")
Back to Journals » Drug Design, Development and Therapy » Volume 9
Neuroprotective and antioxidant effects of ghrelin in an experimental glaucoma model
Authors Can N, Catak O, Turgut B, Demir T, Ilhan N , Kuloglu T, Ozercan IH
Received 18 February 2015
Accepted for publication 23 March 2015
Published 2 June 2015 Volume 2015:9 Pages 2819—2829
DOI https://doi.org/10.2147/DDDT.S83067
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Shu-Feng Zhou
Nagehan Can,1 Onur Catak,2 Burak Turgut,2 Tamer Demir,2 Nevin Ilhan,3 Tuncay Kuloglu,4 Ibrahim Hanifi Ozercan5
1Department of Ophthalmology, Elaziğ Training and Research Hospital, 2Department of Ophthalmology, 3Department of Biochemistry, 4Department of Histology and Embryology, 5Department of Pathology, School of Medicine, Firat University, Elaziğ, Turkey
Abstract: Damage to retinal ganglion cells due to elevation of intraocular pressure (IOP) is responsible for vision loss in glaucoma. Given that loss of these cells is irreversible, neuroprotection is crucial in the treatment of glaucoma. In this study, we investigated the possible antioxidant and neuroprotective effects of ghrelin on the retina in an experimental glaucoma model. Twenty-one Sprague–Dawley rats were randomly assigned to three groups comprising seven rats each. The rats in the control group were not operated on and did not receive any treatment. In all rats in the other groups, IOP was increased by cauterization of the limbal veins. After creation of the IOP increase, saline 1 mL/kg or ghrelin 40 µg/kg was administered intraperitoneally every day for 14 days in the vehicle control group and ghrelin groups, respectively. On day 14 of the study, the eyes were enucleated. Levels of malondialdehyde (MDA), nitric oxide (NO), and nitric oxide synthase-2 (NOS2) in anterior chamber fluid were measured. The retinas were subjected to immunohistochemistry staining for glial fibrillary acidic protein (GFAP), S-100, and vimentin expression. Mean levels of MDA, NO, and NOS2 in the aqueous humor were higher in the vehicle control group than in the control group (P<0.05). Mean levels of MDA, NO, and NOS2 in the ghrelin group did not show a significant increase compared with levels in the control group (P>0.05). Retinal TUNEL and immunohistochemistry staining in the vehicle control group showed an increase in apoptosis and expression of GFAP, S-100, and vimentin compared with the control group (P<0.05). In the ghrelin group, apoptosis and expression of GFAP, S-100, and vimentin was significantly lower than in the vehicle control group (P<0.05). This study suggests that ghrelin has antioxidant and neuroprotective effects on the retina in an experimental glaucoma model.
Keywords: experimental glaucoma, ganglion cells, ghrelin, neuroprotective, antioxidant
Introduction
Glaucoma is a multifactorial, progressive optic neuropathy characterized by death of retinal ganglion cells (RGCs), visual field loss, and excavation of the optic nerve head. Elevated intraocular pressure (IOP) is considered to be one of the important factors in initiation or progression of glaucoma and in the loss of RGCs. Characteristic glaucomatous changes in the retinal layers are thinning in the retinal nerve fiber layer and a decrease in the number of RGCs.1 RGCs are the retinal cells that are most sensitive to IOP elevation, and damage to these cells is responsible for vision loss in glaucoma. Given that loss of RGCs is irreversible, neuroprotection is crucial in the treatment of glaucoma.2
Ghrelin, first described by Kojima et al is a hormone with a polypeptide structure and is synthesized by a number of types of tissue and by many inflammatory cells, in particular by enteroendocrine cells.3–5 Ghrelin also has antioxidant properties,6,7 and induces appetite, lipogenesis, and secretion of growth hormone. Studies in rat eyes have demonstrated the presence of ghrelin mRNA in the anterior chamber. Rocha-Sousa et al also found ghrelin in the anterior chamber of the human eye.8 In another study, Katsanos et al established that ghrelin levels in the anterior chamber of patients with glaucoma were significantly lower than levels in controls.9 Erşahin et al showed that ghrelin had neuroprotective effects in rats with induced oxidative brain damage.10 Thus, we believe that ghrelin could have neuroprotective effects in retinas with glaucomatous damage. A search of the PubMed database did not reveal any previous research concerning the neuroprotective effects of ghrelin in an experimental glaucoma model. In this study, we investigated the potential neuroprotective and antioxidant effects of ghrelin on the retina in a rat model of glaucoma.
Materials and methods
Animals and study ethics
The study included 21 Sprague–Dawley rats of mean weight 250 g and aged 2–3 months. Throughout the study, the rats were maintained in the experimental research center at Firat University. The animals were housed in wire-bottomed cages at room temperature and on a 12-hour light–dark cycle. All were fed with standard rat chow, but were given only water 12 hours before surgery.
With approval from the Firat University Ethics Committee, Elaziğ, Turkey, the study was carried out using one eye from each animal. All procedures were performed with strict adherence to the guidelines for animal care and experimentation as prepared by the Association for Research in Vision and Ophthalmology and Guidelines for the Housing of Rats in Scientific Institutions.
Groups
The rats were randomly assigned to three groups, with seven rats in each group. Group 1 (controls) included rats that were not operated on and did not receive any treatment. Group 2 (vehicle controls) included rats in which induction of an IOP increase was performed and which received saline 1 mL/kg via the intraperitoneal route each day for 14 days. Group 3 (ghrelin group) included rats in which induction of an IOP increase was performed and which received ghrelin 40 μg/kg via the intraperitoneal route each day for 14 days. On day 14, the eyes were enucleated after induction of analgesia and anesthesia.
Anesthetic technique
The rats were injected with a combination of intramuscular ketamine hydrochloride 50 mg/kg (Ketalar, Eczacibaşi, Turkey) and xylazine hydrochloride 5 mg/kg (Rompun, Bayer, Turkey) to induce anesthesia and analgesia. Proparacaine hydrochloride 1% was administered as a topical anesthetic to both eyes of each animal prior to surgical intervention.
Induction of IOP elevation
After induction of anesthesia and analgesia, the episcleral veins, including the three branches rooted from the limbal veins, with the exception of one placed at the nasal quadrant, were cauterized using unipolar ophthalmic cautery, and ocular hypertension was induced. The eyes were then washed with saline, and antibiotic drops were administered.
Intraocular pressure measurement
A Tono-Pen tonometer was used to perform accurate, repeatable, and noninvasive IOP measurements in the rats. On average, ten measurements with a percent age error of less than 5% were obtained. IOP measurements were performed before surgery and on postoperative days 5 and 10, following instillation of 1% proparacaine hydrochloride.
Histopathologic preparation
Analgesia and anesthesia were administered to the animals before enucleating the eyes. Samples of aqueous humor from the eyes of the sacrificed rats were obtained by aspiration using a 27-gauge needle, and sent to the biochemistry laboratory to measure malondialdehyde (MDA) and nitric oxide (NO) levels. Iris and ciliary body samples were also obtained and sent to the biochemistry laboratory for measurement of nitric oxide synthase-2 (NOS2) levels. Retinal and optic nerve dissection was performed on the remaining posterior segments of the globe and the specimens were sent to the pathology laboratory to determine expression of glial fibrillary acidic protein (GFAP), vimentin, and S-100 proteins by immunohistochemistry and expression of apoptosis by TUNEL (terminal deoxyribonucleotidyl transferase-mediated dUTP-biotin end labeling) assay.
Determination of MDA, NO, and NOS2 levels
Equal amounts of aqueous humor were obtained from the anterior chamber of each eye. Levels of MDA, as an indicator of lipid peroxidation, were analyzed using an MDA kit (Immuchrom GmbH, Hessen, Germany) with high performance liquid chromatography. After transforming the MDA into fluorescent products with a derivatization reagent, the reaction solution was added to achieve an optimum pH level. MDA-generated fluorescence was measured in the isocratic high performance liquid chromatography system with a spectrofluorometer detector at 553 nm (emission) and 515 nm (excitation). MDA levels in the samples were given as μmol/L.
NO levels were determined using an enzyme-linked immunosorbent assay device with a nitrite/nitrate calorimetric assay kit (Cayman Chemicals, Ann Arbor, MI, USA).
For Western blot analysis, frozen ciliary samples were weighed and dissolved in RIPA lysis buffer containing protease and phosphatase inhibitors. First, 3 mL of RIPA lysis buffer was used per gram of tissue. Then, 10 μL from the phenylmethylsulfonyl fluoride, 10 μL from sodium orthovanadate, and 10 μL from protease inhibitor cocktail were added per gram of tissue. All procedures were carried out at 4°C; in accordance with the manufacturer’s instructions, a Bullet Blender tissue homogenizer (Next Advanced Inc, Averill Park, NY, USA) was then used to complete the homogenization process. Homogenized samples were centrifuged at 4°C and 10,000× g for 10 minutes to obtain the supernatant and then recentrifuged to form a clear lysate. Samples were stored at −80°C. The amount of protein in the samples was determined with a Qubit fluorometer using a Quant-iT™ protein kit. A chromogenic detection method was used to load 50 μg of protein into the protein gel to prepare the samples. In addition to the sample added to the microcentrifuge tube, 2.5 μL of NuPAGE® lithium dodecyl sulfate sample buffer (4×) and 1 μL of NuPAGE reducing agent (10×) were added and denatured at 70°C for 10 minutes. The denatured sample was chilled on ice. SeeBlue® Plus2 was added to the first well in the Surelock X Cell vertical gel system (Invitrogen, Thermo Fisher Scientific, Waltham, MA, USA), and the prepared samples were loaded into the other wells. The electrophoresis process was complete after conducting the gel at 100 V for 35 minutes. After electrophoresis, gel casette was opened and gel was transferred into distilled water carefully. The proteins were transferred to PVDF membranes using an iBlot® dry blotting system to complete the blotting process, and then incubated for 1 hour in a 1:200 dilution of the anti-NOS2 polyclonal antibody (rabbit IgG, obtained from Cruz Biotechnology, Santa Cruz, CA) at 4°C Membranes were washed with antibody wash solution and then incubated for 30 minutes with prepared secondary antibody solution. The membranes were washed, and the protein bands were visualized by chromogenic substrates.
TUNEL and retinal immunohistochemistry staining
Apoptotic cell screening
Sections 5 μm thick were taken from the paraffin blocks and placed on slides with polylysine. In accordance with the manufacturer’s instructions, an ApopTag® Plus Peroxidase in situ apoptosis detection kit (Chemicon, Temecula, CA, USA) was used to identify apoptotic cells. Tissue deparaffinized with xylene was put through a series of graded alcohols for dehydration and then washed with phosphate-buffered saline. The tissue that was incubated with 0.05% proteinase K for 10 minutes was incubated with 3% hydrogen peroxide to prevent the activity of endogenous peroxidase. After washing, the tissue was incubated for 6 minutes with equilibration buffer and for 60 minutes with a 3°C moist-setting operation solution (70%/30% TdT mu l enzyme reaction buffer). The tissue was held in stop/wash buffer for 10 minutes and then incubated with antidigoxigenin peroxidase. Apoptotic cells were visualized using a diaminobenzidine substrate. The sections were cross-stained with Harris hematoxylin. Breast tissue was used as a positive control. Reaction buffer was used instead of TdT enzyme in the negative control tissue. The preparations were observed using a research microscope (BX50, Olympus, Tokyo, Japan) and photographed. In the TUNEL assay, nuclei stained blue with Harris hematoxylin were considered normal, and cells with brown nuclear staining were deemed apoptotic. The assessment of TUNEL staining was performed based on the extent of the staining of apoptotic cells. The extent of TUNEL staining was scored semiquantitatively as 0 (no), 1 (light), 2 (medium), and 3 (intense).
GFAP immunohistochemistry staining
Sections (4 μm thick) that passed through the retina and optic disc were prepared from paraffin blocks for immunohistochemical staining. The sections were stained using a GFAP kit (Lifespan BioSciences, Seattle, WA, USA) with an automatic immunohistochemistry staining instrument (Benchmark XT, Ventana Medical Systems, Oro Valley, AZ, USA). The preparations were covered with special sealing material and randomly examined with an Olympus light microscope. Using an Olympus micrograph attachment, photographs were taken of the tissue at a magnification of 40×. Nuclear positivity was evaluated as weak (+), moderate (++), or strong (+++).
S-100 immunohistochemical staining
Sections (4 μm thick) that passed through the retina and optic disc were prepared from paraffin blocks for immunohistochemical staining to show S-100 immune staining patterns. The sections were stained with an S-100 kit (Lifespan BioSciences) using an automatic immunohistochemistry staining device (Ventana Medical Systems). The preparations were covered with special sealing material and randomly examined with an Olympus light microscope. Using an Olympus microscope photographic attachment, photographs were taken of the tissue at a magnification of 40×. Nuclear positivity was evaluated as weak (+), moderate (++), or strong (+++).
Vimentin immunohistochemistry staining
Sections (4 μm thick) that passed through the retina and optic disc were prepared from paraffin blocks for immunohistochemical staining. The sections were stained using a vimentin kit (Lifespan BioSciences) with an automatic immunohistochemistry staining device (Ventana Medical Systems). The preparations were covered with special sealing material and randomly examined with an Olympus light microscope. Using an Olympus micrographic attachment, photographs were taken of the tissue at a magnification of 40×. Nuclear positivity was evaluated as weak (+), moderate (++), or strong (+++).
Statistical analysis
The statistical analysis was performed using Statistical Package for the Social Sciences version 16.0 software (SPSS Inc, Chicago, IL, USA). All data are presented as the mean ± standard deviation. P<0.05 was considered to be statistically significant. For data conforming to a normal distribution, parametric single-direction variance analysis and Tukey’s post hoc test were applied. For data that did not conform to a normal distribution, the nonparametric Kruskal–Wallis analysis of variance was used, followed by the Mann–Whitney U-test. In addition, the Wilcoxon test for related variables was applied.
Results
Intraocular pressures
The mean IOPs in the study groups on days 0, 5, and 10 are shown in Table 1. In the vehicle control and ghrelin groups, the increase in IOP following cauterization was statistically significant (P<0.05, see Table 1). The change in IOP in the control group was not statistically significant (P>0.05). The increase in IOP in the vehicle control group was statistically significant when compared with the control group (P<0.05). In the vehicle control group, the differences between preoperative IOP and the IOP on postoperative days 5 and 10 were statistically significant (P<0.05).
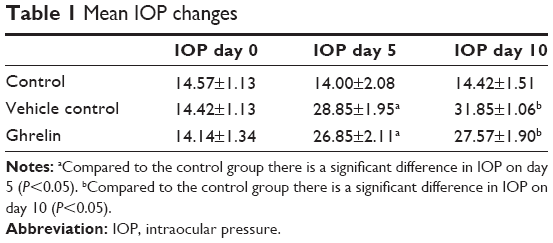 | Table 1 Mean IOP changes |
MDA levels
Mean MDA levels in aqueous humor for the study groups are shown in Table 2. Mean MDA levels in the vehicle control group were significantly higher (P<0.01) than in the control group. Mean MDA levels in the ghrelin group were significantly lower than in the vehicle control group (P<0.01). When mean MDA levels in the ghrelin and control groups were compared, there was no statistically significant difference between the two groups (P>0.05).
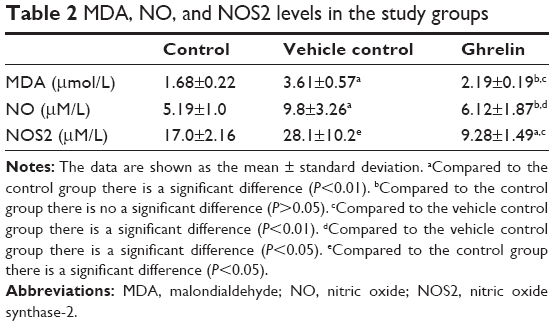 | Table 2 MDA, NO, and NOS2 levels in the study groups |
NO levels
Mean NO levels in aqueous humor for the study groups are shown in Table 2. Mean NO levels in the vehicle control group were significantly increased (P<0.01) compared with the control group. There was no significant difference in NO levels between the ghrelin group and the control group (P>0.05). The mean NO level in the ghrelin group was significantly lower than that in the vehicle control group (P<0.05).
NOS2 levels
Mean NOS2 levels in the study groups are shown in Table 2. The mean NOS2 level in the vehicle control group was significantly higher than that in the control group (P<0.05). However, the mean NOS2 level in the ghrelin group was significantly lower than that in the control group (P<0.01). The mean NOS2 level in the ghrelin group was significantly lower than that in the vehicle control group (P<0.01).
Retinal immunohistochemistry and TUNEL staining
Apoptosis
Microphotographs showing TUNEL staining for retinal apoptosis from an animal in each study group are shown in Figure 1. TUNEL positivity was observed to be weak in the control group (Figure 1A). When compared with the control group, TUNEL positivity in the vehicle control group was strong (Figure 1B). More apoptotic cells were seen in the outer and inner plexiform layers (OPL and IPL, respectively). The ghrelin group showed significantly decreased apoptosis compared with the vehicle control group, however, retinal apoptosis in the ghrelin group was close to that in the control group (Figure 1C). When retinal sections were examined, TUNEL-positive RGC layers were observed in the IPL and inner nuclear layer (INL). Compared with the control group, TUNEL staining was significantly increased in the vehicle control group (P<0.05). In the group treated with ghrelin, TUNEL positivity was significantly decreased (P<0.05).
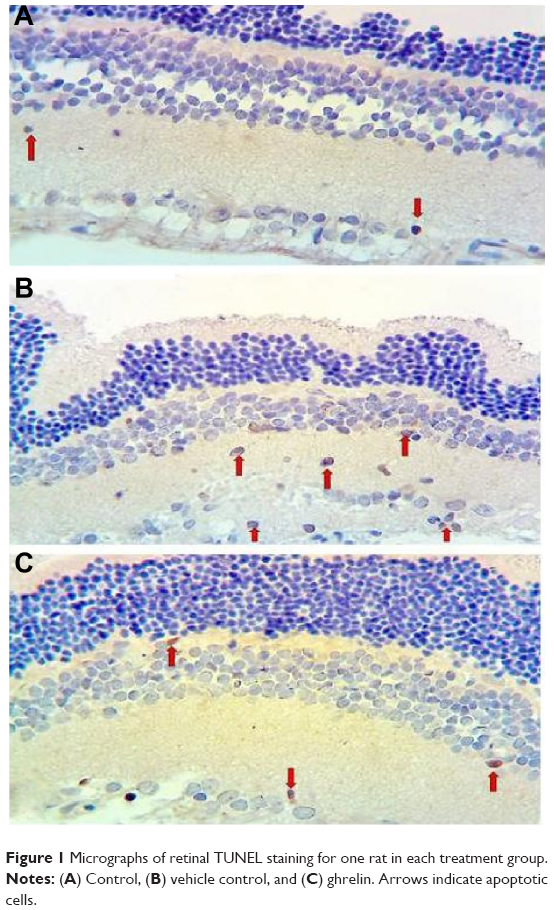 | Figure 1 Micrographs of retinal TUNEL staining for one rat in each treatment group. |
Retinal hematoxylin and eosin staining
Retinal thinning under pressure as a result of increased IOP can be observed on light microscopy in a retina stained with hematoxylin and eosin. Thinning was observed particularly in the INL. In the sections of samples taken from the control group, there is a visible increase in thickness in the INL (Figure 2A). INL thinning in the vehicle control group is marked in comparison with that in the control group (Figure 2B). Staining in the ghrelin group was similar to that in the control group (Figure 2C).
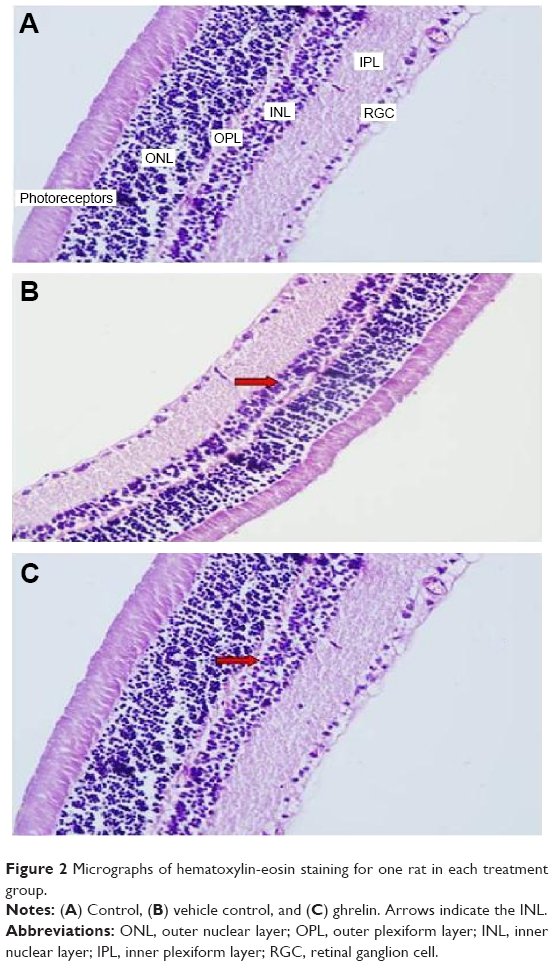 | Figure 2 Micrographs of hematoxylin-eosin staining for one rat in each treatment group. |
Retinal GFAP staining
Immunohistochemistry staining for retinal GFAP on light microscopy is shown for a sample from each group (Figure 3). It is noteworthy that both Müller cells (MCs) and MC processes showed GFAP immunoreactivity. GFAP positivity was observed to be +1 in the control group (Figure 3A). When compared with the control group, the vehicle control group (Figure 3B) had a significant increase in retinal GFAP immunoreactivity, which was observed to be +++ (P<0.05). Staining in the ghrelin group was significantly lower than that in the vehicle control group (P<0.05), and was close to the control group, with a prevalence of ++ observed (Figure 3C). In the control group, retinal staining did not progress to the outer segments; in addition, there was a small amount of staining detected in the RGC, IPL, INL, and OPL. In the vehicle control animals, in which glaucoma was induced, GFAP expression was increased significantly (P<0.05) and this increase was generally concentrated in the IPL (Figure 3B). It is noteworthy that MCs and MC processes were concentrated around the RGC, INL, OPL, and outer nuclear layer (ONL) in this group (Figure 3B). Immunoreactive GFAP cells and processes were observed throughout all retinal layers in the vehicle control group. The ONL in these cells was seen to extend into the outer segment and even appear in the photoreceptor layer (Figure 3B). In the ghrelin group, staining was more similar to that in the control group (Figure 3C) and did not reach the photoreceptor layer. Immunoreactive GFAP cells in the ghrelin group were observed to extend out to RGCs and the IPL, INL, and OPL.
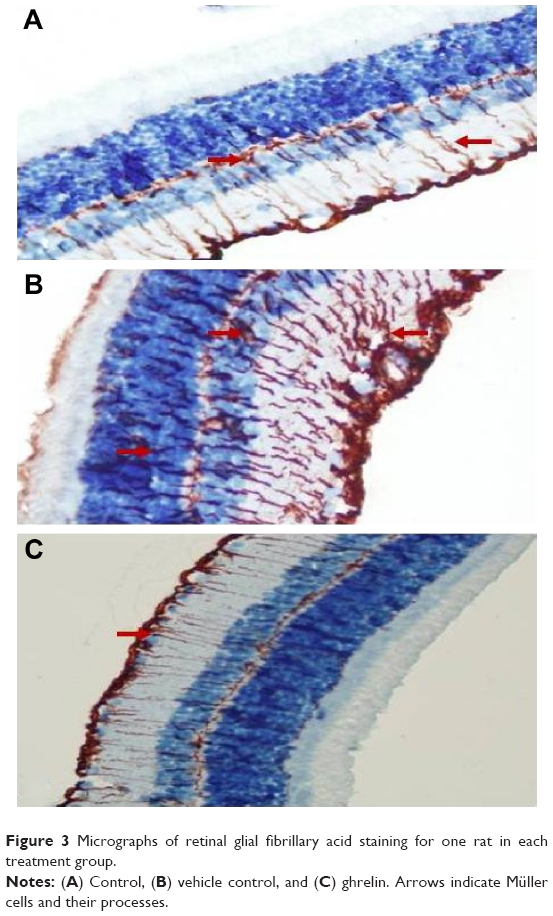 | Figure 3 Micrographs of retinal glial fibrillary acid staining for one rat in each treatment group. |
Retinal S-100 staining
Immunohistochemistry staining for retinal GS-100 on light microscopy is shown for a sample from each group. MCs showed S-100 immunoreactivity. In samples taken from the control group, S-100 expression was observed in MCs from the INL (Figure 4A). Of note, there was a small amount of S-100 staining in the RGC layer. S-100 staining in the control group was evaluated as +1.
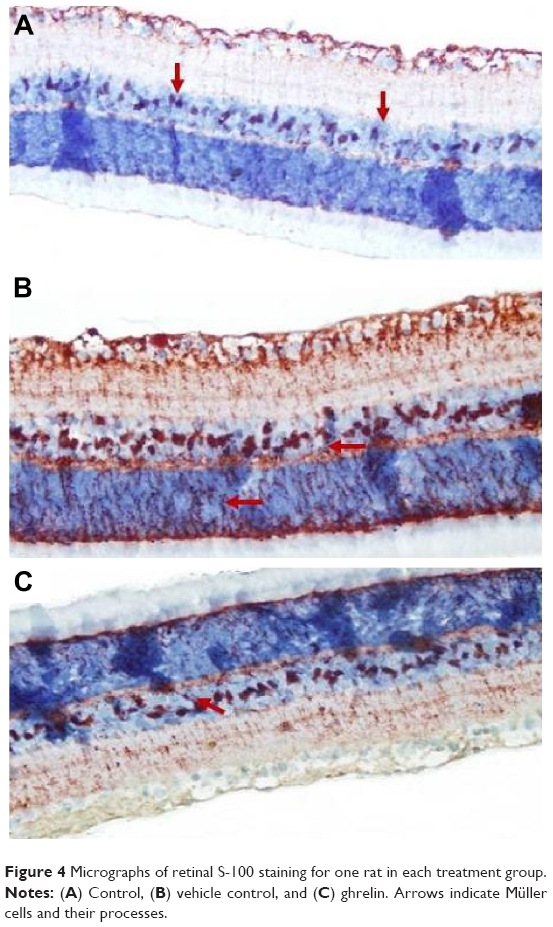 | Figure 4 Micrographs of retinal S-100 staining for one rat in each treatment group. |
There was a statistically significant increase in S-100 expression (prevalence +++) in the vehicle control group when compared with the control group (P<0.05). In the INL, there was not only an increase in MCs, but also immunoreactivity of S-100 extending to the IPL and ONL (Figure 4B).
A statistically significant increase (++) in S-100 immunoreactivity of MCs in the INL was observed in the ghrelin group (P<0.05). Less staining was seen in the IPL and ONL (Figure 4C).
Retinal vimentin staining
Immunohistochemistry staining for retinal vimentin on light microscopy is shown for a sample from each group, and is noteworthy in the MCs and MC processes. In the control group, retinal vimentin expression was observed in RGCs and in the IPL and OPL. Vimentin expression was also seen in the MC processes of the INL, in which the prevalence of immunohistochemistry staining for vimentin was +1 (Figure 5A). Compared with the control group, the prevalence of immunohistochemistry staining for retinal vimentin (+++) was significantly increased in the vehicle control group (Figure 5B).
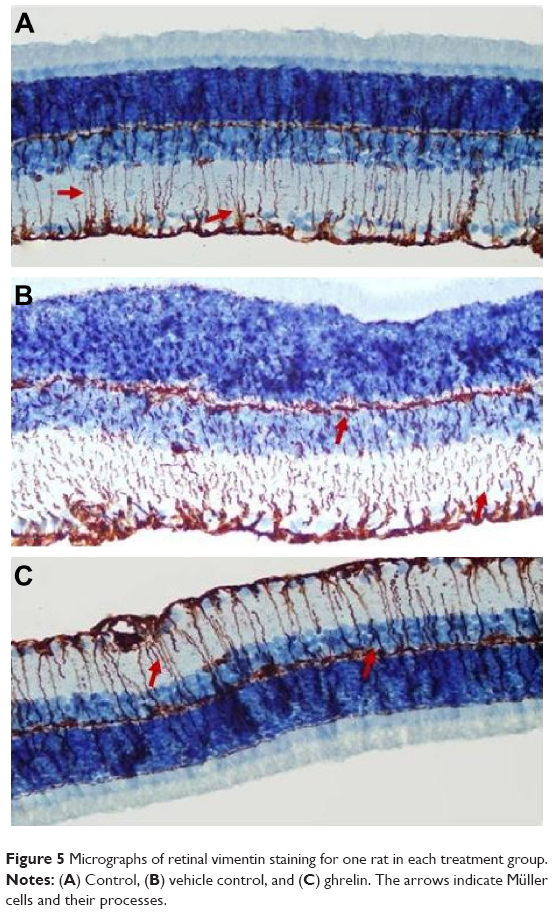 | Figure 5 Micrographs of retinal vimentin staining for one rat in each treatment group. |
The IPL, MCs, and MC processes in the INL showed increased expression of vimentin in the vehicle control group; vimentin expression in the OPL was also significantly higher than in the control group. In the vehicle control group, vimentin was observed to be increased in the ONL.
When vimentin expression in the ghrelin group (Figure 5C) was evaluated, a significant decrease was seen when compared with the vehicle control group (P<0.05). Similar to the control group, immunohistochemistry staining for retinal vimentin in these groups was evaluated to be +1. Vimentin expression of MCs in the ghrelin group was found to be in the RGCs, IPL, and INL. Vimentin expression in the OPL was also observed.
Discussion
Worldwide, glaucoma is the third most common cause of vision loss following cataract and age-related macular degeneration. Vision loss due to glaucoma is a result of deterioration of RGCs and their axons. Although it has been determined that high IOP is one of the risk factors for the disease, the degeneration mechanism is unclear. Given that vision loss occurs in some glaucoma patients despite control of IOP progression, research has been focused on neuroprotective treatment.11
It is well known that the pathologic changes in glaucoma are caused by damage due to oxidative stress and lipid peroxidation.12,13 Evaluation of lipid peroxidation is very useful in determining in vivo oxidative stress.14 The level of MDA is an indicator of lipid peroxidation. In patients with glaucoma, it has been found that lipid peroxidation products and MDA levels in aqueous humor and blood samples are present in significantly higher concentrations.15–18 Similarly, a study using a rat glaucoma model demonstrated that MDA levels were higher both in vitreous and in retina.14,17 Therefore, in diseases like glaucoma that progress to serious RGC damage and severe vision loss, if increased oxidative stress in intraocular tissue can be prevented and an antioxidative defense system can be developed to maintain the oxidant–antioxidant balance, the vision loss and RGC damage could be prevented.19 In our study, we observed that MDA levels were increased in the vehicle control group. However, we found that MDA levels in the ghrelin group are decreased. The decrease in MDA levels indicates decreased lipid peroxidation and reduced oxidative-nitrosative damage. The low levels of MDA in aqueous humor samples from the ghrelin group support our findings about the prevention of glaucomatous damage by ghrelin treatment.
It is well known that oxidative and nitrosative stress plays a role in the development of glaucomatous optic nerve damage. The finding of NOS2 in the iris ciliary body, retina, and optic disc in recent research suggests that NO might play a role in the pathogenesis of glaucoma.20 NO is an important mediator that serves to regulate IOP and modulates ocular blood flow. It is synthesized via the NOS enzyme.21 The NOS2 form is expressed in response to immunologic and inflammatory stimulation, and the NO made by this enzyme is pathological.22,23 Intraocular synthesis of NO has been studied in the past 5 years, and the presence of all forms of the NOS enzyme in the eye has been shown.24 The effects of NO, such as vasodilatation and neurotransmission, are established via cyclic guanosine monophosphate by means of the guanylate cyclase enzyme.25 In experimental glaucoma models in animals, elevated IOP increases apoptosis in cells, stimulates expression of NOS2, and leads to nitration of protein.26 Studies in glaucomatous optic discs in rats and humans have shown the presence of NOS2 on nitrotyrosine staining and demonstrated the role of NO in glaucomatous optic neuropathy.13,20 In clinical situations accompanied by ocular degeneration, nitrative stress worsens the disease course and increases NOS2 expression, pointing to the role of NO in ocular pathologies such as glaucoma.25 These data indicate that reactive nitrogen types may contribute to the death of RGCs associated with IOP.21,24
The main mechanism of vision loss in glaucoma is weakening of the INL and apoptosis of RGCs, which causes axonal loss in the optic nerve.27 It has been reported that these cytotoxic and apoptotic effects are stimulated by NO in macrophages, astrocytes, and neuronal cells. A study by Aslan et al demonstrated that elevated IOP increases expression of NOS2.23 It has also been shown that inhibition of NOS2 protects against the degeneration caused by glaucoma in RGCs.28 It is known that ghrelin indirectly inhibits expression of NOS2 in gastric mucosal cells.29 An ischemia-reperfusion model in rats demonstrated that ghrelin is protective against damage due to an increase in NO.30 In our study, the ghrelin group had low levels of NOS2 when compared with the vehicle control group, which is consistent with the literature.
NO assignment can be used as a diagnostic marker of oxidants that generate from NO, both in humans and animals. In cases of elevated IOP, expression of NOS2 increases retinal protein nitration and apoptosis.23 Pharmacologic studies in a rat model of chronic glaucoma showed that aminoguanidine with inhibition of NOS2 supports neuronal protection in RGCs.28 Elevated levels of NOS2 observed in glaucoma cases suggests that NOS2 may contribute to the RGC death associated with IOP.21,24 Cytotoxicity and apoptosis mediated by NO has been reported in macrophages, astrocytes, and neuronal cells.31–33 Although the mechanism for NO-mediated apoptosis is still unclear, it may be the result of activation of p53, leading to DNA damage.34 Erdurmuş et al found that serum NO levels are significantly higher in patients with pseudoexfoliation glaucoma and those with primary open angle glaucoma.15 In our study, we found that NO levels in the vehicle control group were higher than those in the control group. However, NO levels in the ghrelin group were significantly lower than those in the vehicle control group, and there was no significant difference in NO levels between the control and ghrelin groups. Many previous studies have shown that ghrelin reduces NO levels in different types of tissue.35,36 In our study, we observed that NO levels were lower in aqueous humor, which is consistent with the literature. The finding of low levels of NO (which had an important role in RGC death) in the ghrelin group demonstrates that glaucomatous damage was less in this group.
These hypotheses are the blockage of axoplasmic flow and consequently the withdrawal of the neurotrophins, the increase of intravitreal glutamate concentration, and ocular vasospasm.37 Recent TUNEL findings in rats with elevated IOP support apoptotic cell death. In a study by Gross et al, it was reported that TUNEL-positive cells were seen in the RGC layer, and that they were rarely in the control group compared to the experiment group.38 In previous studies of the sequence of normal retinal tissue, histogenetic cell death has been shown to start at the RGC layer, and progress toward the INL and ONL.39 Similarly, in the sequence of death due to glaucomatous damage, IOP increased as a result of axonal degeneration, microglial activation in the tissue, secretion of tumor necrosis factor-a, secretion of cytokines, and activation of the complement pathway, culminating in death of RGCs.40,41 In our study, microscopic DNA fragmentation, the most defining feature of apoptosis, was performed to determine apoptosis in the retinal layers. In our study, in accordance with the literature, apoptosis determined by the TUNEL method was shown to be less in the control and ghrelin groups, and more in the vehicle control group, so ghrelin might have protective effects on RGCs.
Astrocytes, MCs, and microglial cells have both protective and destructive roles in the retina.42,43 It has also been demonstrated that MCs can recognize various neuronal signals and actively modulate the levels of some ions such as K+ and H+, and some neurotransmitters including glutamate and gamma aminobutyric acid, in the extracellular space in the retina.44 It is also claimed that microglia are sensitive to potassium conductance and play an important role in processes such as production of aggressive oxygen radicals and secretion of glutamate, which may have an effect on the pathophysiologic mechanism of the cell death in glaucoma.44 An experimental study of the role of retinal glial cells in a rat glaucoma model reported that macroglia such as astrocytes and MCs might be involved in the pathophysiology of RGC death, and that activated microglial/phagocytic cells have an important role in modulating the changes in glaucomatous opti nerve heads.45 A similar report suggests that glial cells were activated by increased IOP and that reactivity of these cells may be associated with neuronal degeneration in the glaucomatous retina.46 The same study suggests that glial cell activation increased in order to clean the products from neurodegeneration. It has been determined that the GFAP levels taken in MC, which is the retina’s main glial cell, are significantly higher in many retinal pathologies, such as glaucoma.47 It is therefore believed that GFAP-positive immunostaining of MCs is a reliable marker for both acute and chronic pathology.46 In a study by Woldemussi et al47 formation of GFAP in MCs in response to IOP elevation was found as early as day 4. Increased expression of GFAP in glaucoma is well known, and our finding that GFAP was elevated in the vehicle control group and decreased in the ghrelin group is consistent with the literature.
Vimentin, a primary member of the intermediate filamentous family of proteins, comprises a cytoskeleton with microfilaments and microtubules in eukaryotic cells.48 Previous experimental glaucoma models have shown that vimentin in MCs and astrocytes increases damage to the retinal nerve fiber layer and that the increase of glutamine, a marker of glutamate metabolism, is connected to vimentin level.49–51 Carter-Dawson et al reported that the increase in glutamine in MCs was not a consequence of their loss and that MC function in the glutamate-glutamine cycle continued in glaucomatous eyes.50 Accordingly, it has been concluded that the immune positivity for vimentin seen to increase in the early period may drop due to the decrease in vitreous concentration of glutamate in the chronic period.51 There is some suggestion that tumor necrosis factor-a released from astrocytes and vimentin from RGC is generated as a response to glaucomatous damage triggered by NO and other cytokines.52 In our study, only responses in the early period were examined, and in this period the vehicle control group showed increased expression of vimentin when compared with the control group. It was observed that vimentin immunoreactivity in MCs decreased in the ghrelin group.
S-100 protein is a specific marker of retinal MCs in adult mammals and it acts intracellularly as a calcium ion-signaling or a calcium ion-buffering protein. Phosphorylation of S-100 protein plays an important role in many intracellular activities, including enzyme activity, the dynamics of the building blocks of the cytoskeleton, and protection of cells against oxidative damage.53 It also has a role in chemoattraction of leukocytes in the extracellular field and activation of macrophages.54 S-100 protein has both protective and destructive effects, depending on extracellular and intracellular activity. Previous studies in an experimental rat glaucoma model have shown that S-100, which also plays a role in metabolic activity, is a marker of MC damage in the eye.45,55 Our data are similar to those in the literature. Compared with our control group, the vehicle control group showed increased S-100 expression, particularly in MCs in the INL. However, in the group treated with ghrelin, S-100 immunoreactivity decreased.
First discovered in 1999 by Kojima et al ghrelin is basically a hormone with a 28-amino acid lipopeptide structure and is excreted by the fundus of the stomach. These hormones are also synthesized in the hypothalamus, pituitary gland, salivary gland, thyroid gland, small intestine, kidneys, heart, pancreas, central nervous system, lung, placenta, gonads, immunologic system, breasts, and teeth.3,4 It is known that ghrelin affects many systems, including growth hormone, adrenocorticotropic hormone, prolactin secretion, nutrition, gastric acid secretion, gastric motility, and cell proliferation. The anti-inflammatory and antioxidant effects of ghrelin have been shown previously.56,57 In addition to inhibiting lipid peroxidation, it increases catalase, glutathione peroxidase, and superoxide dismutase enzyme activity.58 By inhibiting apoptotic stimuli, ghrelin shows a proactive effect in many cells, including adipocytes, osteoblasts, cardiomyocytes, and endothelial cells.59–61 It has been reported that, in low doses, ghrelin prevents cell death by inhibition of apoptosis in hypothalamic neuronal cells.62 The proactive effects of ghrelin on cells have been shown in models of ischemia-reperfusion.30,63 The neuroprotective activity of ghrelin is occur via the growth hormone secretagogue (GHS) receptor upon activation of GHS-R1a.59
In rat models of Parkinson’s disease, it has been reported that intraperitoneal ghrelin injections reduced dopamine cell loss, and that this effect might be associated with reorganization of Bcl-2 and Bax molecules.64,65 Increased activation of microglia, which contributes to Parkinson’s disease, was inhibited after injection of ghrelin.65 The effect of ghrelin on neuroprotection is achieved by mitochondrial biogenesis, reorganization of proteins in the mitochondrial respiratory chain, and increased suppression of reactive oxygen species.66
The effect of ghrelin on the eye has not been clearly established as yet. In studies done in rat eyes, mRNA for ghrelin has been found in the anterior chamber.8 In a study conducted by Rocha-Sousa et al, it was reported that ghrelin was identified in humor aqueous which filled the human anterior chamber.8 Ghrelin crosses the blood–brain barrier easily, so can reach the ocular tissue. Determination of mRNA for ghrelin in studies performed in rat also demonstrates that ghrelin can be present locally in the human eye. Studies by Katsanos et al showed that ghrelin levels in the anterior chamber in patients with glaucoma were significantly lower than those in a control group.9 Following on from these studies, ghrelin was used in experimental glaucoma models. Ghrelin has a protective effect on RGCs, and can attenuate the harmful effects of glaucomatous damage. In vivo and in vitro studies in recent years have reported that the neuroprotective activity of ghrelin is increased in models of ischemic stroke. In rat models of ischemia-reperfusion, intraperitoneal or intravenous administration of ghrelin had a significant neuroprotective effect, reducing the infarct volume in the brain and decreasing cell death.62,67
Conclusion
To the best of our knowledge, there have been no previous reports in the literature on the impact of ghrelin on oxidative damage in glaucoma. Although the small number of experimental animals used and the lack of retrograde labeling of RGCs and determination of RGC survival can be regarded as the main limitations of this study, our findings suggest that ghrelin had antioxidant and neuroprotective effects on the retina in an experimental model of glaucoma.
Acknowledgment
This work was funded by an unrestricted grant from the Firat University Scientific Research Unit.
Author contributions
NC and TD performed the study; NC, NI, IHO, and TK collected the data; BT and OC translated, typed, prepared, edited and reviewed the manuscript; NC and TD performed the statistical analysis; and all authors approved the final draft of the manuscript. All authors contributed toward data analysis, drafting and revising the paper, and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Quigley HA. Glaucoma: macrocosm to microcosm the Friedenwald lecture. Invest Ophthalmol Vis Sci. 2005;46:2662–2670. | ||
Shields MB. Textbook of Glaucoma. 4th ed. Baltimore, MD, USA: Williams and Wilkins; 1998. | ||
Kojima M, Hosoda H, Date Y. Ghrelin is a growth-hormone-releasing acylated peptide from stomach. Nature. 1999;402:656–660. | ||
Kojima M, Kangawa K. Ghrelin: structure and function. Physiol Rev. 2005;85:495–522. | ||
Aydin S, Ozkan Y, Caylak E, et al. Ghrelin and its biochemical functions. Turkiye Klinikleri J Med Sci. 2006;26:272–283. | ||
Wren AM, Seal LJ, Cohen MA, et al. Ghrelin enhances appetite and increases food intake in humans. J Clin Endocrinol Metab. 2001;86:5992–5994. | ||
Tschop M, Smiley DL, Heiman ML. Ghrelin induces adiposity in rodents. Nature. 2000;407:908–913. | ||
Rocha-Sousa A, Saraiva J, Henriques-Coelho T, et al. Ghrelin as a novel locally produced relaxing peptide of the iris sphincter and dilator muscles. Exp Eye Res. 2006;83:1179–1187. | ||
Katsanos A, Dastiridou A, Georgoulias P, et al. Plasma and aqueous humour levels of ghrelin in open-angle glaucoma patients. Clin Experiment Ophthalmol. 2011;39:324–329. | ||
Erşahin M, Toklu HZ, Erzik C, et al. The antinflammatory and neuroprotective effects of ghrelin in subarachnoid hemorrhage-induced oxidative brain damage in rats. J Neurotrauma. 2010;27:1143–1155. | ||
Naskar R, Wissing M, Thanos S. Detection of early neuron degeneration and accompanying microglial reponses in the retina of a rat model of glaucoma. Invest Ophthalmol Vis Sci. 2002;43:2962–2968. | ||
Moreno MC, Campanelli J, Sande P, Sanez DA, Keller MI. Retinal oxidative stress induced by high intraocular pressure. Free Radic Biol Med. 2004;37:803–812. | ||
Tezel G, Yang X, Cai J. Proteomic identification of oxidatively modified retinal proteins in a chronic pressure-induced rat model of glaucoma. Invest Ophthalmol Vis Sci. 2005;46:3177–3187. | ||
Ko ML, Peng PH, Ma MC, Ritch R, Chen CF. Dynamic changes in reactive oxygen species and antioxidant levels in retinas in experimental glaucoma. Free Radic Biol Med. 2005;39:365–373. | ||
Erdurmuş M, Yağci R, Atiş Ö, Karadağ R, Akbaş A, Hepşen IF. Antioxidant status and oxidative stress in primary open angle glaucoma and pseudoexfoliative glaucoma. Curr Eye Res. 2011;36:713–718. | ||
Babizhayev MA, Bunin A. Lipid peroxidation in open-angle glaucoma. Acta Ophthalmol. 1989;67:371–377. | ||
Yucel I, Akar Y, Yucel G, Çiftçioğlu MA, Keles N, Aslan M. Effect of hypercholesterolemia on inducible nitric oxide synthase expression in a rat model of elevated intraocular pressure. Vision Res. 2005;45:1107–1114. | ||
Ghanem AA, Arafa LF, El-Baz A. Oxidative stress markers in patients with primary open-angle glaucoma. Curr Eye Res. 2010;35:295–301. | ||
Ferreira SM, Lerner SF, Brunzini R, Reides CG, Evelson PA, Llesuy SF. Time course changes of oxidative stress markers in a rat experimental glaucoma model. Invest Ophthalmol Vis Sci. 2010;51:4635–4640. | ||
Pang IH, Johnson EC, Jia L, et al. Evaluation of inducible nitric oxide synthase in glaucomatous optic neuropathy and pressure-induced optic nerve damage. Invest Ophthalmol Vis Sci. 2005;46:1313–1321. | ||
Liu BA, Neufeld H. Expression of nitric oxide synthase-2 (NOS-2) in reactive astrocytes of the human glaucomatous optic nerve head. Glia. 2000;30:178–186. | ||
Petros A, Lamb G, Leone A, Moncada S, Bennett D, Vallance P. Effects of a nitric oxide synthase inhibitor in humans with septic shock. Cardiovasc Res. 1994;28:34–39. | ||
Aslan M, Yucel I, Akar Y, Yucel G, Çiftçioğlu MA, Şanlioğlu S. Nitrotyrosine formation and apoptosis in rat models of ocular injury. Free Radic Res. 2006;40:147–153. | ||
Shareef S, Sawada A, Neufeld AH. Isoforms of nitric oxide synthase in the optic nerves of rat eyes with chronic moderately elevated intraocular pressure. Invest Ophthalmol Vis Sci. 1999;40:2884–2891. | ||
Goureau O, Bellot J, Thillaye B, Courtois Y, Kozak Y. Increased nitric oxide production in endotoxin-induced uveitis. Reduction of uveitis by an inhibitor of nitric oxide synthase. J Immunol. 1995;154:6518–6523. | ||
Sacca SC, Izzotti A, Rossi P, Traverso C. Glaucomatous outflow pathway and oxidative stress. Exp Eye Res. 2007;84:389–399. | ||
Fechtner RD, Weinreb RN. Mechanisms of optic nerve damage in primary open angle glaucoma. Surv Ophthalmol. 1994;39:23–42. | ||
Neufeld AH, Sawada A, Becker B. Inhibition of nitric-oxide synthase 2 by aminoguanidine provides neuroprotection of retinal ganglion cells in a rat model of chronic glaucoma. Proc Natl Acad Sci U S A. 1999;96: 9944–9948. | ||
Slomiany BL, Slomiany A. Helicobacter pylori induces disturbances in gastric mucosal Akt activation through inducible nitric oxide synthase-dependent s-nitrosylation: effect of ghrelin. Gastroenterology. 2011;308:727–728. | ||
Konturek PC, Brzozowski T, Walter B, Burnat G, Hess T, Hahn EG. Ghrelin induced gastroprotection against ischemia-reperfusion injury involves an activation of sensory afferent nerves and hyperemia mediated by nitric oxide. Eur J Pharmacol. 2006;536:171–181. | ||
Sarih M, Souvannavong V, Adam A. Nitric oxide synthase induces macrophage death by apoptosis. Biochem Biophys Res Commun. 1993;191:503–508. | ||
Hu J, Van Eldik LJ. S100 beta induces apoptotic cell death in cultured astrocytes via a nitric oxide-dependent pathway. Biochim Biophys Acta. 1996;1313:239–245. | ||
Heneka MT, Loschmann PA, Gleichmann M, et al. Induction of nitric oxide synthase and nitric oxide-mediated apoptosis in neuronal PC12 cells after stimulation with tumor necrosis factor-alpha/lipopolysaccharide. J Neurochem. 1998;71:88–94. | ||
Kim YM, Bombeck CA, Billiar TR. Nitric oxide as a bifunctional regulator of apoptosis. Circ Res. 1999;84:253–256. | ||
Aslan A, Yildirim M, Ayyildiz M, Güven A, Ağar E. The role of nitric oxide in the inhibitory effect of ghrelin against penicillin-induced epileptiform activity in rat. Neuropeptides. 2009;43:295–302. | ||
Alderton WK, Cooper CE, Knowles RG. Nitric oxide synthases: structure, function and inhibition. Biochem J. 2001;357:593–615. | ||
Levin LA, Louhab A. Apoptosis of retinal ganglion cells in anterior ischemic optic neuropathy. Arch Ophthalmol. 1996;114:488–491. | ||
Gross RL, Ji J, Chang P, Pennesi ME. A Mouse model of elevated intraocular pressure: retina and optic nerve findings. Trans Am Ophthalmol Soc. 2003;101:163–169. | ||
Beazley LD, Perry VH, Baker B, Darby JE. An investigation into the role of ganglion cells in the regulation of division and death of other retinal cells. Dev Brain Res. 1987;33:169–184. | ||
Ju KR, Kim HS, Kim JH, Lee NY, Park CK. Retinal glial cell responses and Fas/FasL activation in rats with chronic ocular hypertension. Brain Res. 2006;1122:209–221. | ||
Kuehn MH, Kim CY, Ostojic J, et al. Retinal synthesis and deposition of complement components induced by ocular hypertension. Exp Eye Res. 2006;83:620–628. | ||
Battisti WP, Wang J, Bozek K. Macrophages, microglia, and astrocytes are rapidly activated after crush injury of the goldfish optic nerve: a light and electron microscopic analysis. J Comp Neurol. 1995;354:306–320. | ||
Hughes EH, Schlichtenbrede FC, Murphy CC, et al. Minocycline delays photoreceptor death in the rds mouse through a microglia-independent mechanism. Exp Eye Res. 2004;78:1077–1084. | ||
Newman E, Reichenbach A. The Muller cell: a functional element of the retina. Trends Neurosci. 1996;19:307–312. | ||
Lam TT, Kwong JMK, Tso OM. Early glial responses after acute elevated intraocular pressure in rats. Invest Ophthalmol Vis Sci. 2003;44:638–645. | ||
Wang X, Tay SS, Ng YK. An immunohistochemical study of neuronal and glial cell reactions in retina of rats with experimental glaucoma. Exp Brain Res. 2000;132:476–484. | ||
Woldemussie E, Wijono M, Ruiz G. Müller cell response to laser-induced increase in intraocular pressure in rats. Glia. 2004;47:109–119. | ||
Dabbs DJ. Diagnostic Immunohistochemistry. Philadelphia, PA, USA: Saunders Elsevier; 2006. | ||
Hernandez M, Rodriguez FD, Sharma SC, Vecino E. Immunohistochemical changes in rat retinas at various time periods of elevated intraocular pressure. Mol Vis. 2009;15:2696–2709. | ||
Carter-Dawson L, Shen F, Harwerth R, Smith EL III, Crawford ML, Chuang A. Glutamine immunoreactivity in Muller cells of monkey eyes with experimental glaucoma. Exp Eye Res. 1998;66:537–545. | ||
Dreyer EB, Zurakowski D, Schumer RA, Podos SM, Lipton SA. Elevated glutamate levels in the vitreous body of human and monkeys with glaucoma. Arch Ophthalmol. 1996;114:299–305. | ||
Tezel G, Wax MB. Increased production of tumor necrosis factor-alpha by glial cells exposed to simulated ischemia or elevated hydrostatic pressure induces apoptosis in cocultured retinal ganglion cells. J Neurosci. 2000;20:8693–8700. | ||
Donato R. S100: a multigenic family of calcium-modulated proteins of the EF-hand type with intracellular and extracellular functional roles. Int J Biochem Cell Biol. 2001;33:637–668. | ||
Barger SW, Van Eldik LJ, Mattson MP. S100 beta protects hippocampal neurons from damage induced by glucose deprivation. Brain Res. 1995;677:167–170. | ||
Sommer I, Lagenaur C, Schachner M. Recognition of Bergmann glial and ependymal cells in the mouse nervous system by monoclonal antibody. J Cell Biol. 1981;90:448–458. | ||
Dixit VD, Schaffer EM, Pyle RS, et al. Ghrelin inhibits leptin- and activation-induced proinflammatory cytokine expression by human monocytes and T cells. J Clin Invest. 2004;114:57–66. | ||
Brzozowski T, Konturek PC, Konturek SJ, et al. Exogenous and endogenous ghrelin in gastroprotection against stress-induced gastric damage. Regul Pept. 2004;120:39–51. | ||
Obay BD, Taşdemir E, Tümer C, Bilgin HM, Atmaca M. Dose dependent effects of ghrelin on pentylenetetrazole-induced oxidative stress in a rat seizure model. Peptides. 2008;29:448–455. | ||
Kim MS, Yoon CY, Jang PG, Park SJ. The mitogenic and antiapoptotic actions of ghrelin in 3T3-L1 adipocytes. Mol Endocrinol. 2004;18:2291–2301. | ||
Kim S, Her SJ, Park SJ. Ghrelin stimulate proliferation and differentiation and inhibits apoptosis in osteoblastic MC3T3-E1 cells. Bone. 2005;37:359–369. | ||
Baldanzi G, Filigheddu N, Cutrupi S, Catapano F, Bonissoni S. Ghrelin and des acyl ghrelin inhibit cell death in cardiomyocytes and endothelial cells through ERK1/2 and PI 3-kinase/AKT. J Cell Biol. 2006;159:1029–1037. | ||
Chung H, Kim E, Lee DH, Seo S, Ju S, Lee D. Ghrelin inhibits apoptosis in hypothalamic neuronal cells during oxygen-glucose deprivation. Endocrinology. 2007;148:148–159. | ||
Chang L, Ren Y, Liu X, Li WG, Yang J, Geng B. Protective effects of ghrelin on ischemia/reperfusion injury in the isolated rat heart. J Cardiovasc Pharmacol. 2004;43:165–170. | ||
Andrews ZB, Eroin D, Beiler R, Liu ZW, Abizaid A. Ghrelin promotes and protects nigrostriatal dopamine function via UCP2-dependent mitochondrial mechanism. J Neurosci. 2009;29:14057–14065. | ||
Jiang H, Li LJ, Wang J, Xie JX. Ghrelin antagonizes MPTP-induced neurotoxicity to the dopaminergic neurons in mouse substantia nigra. Exp Neurol. 2008;212:532–537. | ||
Andrews ZB. The extra hypothalamic actions ghrelin on neuronal function. Trends Neurosci. 2011;34:31–40. | ||
Hwang S, Moon M, Kim S, Hwang L, Ahn KJ, Park S. Neuroprotective effect of ghrelin is associated with decreased expression of prostate apoptosis response. Endocrinol J. 2009;56:609–617. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.