")
Back to Journals » Drug Design, Development and Therapy » Volume 13
Neuroprotective potential of ketamine prevents developing brain structure impairment and alteration of neurocognitive function induced via isoflurane through the PI3K/AKT/GSK-3β pathway
Authors Wang R, Zhang Z, Kumar M, Xu G, Zhang M
Received 25 September 2018
Accepted for publication 20 November 2018
Published 4 February 2019 Volume 2019:13 Pages 501—512
DOI https://doi.org/10.2147/DDDT.S188636
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Anastasios Lymperopoulos
Ruiwei Wang,1,* Zihao Zhang,2,* Mukesh Kumar,3 Guangming Xu,4 Mengyuan Zhang1
1Department of Anesthesiology, Shandong Provincial Hospital Affiliated to Shandong University, Jinan, Shandong Province 250021, People’s Republic of China; 2Department of Clinical Medicine, Nanchang University, Nanchang, Jiangxi Province 330031, People’s Republic of China; 3Radhagovind College, Moradabad 204411, India; 4Department of Neurosurgery, Shandong Provincial Hospital Affiliated to Shandong University, Jinan, Shandong Province 250021, People’s Republic of China
*These authors contributed equally to this work
Background: The aim of the current experimental study was to scrutinize the neuroprotective effect of ketamine on the isoflurane (iso)-induced cognitive dysfunction in rats via phosphoinositide 3 kinase (PI3K)/protein kinase B (AKT)/glycogen synthase kinase 3β (GSK-3β) pathway.
Materials and methods: Sprague-Dawley rats were used for the current experimental study. The rats were divided into six groups and rats were treated with ketamine and memantine. For the estimation of cognitive function study, we used the Morris water test. Pro-inflammatory cytokines such as IL-1β, IL-6, tumor necrosis factor-α (TNF-α), and caspase-6; the antioxidant parameters malondialdehyde, glutathione, superoxide dismutase, catalase, and protein carbonyl; acetylcholinesterase, amyloid β, and brain-derived neurotrophic factor were estimated, respectively. The protein expression of AKT, GSK-3β, p21WAF1/CIP1, and p53 was also estimated, respectively.
Results: Ketamine significantly enhanced cognitive function and showed anti-inflammatory and antioxidant effects, and exhibited the neuroprotective effect of ketamine against the isoflurane-induced cognitive impairment. Additionally, ketamine significantly (P<0.005) suppressed IL-1β, TNF-α, IL-6, caspase-6 and p21WAF1/CIP1, p53 expression and up-regulated the PI3K/AKT/GSK-3β expression in the group of iso-induced rats.
Conclusion: We can conclude that ketamine prevented the cognitive impairment induced by isoflurane anesthesia through anti-apoptotic, anti-inflammatory, and antioxidant effects via the PI3K/AKT/GSK-3β pathway.
Keywords: ketamine, isoflurane, neuroinflammatory, PI3K/AKT/GSK-3β pathway, cognitive impairment
Background
Alzheimer’s disease (AD) is considered the most common form of dementia. The common pathophysiological features of AD is reducing visual skills, alteration of personality, and progressive loss of memory and cognition.1 β-Amyloid (Aβ) generation in the brain regions, especially cerebral cortex and hippocampus, are the chief neuropathological feature of an AD.2 Aβ triggers the inflammatory reaction, which likely starts as a host defense response to the destruction of brain tissue and latterly contributes to neuronal damage.1 During AD, the activated microglia promote the neurodegenerative process via secretion of pro-inflammatory cytokines such as tumor necrosis factor-α (TNF-α), IL-6, and IL-1β, which start the neuronal cell-damage and eventual death.3
Moreover, these inflammatory mediators nearby in AD lesions are thought to increase the key events of the pathological cascade that result in the enhancement of Aβ production, which further activates the microglia cells.4 Recently, researchers have been targeting Aβ production to cure AD via restoration of the cholinesterase inhibitors such as rivastigmine and donepezil or regulating neuroinflammation with the use of COX-2 inhibitors and NSAIDs such as indomethacin and ibuprofen.5 But, neuroinflammatory and NSAID drugs have their own side- and toxic-effects such as renal toxicity, gastrointestinal bleeding, nausea, and liver toxicity. So currently, researchers are targeting this inflammatory reaction to reduce AD.3
Neuroinflammation is involved in the activation of microglial cells and participation of neurons and astrocytes.6 Various previous published research papers suggest that neuroinflammation plays an important role in neurodegenerative diseases such as AD.7,8 Particularly in the field of AD research, a recent investigation suggests that IL-1β, IL-6, and TNF-α trigger neuroinflammation and further destruction of cognitive function.9,10 Some investigations have suggested that isoflurane increases the level of IL-6, IL-1β, and TNF-α in vivo, which possibly underlies the cognitive disorder after isoflurane use.9–11 Whereas the mechanism connected with isoflurane (iso)-induced neuroinflammatory response is unclear.
Glycogen synthase kinase-3 (GSK-3) is uniformly active serine/threonine kinase, and is commonly found in metazoan organisms.12 Two isoforms of GSH-3 such as GSK-3α and GSK-3β are mostly found in mammals.13 Research suggests that GSK-3β mostly presents in the thalamus, hippocampus and striatum region of brain.14 It is believed that GSK-3β, a significant component of the signaling pathway, which plays a significant role in the various cellular pathways and regulates the numerous cellular processes ranging from glycogen metabolism to neuronal polarity and cell survival.15 Moreover, GSK-3β is also activated by nuclear factor κB (NF-κB), which arbitrates the gene expression of pro-inflammatory cytokines involved in the neuroinflammatory reaction.16 Various researchers have suggested that the intravenous general anesthetics not only show excellent analgesic effects but they also show no side effects during respiration.17 Due to their specific protective effects and fewer side effects, they are commonly used in clinical surgical treatment. In recent years, ketamine has shown anti-inflammatory effects as it can reduce pro-inflammatory cytokines.18,19 It is believed that ketamine shows inhibitory effects on cytokines and has an antioxidant potential. Ketamine inhibits iso-induced neuroapoptosis via phosphatidylinositol 3-kinase (PI3K)/protein kinase B (Akt) pathway is unclear. So, in this current study, we attempted to scrutinize the neuroprotective effects of ketamine on iso-induced neuroinflammation via the PI3K/AKT/GSK-3β pathway.
Materials and methods
Chemical
Rabbit monoclonal antibodies against acetyl-NF-κB p65 (catalog # PA5-17264), GSK3β (catalog # 710132), and mouse monoclonal antibody against Lamin B and β-actin (catalog # MA1-140) were procured from Thermo Fisher Scientific (Waltham, MA, USA). Anti-rabbit IgG (catalog # A0545), horseradish peroxidase-conjugated goat anti-mouse IgG, and secondary antibodies were purchased from Sigma-Aldrich (St Louis, MO, USA). Interleukin-6 kit (catalog # H007), interleukin-1β (catalog # H002), glutathione (catalog # A006-2), malondialdehyde (catalog # A003-1), and caspase-3 (catalog # H076) were purchased from the Nanjing Jiancheng Bioengineering Institute (Nanjing, People’s Republic of China).
Experimental animals
For the current experimental study, Sprague-Dawley rats (300–350 g, both sexes) were used. The rats were procured from the institutional animal house and kept in standard conditions (22°C±5°C; 12 hours dark/light cycle) and received the standard diet pellets and water ad libitum. The current experimental study was performed according to the NIH Guide for the Care and Use of Laboratory Animals. The whole experimental protocol was approved by the Institutional Ethical Committee of Shandong University (SU/18/02/009).
Preclinical study
Aβ (1–42) model
Injection of Aβ (1–42) (soluble in glacial acetic acid) was used to induce the peptide aggregation. All rats were randomly divided into six groups and each group contained ten rats: Gp-I: normal control received vehicle only; Gp-II: Aβ (1–42) received only; Gp-III: Aβ (1–42) control group rats received ketamine (2.5 mg/kg); Gp-IV: Aβ (1–42) control group rats received ketamine (5 mg/kg); Gp-V: Aβ (1–42) control group rats received ketamine (10 mg/kg) and Gp-VI: Aβ (1–42) control group rats received celebrex (10 mg/kg). The rats received the above-discussed treatment for 70 days. At 60 days, all the rats in Gps III–VI were injected with Aβ (1–42) peptide (1 μL into the lateral ventricle of each rat).20
Novel object recognition (NOR)
The previously reported method of Bhatt et al21 was used for the estimation of NOR. The current model constituted of three sessions in which training, retention, and habituation were performed. In the first session, the rats explored the box (individually), which was empty of any objects, for 5 minutes. During the training session, two novel objects were introduced, each kept at a different location in the box and the rats were again allowed to explore the box for 5 minutes. A rat was considered to be exploring an object when its head was facing the object or it was sniffing or touching the object and the time spent during the exploring was recorded. After completing the training session of rats, the rats were returned to the home cage and in the last and final session, one object was successfully changed with the novel object. The rats further explored during the training session for 24 hours after the 5-minute training session, each rat was kept in the box to explore for 5 minutes, and the time spent exploring was recorded. The capability to distinguish the novel objects was scrutinized by a discrimination index using the following formula:
|
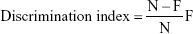
This showed the difference in time spent exploring between the novel (N) and familiar object (F), exacted for total time spent exploring both objects.
Iso-induced model
The rats were divided into six groups and each group contained six rats: Gp-I: normal control received only vehicle; Gp-II: Isoflurane control (15 μg/5 μL); Gps-III–V received isoflurane + ketamine (2.5, 5, and 10 mg/kg); and Gp-VI Isoflurane + memantine respectively. The experimental animals were intracerebroventricularly (icv) infused with either isoflurane (15 μL) or artificial cerebrospinal fluid. The experimental animals were further used for neurochemical and neurobehavioral assessment. The intracerebroventricular and surgical procedure was used for the administration of isoflurane.22
Experimental animals were anesthetized by chloral hydrate and stereotaxic apparatus was fixed on the animal’s head in a frame while a midline sagittal incision was cut into the scalp of the animals. The skull of the animal was drilled for placing the cannula and closed via using suture. In the normal control group rats, the surgery was identical including drilling the hole in the brain and placing the cannula.
Post-operative procedures
After the surgery, special care, and aseptic conditions were maintained and a special diet was provided to the animals. For the prevention of sepsis, the injection of gentamicin (5 mg/kg) was used for 3 days. After that, the experimental study was started.
Behavioral experimental study
Morris method
The Morris water test was performed for the determination of memory and learning activity in rats by using the reported protocol of Morris and minor modification of Falls et al23 method. The Morris water test comprises a circular pool having 60 cm in height and 180 cm in diameter and is filled with water. Four edges of the pool were designed as N (North), E (East), S (South), and W (West) quadrants and rats were kept in this quadrant during the retention and acquisition phase. During the first acquisition session (Session 1), an 8-cm black colored platform was kept 1 cm above the surface and the rats were allowed to find its position. In the next two sessions (2 and 3) the same platform was used but the surface was kept 1 cm below the water and again the rats were allowed to find its position. While all the rats were trained to mount the platform they ultimately cultured the spatial position of the platform in any of quadrant. On the other hand, in learning phase session 1, the rats were trained on the 14th day of the treatment consisting of four trials in a day interspaced by 5 minutes for each rat. In the current model, we provide the maximum time of 120 seconds to locate the hidden platform and allowed the rats to stay on the hidden platform for 30 seconds. If the animal failed to locate the hidden platform during the 120 seconds, they were kept on the platform during the session 1. We also estimated the Escape latency time for each session.
Probe trial
For the probe trial, on the last day of the training session, the platform was successfully removed from the pool and the rats were allowed to swim freely in the pool (120 seconds). The time spent by rats to reach the target quadrant was compared among the different groups.
Passive avoidance paradigm
For the estimation of memory and learning capability of the rats, we used the passive avoidance paradigm model with minor modification.24 Briefly, the method was carried out by placing the animal in a shuttle box (having two compartments) and having one guillotine door for separating both compartments. The rats were kept in the light chamber for 30 seconds (acclimatization time period) and after the 30 seconds the guillotine door opened. When the rats entered into the dark room the guillotine door closed and low-intensity foot shock was given to the rats for 10 seconds. The current experiment was performed for a maximum 270 seconds and the first trial was termed as acquisition followed by retention I, II, and III interspaced by 24 hours for each and every trial and the latency time was calculated.
Neurochemical and antioxidant parameters
Neurochemical parameters such as acetylcholinesterase (AChE) activity, protein carbonyl and antioxidant parameters such as lipid peroxidation (LPO), superoxide dismutase (SOD), glutathione (GSH), and catalase (CAT) were estimated using the previously reported method with minor modification.25
Western blotting
The hippocampus region of the brain was homogenized with RIPA buffer solution, which contained the phosphatase inhibitors and protease inhibitor mixture. BCA protein kits were used for the estimation of total protein concentration. The tris buffer solution was used for blocking the immunoblots and then this was incubated overnight with primary antibodies. The following antibodies such as Histone 3, Ac-H3K9, Histone 4, Ac-H4K12, histone deacetylase 1 (HDAC1), β-actin, histone deacetylase 2 (HDAC2), brain-derived neurotrophic factor (BDNF), pro-BDNF, CREB binding protein (CBP), CAMK II, p-CREB, phospho (p)-CAMK II, p-ERK, ERK, CREB, p-p65, TrKB, p-IκBα, p65, and IκBα were used.
Chromatin immunoprecipitation (ChIP)
The ChIP extraction method was performed according to the previously published method with minor modification.26 Briefly, a minced hippocampus tissue portion was homogenized in lysis buffer and centrifuged for 5 minutes at 5,500 × g after the homogenate was sonicated on ice to break down the chromatin into fragments (200–500 bp). Now, the sheared chromatin solution was immunoprecipitated at 4°C with H3 acetylated and H4 acetylated antibodies at Lys9 (Abcam, Cambridge, MA, USA) and H4 acetylated at Lys12 (Abcam). At the end of the incubation step, precipitate contained a DNA–protein complex.
Quantitative real time-PCR
Briefly, the PCR reaction mixture, which contains the cDNA (2 μL) and 5 mmol/L solution reverse and forward primers added in each wall and SYBR green PCR Master Mix was added making the final volume 25.2 μL and was then incubated for 10 minutes at 97°C. The relative expressions in the hippocampus region of all the animals in all the groups were estimated by using the manufacturer’s instruction.
Pro-inflammatory cytokines and inflammatory mediators
The pro-inflammatory cytokines such as IL-6, TNF-α, and IL-10, IL-1β and caspase-3 were estimated using the manufacturer’s instructions from Nanjing Jiancheng Bioengineering Institute.
Statistical method
For the current experimental study, all the behavioral data were presented as mean ± SEM and analyzed via two-way ANOVA. For the estimation of biochemical estimation, one-way ANOVA is used. Tukey’s test with P<0.05 was considered significant for post hoc comparisons between groups.
Results
Effect on the Morris water maze test
Figure 1 shows the effect of ketamine on the Morris water maze test. The normal control group rats were able to find the platform quickly. Animals treated with isoflurane demonstrated the higher escape latencies of around 65 seconds as compared to the escape latency of the normal control treated animals. The normal control group rats were six times quicker compared to the isoflurane group rats, indicating memory dysfunction in response to isoflurane. Iso-induced group rats treated with ketamine significantly (P<0.05) reduce the escape latency in a dose-dependent manner as compared to isoflurane-treated rats. The animals showed the ability to find the hidden platform inside the quadrant water pool after ketamine treatment. After 21 days of treatment, the ketamine-treated group rats showed less time for escape latency and suggest the reversal of behavioral abnormalities. Likewise, the memantine-treated group rats showed similar results.
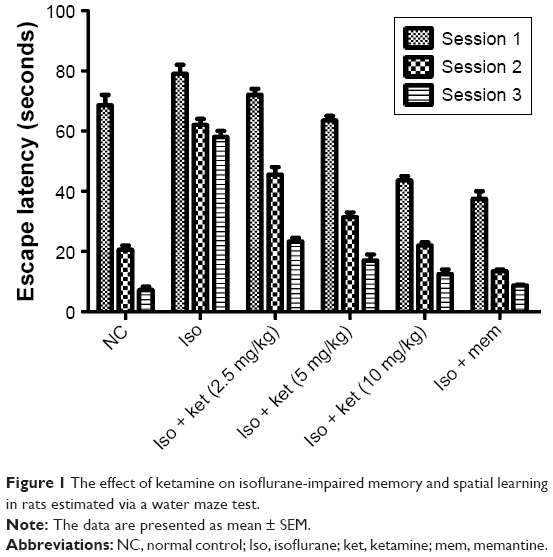 | Figure 1 The effect of ketamine on isoflurane-impaired memory and spatial learning in rats estimated via a water maze test. |
Platform quadrant time spent analysis
The probe trial was used for the estimation of time spent in the platform quadrant. Iso-induced group rats showed 20 seconds for latency time in the probe trial data analysis study which exhibited a significant (P<0.05) down-regulation toward the target quadrant. The ketamine-treated group rats showed increased quadrant time spent as compared to the iso-induced animals. Ketamine (10 mg/kg)-treated group rats took 62 seconds which was almost double compared to the iso-induced group rats. A similar result was found in the memantine-treated group rats (Figure 2).
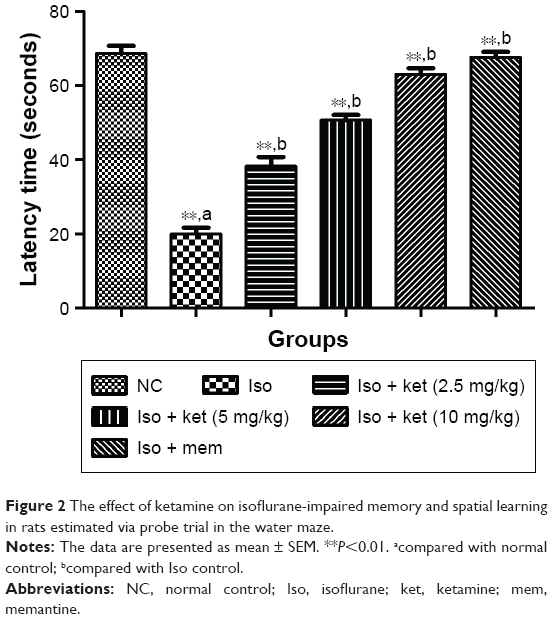 | Figure 2 The effect of ketamine on isoflurane-impaired memory and spatial learning in rats estimated via probe trial in the water maze. |
Passive avoidance paradigm
Figure 3 demonstrates the effect of ketamine on learning and memory activity. Iso-induced brain damage was evident by consistent impairment in the learning and memory activity of rats as compared with normal control group rats. The third retention trial showed the reduced transfer latency time (TLT) as a comparison to acquisition trial TLT from the normal control group to isoflurane group rats. Figure 3 shows that the TLT in normal control and the memantine-treated group rats showed a significant enhancement in the retention trials as compared to the acquisition trials. On the other hand, the ketamine-treated group rats showed an increase in TLT in the retention trials as comparison to acquisition trial TLT.
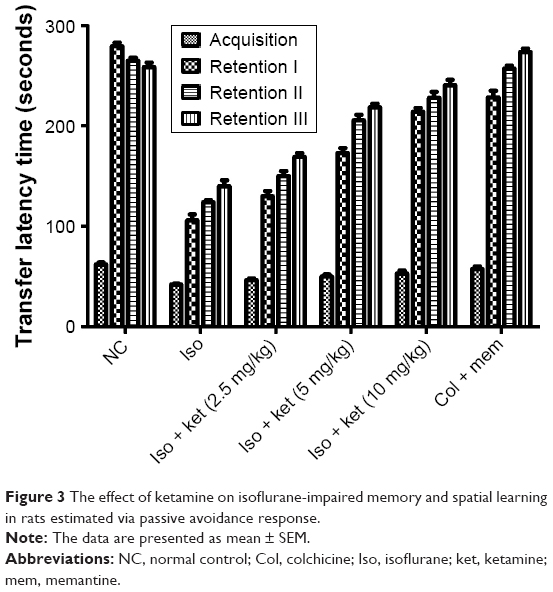 | Figure 3 The effect of ketamine on isoflurane-impaired memory and spatial learning in rats estimated via passive avoidance response. |
Effect on Aβ and BDNF
Figure 4 shows the effect of ketamine on the Aβ peptide protein. Normal control group rats exhibited the Aβ peptide protein (20.03±1.83) and iso-induced rats showed the Aβ peptide protein (85.23±4.34). Concentration-dependent treatment of the ketamine-treated group rats demonstrated the reduction in the level of Aβ peptide protein (Figure 4A).
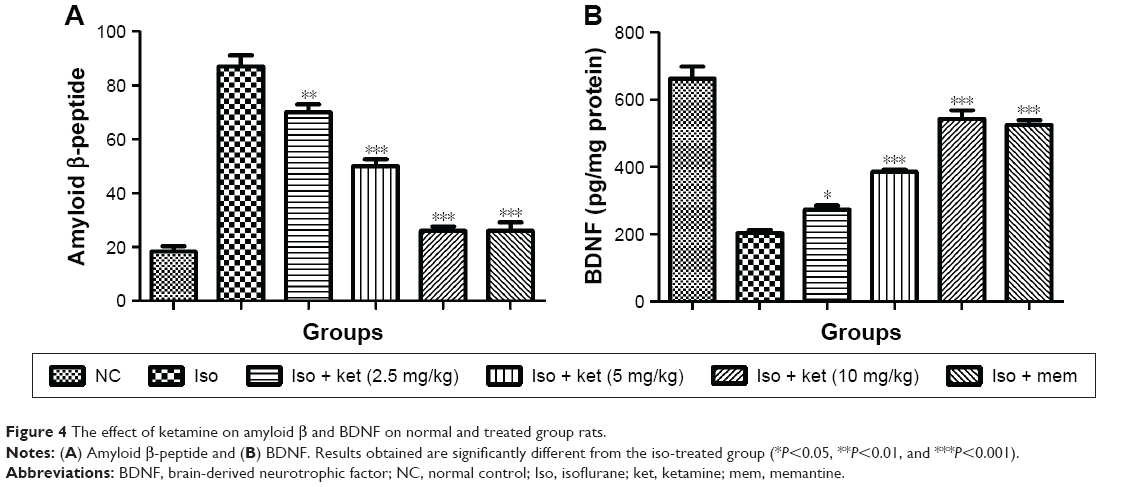 | Figure 4 The effect of ketamine on amyloid β and BDNF on normal and treated group rats. |
An opposite trend was observed in the BDNF content. The normal control group rats showed a BDNF level of 643.34±18.76 but the iso-induced group rats showed a reduced BDNF level of 200.9±12.34 and dose-dependent treatment with ketamine showed an increased level of BDNF, respectively (Figure 4B).
Effect on protein carbonyl content
Figure 5 shows the effect of protein carbonyl on the normal and treated group of rats. The protein carbonyl content is the essential marker to estimate the oxidative stress injury due to protein oxidation. Isoflurane caused an increased level of protein carbonyl in the hippocampus region of the brain. Normal control group rats showed a protein carbonyl level of 22.2±1.82 nmol/mg protein and iso-induced group rats showed an increased protein carbonyl level (58.52±3.43 nmol/mg protein), respectively. Concentration-dependent treatment of ketamine showed a reduction in the protein carbonyl level as compared to the iso-induced group rats (Figure 5).
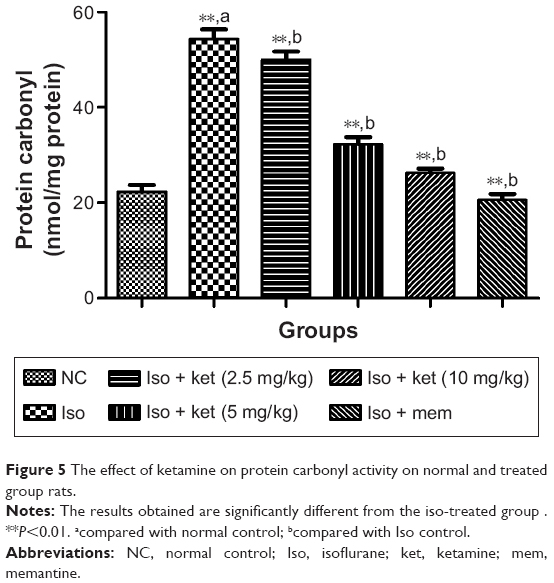 | Figure 5 The effect of ketamine on protein carbonyl activity on normal and treated group rats. |
An opposite trend was observed in the AChE level. Normal control group rats showed an increased level of AChE and iso-induced group rats showed a reduced level of AChE at the end of the experimental study. Concentration-dependent treatment of ketamine showed a reduced level of AChE activity (Figure 6).
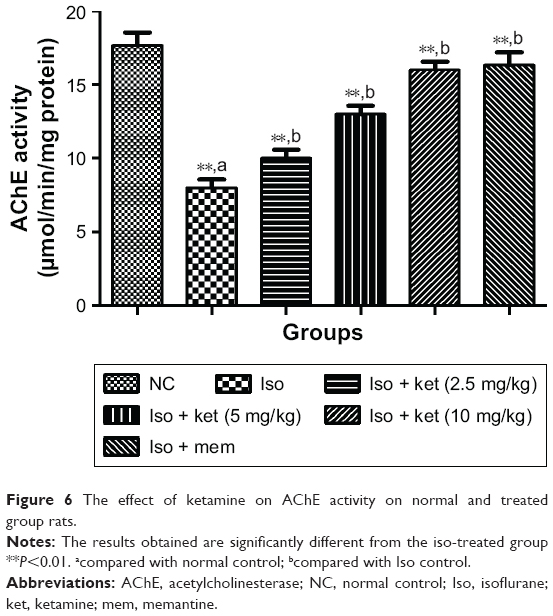 | Figure 6 The effect of ketamine on AChE activity on normal and treated group rats. |
Effect on antioxidant parameters
It is believed that the LPO level is the key marker of oxidative degradation of lipid in the cell membrane, the result of which leads to cellular damage. Isoflurane-induced rats showed an increased level of malondialdehyde (MDA) (an LPO parameter) (36.54±2.93) as compared to normal control (23.43±1.28). Concentration-dependent treatment of ketamine exhibited a reduction of the MDA level as compared to the iso-induced group rats (Figure 7).
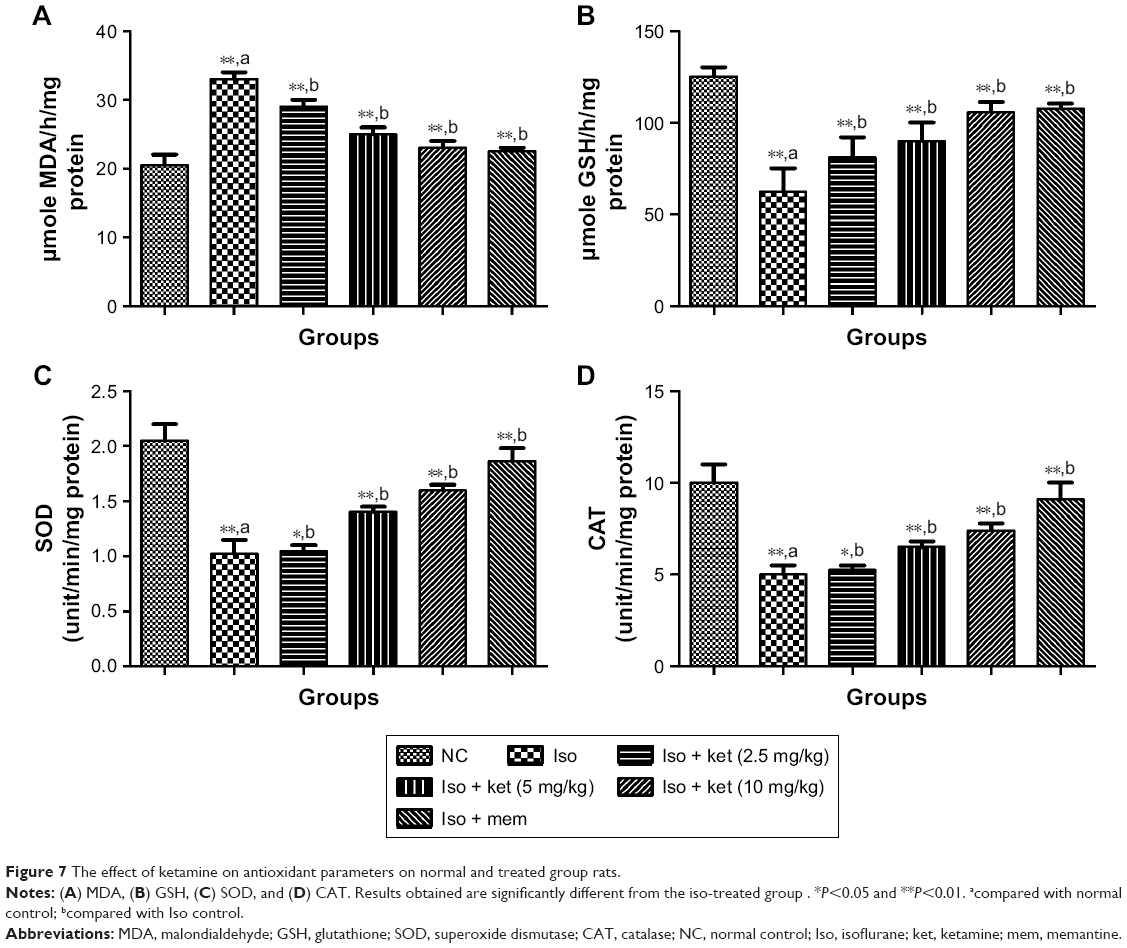 | Figure 7 The effect of ketamine on antioxidant parameters on normal and treated group rats. |
An opposite trend was observed in the levels of SOD, GSH, and CAT in that they were significantly (P<0.05) decreased in the iso-induced group rats and dose-dependent treatment of ketamine improved the level as compared to iso-induced group rats.
Effect on pro-inflammatory and inflammatory mediators
Inflammation plays an important role in the expansion of disease. For the estimation of neuroinflammation, pro-inflammatory and inflammatory mediators were estimated. Pro-inflammatory cytokines such as IL-1β, IL-6, and TNF-α were estimated. Iso-induced group rats showed increased levels of IL-1β (8.91±0.73) (Figure 8A), IL-6 (6.43±0.35) (Figure 8B), and TNF-α (22.12±2.93) (Figure 8C) as compared to normal control IL-1β (2.5±0.21), IL-6 (2.1±0.14), and TNF-α (5.2±0.23), respectively. Ketamine showed a reduction of IL-1β (3.9±0.12), IL-6 (3.2±0.15), and TNF-α (8.4±0.83) at a dose of 10 mg/kg.
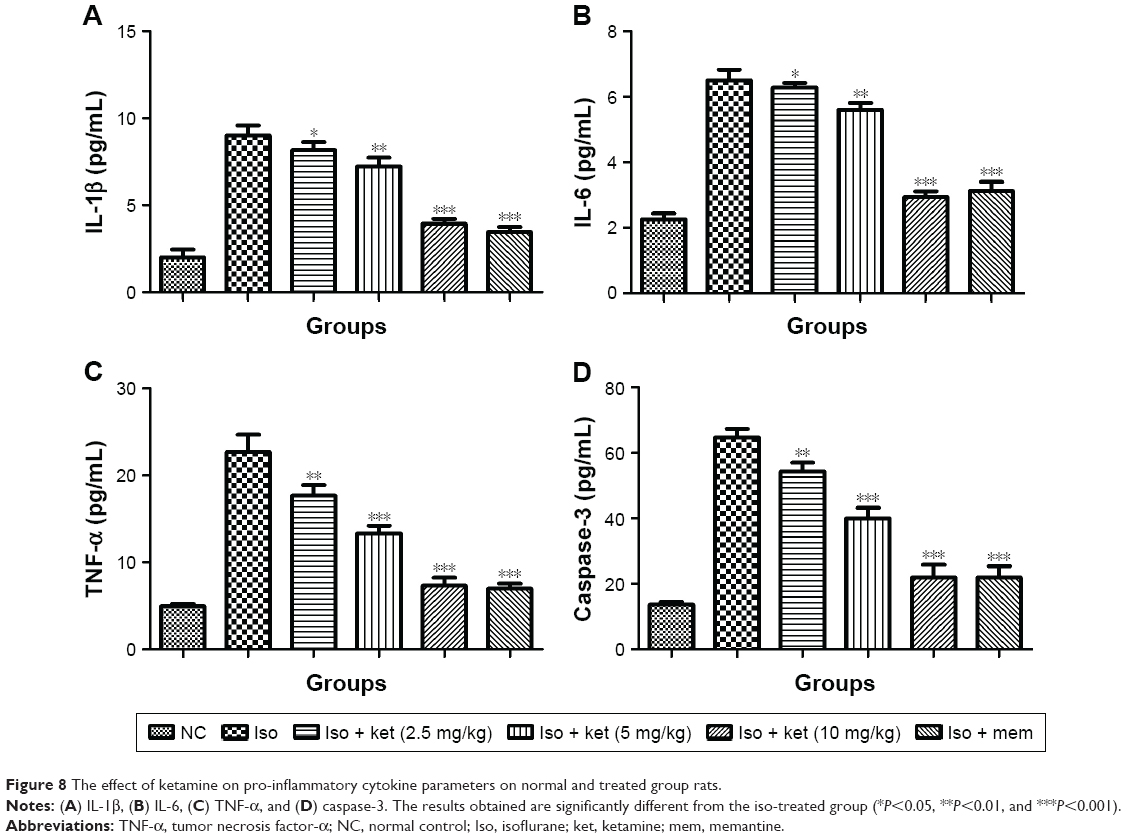 | Figure 8 The effect of ketamine on pro-inflammatory cytokine parameters on normal and treated group rats. |
A similar trend was observed in the caspase-3 level. The iso-induced group rats showed a caspase-3 level of 62.34±03.92 and the ketamine (10 mg/kg)-treated group showed levels of 22.45±1.84 (Figure 8D).
Effect on iso-induced histone de-acetylation
The previous research suggests that the histone acetylation is dysregulated in the hippocampus region of the brain of animals undergoing exposure to isoflurane. In the current study, we exposed the animals for a short time. For the confirmation of the same, we estimated the histone H3 lysine 9 (Ac-H3K9) and histone H4 lysine 12 (Ac-H4K12) in the hippocampus region of the rat after the isoflurane exposure. As presented in Figure 9A and B, rats exposed to the isoflurane showed less Ac-H3K9 and Ac-H4K12 concentration as compared to iso-induced group rats. The ketamine-treated group rats showed a reduction of histone deacetylation induced via isoflurane in a dose-dependent manner.
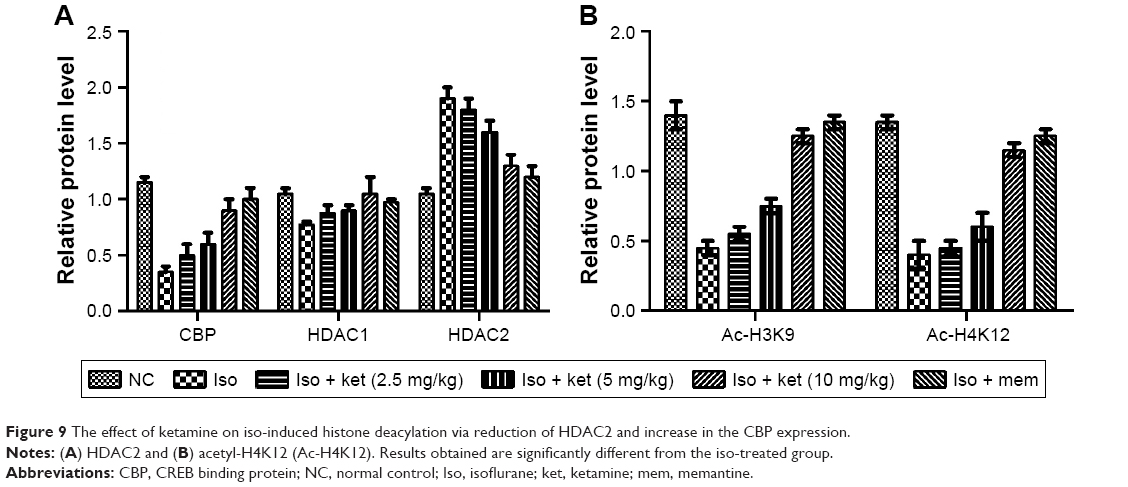 | Figure 9 The effect of ketamine on iso-induced histone deacylation via reduction of HDAC2 and increase in the CBP expression. |
For the confirmation, alteration in the histone acetylation, we examined that histone acetylation enzymes such as HDAC1, HDAC2, and lysine acetyltransferase CBP. As presented in Figure 9A and B, we observed that HDAC1 level was not altered by isoflurane and ketamine treatment. Likewise, the HDAC2 level was increased and dose-dependent treatment of ketamine.
Reduced level of HDAC2 and increased level of CBP were observed in the ketamine treatment. Our results clearly showed that the isoflurane-induced histone deacetylation due to reduction of CBP and increased HDAC2 and the dose-dependent treatment of ketamine significantly altered the effect of iso-induced histone acetylation.
Neuroprotective effect of ketamine on p-AKT/AKT, GSK-3β, p21WAF1/CIP1, and p53
Figure 10A shows the effect of ketamine on the p-AKT/AKT level of the different groups of rats. Iso-induced group rats showed a reduction in the level of p-AKT/AKT and dose-dependent treatment of ketamine exhibited the enhanced level of p-AKT/AKT.
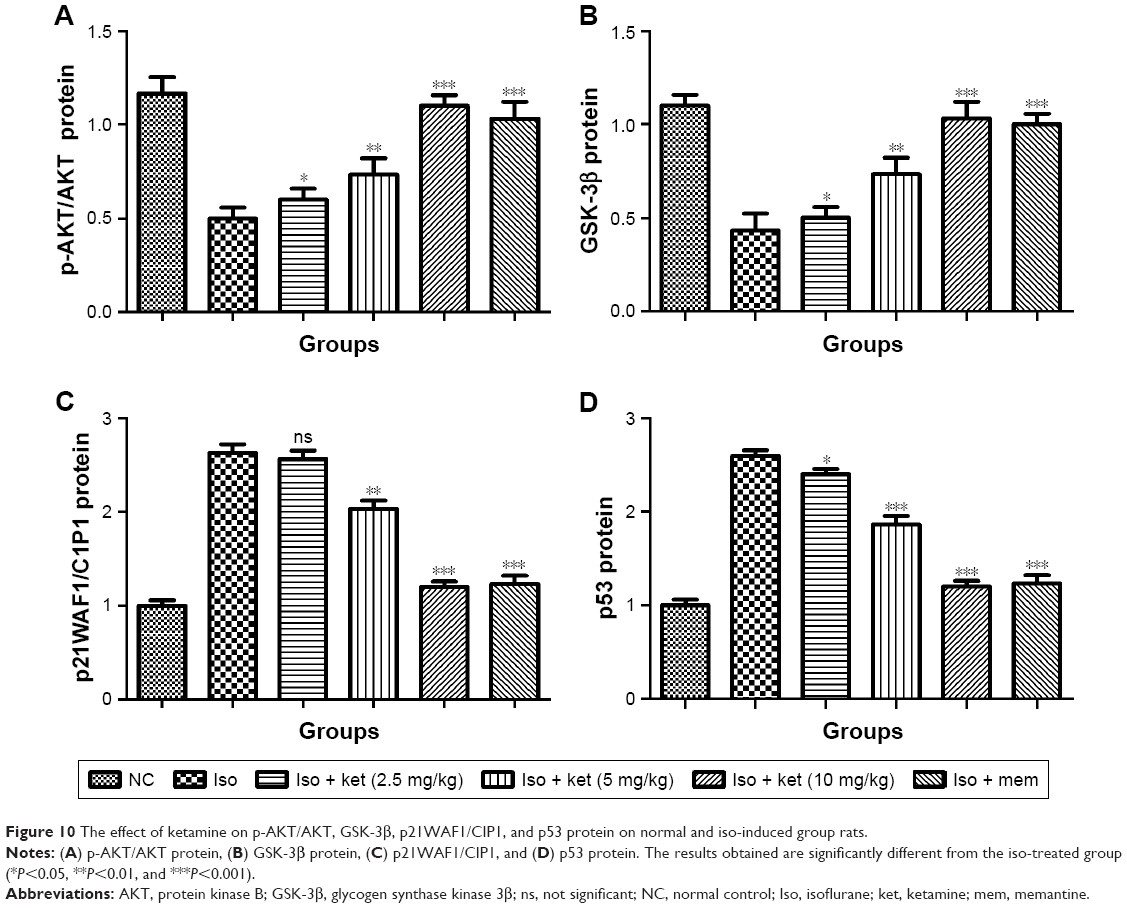 | Figure 10 The effect of ketamine on p-AKT/AKT, GSK-3β, p21WAF1/CIP1, and p53 protein on normal and iso-induced group rats. |
A similar result was observed in the expression of GSK 3β. Iso-induced group rats showed a suppressed level of GSK 3β and ketamine exhibited up-regulation in the expression of GSK 3β (Figure 10B).
An opposite trend was observed in the p21WAF1/CIP1. Iso-induced group rats demonstrated a higher expression of p21WAF1/CIP1 and dose-dependent treatment of ketamine showed a reduction in the level of p21WAF1/CIP1 (Figure 10C).
Iso group rats showed an increased level of p53 protein and dose-dependent treatment of ketamine significantly (P<0.001) reduced the expression of p53 protein (Figure 10D).
Discussion
AD is considered a neurodegenerative disease characterized via progressive deterioration of the hippocampal region of the brain and cortical neurons that start the destruction of cognitive and memory ability.27,28 Neurodegeneration in the neocortex and hippocampus is linked with spatial memory destruction.29 Various researchers have suggested that a deficiency in AChE plays a critical role in the pathogenesis of AD and its symptoms.30,31 Various researchers have suggested that it most commonly induces the symptom of dementia and its incidence increases with age.30–32 The first clinical symptoms of AD are the destruction of short-term memory and when the condition progresses additional cognitive capability is impaired such as the capacity to calculate and use common tools and objects. According to a report by Prince et al,33 the 2010 figure of 35.6 million individual cases of dementia worldwide will increase to almost double (65.7 million) by 2030 with almost four times more (115.4 million) cases by 2050.34,35 Almost, two-thirds of people with dementia live in lower- and middle-income countries. Inflammation plays an important role in the pathogenesis of AD and additionally accumulation of ROS and oxidative stress starts the reduction of endogenous antioxidant levels and are implicated in AD etiology. It is believed that oxidative stress plays a crucial role in the pathogenesis of AD and precedes prominent neuropathological alteration.36,37
Various researchers have targeted BDNF to find the best neurological treatment for AD. BDNF is a significant marker in the pathophysiology of neurological disease.38,39 The rodent model is an important tool for elucidation of the pathophysiology of AD. One of the best models of AD is the iso-induced AD model in which a central injection of isoflurane is made into the lateral ventricles, which are considered to exhibit a similar sporadic dementia to AD in humans.40 Therefore, the aim of the current study was to explore the neuroprotective effect of ketamine in an iso-induced AD model. On the contrary, Aβ peptide significantly increased in the hippocampal tissue in the iso-induced group and dose-dependent treatment of ketamine reduced the Aβ peptide in the hippocampal tissue due to clearance of Aβ peptide and contributes as the neuroprotective action of ketamine.
Free radical/oxidative stress plays an important role in the expansion of neuroinflammation and endogenous antioxidant systems.41 First line antioxidants such as SOD and CAT protect cell damage by scavenging the superoxide and hydrogen peroxide free radicals. On the other hand, GSH (endogenous) antioxidant enzymes are present in a reduced form in the cells. Normally, it reacts with the free radicals and stops the generation of harmful hydroxyl radicals. Iso-induced group rats showed a reduced level of GSH and GST and showed an increased level of free radical generation and oxidative stress. Ketamine treatment restores GSH and GST levels to normal.
Several researchers have suggested that the overproduction of pro-inflammatory cytokines such as IL-1β, IL-6, and TNF-α play a significant role in impairing cognitive function in the brain.9,42 Pro-inflammatory cytokines, such as TNF-α, start secreting after surgery. TNF-α also boosts the level of IL-1β in the production of the central nervous system. The increased level of IL-1β in the hippocampus region has been shown to lead the interference of long-term potentiating that underlies cognitive impairment. The increased level of IL-1β in the hippocampus region of the brain is linked with the reduction of long-term potentiating which finally induces the dysfunction of synaptic plasticity.43 It also inhibits the expression of BDNF mRNA in the CA1 and CA2, which are indirectly down-regulated by LTP and further induce cognitive function. It is believed that an increased level of IL-1β and TNF-α in the hippocampus are also responsible for postoperative cognitive dysfunction.44 Our experimental study showed that isoflurane leads to neurotoxicity connected with cognitive dysfunction in rats.
Several researchers believe that during injury the p53 gene is activated, and the expression of p53 mediates cell cycle arrest via activating a series of cells or via joining the single or double DNA stand to induce self-protection of DNA in the cell, it is generally located upstream and effects via DNA damage in cells.45–47 On the other hand, p21WAF1/CIP1 is the significant gene in maintaining the cell cycle via joining and controlling the cyclin-dependent kinase. During cell cycle arrest, p21WAF1/CIP1 is going to suppress p53-mediated apoptosis and also enhances cell apoptosis. In the current study, we found that ketamine reduced p21WAF1/CIP1 expression in iso-induced rats.
Caspase-3 (protease) is the key maker of apoptosis, widely used as an apoptotic detection indicator. Isoflurane-treated rats showed an increase in brain cell apoptosis, and prolonged treatment of anesthesia increased the number of apoptotic cells.48,49 The result of the current experimental study showed that a causal association between the drug treatment and brain cell apoptosis. Of note, ketamine exerts an anti-apoptotic effect in a concentration-dependent manner and reduced caspase-3 expression in iso-induced control group rats. In the current experimental study, ketamine reduced caspase-3 and neuroinflammation in rats with cerebral injury. Several researchers have suggested that the PI3K/AKT signaling pathway is a significant target for membrane receptor signal transduction into cells and it also suppresses cell apoptosis.50,51 GSK-3β (multifunctional serine/threonine protein kinase) involved in apoptosis, cell proliferation, and differentiation with the exception of glycometabolism. It is believed that the PI3K/AKT pathway is involved in the regulation of several gene expressions such as myeloid cell leukemia-1 (Mcl-1) in several types of tumor cells.52 AKT can phosphorylate B cell lymphoma-linked X protein and produces Mcl-1 in cytoplasm and heterodimer with Bcl, leading to a reduction of cell apoptosis and mitochondrial membrane translocation. GSK-3β can phosphorylate Mcl-1 to reduce the apoptosis induction and boost ubiquitination of Mcl-1. Additionally, AKT showed the effects of Mcl-1 stability via negative regulation of GSK-3β to suppress apoptosis. On the basis of this result, we can say that ketamine activated the PI3K/AKT/GSK-3β pathway in iso-induced rats.
Conclusion
Our study showed that ketamine exhibited a neuroprotective effect on isoflurane-induced cognitive impairment in rats via anti-inflammatory, antioxidant, and anti-apoptotic effects mediated via the PI3K/AKT/GSK-3β pathway in aging rats.
Disclosure
The authors report no conflicts of interest in this work.
References
Sanabria-Castro A, Alvarado-Echeverría I, Monge-Bonilla C. Molecular pathogenesis of Alzheimer’s disease: an update. Ann Neurosci. 2017;24(1):46–54. | ||
Mattson MP, Partin J, Begley JG. Amyloid beta-peptide induces apoptosis-related events in synapses and dendrites. Brain Res. 1998;807(1–2):167–176. | ||
Rubio-Perez JM, Morillas-Ruiz JM. A review: inflammatory process in Alzheimer’s disease, role of cytokines. Scientific World Journal. 2012:756357. | ||
Daniele SG, Béraud D, Davenport C, Cheng K, Yin H, Maguire-Zeiss KA. Activation of MyD88-dependent TLR1/2 signaling by misfolded α-synuclein, a protein linked to neurodegenerative disorders. Sci Signal. 2015;8(376):ra45. | ||
Ha SK, Moon E, Ju MS, Kim DH, Ryu JH, Oh MS, Kim SY. 6-Shogaol, a ginger product, modulates neuroinflammation: a new approach to neuroprotection. Neuropharmacology. 2012;63(2):211–223. | ||
González H, Elgueta D, Montoya A, Pacheco R. Neuroimmune regulation of microglial activity involved in neuroinflammation and neurodegenerative diseases. J Neuroimmunol. 2014;274(1–2):1–13. | ||
Dobos N, Korf J, Luiten PG, Eisel UL. Neuroinflammation in Alzheimer’s disease and major depression. Biol Psychiatry. 2010;67(6):503–504. | ||
Baj T, Seth R. Role of curcumin in regulation of TNF-α mediated brain inflammatory responses. Recent Pat Inflamm Allergy Drug Discov. 2018;12(1):69–77. | ||
Singhal G, Jaehne EJ, Corrigan F, Toben C, Baune BT. Inflammasomes in neuroinflammation and changes in brain function: a focused review. Front Neurosci. 2014;8:315. | ||
Cao XZ, Ma H, Wang JK, et al. Postoperative cognitive deficits and neuroinflammation in the hippocampus triggered by surgical trauma are exacerbated in aged rats. Prog Neuropsychopharmacol Biol Psychiatry. 2010;34(8):1426–1432. | ||
More SV, Kumar H, Kim IS, Song S-Y, Choi D-K. Cellular and molecular mediators of neuroinflammation in the pathogenesis of Parkinson’s disease. Mediators Inflamm. 2013;2013:952375. | ||
Meijer L, Flajolet M, Greengard P. Pharmacological inhibitors of glycogen synthase kinase 3. Trends Pharmacol Sci. 2004;25(9):471–480. | ||
Yao HB, Shaw PC, Wong CC, Wan DC. Expression of glycogen synthase kinase-3 isoforms in mouse tissues and their transcription in the brain. J Chem Neuroanat. 2002;23(4):291–297. | ||
Azoulay-Alfaguter I, Yaffe Y, Licht-Murava A, et al. Distinct molecular regulation of glycogen synthase kinase-3alpha isozyme controlled by its N-terminal region: functional role in calcium/calpain signaling. J Biol Chem. 2011;286(15):13470–13480. | ||
Li DW, Liu ZQ, Chen W, Yao M, Li GR. Association of glycogen synthase kinase-3β with Parkinson’s disease (review). Mol Med Rep. 2014;9(6):2043–2050. | ||
Shih RH, Wang CY, Yang CM. NF-kappaB signaling pathways in neurological inflammation: a mini review. Front Mol Neurosci. 2015;8:77. | ||
Mazoit JX, Dalens BJ. Pharmacokinetics of local anaesthetics in infants and children. Clin Pharmacokinet. 2004;43(1):17–32. | ||
Auriel E, Regev K, Korczyn AD. Nonsteroidal anti-inflammatory drugs exposure and the central nervous system. Handb Clin Neurol. 2014;119:577–584. | ||
Gao M, Rejaei D, Liu H. Ketamine use in current clinical practice. Acta Pharmacol Sin. 2016;37(7):865–872. | ||
Schmidt M, Sachse C, Richter W, Xu C, Fändrich M, Grigorieff N. Comparison of Alzheimer Abeta(1-40) and Abeta(1-42) amyloid fibrils reveals similar protofilament structures. Proc Natl Acad Sci U S A. 2009;106(47):19813–19818. | ||
Bhatt PC, Pathak S, Kumar V, Panda BP. Attenuation of neurobehavioral and neurochemical abnormalities in animal model of cognitive deficits of Alzheimer’s disease by fermented soybean nanonutraceutical. Inflammopharmacology. 2018;26(1):105–118. | ||
Zhang J, Dong Y, Xu Z, et al. 2-Deoxy-D-glucose attenuates isoflurane-induced cytotoxicity in an in vitro cell culture model of H4 human neuroglioma cells. Anesth Analg. 2011;113(6):1468–1475. | ||
Falls N, Singh D, Anwar F, Verma A, Kumar V. Amelioration of neurodegeneration and cognitive impairment by lemon oil in experimental model of stressed mice. Biomed Pharmacother. 2018;106:575–583. | ||
Newman JP, Kosson DS. Passive avoidance learning in psychopathic and nonpsychopathic offenders. J Abnorm Psychol. 1986;95(3):252–256. | ||
Weydert CJ, Cullen JJ. Measurement of superoxide dismutase, catalase and glutathione peroxidase in cultured cells and tissue. Nat Protoc. 2010;5(1):51–66. | ||
Haring M, Offermann S, Danker T, Horst I, Peterhansel C, Stam M. Chromatin immunoprecipitation: optimization, quantitative analysis and data normalization. Plant Methods. 2007;3:11. | ||
Ramos Bernardes da Silva Filho S, Oliveira Barbosa JH, Rondinoni C, et al. Neuro-degeneration profile of Alzheimer’s patients: a brain morphometry study. Neuroimage Clin. 2017;15:15–24. | ||
Salek RM, Xia J, Innes A, et al. A metabolomic study of the CRND8 transgenic mouse model of Alzheimer’s disease. Neurochem Int. 2010;56(8):937–947. | ||
Rudnitskaya EA, Muraleva NA, Maksimova KY, Kiseleva E, Kolosova NG, Stefanova NA. Melatonin attenuates memory impairment, amyloid-β accumulation, and neurodegeneration in a rat model of sporadic Alzheimer’s disease. J Alzheimers Dis. 2015;47(1):103–116. | ||
Maccioni RB, Rojo LE, Fernández JA, Kuljis RO. The role of neuroimmunomodulation in Alzheimer’s disease. Ann N Y Acad Sci. 2009;1153:240–246. | ||
Tansey MG, Goldberg MS. Neuroinflammation in Parkinson’s disease: its role in neuronal death and implications for therapeutic intervention. Neurobiol Dis. 2010;37(3):510–518. | ||
Hsiao HY, Chen YC, Chen HM, Tu PH, Chern Y. A critical role of astrocyte-mediated nuclear factor-κB-dependent inflammation in Huntington’s disease. Hum Mol Genet. 2013;22(9):1826–1842. | ||
Prince M, Bryce R, Albanese E, Wimo A, Ribeiro W, Ferri CP. The global prevalence of dementia: a systematic review and metaanalysis. Alzheimers Dement. 2013;9(1):63–75. | ||
Prince M, Comas-Herrera A, Knapp M, Guerchet M, Karagiannidou M. World Alzheimer Report 2016: Improving Healthcare for People Living with Dementia. Coverage, Quality and Costs now and in the Future. Commissioned Report by King’s College, London, UK. London: Alzheimer’s Disease International; 2016:131. | ||
Javier F, Cobos M, del Mar MM. A review of psychological intervention in Alzheimer’s disease. Int J Psychol Psychol Ther. 2012;12(3):373–388. | ||
Ivanov AV, Bartosch B, Isaguliants MG. Oxidative stress in infection and consequent disease. Oxid Med Cell Longev. 2017;2017:3496043. | ||
Castillo WO, Aristizabal-Pachon AF. Galantamine protects against beta amyloid peptide-induced DNA damage in a model for Alzheimer’s disease. Neural Regen Res. 2017;12(6):916–917. | ||
Diniz BS, Teixeira AL. Brain-derived neurotrophic factor and Alzheimer’s disease: physiopathology and beyond. Neuromolecular Med. 2011;13(4):217–222. | ||
Fumagalli F, Racagni G, Riva MA. The expanding role of BDNF: a therapeutic target for Alzheimer’s disease? Pharmacogenomics J. 2006;6(1):8–15. | ||
Jebelli JD, Piers TM. Amyloid-β oligomers unveil a novel primate model of sporadic Alzheimer’s disease. Front Neurosci. 2015;9:47. | ||
Pisoschi AM, Pop A. The role of antioxidants in the chemistry of oxidative stress: a review. Eur J Med Chem. 2015;97:55–74. | ||
Stenvinkel P, Ketteler M, Johnson RJ, et al. IL-10, IL-6, and TNF-alpha: central factors in the altered cytokine network of uremia – the good, the bad, and the ugly. Kidney Int. 2005;67(4):1216–1233. | ||
O’Connor JJ. Neuroimmunology and synaptic function. Neuropharmacology. 2015;96(Pt A):1–2. | ||
Ludwig A. Mechanisms of KCC2 Upregulation during Development [dissertation]. Helsinki: University of Helsinki; 2008. | ||
Chen J. The cell-cycle arrest and apoptotic functions of p53 in tumor initiation and progression cold spring harb perspect med. 2016;6(3):a026104. | ||
Noteborn MH. Chicken anemia virus induced apoptosis: underlying molecular mechanisms. Vet Microbiol. 2004;98(2):89–94. | ||
Chang WL, Chapkin RS, Lupton JR. Fish oil blocks azoxymethane-induced rat colon tumorigenesis by increasing cell differentiation and apoptosis rather than decreasing cell proliferation. J Nutr. 1998;128(3):491–497. | ||
Rodríguez-Feo JA, Fortes J, Aceituno E, et al. Doxazosin modifies Bcl-2 and Bax protein expression in the left ventricle of spontaneously hypertensive rats. J Hypertens. 2000;18(3):307–315. | ||
Paraiso KHT, Van Der Kooi K, Messina JL, Smalley KSM. Measurement of constitutive MAPK and PI3K/AKT signaling activity in human cancer cell lines. Methods Enzymol. 2010;484:549–567. | ||
Porta C, Paglino C, Mosca A. Targeting PI3K/Akt/mTOR signaling in cancer. Front Oncol. 2014;4:64. | ||
Zhang Y, Zhang Z, Wang H, et al. Neuroprotective effect of ginsenoside Rg1 prevents cognitive impairment induced by isoflurane anesthesia in aged rats via antioxidant, anti-inflammatory and anti-apoptotic effects mediated by the PI3K/AKT/GSK-3β pathway. Mol Med Rep. 2016;14(3):2778–2784. | ||
Spokoini R, Kfir-Erenfeld S, Yefenof E, Sionov RV. Glycogen synthase kinase-3 plays a central role in mediating glucocorticoid-induced apoptosis. Mol Endocrinol. 2010;24(6):1136–1150. | ||
Altman FP. Studies on the reduction of tetrazodium salts – III. The products of chemical and enzymic reduction. Histochemistry. 1974;38(2):155–171. | ||
Zhang X, Jiang D, Jiang W, Zhao M, Gan J. Role of TLR4-Mediated PI3K/AKT/GSK-3b signaling pathway in apoptosis of rat hepatocytes. Biomed Res Int. 2015;2015:631326. | ||
Kennaway NG, Carrero-Valenzuela RD, Ewart G, et al. Isoforms of mammalian cytochrome c oxidase: correlation with human cytochrome c oxidase deficiency. Pediatr Res. 1990;28(5):529–535. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.