")
Back to Archived Journals » Orphan Drugs: Research and Reviews » Volume 7
Nitisinone: a review
Authors Aktuglu-Zeybek AC, Zubarioglu T
Received 27 May 2016
Accepted for publication 3 November 2016
Published 31 January 2017 Volume 2017:7 Pages 25—35
DOI https://doi.org/10.2147/ODRR.S92995
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Lise Aagaard
A Cigdem Aktuglu-Zeybek, Tanyel Zubarioglu
Department of Pediatrics, Division of Nutrition and Metabolism, Cerrahpasa Medical Faculty, Istanbul University, Kocamustafapasa Fatih, Istanbul, Turkey
Abstract: Nitisinone (2-[2-nitro-4-trifluoromethylbenzoyl]cyclohexane-1,3-dione), an effective triketone herbicide, is a potent inhibitor of 4-hydroxyphenylpyruvate dioxygenase, the second enzyme in the tyrosine catabolic pathway. Since 1992, the drug has become an effective pharmacological treatment for hereditary tyrosinemia type 1 (HT1). Nitisinone can prevent the development of liver disease, reverse and prevent the renal tubular dysfunction, severe neurological crisis and cardiomyopathy and significantly reduce the risk of developing hepatocellular carcinoma in HT1 patients. Its mode of action, with few side effects reported, make the drug a potential candidate for the treatment of other disorders of tyrosine metabolism, including alkaptonuria, with successful reduction in homogentisic acid production to prevent long-term complications. Nitisinone could also be a promising agent in the treatment of tumors with active tyrosine metabolic pathways. In this review, we discuss the effects of nitisinone for various tyrosine pathway disorders.
Keywords: nitisinone, hereditary tyrosinemia type 1, alkaptonuria, tyrosine
Introduction
The development of nitisinone (2-[2-nitro-4-trifluoromethylbenzoyl]cyclohexane-1,3-dione, NTBC) has dramatically changed the natural course of hereditary tyrosinemia type 1 (HT1). Discovery of its mode of action, with a few side effects reported, resulted in clinical use of this drug in treating various clinical disorders in the tyrosine (Tyr) pathway. In this review, we focus on the mode of action and clinical effects of nitisinone for various Tyr pathway disorders.
The history of nitisinone
Nitisinone is a triketone herbicide derived from leptospermone produced by the bottlebrush plant.1 It was first discovered by Zeneca Agrochemicals in 1982.1 The members of this family occur naturally in plant oils. Treating plants with triketone herbicides causes bleaching symptomatology due to reduced chlorophyll and carotenoid synthesis and prevents the growth of competing plants around.2 Nitisinone, despite belonging to this wide family, revealed a novel mechanism of action by inhibiting the enzyme 4-hydroxyphenyl pyruvate dioxygenase (4-HPPD) which catalyzes the conversion of 4-hydroxyphenyl pyruvate (4-HPP) to homogentisic acid (HGA) as part of the Tyr degradation pathway (Figure 1).3–5 The same mechanism was also confirmed in animal toxicology studies in the rat.6 Inhibition of 4-HPPD led to a profound increase in plasma Tyr levels in rats and corneal lesions similar to those detected in tyrosinemia type-2 patients.6 This discovery led to the hypothesis that the drug could decrease the production of toxic metabolites (fumarylacetoacetate [FAA], maloylacetoacetate [MAA] and succinylacetone [SA]) seen in HT1 by effective blockage of the Tyr catabolic pathway. In 1992, Lindstedt et al presented a new principle for treatment of HT1 based on the inhibition of 4-HPPD with nitisinone.7 The first patient showed a dramatic response to treatment with an immediate biochemical response. SA excretion was reduced with a dose of 0.05 mg/kg/day of nitisinone as early as 12 hours after the initial dose and became undetectable as the dose was increased.8 Upon marked improvement in clinical conditions and biochemical response, four more patients were also treated with nitisinone with similar response during 7–9 months of follow-up.7 Finally, the Gothenburg group9 coordinated an international study of nitisinone treatment in HT1.
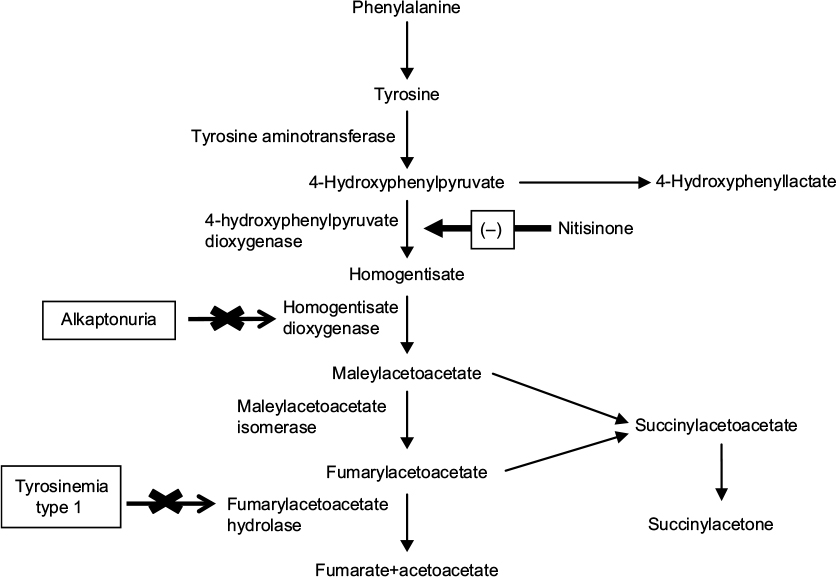 | Figure 1 Tyrosine metabolism and nitisinone. |
The drug has received approval from both the US FDA and the EU Drug Agency in January 2002 and February 2005, respectively, without formal phase II, III, and IV clinical trials,9–11 and is marketed as Orfadin® (Swedish Orphan Biovitrum, Stockholm, Sweden).
Pharmacological properties and mechanism of action
The structure of nitisinone has some similarities to 4-HPP which is a substrate for 4-HPPD. The drug is a potent time-dependent (tight-binding) reversible inhibitor of mouse, rat, and human liver 4-HPPD in vitro.3–6 4-HPPD is a non-heme Fe(II) enzyme that catalyzes the second step in the Tyr degradation pathway, involving the conversion of 4-HPP to HGA (Figure 1).5 The binding of nitisinone to HPPD‚ Fe(II), was shown to occur in two phases and comprise three steps: a reversible binding proximal to the active site of metal ion, bidentate association with the Fe+2 center and an irreversible deprotonation of the enol.4 Hanauske-Abel et al also suggested a tetradentate association, obtained from crystallographic and computational data on nitisinone binding.12
Studies on the rat liver cytosol extracts by Ellis and Laschi revealed an effective time-dependent reversible inhibition of liver HPPD with median inhibitory concentrations (IC50) of 40 and 173 nM and a very slow dissociation of the NTBC–enzyme complex.6,13 A single oral dose of 0.1 mg/kg nitisinone produced complete inhibition of hepatic HPPD within 1 hour of administration and only 40%–50% of the enzyme returned normal 4 days after dosing. Thus, the interaction of nitisinone with HPPD is characterized by a rapid inhibition step to form an enzyme–inhibitor complex that can dissociate slowly with recovery of enzyme activity.12 Analysis of the key hepatic enzymes involved in Tyr catabolism in rodents showed despite inhibition of HPPD soon after dosing, the activity of tyrosine aminotransferase (TAT) in the liver was induced by about twofold presumably due to enzyme induction and homogentisic acid oxidase (HGO) was not affected.14 There is no information about the effects of nitisinone on renal HPPD or the kinetics of HPPD inhibition in human liver or kidney tissue.
The HPPD affinity was higher in humans than in rats with an IC50 at 5 nM on purified human enzyme.2 In 10 healthy male volunteers aged 19–39 years, peak plasma concentrations were achieved rapidly following a single oral dose of 1 mg/kg body weight (BW) with a half-life of approximately 54 hours.15 Pharmacokinetic studies in 9 infants with HT1 over a period of up to 24 hours also revealed that the administration of a single daily dose results in therapeutic drug levels and suppression of SA formation.16 Nitisinone was rapidly and completely absorbed after oral administration and had a terminal half-life of about 9 hours in rats.17 Absorption kinetics were also studied in either the liquid or capsule form in healthy volunteers, and the median time to reach the maximum plasma concentration was 15 minutes for the liquid and 3 hours for the capsule form.15 The capsule forms should be refrigerated at a temperature range of 2°C–8°C because of heat instability, while the liquid forms should be stored and refrigerated between 2°C and 8°C prior to first use and at room temperature after first opening. For patients who have difficulty swallowing the capsules and for whom oral suspension is not suitable, opening the capsules and suspending the contents in a small amount of water, formula, or apple sauce immediately before use are recommended.17 There are no pharmacokinetic studies on the effect of food on absorption nor on plasma protein binding.
Following oral administration of radiolabeled nitisinone in rats, the bioavailability was found to be high (>90%) with multiple organ distribution.18 Administration of a single oral dose of [14C]-nitisinone in rats (0.3 µmol/kg [0.1 mg/kg] or 30 µmol/kg [10 mg/kg]) and in mice (30 µmol/kg [10 mg/kg]) led to selective retention of radiolabeled nitisinone in the liver (>90% associated reversibly with the cytosol fraction), in kidneys, and to a lesser extent in the Harderian gland.13,18 Radioactivity was detected up to 7 days in these organs.
The route of elimination of nitisinone has been investigated in both animals and man.3,15,18–20 In an autoradiographic study in rats, nitisinone and two hydroxylated metabolites were found to be excreted in the urine.19 Animal and human studies indicate that nitisinone is excreted through both the feces and the urine at approximately equal levels.19,20 The tentative identification of hydroxylated metabolites and 2-nitro-4-trifluoromethylbenzoic acid in urine points to the involvement of the cytochrome P450 (CYP) system in the metabolism of nitisinone.21 Investigations on the potential for drug–drug interactions between nitisinone and other medicines that induce or inhibit CYP activity have been studied in vitro on human microsomes.21 In summary, with the exception of CYP2C9 inhibition, nitisinone was found to have little effect on human CYP enzymes as either an inhibitor or an inducer.
The inhibition of HPPD leads to a marked increase in plasma Tyr. Administration of a single dose of nitisinone in rats resulted in a marked increase in Tyr level in both plasma (about 2,500 nmol/mL) and ocular fluids (3,500–4,000 nmol/mL).14,18 Tyrosinemia was both time and dose dependent with a maximum Tyr level at 24 hours after dosing. Plasma Tyr level returned to normal level by 48 hours, indicating that only a small recovery of the enzyme activity was able to clear Tyr from plasma.3,14,18 On the other hand, it took 3 days to normalize for the larger increase of Tyr in aqueous humor of the eye.3 Human fibroblast cultures treated with nitisinone also revealed clearly that Tyr accumulation during administration of nitisinone appears to be dose-dependent.13
Tyr accumulation in ocular fluids can lead to eye lesions both in human and in some animals.14,18,22 Administration of a dose of 10 mg/kg/day to mice for 6 weeks increased plasma Tyr level to 650 nmol/L and to 2,300 nmol/L in aqueous humor without any eye lesions.14 This finding was thought to be due to higher activity of TAT in mouse liver which is the first enzyme of the Tyr degradation pathway, which can clear Tyr as hydroxyphenyllactic acid and HPP. Also, there is a threshold plasma Tyr level, above which it leads to eye lesions.14,18,22
The first report of nitisinone lethal dose 50% (LD50) values on human primary fibroblasts following both acute (24 hour) and chronic (144 hour) treatment revealed that the short-term LD50 was considered to be >1 μM, while long-term LD50 was about 1 μM.13 A long-term LD50 value lower than the short-term LD50 was reported to support the hypothesis that the toxicity of nitisinone was linked to its mode of action.13,22–25
Nitisinone and hereditary tyrosinemia type 1
Phenylalanine and Tyr, from the diet and endogenous protein turnover in vivo, are used to synthesize new proteins, hormones (eg, thyroxine and catecholamines) and melanin. The majority of Tyr is metabolized to malate, acetoacetate, and products that enter the intermediary metabolism (Figure 1).
HT1 is an autosomal recessive inborn error of Tyr metabolism caused by deficiency of the last enzyme in the Tyr degradation pathway, “FAH”. Reduced activity of FAH leads to the accumulation of upstream metabolites such as FAA and MAA, which in turn are converted to SA.26 Toxic metabolites FAA, MAA, SA and succinylacetoacetate (SAA) are highly reactive electrophilic toxic compounds and bind to sulfhydryl groups. These toxic metabolites are thought to be responsible for progressive hepatic, renal, and neurological findings.26–29 The human FAH complementary DNA has been mapped to chromosome 15q, and various mutations have been identified in the human FAH gene.26,30–34 FAH is mainly expressed in liver and kidney cells, although a basal level of expression has been detected in many tissues.26 No clinical correlation has been detected between genotype and phenotype.26,30–34 The clinical manifestations of HT1 are heterogeneous even within the members of the same family. HT1 can be classified based on the age at symptom onset: acute HT1 (<6 months), subacute HT1 (6–12 months), chronic HT1 (>12 months).35,36 Acute or chronic liver disease, renal dysfunction with Fanconi syndrome and hypophosphatemic rickets, polyneuropathy, and abdominal pain resembling acute porphyria are the main clinical findings.26,32,34–37 Cardiomyopathy and hyperinsulinism have also been described.38–40 Most patients present with the acute and subacute form of the disease characterized by clinical findings of acute–subacute liver failure with poor growth, vomiting, ascites, coagulopathy, hypoglycemia, hypoalbuminemia, and hyperbilirubinemia. Patients with the chronic form have growth failure; renal tubular dysfunction including Fanconi syndrome; and neurologic crises with pain and paresthesias associated with evidence of liver dysfunction, including hepatomegaly, transaminasemia, hyperbilirubinemia, and coagulopathy.8,29,35–37,41 An odor resembling boiled cabbage may be present due to increased methionine metabolites.36,37
The natural course of the disease usually results in death.35 The causes of death are primarily liver failure, hepatocellular carcinoma (HCC) and respiratory failure from neurological crisis.32 The prognosis gets poorer if the presentation is earlier.8,35,41,42 The leading causes of death are fulminant liver failure, porphyria-like neurological crises, and metastatic HCC, especially if left untreated.35 Micro-macronodular cirrhosis and HCC are the long-term complications. HT1 is the inborn error of metabolism with the highest incidence of progression to HCC due to mutagenic effects of accumulated toxic metabolites.26,35
Liver transplantation (LT) was the only choice of treatment and a life-saving procedure, although renal dysfunction remained, as low Tyr diet was not effective in the acute form and had limited success in the chronic form.8,35 Still, LT carries its own risks with a mortality of 10%–15% and side effects associated with direct and indirect costs.43 The introduction of nitisinone in 1992 has radically improved the natural course of the disease. The drug was found to be effective not only in preventing the acute life-threatening complications of the disease (eg, acute hepatic failure and neurological crises) but also in reducing the need for LT, improving the survival and the quality of life in HT1 patients, especially when started in the newborn period.8,33,44 The starting dose is 1–2 mg/kg BW per day.44 The initial dose can be titrated down by monitoring nitisinone levels without risking metabolic control as judged from plasma SA levels and SA excretion.45,46 A few anecdotal patients were reported who did well under doses lower than 1 mg/kg/day. El-Karaksy et al described 4 patients and D’Eufemia et al described one patient for whom doses of 0.55–0.65 mg/kg/day were sufficient to prevent liver dysfunction.47,48
NTBC should be quantified in plasma and blood spot samples to identify the optimal dose for an individual. A column liquid chromatographic method, a liquid chromatographic–tandem mass spectrometry (LC–MS/MS); a capillary electrophoretic method with photometric detection and a high-performance liquid chromatographic–tandem mass spectrometric method have been applied for the detection and quantitative determination of nitisinone in serum, plasma and dried blood spot samples.49–54 Although the mean plasma nitisinone concentration which is theoretically sufficient to produce a 99.9% inhibition of HPPD based on in vitro studies has been determined to be 35 μmol/L, it is quite common that some HT1 patients require higher plasma levels (<70 μmol/L) to achieve complete elimination of urinary SA excretion.8,11 As plasma SA is protein bound, plasma SA level and δ-aminolevulinic acid (ALA) dehydratase inhibition take longer to normalize.8 Different plasma nitisinone levels are proposed to keep urinary SA below the limit of detection. Schlune et al proposed plasma nitisinone levels of 45–50 μmol/L, Herebian et al proposed plasma nitisinone levels of 50–150 μmol/L and Prieto et al proposed plasma nitisinone levels of 15–40 μmol/L.16,49,50
Administration of nitisinone in two divided daily doses is recommended but administration of a single daily dose which is found to be well tolerated and safe, potentially improving compliance with therapy, also results in therapeutic plasma drug levels and effective suppression of SA formation.8,16,45
Effects of nitisinone on the hepatic manifestations of HT1
Infants presenting with acute hepatic dysfunction can rapidly develop severe liver dysfunction with a high risk of mortality.9,35 Nitisinone administration results in a remarkable clinical response, where 90% of the patients respond completely.54,55 SA and δ-ALA decrease within the hours after the first dose of nitisinone.7,8 The majority of infants who present clinically already have established cirrhosis, and treatment could convert them to a stable chronic liver disease.9 In most cases, liver function and coagulopathy improve within 1 week of treatment.7,8,56,57 In those who are unresponsive, the nitisinone dosage should be increased and in those who are unresponsive even after higher doses, LT is indicated.9,45,57 No new acute hepatic exacerbation has been reported after nitisinone initiation.23
Children with chronic tyrosinemia and those with acute/subacute tyrosinemia treated with nitisinone have generally established chronic liver disease and nodular cirrhosis. Nitisinone slows down the progression of chronic hepatic disease.9,11,33 Even radiological regression of liver nodules has also been reported.58 Nitisinone also improves growth, liver function, and portal hypertension in HT1 patients with chronic liver disease.57 The effect of nitisinone on the hepatic findings depends on the drug use on a regular basis. FAH-/- mouse model studies revealed that nitisinone treatment prevented lethal liver dysfunction when started in the neonatal period but discontinuation of this treatment led to disruption of hepatic architecture depending on the length of time.20
The development of HCC is the most important long-term complication of HT1. Although improvement in liver function and portal hypertension can be achieved, nitisinone treatment cannot completely alter abnormal gene expression in the liver and collagen metabolism nor can it reverse established hepatic lesions such as oncogenic mutations, hepatic dysplasia, and fibrosis.12,59–62 Late-treated HT1 patients can develop cirrhosis and HCC with progression in nodules.9,33,56,63,64 Development of HCC mainly affects patients with chronic form or who have been treated after 2 years of age, in whom malign transformation at the microscopic level is thought to have started even by the time nitisinone is started.56,63–65 These patients usually have a slow decrease in α-fetoprotein (AFP) levels without reaching normal levels despite treatment or persistent high levels of AFP.66 Despite these findings, nitisinone decreases the risk of HCC compared to those before nitisinone treatment, as HCC incidence was reported to be 18%–37% among children who have survived past their second birthday.35,67 Early treatment has been shown to decrease the risk of HCC.44,46,56 The earlier the nitisinone treatment is commenced, the lower the risk a patient should develop HCC as the exposure of the liver to the mutagenic effects of FAA and MA is lowered.33 Medium-term follow-up results of the effect of nitisinone treatment on clinical course of HT1 revealed that none of the patients treated from the neonatal period developed detectable liver disease.44 In sharp contrast, all late-treated HT1 patients had chronic liver abnormalities before receiving nitisinone and developed cirrhosis, and some developed HCC.23,44,63,68 Larochelle et al reported that LT was performed in 71% of non-nitisinone treated patients and 26% of late-treated patients. The clinical indications include cirrhosis or cancer, acute hepatic failure, and previous neurologic crisis.33 None of the early-treated patients required LT.33,44 Today, indications for LT are limited to failure to respond to nitisinone or HCC.45,46
Extrahepatic manifestations of HT1
Variable degree of renal dysfunction is detectable in most of the HT1 patients at presentation, ranging from mild tubular dysfunction to chronic renal disease.26 As a result of mutagenic effects of FAA, MAA, and SA on intracellular metabolism of hepatic cells and renal cells, these cells undergo either death of apoptotic cell or adaptive alteration of gene expression.26 Animal models suggest that while FAA is responsible for apoptosis, glomerulosclerosis and interstitial fibrosis, SA is responsible for renal tubular dysfunction.69,70 Renal disease manifests with abnormal renal architecture (size and ultrasonographic echogenicity), tubular disorder (all or some components of Fanconi syndrome: aminoaciduria, phosphaturia, and renal tubular acidosis) and frank nephrocalcinosis.26,46,71 Renal tubular function shows a rapid improvement after initiation of nitisinone therapy and returns to normalcy.64,71–73 Maiorana et al revealed that almost all of the biomarkers of renal tubular dysfunction (including phosphaturia, glycosuria, and urinary β-2 microglobulin) normalized within 2 weeks in all HT1-affected children treated with nitisinone.73 No child redeveloped tubular dysfunction at long-term follow-up after commencing nitisinone therapy.33,64,71,72 On the contrary, long-term outcome in French patients showed that some tubular dysfunction remained in some patients despite nitisinone therapy, although no glomerular filtration abnormality was observed.23 This finding was considered to be influenced by the age at the start of nitisinone therapy as tubular function remains normal in those treated prospectively from infancy.23,33,46 Early nitisinone treatment not only reduces the need for LT but also improves post-transplant renal function.73–75 Glomerular function seems to remain stable on long-term follow-up under nitisinone therapy 74,75
Porphyria-like neurologic crises (PLNCs) and recurrent peripheral neuropathy have been reported in HT1 patients.35,36,76,77 PLNCs are among the major causes of death in untreated HT1 patients.35 These crises include change in mental status, abdominal pain, peripheral neuropathy, and/or respiratory failure requiring mechanical ventilation, which last 1–7 days.76,77 PLNC and neural damage in HT1 patients are caused by inhibition of heme synthesis by SA. SA is the most potent inhibitor of the enzyme 5-ALA dehydratase known, resulting in the increased production of 5-ALA.77 Normalization of SA in blood and correction of the complete inhibition of porphobilinogen synthase (PBGS) in erythrocytes during nitisinone treatment protect from the porphyric crisis.7 PLNCs also respond dramatically to nitisinone therapy.68 Interruption of the treatment can cause relapsing of severe crises that can lead to death in patients with HT1.64,78,79
Cardiomyopathy is a rare complication of HT1 and usually reported at the initial presentation as hypertrophic cardiomyopathy involving hypertrophy of the interventricular septum.39,40 The pathogenesis is poorly understood and is thought to be related to the accumulation of toxic FAA in the cardiac tissues or to the elevated Tyr levels.40 Cardiomyopathy resolves in the patients treated with nitisinone.39,40,80
Nitisinone and alkaptonuria
Alkaptonuria (AKU; OMIM 203500) is a rare autosomal recessive disease caused by a deficiency of homogentisate 1,2-dioxygenase (HGD) that converts intermediate HGA to MAA in the Tyr metabolism pathway (Figure 1). The single-copy human HGD gene maps to chromosome 3q13.33, encompassing 14 exons and encoding a 445-mer protein.81 AKU arises from homozygous or compound heterozygous loss-of-function mutations in the HGD gene, with 115 different human mutations currently identified.81,82 There appears to be no correlation, currently, between the type of mutation and severity of the phenotype.83
Inability to metabolize HGA increases both the circulating concentration and urinary excretion of HGA, with at least 90% of the compound.24,84 Despite increased urinary excretion, it is not sufficient to completely remove it from bodily fluids and tissues. HGA in urine polymerizes to benzoquinone acetic acid which forms a melanin-like polymeric pigment that leads to a characteristic early sign of the disorder: darkening of the urine on standing.85,86 The melanin-like polymeric pigment in vivo also deposits in and binds to connective tissues like cartilage, leading to pathological pigmentation and destructive consequences “ochronosis”.87–89 Ochronosis is a chronic process and clinical symptoms are typically observed from the second decade of life, including severe spondyloarthropathy, ruptures of the ligaments, tendons, and muscles; osteopenia, fractures; valvular heart disease; pigmentation of eyes and ears; central vein occlusion and elevated intraocular pressure; renal, prostatic, gallbladder, and salivary stones and renal insufficiency.83,90–98 Early degenerative ochronotic arthropathy is seen especially in weight-bearing joints, leading to severe pain in spine and large joints such as shoulder, hip, and knee.98 Symptoms worsen from the fourth decade onward with progressive kyphoscoliosis and impaired spinal and thoracic mobility. Hearing loss (40%), cardiac arrhythmias (40%), and congestive cardiac failure (10%) also occur in AKU patients.86 Acute metabolic decompensation, including oxidative hemolysis and/or methemoglobinemia, has also been reported.99,100 There is currently no licensed pharmacological treatment for AKU, except palliative treatments.
Nitisinone inhibits 4-HPPD and has been demonstrated to significantly reduce the serum concentration and urinary excretion of HGA in both mouse models and humans.13,24,25,101–104 The drop in HGA concentrations was rapid, with approximately 60% decrease in circulating HGA within 48 hours on 2 mg dose (unpublished data), and thus is an efficacious treatment.101 Six clinical trials have been undertaken on the use of nitisinone to treat AKU.24,25,86,102–104 The first study was an early dose ranging study in two older women with AKU.102 An open-label, single-center study of 9 AKU patients showed that the dose required to decrease HGA significantly was up to 30-fold lower than that used in treating HT1.86 Administering nitisinone (1.05 mg bid) daily with a regular diet decreased HGA by 95% and increased Tyr by 11-fold in 9 AKU adult patients over a 4-month period.86 In another 3-year single-blind, controlled clinical trial of 30 patients revealed that the nitisinone group (2.1 mg/day) showed a sustained decrease in mean urinary HGA from 5.1 to 0.125 g/day and mean plasma HGA from 5.74 to 0.306 mg/L after treatment.24 Urine and plasma HGA levels decreased by 98% and 95%, respectively. The increased plasma Tyr level did not cause corneal or any other toxicity. About 6 out of 7 patients who received nitisinone for more than 1 week noted decreased pain in their affected joints.
In an international, multicenter, randomized, open-label, nontreatment controlled, parallel-group, dose–response study of 40 patients, a clear dose–response relationship was observed between nitisinone and the urinary excretion of HGA.103 The most efficacious dose was 8 mg daily, compared to 1, 2, and 4 mg daily with the mean reduction of urinary HGA being 98.8% with a daily 8 mg dose.103 Olsson et al also found similar findings in 32 patients with AKU who received 1, 2, 4, or 8 mg nitisinone daily. Nitisinone decreased the urinary excretion of HGA in a concentration-dependent manner, up to serum nitisinone concentrations of about 3 μmol/L.25 This concentration was reached with a dose of 8 mg daily in seven of eight patients with that dose. These patients had a reduction in urinary HGA of at least 99.4%. On the contrary, Gertsman et al revealed a significant decrease in urine HGA level in 7 AKU patients when the nitisinone dose was increased from 2 to 4 mg/day, with no significant changes at higher doses.104
Despite the dosage to treat AKU is lower than that to treat HT1, it still causes accumulation of Tyr over the recommended threshold of 400–500 μmol/L with a large interindividual variability, and long-term monitoring of plasma Tyr levels is needed because the metabolic fate of this is largely unknown in AKU.13,24,25,102–104 It is postulated that treatment with nitisinone should be started as early as possible before renal functions decline and other potentially fatal acute complications develop.99,100
Nitisinone and neuroblastoma
Neuroblastoma, the most common solid neoplasm of childhood outside of the central nervous system, is a neural crest-derived neoplasm. The overall outcome is excellent for children with localized disease. Unfortunately, two-third of the children at the diagnosis have widespread disease with poor outcome, despite all treatment modalities.105,106 As the tumor has a highly active Tyr pathway which produces large amounts of homovalinic acid, vanilmandelic acid (VMA), metanephrines, and other catecholamines, proximal blockade of this pathway is assumed to raise intracellular Tyr, alter the distribution of intracellular catecholamines, and facilitate the differentiation or cytotoxicity of neuroblastoma cells. Interestingly, nitisinone (0.8 mg/kg/day), when used as a single agent, demonstrated antitumor activity sustained for 10 weeks in a child with recurrent stage 4 metastatic neuroblastoma.107 At recurrence, the drug induced a very good partial response when combined with low-dose cyclophosphamide and doxorubicin. This response was sustained for 18 months till he succumbed, during which he was pain-free, attended school, and had an excellent quality of life. The authors suggested that nitisinone may have a role to play in the treatment of other neural crest-derived tumors, which have highly active Tyr metabolic pathways.
Adverse effects of nitisinone
Nitisinone is a well-tolerated drug and the first adverse effect reported was the observation of development of corneal lesions in rats treated with nitisinone, which also resulted in discovery of the drug for treatment of HT1. Hypertyrosinemia is likely to be one of the causes of these deposits and can be prevented by the restriction of Tyr in the diet.3 Nitisinone biochemically switches the enzymatic defect from HT1 to tyrosinemia type 3, inducing elevated Tyr concentrations up to 1,500 mmol/L (normal, 40–90 mmol/L) when dietary treatment is not provided.9 Transient ocular symptoms such as irritation, corneal erosion, and photophobia have been reported in patients who are poorly compliant with the diet.108,109 However, a longitudinal study demonstrated no development of these opacities despite high plasma Tyr concentrations in some cases.110 There seems to be no exact relationship between the plasma Tyr levels and occurrence of the ocular symptoms, and it is difficult to set up a level for Tyr concentration that might be related to corneal lesions.110,111 Some researchers consider predisposition to ocular toxicity existing independent of the peak plasma Tyr concentration and the differences in Tyr concentrations in different body pools.18,24 In the treatment of HT1 with nitisinone, it is recommended that the serum Tyr be kept below 400 μmol/L, by using a Tyr-restricted low protein diet.45,46,48 Nitisinone, even with the lowest dose, 1 mg daily, led to increase in Tyr levels above this limit in AKU patients.24 Two AKU patients have been reported to develop reversible keratopathy due to nitisinone therapy.24,112
With regard to the effects on the central nervous system, nitisinone is reported to have low blood-to-brain permeability in rats, and based on the results of the studies on the cerebrospinal fluid concentrations of nitisinone after its administration to patients with HT1, its blood-to-brain permeability is considered low in humans too.112 In the NTBC clinical study in patients with HT1, the following events were reported: convulsions (3 of 291 subjects), hyperkinesia (2 of 291 subjects), headache (1 of 291 subjects), hypokinesia (1 of 291 subjects), and somnolence (1 of 291 subjects).113 However, it is not known if nitisinone has direct effects on these events. HT1 patients treated with nitisinone are found to be at risk of developing impaired cognitive function and a total intelligence quotient (IQ) score below the average despite a protein-restricted diet.114,115 They were found to have 20 IQ points lower when compared to their unaffected siblings.114 The lower IQ may be an unwanted side effect of treatment. In support, a significant decrease in IQ was noted in a subset of patients in whom IQ was regularly tested.114 Most likely, nitisinone affects cognitive function indirectly, by inducing profoundly elevated plasma Tyr levels. In addition, attention deficit; decreased ability to verbal reasoning, comprehension, and verbal expression; and school difficulties are described in HT1 patients treated with nitisinone.116 This finding is thought to be connected with the variation of the plasma Tyr level, and therapeutic trials to stabilize the Tyr level could alleviate the difficulties in focusing attention. Despite these neurological findings in HT1 patients, none of the patients with AKU developed neurological complications after being treated with nitisinone.13,24,25,102–104
Still, all patients taking nitisinone should adhere to a phenylalanine- and Tyr-restricted diet to minimize the tyrosinemia produced by the compound.
Mild gastrointestinal system complaints (diarrhea, enanthema, gastroenteritis), transient thrombocytopenia, and leucopenia have also been reported but this has rarely necessitated discontinuation of therapy.56 There was no bleeding or infection because of the low platelet and leucocyte counts.
The potential of nitisinone for undesired pharmacological effects and pharmacodynamic interactions with other medicines is still unknown, as the general receptor binding profile of nitisinone has not yet been established.9
Three successful pregnancies carried to term by three unrelated HT1 patients maintained with nitisinone have been reported till now. All pregnancies were uneventful and there was no evidence of NTBC-induced harm to the developing fetus.118–120
Conclusion
Nitisinone is the licensed, well-tolerated treatment for HT1 based on the inhibition of 4-HPPD in the Tyr degradation pathway. The drug effectively prevents acute hepatic insufficiency, neurologic crises and cardiomyopathy, improves liver functions, but cirrhosis with high grade dysplasia or hepatocarcinoma still develops, especially in some late-treated patients. Furthermore, normal liver function can be achieved in children with HT1 detected by newborn screening and early treated within a month of birth. The combination of neonatal screening plus early treatment is believed to be the best medical treatment of choice for HT1. Inhibition of the Tyr degradation pathway at 4-HPPD level also strongly increases the hope for applying the treatment of other diseases in this pathway like AKU.
Disclosure
The authors report no conflicts of interest in this work.
References
Michaeley WJ, Kratz GW. Certain 2-(2-substituted benzyl)-1,3-cyclohexanediones. European Patent Application 0135191. 1986. | ||
Prisbylla MP, Onisko BC, Shribbs JM, et al. The novel mechanism of action of the herbicidal triketones. In: Proceedings of the Brighton Crop Protection Conference-Weeds, London, UK. 1993;2:731–738. | ||
Lock EA, Ellis MK, Gaskin P, et al. From toxicological problem to therapeutic use: the discovery of the mode of action of 2-(2-nitro-4-trifluoromethylbenzoyl)-1,3-cyclohexanedione (NTBC), its toxicology and development as a drug. J Inherit Metab Dis. 1998;21(5):498–506. | ||
Schulz A, Ort O, Beyer P, Kleinig H. SC-0051, a 2-benzoyl-cyclohexane-1,3-dione bleaching herbicide, is a potent inhibitor of the enzyme p-hydroxyphenylpyruvate dioxygenase. FEBS Lett. 1993;318(2):162–166. | ||
Kavana M, Moran GR. Interaction of (4-hydroxyphenyl) pyruvate dioxygenase with the specific inhibitor 2-[2-nitro-4-(trifluoromethyl)benzoyl]-1,3-cyclohexanedione. Biochemistry. 2003;42(34):10238–10245. | ||
Ellis MK, Whitfield AC, Gowans LA, et al. Inhibition of 4-hydroxy-phenylpyruvate dioxygenase by 2-(2-nitro-4-trifluoromethylbenzoyl)-cyclohexane-1,3-dione and 2-(2-chloro-4-methanesulfonylbenzoyl)-cyclohexane-1,3-dione. Toxicol Appl Pharmacol. 1995;133(1):12–19. | ||
Lindstedt S, Holme E, Lock EA, Hjalmarson O, Strandvik B. Treatment of hereditary tyrosinaemia type I by inhibition of 4-hydroxyphenylpyruvate dioxygenase. Lancet. 1992;340(8823):813–817. | ||
Holme E, Lindstedt S. Tyrosinemia type I and NTBC (2-(2-nitro-4-trifluoromethylbenzoyl)-1,3-cyclohexanedione). J Inherit Metab Dis. 1998;21(5):507–517. | ||
McKiernan PJ. Nitisinone in the treatment of hereditary tyrosinemia type 1. Drugs. 2006;66(6):743–750. | ||
Drug to treat rare pediatric liver disease. FDA Consum. 2002;36(2):5. | ||
Santra S, Baumann U. Experience of nitisinone for the pharmacological treatment of hereditary tyrosinaemia type 1. Expert Opin Pharmacother. 2008;9(7):1229–1236. | ||
Hanauske-Abel HM, Popowicz A, Remotti H, Newfield RS, Levy J. TyrosinemiaI, a model for human diseases mediated by 2-oxoacid-utilizing dioxygenases: hepatotoxin suppression by NTBC does not normalize hepatic collagen metabolism. J Pediatr Gastroenterol Nutr. 2002;35(1):73–78. | ||
Laschi M, Bernardini G, Dreassi E, et al. Inhibition of para-hydroxyphenylpyruvate dioxygenase by analogues of the herbicide nitisinone as a strategy to decrease homogentisic acid levels, the causative agent of alkaptonuria. Chem Med Chem. 2016;11(7):674678. | ||
Lock EA, Gaskin P, Ellis MK, McLean Provan W, Robinson M, Smith LL. Tissue distribution of 2-(2-nitro-4-trifluoromethylbenzoyl)-cyclohexane-1,3-dione (NTBC) and its effect on enzymes involved in tyrosine catabolism in the mouse. Toxicology. 2000;144(1–3):179–187. | ||
Hall MG, Wilks MF, Provan WM, Eksborg S, Lumholtz B. Pharmacokinetics and pharmacodynamics of NTBC (2-(2-nitro-4-fluoromethylbenzoyl)-1,3-cyclohexanedione) and mesotrione, inhibitors of 4-hydroxyphenyl pyruvate dioxygenase (HPPD) following a single dose to healthy male volunteers. Br J Clin Pharmacol. 2001;52(2):169–177. | ||
Schlune A, Thimm E, Herebian D, Spiekerkoetter U. Single dose NTBC-treatment of hereditary tyrosinemia type I. J Inherit Metab Dis. 2012;35(5):831–836. | ||
Orfadin nitisinone (n.d.). Available from: http://orfadin.com/wp-content/uploads/2016/06/prescribing_information.pdf. Accessed September 3, 2016. | ||
Lock EA, Gaskin P, Ellis MK, et al. Tissue distribution of 2-(2-nitro-4-trifluoromethylbenzoyl)cyclohexane-1-3-dione (NTBC): effect on enzymes involved in tyrosine catabolism and relevance to ocular toxicity in the rat. Toxicol Appl Pharmacol. 1996;141(2):439–447. | ||
Szczeciński P, Lamparska D, Gryff-Keller A, Gradowska W. Identification of 2-[2-nitro-4-(trifluoromethyl)benzoyl]-cyclohexane-1,3-dione metabolites in urine of patients suffering from tyrosinemia type I with the use of 1H and 19F NMR spectroscopy. Acta Biochim Pol. 2008;55(4):749–752. | ||
Grompe M, Lindstedt S, al-Dhalimy M, et al. Pharmacological correction of neonatal lethal hepatic dysfunction in a murine model of hereditary tyrosinaemia type I. Nat Genet. 1995;10(4):453–460. | ||
Neat JN, Wolff A, K F, et al. In vitro inhibition and induction of human liver cytochrome P450 enzymes by NTBC and its metabolism in human liver microsomes. Drug Metab Rev. 2010;42(Supp 1):115–116. | ||
Lock EA, Gaskin P, Ellis M, Provan WM, Smith LL. Tyrosinemia produced by 2-(2-nitro-4-trifluoromethylbenzoyl)-cyclohexane-1,3-dione (NTBC) in experimental animals and its relationship to corneal injury. Toxicol Appl Pharmacol. 2006;215(1):9–16. | ||
Masurel-Paulet A, Poggi-Bach J, Rolland MO, et al. NTBC treatment in tyrosinaemia type I: long-term outcome in French patients. J Inherit Metab Dis. 2008;31(1):81–87. | ||
Introne WJ, Perry MB, Troendle J, et al. A 3-year randomized therapeutic trial of nitisinone in alkaptonuria. Mol Genet Metab. 2011;103(4):307–314. | ||
Olsson B, Cox TF, Psarelli EE, et al. Relationship between serum concentrations of nitisinone and its effect on homogentisic acid and tyrosine in patients with alkaptonuria. JIMD Rep. 2015;24:21–27. | ||
Chakrapani A, Gissen P, McKiernan P. Disorders of tyrosine metabolism. In: Saudubray JM, van den Berghe G, Walter JH, editors. Inborn Metabolic Diseases, 5th ed. Heidelberg: Springer; 2012:265–276. | ||
Sassa S, Kappas A. Hereditary tyrosinemia and the heme biosynthetic pathway. Profound inhibition of delta-aminolevulinic acid dehydratase activity by succinylacetone. J Clin Invest. 1983;71(3):625–634. | ||
Jorquera R, Tanguay RM. Fumarylacetoacetate, the metabolite accumulating in hereditary tyrosinemia, activates the ERK pathway and induces mitotic abnormalties and genomic instability. Hum Mol Genet. 2001;10(17):1741–1752. | ||
Jonas MM, Perez-Atayde AR. Liver disease in infancy and childhood. In: Schiff ER, Sorrell MF, Maddrey WC, editors. Schiff’s Diseases of Liver, 10th ed. Philadelphia: Lippincott Williams & Wilkins; 2007:1307–1347. | ||
Bergman AJ, van den Berg IE, Brink W, Poll-The BT, Ploos van Amstel JK, Berger R. Spectrum of mutations in the fumarylacetoacetate hydrolase gene of tyrosinemia type 1 patients in northwestern Europe and Mediterranean countries. Hum Mutat. 1998;12(1):19–26. | ||
Arranz JA, Piñol F, Kozak L, et al. Splicing mutations, mainly IVS6-1(G≥T), account for 70% of fumarylacetoacetate hydrolase (FAH) gene alterations, including 7 novel mutations, in a survey of 29 tyrosinemia type I patients. Hum Mutat. 2002;20(3):180–188. | ||
Dursun A, Ozgül RK, Sivri S, et al. Mutation spectrum of fumarylacetoacetase gene and clinical aspects of tyrosinemia type I disease. JIMD Rep. 2011;1:17–21. | ||
Larochelle J, Alvarez F, Bussières JF, et al. Effect of nitisinone (NTBC) treatment on the clinical course of hepatorenal tyrosinemia in Québec. Mol Genet Metab. 2012;107(1–2):49–54. | ||
Angileri F, Bergeron A, Morrow G, et al. Geographical and ethnic distribution of mutations of the fumarylacetoacetate hydrolase gene in hereditary tyrosinemia type 1. JIMD Rep. 2015;19:43–58. | ||
van Spronsen FJ, Thomasse Y, Smit GP, et al. Hereditary tyrosinemia type I: a new clinical classification with difference in prognosis on dietary treatment. Hepatology. 1994;20(5):1187–1191. | ||
Russo PA, Mitchell GA, Tanguay RM. Tyrosinemia: a review. Pediatr Dev Pathol. 2001;4(3):212–221. | ||
Mitchell G, Russo PA, Dubois J, Alverez F. Tyrosinemia. In: Suchy FJ, Sokol RJ, Balistreri WF, editors. Liver Disease in Children. 3rd ed. New York: Cambridge University Press; 2007:694–713. | ||
Baumann U, Preece MA, Green A, Kelly DA, McKiernan PJ. Hyperinsulinism in tyrosinaemia type I. J Inherit Metab Dis. 2005;28(2):131–135. | ||
Arora N, Stumper O, Wright J, Kelly DA, Mckiernan P. Cardiomyopathy in tyrosinaemia type 1 is common but usually benign. J Inherit Metab Dis. 2006;29(1):54–57. | ||
Mohamed S, Kambal MA, Al Jurayyan NA, et al. Tyrosinemia type 1: a rare and forgotten cause of reversible hypertrophic cardiomyopathy in infancy. BMC Res Notes. 2013;6:362. | ||
Squires JE, Heubi JE. Metabolic liver disease: Part 1. In: Murray KF, Simon H, editors. Diseases of the Liver in Children, Evaluation and Management. Heidelberg: Springer; 2013:151–188. | ||
Fernández-Lainez C, Ibarra-González I, Belmont-Martínez L, Monroy-Santoyo S, Guillén-López S, Vela-Amieva M. Tyrosinemia type I: clinical and biochemical analysis of patients in Mexico. Ann Hepatol. 2014;13(2):265–272. | ||
Simoncelli M, Samson J, Bussières JF, et al. Cost-consequence analysis of nitisinone for treatment of tyrosinemia type I. Can J Hosp Pharm. 2015;68(3):210–217. | ||
McKiernan PJ, Preece MA, Chakrapani A. Outcome of children with hereditary tyrosinaemia following newborn screening. Arch Dis Child. 2015;100(8):738–741. | ||
de Laet C, Dionisi-Vici C, Leonard JV, et al. Recommendations for the management of tyrosinaemia type 1. Orphanet J Rare Dis. 2013;8:8. | ||
Mayorandan S, Meyer U, Gokcay G, et al. Cross-sectional study of 168 patients with hepatorenal tyrosinaemia and implications for clinical practice. Orphanet J Rare Dis. 2014;9:107. | ||
El-Karaksy H, Rashed M, El-Sayed R, et al. Clinical practice. NTBC therapy for tyrosinemia type 1: how much is enough? Eur J Pediatr. 2010;169(6):689–693. | ||
D’Eufemia P, Celli M, Tetti M, Finocchiaro R. Tyrosinemia type I: long-term outcome in a patient treated with doses of NTBC lower than recommended. Eur J Pediatr. 2011;170(6):819. | ||
Herebian D, Spiekerkötter U, Lamshöft M, Thimm E, Laryea M, Mayatepek E. Liquid chromatography tandem mass spectrometry method for the quantitation of NTBC (2-(nitro-4-trifluoromethylbenzoyl)1,3-cyclohexanedione) in plasma of tyrosinemia type 1 patients. J Chromatogr B Analyt Technol Biomed Life Sci. 2009;877(14–15):1453–1459. | ||
Prieto JA, Andrade F, Lage S, Aldámiz-Echevarría L. Comparison of plasma and dry blood spots as samples for the determination of nitisinone (NTBC) by high-performance liquid chromatography-tandem mass spectrometry. Study of the stability of the samples at different temperatures. J Chromatogr B Analyt Technol Biomed Life Sci. 2011;879(11–12):671–676. | ||
Sander J, Janzen N, Terhardt M et al. Monitoring tyrosinaemia type I: blood spot test for nitisinone (NTBC). Clin Chim Acta. 2011;412(1–2):134–138. | ||
Bielenstein M, Astner L, Ekberg S. Determination of 2-(2-nitro-4-trifluoromethylbenzoyl)-1,3-cyclohexanedione in plasma by direct injection into a coupled column liquid chromatographic system. J Chromatogr B Biomed Sci Appl. 1999;730(2):177–182. | ||
Cansever MS, Aktuğlu-Zeybek AC, Erim FB. Determination of NTBC in serum samples from patients with hereditary tyrosinemia type I by capillary electrophoresis. Talanta. 2010;80(5):1846–1848. | ||
Barkaoui E, Debray D, Habès D, Ogier H, Bernard O. Évolution favorable sous traitement par NTBC de l’insuffisance hépatique aiguë révélatrice de la tyrosinémie héréditaire de type I [Favorable outcome of treatment with NTBC of acute liver insufficiency disclosing hereditary tyrosinemia type I]. Arch Pediatr. 1999;6(5):540–544. French. | ||
Joshi SN, Venugopalan P. Experience with NTBC therapy in hereditary tyrosinaemia type I: an alternative to liver transplantation. Ann Trop Paediatr. 2004;24(3):259–265. | ||
Holme E, Lindstedt S. Nontransplant treatment of tyrosinemia. Clin Liver Dis. 2000;4(4):805–814. | ||
Mohan N, McKiernan P, Preece MA, et al. Indications and outcome of liver transplantation in tyrosinaemia type 1. Eur J Pediatr. 1999;158(Suppl 2):S49–S54. | ||
El-Karaksy H, Fahmy M, El-Raziky M, et al. Hereditary tyrosinemia type 1 from a single center in Egypt: clinical study of 22 cases. World J Pediatr. 2011;7(3):224–231. | ||
Luijerink MC, Jacobs SM, van Beurden EA, et al. Extensive changes in liver gene expression induced by hereditary tyrosinemia type I are not normalized by treatment with 2-(2-nitro-4-trifluoromethylbenzoyl)-1,3-cyclohexanedione (NTBC). J Hepatol. 2003;39(6):901–909. | ||
Al-Dhalimy M, Overturf K, Finegold M, Grompe M. Long-term therapy with NTBC and tyrosine-restricted diet in a murine model of hereditary tyrosinemia type I. Mol Genet Metab. 2002;75(1):38–45. | ||
Dieter MZ, Freshwater SL, Miller ML, Shertzer HG, Dalton TP, Nebert DW. Pharmacological rescue of the 14CoS/14CoS mouse: hepatocyte apoptosis is likely caused by endogenous oxidative stress. Free Radic Biol Med. 2003;35(4):351–367. | ||
Pitkänen S, Salo MK, Vettenranta K, Näntö-Salonen K, Heikinheimo M. Serum type III procollagen in children with type I herediatry tyrosinemia. J Pediatr Gastroenterol Nutr. 1999;29(1):38–41. | ||
van Spronsen FJ, Bijleveld CM, van Maldegem BT, Wijburg FA. Hepatocellular carcinoma in hereditary tyrosinemia type I despite 2-(2 nitro-4-3 trifluoro-methylbenzoyl)-1, 3-cyclohexanedione treatment. J Pediatr Gastroenterol Nutr. 2005;40(1):90–93. | ||
Zeybek AC, Kiykim E, Soyucen E, et al. Hereditary tyrosinemia type 1 in Turkey: twenty year single-center experience. Pediatr Int. 2015;57(2):281–289. | ||
Pérez-Cerdá C, Merinero B, Sanz P, et al. Liver transplantation in nine Spanish patients with tyrosinaemia type I. J Inherit Metab Dis. 1995;18(2):119–122. | ||
van Ginkel WG, Gouw AS, van der Jagt EJ, de Jong KP, Verkade HJ, van Spronsen FJ. Hepatocellular carcinoma in tyrosinemia type 1 without clear increase of AFP. Pediatrics. 2015;135(3):e749–e752. | ||
Weinberg AG, Mize CE, Worthen HG. The occurrence of hepatoma in the chronic form of hereditary tyrosinemia. J Pediatr. 1976;88(3):434–438. | ||
McKiernan PJ. Nitisinone for the treatment of hereditary tyrosinemia type I. Expert Opin Orphan Drugs. 2013;1(6):491–497. | ||
Sun MS, Hattori S, Kubo S, Awata H, Matsuda I, Endo F. A mouse model of renal tubular injury of tyrosinemia type 1: development of de Toni Fanconi syndrome and apoptosis of renal tubular cells in Fah/Hpd double mutant mice. J Am Soc Nephrol. 2000;11(2):291–300. | ||
Spencer PD, Roth KS. Effects of succinylacetone on amino acid uptake in the rat kidney. Biochem Med Metab Biol. 1987;37(1):101–109. | ||
Forget S, Patriquin HB, Dubois J, et al. The kidney in children with tyrosinemia: sonographic, CT and biochemical findings. Pediatr Radiol. 1999;29(2):104–108. | ||
Santra S, Preece MA, Hulton SA, McKiernan PJ. Renal tubular function in children with tyrosinaemia type I treated with nitisinone. J Inherit Metab Dis. 2008;31(3):399–402. | ||
Maiorana A, Malamisura M, Emma F, Boenzi S, Di Ciommo VM, Dionisi-Vici C. Early effect of NTBC on renal tubular dysfunction in hereditary tyrosinemia type 1. Mol Genet Metab. 2014;113(3):188–193. | ||
Pierik LJ, van Spronsen FJ, Bijleveld CM, van Dael CM. Renal function in tyrosinaemia type I after liver transplantation: a long-term follow-up. J Inherit Metab Dis. 2005;28(6):871–876. | ||
Bartlett DC, Lloyd C, McKiernan PJ, Newsome PN. Early nitisinone treatment reduces the need for liver transplantation in children with tyrosinaemia type 1 and improves post-transplant renal function. J Inherit Metab Dis. 2014;37(5):745–752. | ||
Gibbs TC, Payan J, Brett EM, Lindstedt S, Holme E, Clayton PT. Peripheral neuropathy as the presenting feature of tyrosinaemia type I and effectively treated with an inhibitor of 4-hydroxyphenylpyruvate dioxygenase. J Neurol Neurosurg Psychiatry. 1993;56(10):1129–1132. | ||
Mitchell G, Larochelle J, Lambert M, et al. Neurologic crises in hereditary tyrosinemia. N Engl J Med. 1990;322(7):432–437. | ||
Schlump JU, Perot C, Ketteler K, et al. Severe neurological crisis in a patient with hereditary tyrosinaemia type I after interruption of NTBC treatment. J Inherit Metab Dis. 2008;31(Suppl 2):S223–S225. | ||
Önenli Mungan N, Yıldızdaş D, Kör D, et al. Tyrosinemia type 1 and irreversible neurologic crisis after one month discontinuation of nitisone. Metab Brain Dis. 2016;31(5):1181–1183. | ||
André N, Roquelaure B, Jubin V, Ovaert C. Successful treatment of severe cardiomyopathy with NTBC in a child with tyrosinaemia type I. J Inherit Metab Dis. 2005;28(1):103–106. | ||
Fernández-Cañón JM, Granadino B, Beltrán-Valero de Bernabé D, et al. The molecular basis of alkaptonuria. Nat Genet. 1996;14(1):19–24. | ||
Zatkova A. An update on molecular genetics of Alkaptonuria (AKU). J Inherit Metab Dis. 2011;34(6):1127–1136. | ||
Nemethova M, Radvanszky J, Kadasi L, et al. Twelve novel HGD gene variants identified in 99 alkaptonuria patients: focus on “black bone disease” in Italy. Eur J Hum Genet. 2016;24(1):66–72. | ||
Lustberg TJ, Schulman JD, Seegmiller JE. Metabolic fate of homogentisic acid-1–14 C (HGA) in alkaptonuria and effectiveness of ascorbic acid in preventing experimental ochronosis. Arthritis Rheum. 1969;12:678. | ||
Peker E, Yonden Z, Sogut S. From darkening urine to early diagnosis of alkaptonuria. Indian J Dermatol Venereol Leprol. 2008;74(6):700. | ||
Phornphutkul C, Introne WJ, Perry MB, et al. Natural history of alkaptonuria. N Engl J Med. 2002;347(26):2111–2121. | ||
Milch RA. Studies of alcaptonuria: mechanisms of swelling of homogentisic acid-collagen preparations. Arthritis Rheum. 1961;4:253–267. | ||
Zannoni VG, Lomtevas N, Goldfinger S. Oxidation of homogentisic acid to ochronotic pigment in connective tissue. Biochim Biophys Acta. 1969;177(1):94–105. | ||
Zannoni VG, Malawista SE, La Du BN. Studies on ochronosis. II. Studies on benzoquinoneacetic acid, a probable intermediate in the connective tissue pigmentation of alcaptonuria. Arthritis Rheum. 1962;5:547–556. | ||
Ranganath LR, Cox TF. Natural history of alkaptonuria revisited: analyses based on scoring systems. J Inherit Metab Dis. 2011;34(6):1141–1151. | ||
Aquaron R. Alkaptonuria: a very rare metabolic disorder. Indian J Biochem Biophys. 2013;50(5):339–44. | ||
Lindner M, Bertelmann T. On the ocular findings in ochronosis: a systemic review of the literature. BMC Opthalmol. 2014;14:12. | ||
Fisher AA, Davis MW. Alkaptonuric ochronosis with aortic valve and joint replacements and femoral fracture: a case report and literature review. Clin Med Res. 2004;2(4):209–215. | ||
Taylor AM, Wilson PJ, Ingrams DR, Helliwell TR, Gallagher JA, Ranganath LR. Calculi and intracellular ochronosis in the submandibular tissues from a patient with alkaptonuria. J Clin Pathol. 2010;63(2):186–188. | ||
Keller JM, Macaulay W, Nercessian OA, Jaffe IA. New developments in ochronosis: review of the literature. Rheumatol Int. 2005;25(2):81–85. | ||
Manoj Kumar RV, Rajasekaran S. Spontaneous tendon ruptures in alkaptonuria. J Bone Joint Surg Br. 2003;85(6):883–886. | ||
Introne WJ, Phornphutkul C, Bernardini I, McLaughlin K, Fitzpatrick D, Gahl WA. Exacerbation of the ochronosis of alkaptonuria due to renal insufficiency and improvement after renal transplantation. Mol Genet Metab. 2002;77(1–2):136–142. | ||
Gil JA, Wawrzynski J, Waryasz GR. Orthopedic manifestations of ochronosis: pathophysiology, presentation, diagnosis, and management. Am J Med. 2016;129(5):536.e1–e6. | ||
Mullan A, Cocker D, Taylor G, Millar C, Ranganath L. Fatal oxidative haemolysis and methaemoglobinaemia in a patient with alkaptonuria and acute kidney injury. Clin Kidney J. 2015;8(1):109–112. | ||
Davison AS, Milan AM, Gallagher JA, Ranganath LR. Acute fatal metabolic complications in alkaptonuria. J Inherit Metab Dis. 2016;39(2):203–210. | ||
Preston AJ, Keenan CM, Sutherland H, et al. Ochronotic osteoarthropathy in a mouse model of alkaptonuria, and its inhibition by nitisinone. Ann Rheum Dis. 2014;73(1):284–289. | ||
Suwannarat P, O’Brien K, Perry MB, et al. Use of nitisinone in patients with alkaptonuria. Metabolism. 2005;54(6):719–728. | ||
Ranganath LR, Milan AM, Hughes AT, et al. Suitability of nitisinone in alkaptonuria 1 (SONIA 1): an international, multicentre, randomised, open-label, no-treatment controlled, parallel-group, dose–response study to investigate the effect of once daily nitisinone on 24-h urinary homogentisic acid excretion in patients with alkaptonuria after 4 weeks of treatment. Ann Rheum Dis. 2016;75(2):362–367. | ||
Gertsman I, Barshop BA, Panyard-Davis J, Gangoiti JA, Nyhan WL. Metabolic effects of increasing doses of nitisinone in the treatment of alkaptonuria. JIMD Rep. 2015;24:13–20. | ||
Bowman LC, Hancock ML, Santana VM, et al. Impact of intensified therapy on clinical outcome in infants and children with neuroblastoma: the St. Jude Children’s Research Hospital experience, 1962 to 1998. J Clin Oncol. 1991;9(9):1599–1608. | ||
Peinemann F, van Dalen EC, Berthold F. Rapid COJEC induction therapy for high-risk neuroblastoma patients—cochrane review. Klin Padiatr. 2016;228(3):130–134. | ||
Kobrinsky NL, Sjolander DE. Response of metastatic recurrent neuroblastoma to nitisinone: a modulator of tyrosine metabolism. Pediatr Blood Cancer. 2006;46(4):517–520. | ||
Ahmad S, Teckman JH, Lueder GT. Corneal opacities associated with NTBC treatment. Am J Ophthalmol. 2002;134(2):266–268. | ||
Gissen P, Preece MA, Willshaw HA, McKiernan PJ. Ophthalmic follow-up of patients with tyrosinaemia type I on NTBC. J Inherit Metab Dis. 2003;26(1):13–16. | ||
Gulmez Sevim D, Gumus K, Cavanagh HD. Corneal pseudodendritic lesions masquerading as herpetic keratitis in a patient with tyrosinemia type I. Eye Contact Lens. Epub 2015 Aug 28. | ||
Schauwvlieghe PP, Jaeken J, Kestelyn P, Claerhout I. Confocal microscopy of corneal crystals in a patient with hereditary tyrosinemia type I, treated with NTBC. Cornea. 2013;32(1):91–94. | ||
Stewart RM, Briggs MC, Jarvis JC, Gallagher JA, Ranganath L. Reversible keratopathy due to hypertyrosinaemia following intermittent low-dose nitisinone in alkaptonuria: a case report. JIMD Rep. 2014;17:1–6. | ||
Thimm E, Herebian D, Assmann B, Klee D, Mayatepek E, Spiekerkoetter U. Increase of CSF tyrosine and impaired serotonin turnover in tyrosinemia type I. Mol Genet Metab. 2011;102(2):122–125. | ||
European Public Assessment Report for Orfadin, European Medicines Agency (EMEA) 2005, Scientific Report Doc. Ref. EMA/823169/2009/EMA/H/C/555: Available from: http://www.eespof.gr/sites/default/files/Orfadin-ENG.pdf. Accessed September 15, 2016. | ||
Bendadi F, de Koning TJ, Visser G, et al. Impaired cognitive functioning in patients with tyrosinemia type I receiving nitisinone. J Pediatr. 2014;164(2):398–401. | ||
Thimm E, Richter-Werkle R, Kamp G, et al. Neurocognitive outcome in patients with hypertyrosinemia type I after long-term treatment with NTBC. J Inherit Metab Dis. 2012;35(2):263–268. | ||
Pohorecka M, Biernacka M, Jakubowska-Winecka A, et al. Behavioral and intellectual functioning in patients with tyrosinemia type I. Pediatr Endocrinol Diabetes Metab. 2012;18(3):96–100. | ||
Vanclooster A, Devlieger R, Meersseman W, et al. Pregnancy during nitisinone treatment for tyrosinaemia type I: first human experience. JIMD Rep. 2012;5:27–33. | ||
Garcia Segarra N, Roche S, Imbard A, et al. Maternal and fetal tyrosinemia type I. J Inherit Metab Dis. 2010;33(Suppl 3):S507–S510. | ||
Kassel R, Sprietsma L, Rudnick DA. Pregnancy in an NTBC-treated patient with hereditary tyrosinemia type I. J Pediatr Gastroenterol Nutr. 2015;60(1):e5–e7. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.