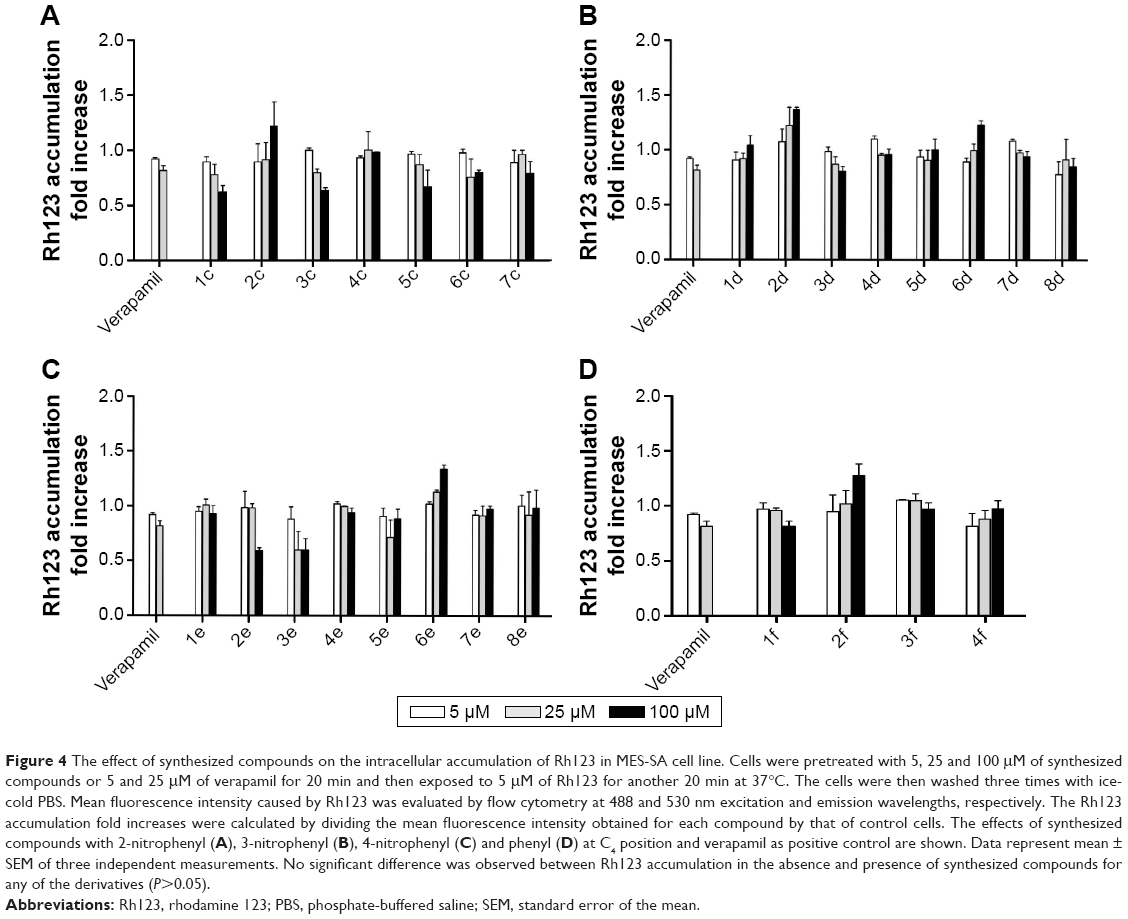 | Figure 4 The effect of synthesized compounds on the intracellular accumulation of Rh123 in MES-SA cell line. Cells were pretreated with 5, 25 and 100 μM of synthesized compounds or 5 and 25 μM of verapamil for 20 min and then exposed to 5 μM of Rh123 for another 20 min at 37°C. The cells were then washed three times with ice-cold PBS. Mean fluorescence intensity caused by Rh123 was evaluated by flow cytometry at 488 and 530 nm excitation and emission wavelengths, respectively. The Rh123 accumulation fold increases were calculated by dividing the mean fluorescence intensity obtained for each compound by that of control cells. The effects of synthesized compounds with 2-nitrophenyl (A), 3-nitrophenyl (B), 4-nitrophenyl (C) and phenyl (D) at C4 position and verapamil as positive control are shown. Data represent mean ± SEM of three independent measurements. No significant difference was observed between Rh123 accumulation in the absence and presence of synthesized compounds for any of the derivatives (P>0.05). |
Determination of chemosensitization in resistant cells induced by synthesized compounds
Effects of synthesized compounds on the alteration of MES-SA/Dx5’s sensitivity to DXR were evaluated by MTT assay, in which cell viability is measured spectrophotometrically by the amount of reduced formazan. For this purpose, the most potent compounds were selected based on the results of Rh123 accumulation assay. To select a nontoxic concentration (cell survival >85%), cytotoxicity assay was performed on P-gp-expressing MES-SA/Dx5 cell line. On the basis of the obtained results, maximum concentration was set to 25 μM (data not shown). The effects of coadministration of synthesized compounds and DXR were then studied in these cells. The percent reduction of DXR’s IC50 was calculated (Figure 5). DXR showed a relative low activity in the assay of cell survival (IC50: 2.7 μM). The compounds 1c and 3c at 25 μM decreased the DXR’s IC50 by 47.7% and 69.8%, respectively, and did not show any considerable chemosensitization effect. Compound 2f, which was the most potent compound in the Rh123 accumulation assay, showed a moderate activity in this assay. The most potent compound 2c bearing 2-nitrophenyl at C4 and 4-chlorophenycarboxamide at C3 showed 92.3% and 99.2% decrease in the IC50 of DXR at 10 and 25 μM, respectively. The other 2-nitrophenyl-containing compound 5c with 4-methoxyphenyl substitute at C3 also resulted in 70.1% and 88.7% decrease in the IC50 of DXR at 10 and 25 μM, respectively. The other tested compounds were able to induce a statistically significant decrease in the IC50 of DXR at 10 and 25 μM, and a good correlation was observed between the results of Rh123 accumulation assay and DXR uptake in resistant cells.
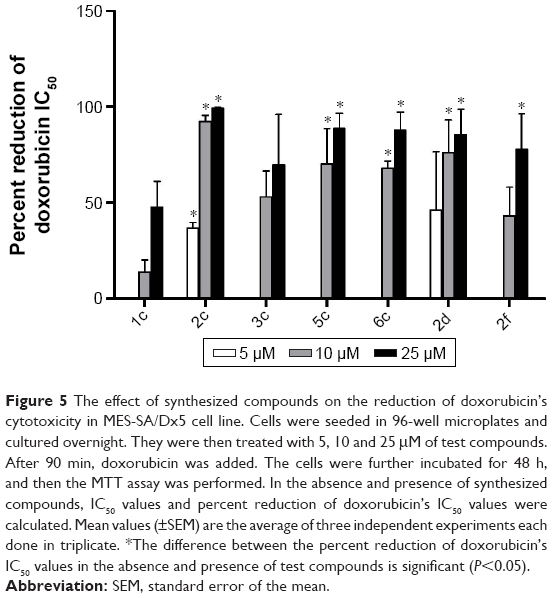 | Figure 5 The effect of synthesized compounds on the reduction of doxorubicin’s cytotoxicity in MES-SA/Dx5 cell line. Cells were seeded in 96-well microplates and cultured overnight. They were then treated with 5, 10 and 25 μM of test compounds. After 90 min, doxorubicin was added. The cells were further incubated for 48 h, and then the MTT assay was performed. In the absence and presence of synthesized compounds, IC50 values and percent reduction of doxorubicin’s IC50 values were calculated. Mean values (±SEM) are the average of three independent experiments each done in triplicate. *The difference between the percent reduction of doxorubicin’s IC50 values in the absence and presence of test compounds is significant (P<0.05). |
Cytotoxicity against HEK293 cells
In order to determine the effect of synthesized compounds on HEK293 (human embryonic kidney) cells used as a normal cell line, the MTT assay was performed. The cells were treated with the test compounds at concentrations of 5, 10 and 25 μM for 48 h. The obtained results (Figure 6) revealed that all compounds exhibited no cytotoxicity against normal cells (viability >80%) except compound 2c which showed 54.9% viability at 25 μM.
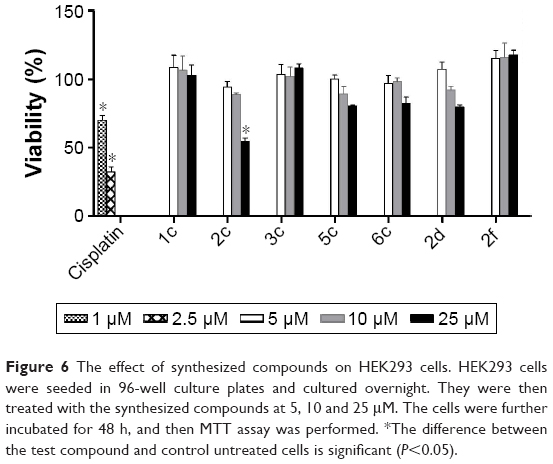 | Figure 6 The effect of synthesized compounds on HEK293 cells. HEK293 cells were seeded in 96-well culture plates and cultured overnight. They were then treated with the synthesized compounds at 5, 10 and 25 μM. The cells were further incubated for 48 h, and then MTT assay was performed. *The difference between the test compound and control untreated cells is significant (P<0.05). |
Computational approach
Different studies have been focused on the elucidation of the mechanism of action of synthesized DHPs on P-gp and identification of possible binding sites of DHPs. To this purpose, domain mapping using photoaffinity labeling along with immunoprecipitation of different DHP derivatives has confirmed that the binding site is located near the N-terminal nucleotide-binding domain of P-gp.32 Ferry et al proposed that DHP-binding site that is located in the cytoplasmic region of P-gp comprises residues 491–525.33 Other investigators have used a dexniguldipine-HCl ([3H] B9209-005) to probe the DHPs’ binding site showing that residues 468–527 are the ligand-interacting region of the protein. These findings show that DHP derivatives probably bind to the nucleotide-binding domain of P-gp, inhibiting the action of this efflux pump.34
In order to explore the structural requirements of 5-oxo-hexahydroquinoline derivatives which determine their interaction with P-gp, we chose the most favorite pose of one of the most potent and nontoxic compounds 5c from docking study and placed it in the proposed active site of homology-modeled P-gp. The crystal structure of murine P-gp (PDB ID: 3G5U, chain A, resolution: 3.8 Å) served as a template to create homology models of human P-gp (UNIPORT ID: P08183). The alignment of human P-gp and mouse P-gp sequences shows 87% identity. The QMEAN Z-score of the obtained models ranged from −2.35 (best) to −4.14. All models obtained from GA341 showed the highest possible value of 1. The derived backbone conformation was inspected by Ramachandran plot which showed that 83.8% of the residues lie in most favored, 12.1% in additionally allowed, 3.3% in generously allowed and 0.8% in outlier regions. These results indicate that the phi and psi backbone dihedral angles in the obtained model are precise.
We incorporated our results into the model for the P-gp ligand–receptor interaction that is shown in Figure 7. This configuration is the midpoint structure of most populated cluster obtained by clustering analysis of MD trajectories (data not shown). Clustering was performed using Cα backbone atoms, least squares alignment and the Gromos algorithm35 (cutoff: 0.15 nm) by g_cluster module as implemented in Gromacs 4.6.5. As shown in Figure 7B, cyclohexanone moiety of the ligand participates in hydrophobic interactions with the side chains of Arg905, Phe904 and Val472.36 There is another hydrophobic interaction that occurs between N-(4-methoxyphenyl) part of ligand and side chain of Val478 as well as phenyl moiety of Tyr490. In order to investigate hydrogen-bonding profile of ligand and receptor, we used g_h bond module implemented in Gromacs 4.6.5 with H-bond forming a distance of 3.5 Å and an angle of 120°. Table 3 includes all information about hydrogen-bonding pattern obtained during 10 ns MD simulation. The analysis revealed that compound 5c forms eleven average H-bonds sharing nine H-bonds with Ser909, Thr911, Arg547, Arg543 and Ser474 as acceptors. Oxygen atom of N-(4-methoxyphenyl) carboxamide pendant at C3 formed the most stable H-bond with Arg543 with occupancy of 81% (some interactions are not shown for clarity). This is in good agreement with our SAR studies which proves that the N-(4-methoxyphenyl) moiety increases the MDR reversal effect of this ligand. Further analysis of ligand interaction with active site residues revealed a π–cation interaction between nitrogen atom of Arg543 and 2-nitrophenyl ring. In summary, we here propose 3D models of complexes of P-gp with a promising 5-oxo-hexahydroquinoline reversal agent that is consistent with our SAR studies.
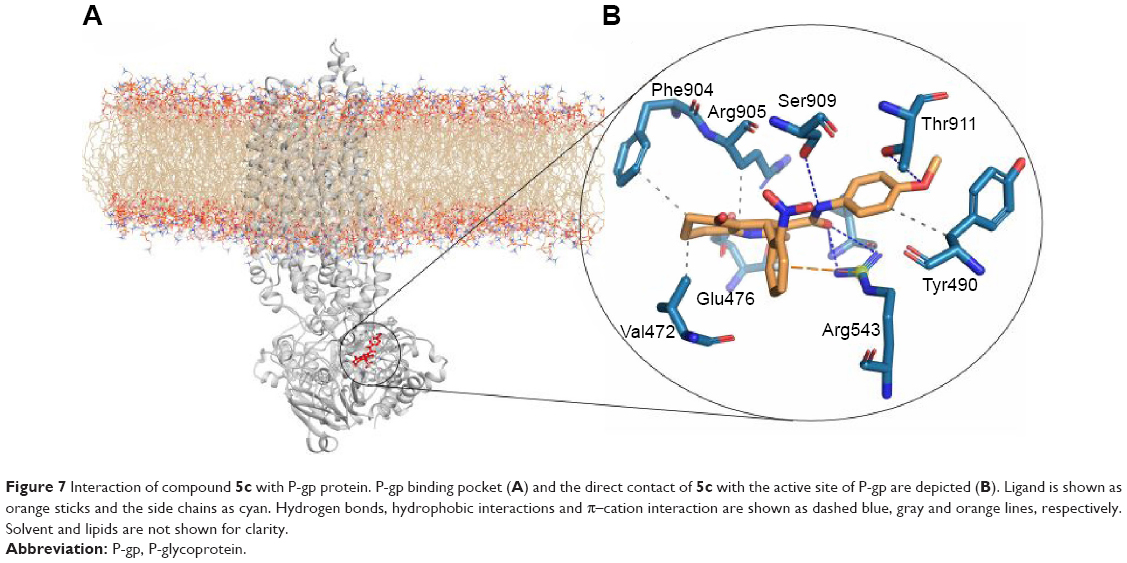 | Figure 7 Interaction of compound 5c with P-gp protein. P-gp binding pocket (A) and the direct contact of 5c with the active site of P-gp are depicted (B). Ligand is shown as orange sticks and the side chains as cyan. Hydrogen bonds, hydrophobic interactions and π–cation interaction are shown as dashed blue, gray and orange lines, respectively. Solvent and lipids are not shown for clarity. |
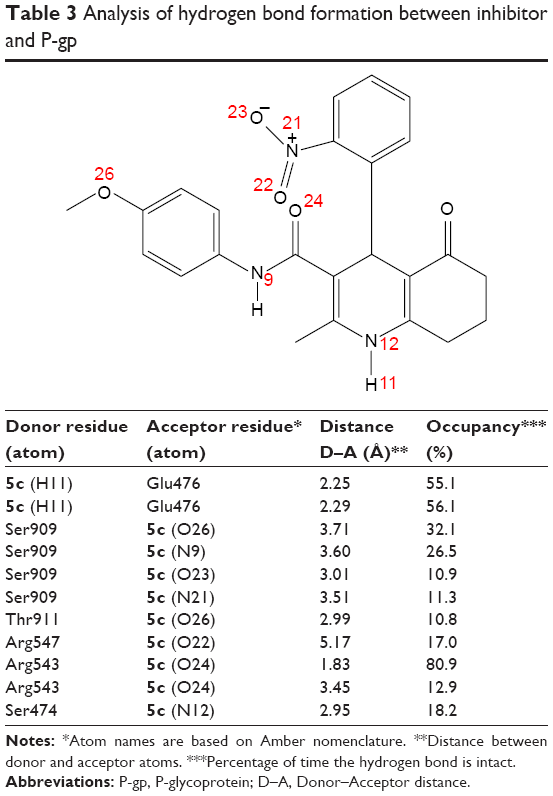 | Table 3 Analysis of hydrogen bond formation between inhibitor and P-gp |
Experimental section
Chemistry
All melting points were taken on a hot stage apparatus (Electrothermal, Essex, UK) and were reported uncorrected. IR spectra were recorded with a Perkin-Elmer spectrometer (KBr disk) (PerkinElmer, Waltham, MA, USA). NMR spectra were recorded on a Bruker Avance 300 spectrometer (300 MHz for 1H NMR, 75 MHz for 13C NMR). Mass spectra were obtained with an Agilent spectrometer (9575c inert MSD; Agilent Technologies, Santa Clara, CA, USA). Chromatographic separations were performed on a silica gel column by gravity chromatography (Kieselgel 40, 0.063–0.200 mm; Merck) or flash chromatography (Kieselgel 40, 0.040–0.063 mm; Merck). Yields are given after purification, unless otherwise stated. All compounds were named following IUPAC guidelines as defined by ChemBioDraw Ultra 12.0 software.
3-Oxo-N-substituted phenylbutanamide
Ten milliliters of xylene was added to a mixture of 2,2,6-trimethyl-4H-1,3-dioxin-4-one (6 mmol) and different primary or secondary amines (5 mmol). The reaction mixture was refluxed for 2–4 h. After completion of the reaction as monitored by thin-layer chromatography, the mixture was cooled and transferred to a separating funnel and washed with petroleum ether (30–50 mL). The solid product was collected and dried (Table 2). Compounds 3a and 4a were reported previously.37 N, N-Diethyl acetoacetamide and N, N-dimethyl acetoacetamide were commercially available. The NMR data are reported in the Supplementary materials.
Reagents
All reagents were purchased from Sigma-Aldrich (St Louis, MO, USA) if not specifically stated, and were ultrapure grade. Penicillin/streptomycin was purchased from Invitrogen. FBS, PBS, RPMI 1640 and trypsin were purchased from Biosera. DXR was obtained from EBEWE Pharma. Rh123 was purchased from Sigma-Aldrich.
Cell culture and treatment
Human uterine sarcoma cell line MES-SA and its multidrug-resistant counterpart MES-SA/Dx538 obtained by continuous in vitro exposure to DXR were used in this study. Both cell lines were obtained from Sigma-Aldrich. Cells were grown in RPMI 1640 medium supplemented with 10% heat-inactivated FBS and 1% penicillin/streptomycin at 37°C and in incubators having humidified air containing 5% CO2. Resistant cells were cultured in the presence of 100 nM DXR, which was removed from the media 24 h before each experiment. HEK293 cell line (obtained from Pasteur Institute of Iran) was also used in this study. This cell line was maintained in Dulbecco’s Modified Eagle’s Medium F12 supplemented with 10% FBS, 1% l-glutamine and 1 mL penicillin/streptomycin.
A 0.05% trypsin solution was used to detach the cells from culture flasks. Test compounds and verapamil were prepared in DMSO at 40 mM stock solutions, and DXR was prepared in complete medium at 10 mM. Drugs and test compounds were then diluted with complete culture medium to obtain the final test concentrations. Maximum concentration of DMSO in wells did not exceed 0.25%.
Flow cytometric determination of Rh123 accumulation
Accumulation of intracellular Rh123, a P-gp substrate, inside resistant and nonresistant cells was measured by flow cytometry as an index of P-gp inhibition. A suspension of MES-SA/Dx5 or MES-SA cells was prepared at a density of 5×105 cells/mL in serum-free RPMI 1640. Half of the cell suspensions was placed in 1.5 mL tubes. Different concentrations of target compounds taken from prepared stock solutions in DMSO were added in a volume of 0.4 mL. After 20 min of incubation at 37°C, the P-gp substrate Rh123, was added in a volume of 100 μL at a final concentration of 5 μM. Incubation was continued for another 20 min at 37°C. Later, the cells were centrifuged and washed three times with ice-cold PBS and were resuspended in PBS for measurements. The control cells were treated in the same way without being treated with any inhibitor. A total of 2×104 cells were counted. The fluorescence uptake of Rh123 within a number of cells was determined by an FACSCalibur flow cytometer (Becton Dickinson, Franklin Lakes, NJ, USA). The results are expressed as Rh123 accumulation fold increase, which was defined as geometric mean value of Rh123 fluorescence in the cells treated with the synthesized compounds or verapamil relative to the geometric mean value of the control cells.
Determination of alteration of resistant cells’ sensitivity to DXR by MTT assay
The assessment of DXR’s cytotoxicity was performed on exponentially growing resistant MES-SA/Dx5 cells. Cells were seeded in 96-well plates at a density of 5×104 cells/mL in a final volume of 100 μL for 24 h. They were then treated with 1, 3 or 10 μM DXR in the presence or absence of synthesized compounds at different concentrations (5, 10 or 25 μM) for 90 min. After incubation for 48 h at 37°C, supernatants were carefully discarded, and 80 μL of MTT reagent (0.5 mg/mL) was added to each well, allowing viable cells to reduce the MTT solution to formazan crystals. After 4 h, the supernatants were discarded, and 200 μL DMSO was added to dissolve the formazan crystals. The absorbance of the solution was then read by a microplate reader at a wavelength of 570 nm with background correction at 675 nm. The final results are expressed as percent reduction of DXR’s IC50, which is calculated based on DXR’s IC50 in the absence and presence of synthesized compounds.
Assessment of cytotoxicity against HEK293 cells
HEK293 cells were seeded in 96-well culture plates at a density of 2,000 cells/well and allowed to attach. After 24 h incubation at 37°C in 5% CO2, the culture medium was removed and replaced with fresh medium containing the target compounds at different concentrations and further incubated at 37 CC for 48 h. Afterward, 80 μL of MTT solution (0.5 mg/mL) was added to all wells and incubated for 4 h. The medium was then removed, and 200 μL DMSO was added into each well to dissolve formazan crystals. After another hour of incubation, the mixture was gently shaken for 30 min. The absorbance was measured using a microplate reader at the wavelength of 570 nm. Each concentration was tested in duplicate, and the experiments were repeated three times.
MD simulation study of 5c in complex with human P-gp
In order to better understand the mode of interaction of synthesized compounds with P-gp receptor, MD simulation was applied to the complex of ligand and receptor. We precisely constructed the homology model of P-gp based on the murine P-gp (PDB entry: 3G5U)39 using Clustal Omega program from its website http://www.ebi.ac.uk/Tools/msa/clustalo/. One thousand 3D models of P-gp were generated, using MODELLER 9v2 program,40 and they were evaluated using GA341 method. The models with lowest DOPE score values were selected for further computational studies. The loops were remodeled using the quantum mechanical method in the MODELLER software. QMEAN Z-score from the website http://swissmodel.expasy.org/qmean/cgi/index.cgi41 was employed for checking the stereochemical quality of the selected models.
Afterward, the model was inserted in the POPC phospholipids bilayer and subjected to 80 ns MD simulation to investigate the stability as well as structural and conformational changes during simulation. In this work, one of the most potent inhibitors among all the synthesized ligands was chosen and placed in the active site of the P-gp. Ligand–receptor complex was then inserted in POPC lipid bilayer, and 20 ns MD simulation was performed. Initial configuration of the ligand was acquired via docking procedure. At first, ligand was sketched using MarvinSketch, and then the structure was optimized by density functional theory, B3LYP/3-21 level using ORCA software. Optimized structure of the ligand was then docked to P-gp model, which was obtained previously. The molecular docking was conducted using AutoDock Vina.42,43 The grid box was set at 18×18×18 with a spacing value of 1 Å. The best conformation with the least binding energy and better interacting residues was selected. Full general amber force field44 topology/coordinate files were created using the programs Parmchk and Tleap of the AmberTools package45 to describe van der Waals and bonded parameters for the ligand. Partial atomic charges were then assigned based on the RESP ESP charge Derive Server.46 The AMBER format files of the ligands were converted to the Gromacs format using the ACPYPE python tool.47 The most favorite pose from the docking study was selected as the starting structure for MD simulation.
Statistical analysis
The data, expressed as the mean ± SEM, were analyzed by one-way ANOVA using the SPSS software version 14.0 for Windows. Differences with P<0.05 were considered statistically significant.
Conclusion
Twenty-six novel 5-oxo-hexahydroquinoline derivatives possessing variable activities against MDR cell line MES-SA/Dx5 were designed and synthesized. The MDR reversal profile was evaluated using Rh123 as substrate and verapamil as a reference drug. The most potent compounds were subjected to further evaluation by measurement of MES-SA-Dx5 cells’ sensitivity to DXR by MTT assay. Compounds with 2-nitrophenyl moiety such as 2c, 5c and 6c showed good activity profiles in both tests and proved to be the most interesting molecules for further investigations. The effect of test compounds against HEK293 cell line was investigated at 5, 10 and 25 μM, and no significant cytotoxicity was observed except for compound 2c. In order to understand the structural requirement of 5-oxo-hexahydroquinoline derivatives’ interactions with P-gp, an MD simulation was performed, and the binding interactions of compound 5c in a homology-modeled human P-gp were investigated. The results indicated that stabilization of 5c occurs through different hydrogen bonds and also hydrophobic and arene–π interactions.
Abbreviations
3D, three-dimensional; ABC, ATP-binding cassette; DHPs, dihydropyridines; DMSO, dimethyl sulfoxide; DXR, doxorubicin; FBS, fetal bovine serum; IR, infrared; MD, molecular dynamics; MDR, multidrug resistance; NMR, nuclear magnetic resonance; P-gp, P-glycoprotein; PBS, phosphate-buffered saline; Rh123, rhodamine 123; RPMI, Rosewell Park Memorial Institute; SAR, structure–activity relationship.
Acknowledgment
The authors thank the support of the Vice-Chancellor for Research of Shiraz University of Medical Sciences (grant number: 93-7237). This study was part of the PhD thesis of Omolbanin Shahraki.
Disclosure
The authors report no conflicts of interest in this work.
References
Chang A. Chemotherapy, chemoresistance and the changing treatment landscape for NSCLC. Lung Cancer. 2011;71(1):3–10. | ||
Gottesman MM, Fojo T, Bates SE. Multidrug resistance in cancer: role of ATP-dependent transporters. Nat Rev Cancer. 2002;2(1):48–58. | ||
Juliano RL, Ling V. A surface glycoprotein modulating drug permeability in Chinese hamster ovary cell mutants. Biochim Biophys Acta. 1976;455(1):152–162. | ||
Teodori E, Dei S, Martelli C, Scapecchi S, Gualtieri F. The functions and structure of ABC transporters: implications for the design of new inhibitors of Pgp and MRP1 to control multidrug resistance (MDR). Curr Drug Targets. 2006;7(7):893–909. | ||
Schneider E, Hunke S. ATP-binding-cassette (ABC) transport systems: functional and structural aspects of the ATP-hydrolyzing subunits/domains. FEMS Microbiol Rev. 1998;22(1):1–20. | ||
Yuan H, Li X, Wu J, et al. Strategies to overcome or circumvent P-glycoprotein mediated multidrug resistance. Curr Med Chem. 2008;15(5):470–476. | ||
Ferry DR, Traunecker H, Kerr DJ. Clinical trials of P-glycoprotein reversal in solid tumours. Eur J Cancer. 1996;32A(6):1070–1081. | ||
Lhommé C, Joly F, Walker JL, et al. Phase III study of valspodar (PSC 833) combined with paclitaxel and carboplatin compared with paclitaxel and carboplatin alone in patients with stage IV or suboptimally debulked stage III epithelial ovarian cancer or primary peritoneal cancer. J Clin Oncol. 2008;26(16):2674–2682. | ||
Kuppens IE, Witteveen EO, Jewell RC, et al. A phase I, randomized, open-label, parallel-cohort, dose-finding study of elacridar (GF120918) and oral topotecan in cancer patients. Clin Cancer Res. 2007;13(11):3276–3285. | ||
Robey RW, Shukla S, Finley EM, et al. Inhibition of P-glycoprotein (ABCB1)- and multidrug resistance-associated protein 1 (ABCC1)-mediated transport by the orally administered inhibitor, CBT-1®. Biochem Pharmacol. 2008;75(6):1302–1312. | ||
Ross DD. Modulation of drug resistance transporters as a strategy for treating myelodysplastic syndrome. Best Pract Res Clin Haematol. 2004;17(4):641–651. | ||
Ruff P, Vorobiof DA, Jordaan JP, et al. A randomized, placebo-controlled, double-blind phase 2 study of docetaxel compared to docetaxel plus zosuquidar (LY335979) in women with metastatic or locally recurrent breast cancer who have received one prior chemotherapy regimen. Cancer Chemother Pharmacol. 2009;64(4):763–768. | ||
Cornwell MM, Pastan I, Gottesman MM. Certain calcium channel blockers bind specifically to multidrug-resistant human KB carcinoma membrane vesicles and inhibit drug binding to P-glycoprotein. J Biol Chem. 1987;262(5):2166–2170. | ||
Safa AR, Glover CJ, Sewell JL, Meyers MB, Biedler JL, Felsted RL. Identification of the multidrug resistance-related membrane glycoprotein as an acceptor for calcium channel blockers. J Biol Chem. 1987;262(16):7884–7888. | ||
Nogae I, Kohno K, Kikuchi J, et al. Analysis of structural features of dihydropyridine analogs needed to reverse multidrug resistance and to inhibit photoaffinity labeling of P-glycoprotein. Biochem Pharmacol. 1989;38(3):519–527. | ||
Khoshneviszadeh M, Edraki N, Javidnia K, et al. Synthesis and biological evaluation of some new 1,4-dihydropyridines containing different ester substitute and diethyl carbamoyl group as anti-tubercular agents. Bioorg Med Chem. 2009;17(4):1579–1586. | ||
Razzaghi-Asl N, Firuzi O, Hemmateenejad B, Javidnia K, Edraki N, Miri R. Design and synthesis of novel 3,5-bis-N-(aryl/heteroaryl) carbamoyl-4-aryl-1,4-dihydropyridines as small molecule BACE-1 inhibitors. Bioorg Med Chem. 2013;21(22):6893–6909. | ||
Fassihi A, Azadpour Z, Delbari N, et al. Synthesis and antitubercular activity of novel 4-substituted imidazolyl-2,6-dimethyl-N3,N5-bisaryl-1,4-dihydropyridine-3,5-dicarboxamides. Eur J Med Chem. 2009;44(8):3253–3258. | ||
Sirisha K, Bikshapathi D, Achaiah G, Reddy VM. Synthesis, antibacterial and antimycobacterial activities of some new 4-aryl/heteroaryl-2,6-dimethyl-3,5-bis-N-(aryl)-carbamoyl-1,4-dihydropyridines. Eur J Med Chem. 2011;46(5):1564–1571. | ||
Miri R, Javidnia K, Mirkhani H, et al. Synthesis, QSAR and calcium channel modulator activity of new hexahydroquinoline derivatives containing nitroimidazole. Chem Biol Drug Des. 2007;70(4):329–336. | ||
Miri R, Javidnia K, Sarkarzadeh H, Hemmateenejad B. Synthesis, study of 3D structures, and pharmacological activities of lipophilic nitroimidazolyl-1,4-dihydropyridines as calcium channel antagonist. Bioorg Med Chem. 2006;14(14):4842–4849. | ||
Navidpour L, Shafaroodi H, Miri R, Dehpour AR, Shafiee A. Lipophilic 4-imidazoly-1,4-dihydropyridines: synthesis, calcium channel antagonist activity and protection against pentylenetetrazole-induced seizure. Farmaco. 2004;59(4):261–269. | ||
McKay LI, Cidlowski JA. Molecular control of immune/inflammatory responses: interactions between nuclear factor-κB and steroid receptor-signaling pathways. Endocr Rev. 1999;20(4):435–459. | ||
Miri R, Mehdipour A. Dihydropyridines and atypical MDR: a novel perspective of designing general reversal agents for both typical and atypical MDR. Bioorg Med Chem. 2008;16(18):8329–8334. | ||
Baumert C, Günthel M, Krawczyk S, et al. Development of small-molecule P-gp inhibitors of the N-benzyl 1,4-dihydropyridine type: novel aspects in SAR and bioanalytical evaluation of multidrug resistance (MDR) reversal properties. Bioorg Med Chem. 2013;21(1):166–177. | ||
Shekari F, Sadeghpour H, Javidnia K, et al. Cytotoxic and multidrug resistance reversal activities of novel 1,4-dihydropyridines against human cancer cells. Eur J Pharmacol. 2015;746:233–244. | ||
Firuzi O, Javidnia K, Mansourabadi E, Saso L, Mehdipour AR, Miri R. Reversal of multidrug resistance in cancer cells by novel asymmetrical 1,4-dihydropyridines. Arch Pharm Res. 2013;36(11):1392–1402. | ||
Chen C, Zhou J, Ji C. Quercetin: a potential drug to reverse multidrug resistance. Life Sci. 2010;87(11–12):333–338. | ||
Critchfield JW, Welsh CJ, Phang JM, Yeh GC. Modulation of adriamycin® accumulation and efflux by flavonoids in HCT-15 colon cells: activation of P-glycoprotein as a putative mechanism. Biochem Pharmacol. 1994;48(7):1437–1445. | ||
Zhang S, Yang X, Morris ME. Flavonoids are inhibitors of breast cancer resistance protein (ABCG2)-mediated transport. Mol Pharmacol. 2004;65(5):1208–1216. | ||
Lee JS, Paull K, Alvarez M, et al. Rhodamine efflux patterns predict P-glycoprotein substrates in the National Cancer Institute drug screen. Mol Pharmacol. 1994;46(4):627–638. | ||
Bruggemann EP, Germann UA, Gottesman MM, Pastan I. Two different regions of P-glycoprotein [corrected] are photoaffinity-labeled by azidopine. J Biol Chem. 1989;264(26):15483–15488. | ||
Ferry DR, Russell MA, Cullen MH. P-glycoprotein possesses a 1,4-dihydropyridine-selective drug acceptor site which is alloserically coupled to a vinca-alkaloid-selective binding site. Biochem Biophys Res Commun. 1992;188(1):440–445. | ||
Borchers C, Boer R, Klemm K, et al. Characterization of the dexniguldipine binding site in the multidrug resistance-related transport protein P-glycoprotein by photoaffinity labeling and mass spectrometry. Mol Pharmacol. 2002;61(6):1366–1376. | ||
Daura X, Gademann K, Jaun B, Seebach D, van Gunsteren WF, Mark AE. Peptide folding: when simulation meets experiment. Angew Chem Int Ed Engl. 1999;38(1–2):236–240. | ||
Salentin S, Schreiber S, Haupt VJ, Adasme MF, Schroeder M. PLIP: fully automated protein–ligand interaction profiler. Nucleic Acids Res. 2015;43(W1):W443–W447. | ||
Clemens RJ, Hyatt JA. Acetoacetylation with 2,2,6-trimethyl-4H-1,3-dioxin-4-one: a convenient alternative to diketene. J Org Chem. 1985;50(14):2431–2435. | ||
Harker WG, Sikic BI. Multidrug (pleiotropic) resistance in doxorubicin-selected variants of the human sarcoma cell line MES-SA. Cancer Res. 1985;45(9):4091–4096. | ||
Aller SG, Yu J, Ward A, et al. Structure of P-glycoprotein reveals a molecular basis for poly-specific drug binding. Science. 2009;323(5922):1718–1722. | ||
Eswar N, John B, Mirkovic N, et al. Tools for comparative protein structure modeling and analysis. Nucleic Acids Res. 2003;31(13):3375–3380. | ||
Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr. 1993;26(2):283–291. | ||
Morris GM, Huey R, Lindstrom W, et al. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J Comput Chem. 2009;30(16):2785–2791. | ||
Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31(2):455–461. | ||
Wang J, Wolf RM, Caldwell JW, Kollman PA, Case DA. Development and testing of a general amber force field. J Comput Chem. 2004;25(9):1157–1174. | ||
AMBER [computer program]. Version 2015. San Francisco: University of California; 2015. | ||
Vanquelef E, Simon S, Marquant G, et al. RED Server: a web service for deriving RESP and ESP charges and building force field libraries for new molecules and molecular fragments. Nucleic Acids Res. 2011;39(Web Server issue):W511–W517. | ||
Sousa da Silva AW, Vranken WF. ACPYPE-AnteChamber PYthon Parser interfacE. BMC Res Notes. 2012;5(1):367. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.