")
Back to Journals » Drug Design, Development and Therapy » Volume 9
Navigating into the binding pockets of the HER family protein kinases: discovery of novel EGFR inhibitor as antitumor agent
Authors Liu W, Ning J, Meng W, Hu J, Zhao Y, Liu C, Cai L
Received 24 March 2015
Accepted for publication 17 April 2015
Published 23 July 2015 Volume 2015:9 Pages 3837—3851
DOI https://doi.org/10.2147/DDDT.S85357
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Shu-Feng Zhou
Wei Liu,1,* Jin-Feng Ning,2,* Qing-Wei Meng,1 Jing Hu,1 Yan-Bin Zhao,1 Chao Liu,3 Li Cai1
1The Fourth Department of Medical Oncology, Harbin Medical University Cancer Hospital, 2The Thoracic Surgery Department, Harbin Medical University Cancer Hospital, Harbin, People’s Republic of China; 3General Surgery Department, Mudanjiang Guanliju Central Hospital, Mishan, Heilongjiang Province, People’s Republic of China
*These authors contributed equally to this work
Abstract: The epidermal growth factor receptor (EGFR) family has been validated as a successful antitumor drug target for decades. Known EGFR inhibitors were exposed to distinct drug resistance against the various EGFR mutants within non-small-cell lung cancer (NSCLC), particularly the T790M mutation. Although so far a number of studies have been reported on the development of third-generation EGFR inhibitors for overcoming the resistance issue, the design procedure largely depends on the intuition of medicinal chemists. Here we retrospectively make a detailed analysis of the 42 EGFR family protein crystal complexes deposited in the Protein Data Bank (PDB). Based on the analysis of inhibitor binding modes in the kinase catalytic cleft, we identified a potent EGFR inhibitor (compound A-10) against drug-resistant EGFR through fragment-based drug design. This compound showed at least 30-fold more potency against EGFR T790M than the two control molecules erlotinib and gefitinib in vitro. Moreover, it could exhibit potent HER2 inhibitory activities as well as tumor growth inhibitory activity. Molecular docking studies revealed a structural basis for the increased potency and mutant selectivity of this compound. Compound A-10 may be selected as a promising candidate in further preclinical studies. In addition, our findings could provide a powerful strategy to identify novel selective kinase inhibitors on the basis of detailed kinase–ligand interaction space in the PDB.
Keywords: EGFR, kinase, inhibitor, protein crystal complex, FBDD, erlotinib
Introduction
The epidermal growth factor receptor (EGFR) family plays a central role in signal transduction pathways associated with cell growth, survival, and differentiation.1 It contains four members: EGFR (HER1), HER2, HER3, and HER4, and all four members share high structural homology, mainly composed of an extracellular ligand-binding domain, a single transmembrane domain, and an intracellular tyrosine kinase domain.2 Overexpression of EGFR and HER2 leading to aberrant signaling has been observed in a range of human tumor types, particularly non-small-cell lung cancer (NSCLC).3 Therefore, targeting EGFR and HER2 by small-molecule tyrosine kinase inhibitors (TKIs) is an attractive approach for cancer therapy. The success of the first-generation EGFR TKIs (gefitinib4 and erlotinib,5 Figure 1) triggered the development of a number of anti-EGFR small-molecule agents in the past 10 years. Subsequently, the two known inhibitors, namely lapatinib6 and afatinib7 (Figure 1), as second-generation EGFR TKIs were approved by US Food and Drug Administration (FDA) for the treatment of breast cancer (2007) and NSCLC (2012), respectively.
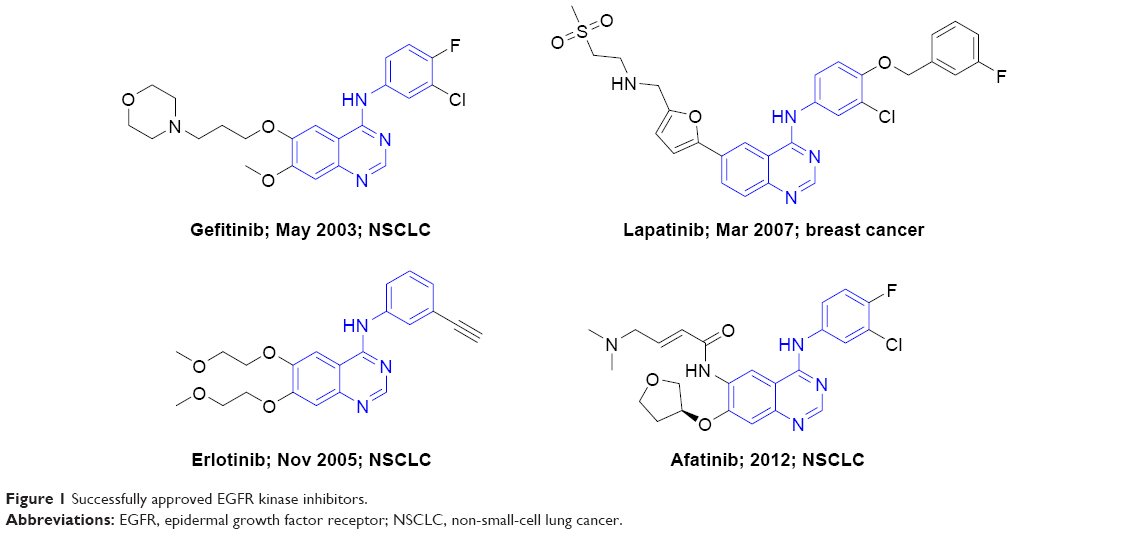 | Figure 1 Successfully approved EGFR kinase inhibitors. |
Activating mutations in EGFR occur in exons 18–21 encoding the kinase domain (Figure 2A). To date, there are dozens of gene mutations reported,8 which could be divided into TKI-sensitive EGFR mutants and TKI-resistant EGFR mutants according to the response to the first-generation EGFR TKIs. Among the sensitive mutations, the two common mutations are in-frame deletions as to the LREA motif of exon 19 (45% of EGFR mutations) and the L858R mutation of exon 21 (40% of EGFR mutations).9,10 The G719 point mutation (G719A, C, or S, about 3% of EGFR mutations) also contributes to increasing the affinity of EGFR to TKIs.11 With regard to the resistant mutants, the T790M mutation in the EGFR gatekeeper residue could be observed in 50% of EGFR-mutant tumors with acquired resistance to erlotinib or gefitinib.12 Besides, small insertions (ins NPG, ins SVQ)13 in exon 20 and three other point mutations (L747S,14 D761Y,15 and T854A16) have been associated with acquired resistance.
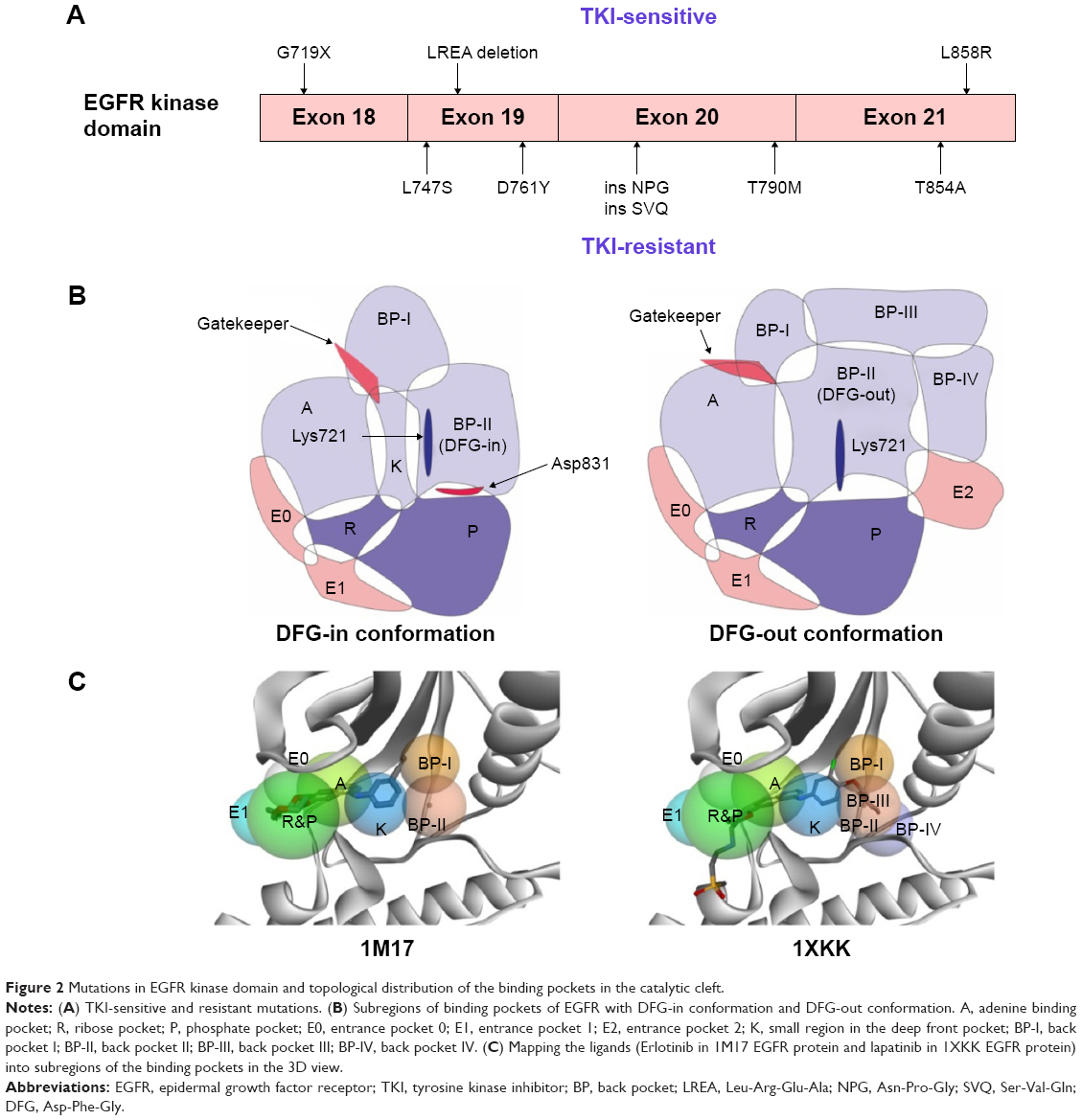 | Figure 2 Mutations in EGFR kinase domain and topological distribution of the binding pockets in the catalytic cleft. |
According to the paradigm of kinase inhibitors, EGFR TKIs can be simply classified into type I and type II inhibitors (Figure 2B and C). Furthermore, whether the acrylamide group (the structure of afatinib, Figure 1) exists within this kind of molecule becomes one criterion that can be used to differentiate the covalent inhibitor. Besides, paying more attention to the inhibitor type and key mutations in the tyrosine kinase domain contributes to overcoming TKI-resistance issue when designing new EGFR TKIs. Recently, there have been many reports on the design and synthesis of novel EGFR small-molecule inhibitors as third-generation EGFR TKIs for overcoming drug resistance.17–19 However, their design protocols appear a bit rough, and the optimization of molecules largely depends on the individual experience of the researcher. With the number of EGFR protein crystal complexes with small molecules increasing every year, we attempted to carry out a detailed analysis of these 42 kinase domain crystal complexes that covered several main EGFR mutants and small-molecule ligands with much structural diversity. Meanwhile, guided by the valuable information provided, we adopted a powerful tactics to develop a novel mutant-selective EGFR kinase inhibitor against EGFR T790M based on fragment-based drug design (FBDD).
Methods
PDB collection and protein structure superposition
All available protein structures in the PDB were downloaded (accessed at 28 May 2014). After careful curation, there were 42 EGFR family protein tyrosine kinase were screened. Many PDB entries possessed 2–4 tyrosine kinase domains, and only one in each entry was retained for superposing. Based on the sequence of wild-type EGFR protein (PDB code: 1M17), the other 41 EGFR protein sequences were aligned together by Discovery Studio 3.5 (Figure 3A). All the structures of kinase domains were superposed based on the sequence alignment above.
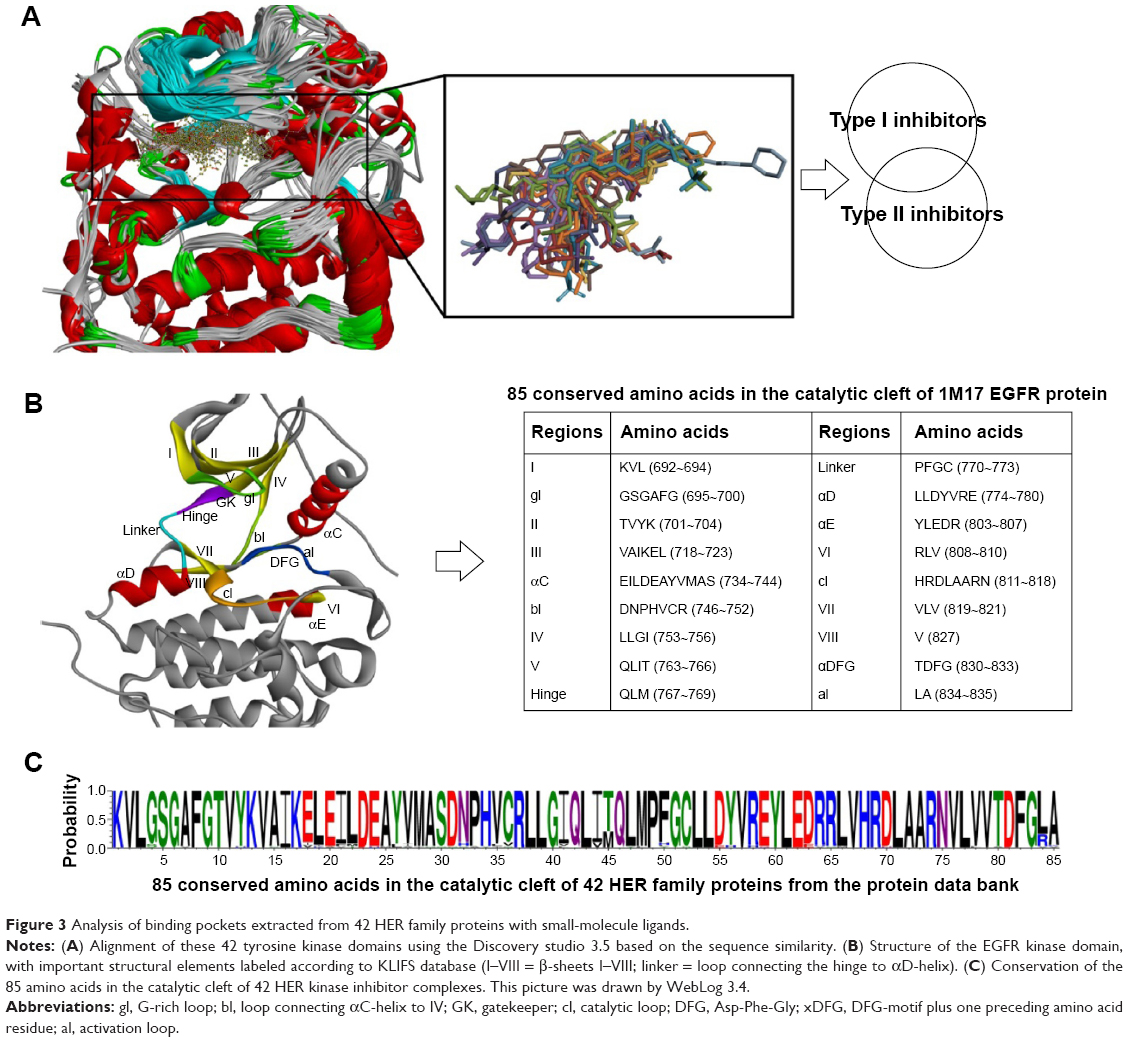 | Figure 3 Analysis of binding pockets extracted from 42 HER family proteins with small-molecule ligands. |
Chemistry general
All chemicals and reagents of analytical grade used were purchased from Aldrich (USA). The melting points (uncorrected) were determined on an XT4MP apparatus (Taike Corp., Beijing, People’s Republic of China). All the 1H NMR spectra were recorded on a Bruker DPX 300 model Spectrometer at 25°C with tetramethylsilane and solvent signals allotted as internal standards, and chemical shifts are reported in ppm (d). ESI-MS spectra were recorded on a Mariner System 5304 mass spectrometer. Elemental analyses were performed on a CHN-O-Rapid instrument and were within 0.4% of the theoretical values. Thin-layer chromatography was performed on glass-backed silica gel sheets (silica gel 60 GF254) and visualized in UV light (254 nm). Column chromatography was performed using silica gel (200–300 mesh) eluting with ethyl acetate (EtOAc) and petroleum ether.
3-Chloro-4-(3-(trifluoromethyl)phenoxy)aniline (4)
This was prepared according to the following procedure. To a solution of dimethylformamide (DMF, 20 mL) was added K2CO3 (2 g), 2-chloro-1-fluoro-4-nitrobenzene (1, 1.75 g, 10.0 mmol), and 3-(trifluoromethyl)phenol (2, 1.62 g, 10.0 mmol) at room temperature, and then gradually heated up to 80°C for 4 hours. Water (60 mL) was then added to the mixture, and the mixture was extracted with EtOAc. The organic layer was washed three times with saturated brine (30 mL), dried over anhydrous Na2SO4, and concentrated in vacuo. The crude product was purified by column chromatography to get compound 3, a light yellow powder, in 80% yield. A mixture of 2-chloro-4-nitro-1-(3-(trifluoromethyl)phenoxy)benzene (3, 1.58 g, 5.0 mmol, dissolved in 20 mL 70% EtOH containing 1 mL AcOH) and Fe (1.5 g) was stirred and heated at 80°C for 6 hours. After cooling down to room temperature, the reaction mixture was alkalinized by the addition of concentrated ammonia (10 mL). The insoluble material was removed by filtration through Celite, and the filtrate was evaporated under reduced pressure. The remaining solution was extracted with EtOAc for column chromatography. The EtOAc layer was collected and purified by silica gel column chromatography (EtOAc:petroleum ether =1:1, v/v) to give amine 4. Yield: 72%, white solid. 1H NMR (CDCl3) δ 7.38 (t, 1H, J =8.0 Hz), 7.23–7.32 (m, 1H), 7.11 (s, 1H), 7.04 (dd, 1H, J =7.8, 2.3 Hz), 6.92 (d, 1H, J =8.6 Hz), 6.79 (d, 1H, J =2.7 Hz), 6.59 (dd, 1H, J =8.6, 2.7 Hz), 3.72 (br s, 2H).
6-Chloro-N-(3-chloro-4-(3-(trifluoromethyl)phenoxy)phenyl)quinazolin-4-amine (7)
This was prepared according to the literature procedure.20 2-Amino-5-chlorobenzonitrile 5 (3.04 g, 20 mmol) was suspended in dimethylformamide dimethyl acetal (10 mL), and the mixture was refluxed for 2 hours. The resulting mixture was cooled to room temperature for 2 hours. The white precipitate in the mixture was filtered, washed with ethyl ether, and dried to give (E)-N′-(4-chloro-2-cyanophenyl)-N,N-dimethylformimidamide 6 (Yield: 90%). A mixture of 6 (2.07 g, 10 mmol, 1.0 equiv) and compound 4 (1.1 equiv) was heated and stirred at reflux in acetic acid (20 mL) for 2 hours. The white precipitate that formed was filtered hot, washed with hot acetic acid and diethyl ether, and dried to give the desired compound 7 (Yield: 83%).
(4-((3-Chloro-4-(3-(trifluoromethyl)phenoxy)phenyl)amino)quinazolin-6-yl)boronic acid (8)
This was prepared as follows.21 A mixture of compound 7 (1.71 g, 3.80 mmol), 1,3-bis(diphenylphosphino)propane (156 mg, 0.38 mmol), 1,3-bis(diphenylphosphino)propane nickel(II) chloride (205 mg, 0.38 mmol), dioxane (8 mL), diisopropylethylamine (1.98 mL, 11.39 mmol), and 4,4,5,5-tetramethyl-1,3,2-dioxaborolane (1.10 mL, 7.59 mmol) was magnetically stirred and heated via microwave irradiation for 30 minutes at 160°C. After cooling to room temperature, the reaction was extracted with dichloromethane (100 mL), and washed three times with saturated aqueous ammonium chloride solution (50 mL ×3). The organic layer was dried over anhydrous Na2SO4 and concentrated in vacuo. The crude product was purified by column chromatography to get compound 8 as a brown solid. MS (ESI): m/z (M+H)+ 460.
6-(2-Aminoethyl)-N-(3-chloro-4-(3-(trifluoromethyl)phenoxy)phenyl)quinazolin-4-amine (9)
A 20 mL conical microwave vial was charged with a magnetic stirring bar, 2-bromoethan-1-amine (122 mg, 1 mmol), compound 8 (460 mg, 1 mmol), cesium carbonate (488 mg, 1.5 mmol), tetrakis(triphenylphosphine)-palladium(0) (90 mg, 0.08 mmol), and dimethoxyethane (10 mL). The reaction mixture was magnetically stirred and heated via microwave irradiation for 30 minutes at 140°C. Upon cooling to room temperature, the reaction was concentrated in vacuo and purified by column chromatography to get compound 9 as a brown solid. MS (ESI): m/z (M+H)+ 459.
N-(2-(4-((3-Chloro-4-(3-(trifluoromethyl)phenoxy)phenyl)amino)quinazolin-6-yl)ethyl)-3-hydroxy-3-methylbutanamide (A-10)
A mixture of 9 (0.92 g, 2 mmol), 3-hydroxy-3-methylbutanoic acid (0.472 g, 4 mmol), 1-(3-Dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDC·HCl) (0.68 g, 3.4 mmol), 1-hydroxybenzotriazole monohydrate (HOBt) (0.52 g, 3.8 mmol), and triethylamine (1 mL) in DMF (10 mL) was stirred at room temperature for 3 days. Water (100 mL) was added to the reaction mixture, and the mixture was extracted with EtOAc (200 mL). The organic layer was washed with water (50 mL) and brine (50 mL), dried over MgSO4, and concentrated in vacuo. The residue was purified by silica gel column chromatography (eluent, EtOAc: petroleum ether =2:1, v/v) to give N-(2-(4-((3-chloro-4-(3-(trifluoromethyl)phenoxy)phenyl)amino)quinazolin-6-yl)ethyl)-3-hydroxy-3-methylbutanamide as a white solid. Yield: 46%. Mp: 197°C–198°C. 1H NMR (400 MHz, DMSO, δ ppm): 10.12 (s, 1H, –NHCO–), 8.41 (s, 1H), 8.16 (s, 1H), 8.01 (d, J =6.72 Hz, 1H), 7.83 (d, J =8.84 Hz, 1H), 7.76 (dd, J1 =11.64 Hz, J2 =11.72 Hz, 1H), 7.38 (dd, J1 =7.24 Hz, J2 =7.24 Hz, 2H), 7.31–7.27 (m, 1H), 7.20 (d, J =4.51 Hz, 1H), 7.14–7.06 (m, 3H), 4.58 (s, 1H, –OH), 3.37 (t, J = 9.07 Hz, 2H), 2.77–2.69 (m, 4H), 1.19 (m, 6H, –CH3). MS (ESI): 559.53 [M+H]+; Anal Calcd for C28H26ClF3N4O3: C, 60.16; H, 4.69; N, 10.02; O, 8.59; Found: C, 60.19; H, 4.49; N, 9.93; O, 8.64.
Biological assay
Cell proliferation assay (cell viability was assessed by MTT assay)
We evaluated the antiproliferative activities of compounds A-10 against A431 (carcinomic human epithelial cell), H1975 (human lung cell line), and MCF-7 (breast cancer) cancer cells. Cell proliferation was determined using the MTT dye (Beyotime Institute of Biotechnology, Haimen, Jiangsu, People’s Republic of China) according to the instructions of the manufacturer. Briefly, 5×103 cells per well were seeded in a 96-well plate, and grown at 37°C for 12 hours. Subsequently, the cells were treated with compound A-10, gefitinib, and erlotinib at increasing concentrations in the presence of 10% fetal bovine serum (FBS) for 24 hours. Afterward, 10 μL MTT dye was added to each well, and the cells were incubated at 37°C for 3–4 hours. Then all the solution in the wells was poured out and 150 μL DMSO was added to every well. The plates were read in a Victor-V multilabel counter (PerkinElmer Inc., Waltham, MA, USA) using the default europium detection protocol. Percent inhibition or GI50 values of compounds were calculated by comparison with DMSO-treated control wells.
HER2 and EGFR kinase assay
The cytoplasmic domain (amino acids 676–1,255) of human HER2 and the cytoplasmic domain (amino acids 669–1,210 containing wild-type or dual T790M/L858R mutations) of human EGFR were expressed as the N-terminal peptide (DYKDDDD)-tagged protein using a baculovirus expression system. The expressed HER2 kinase and EGFR kinase were purified by anti-FLAG M2 affinity gel (Sigma-Aldrich, USA). The HER2 and EGFR kinase assays were performed using radiolabeled [γ-32P] ATP (GE Healthcare, USA) in 96-well plates. The kinase reactions were performed in 50 mmol/L Tris-HCl, pH 7.5, 5 mmol/L MnCl2, 0.01% Tween-20, and 2 mmol/L dithiothreitol (DTT) containing 0.9 μCi of [γ-32P] ATP per reaction, 50 μmol/L ATP, 5 μg/mL poly-Glu-Tyr (4:1), and 0.25 μg/mL purified HER2 or EGFR cytoplasmic domain in a total volume of 50 μL. To measure the IC50 value for enzyme inhibition, the compounds were incubated with the enzyme for 5 minutes prior to the reaction at room temperature. Kinase reactions were initiated by adding ATP. After the kinase reaction was incubated for 10 minutes at room temperature, the reactions were terminated by the addition of 10% (final concentration) trichloroacetic acid. The [γ-32P]-phosphorylated proteins were filtered in a harvest plate (Millipore, USA) with a cell harvester (PerkinElmer) and washed free of [γ-32P] ATP with 3% phosphoric acid. The plates were dried, followed by the addition of 25 μL of MicroScintO (PerkinElmer). Radioactivity was counted by a Topcount scintillation counter (PerkinElmer). IC50 values and 95% confidence intervals were calculated by nonlinear regression analysis.
Molecular docking study
Molecular docking of designed compounds (A-1–B-10) into the three-dimensional X-ray structure of human wild-type EGFR and T790M/L858R EGFR with inactive conformations (PDB code: 3W33 and 3W2R, respectively) was carried out using the Discovery Studio (version 3.5) as implemented through the graphical user interface DS-CDOCKER protocol.22 The three-dimensional structures of the aforementioned compounds were constructed using Chem. 3D ultra 12.0 software [Chemical Structure Drawing Standard; Cambridge Soft corporation, USA (2010)], and then they were energetically minimized by using CHARMM force field. The crystal structures of two HER proteins complex were retrieved from the RCSB PDB (http://www.rcsb.org/pdb/home/home.do). All bound waters and ligands were eliminated from the protein, and the polar hydrogen was added to the proteins. In addition, these 20 small molecules were also evaluated by the Glide23 sp and xp docking protocol implemented in the Schrodinger 2012 suite, respectively. The detailed docking procedure can be found in the Website of Schrodinger Company.
Results and discussion
Investigation into the binding pockets of EGFR proteins
Up to now, there are 42 EGFR family protein tyrosine kinase crystal complexes that are deposited in the Protein Data Bank (PDB, Table 1). We collected them and attempted to give a detailed analysis of the binding pockets and the corresponding ligands in the catalytic cleft. As shown in Table 1, 42 entries (38 entries colored red for EGFR; one entry colored blue for HER2; one entry colored purple for HER3; two entries colored green for HER4) were reported by 24 references in the past 10 years.11,24–46 On the other hand, there were 24 small-molecule ligands with various structural moieties cocrystallized with EGFR family proteins. Lipophilic efficiency (LipE) was a promising parameter in the optimization of the lead compounds, particularly leading to the discovery of PF-04217903, a clinical candidate as a c-MET kinase inhibitor.47 Therefore, we attempted to design novel EGFR inhibitors mainly by means of a modified LipE value (the formula is given in the footnote in Table 1). The AlogP-values of these 24 ligands were calculated by the Schrodinger 2012 suite, and their inhibitory activities toward the corresponding protein tyrosine kinases were extracted either from the reported reference or from the ChEMBL database.
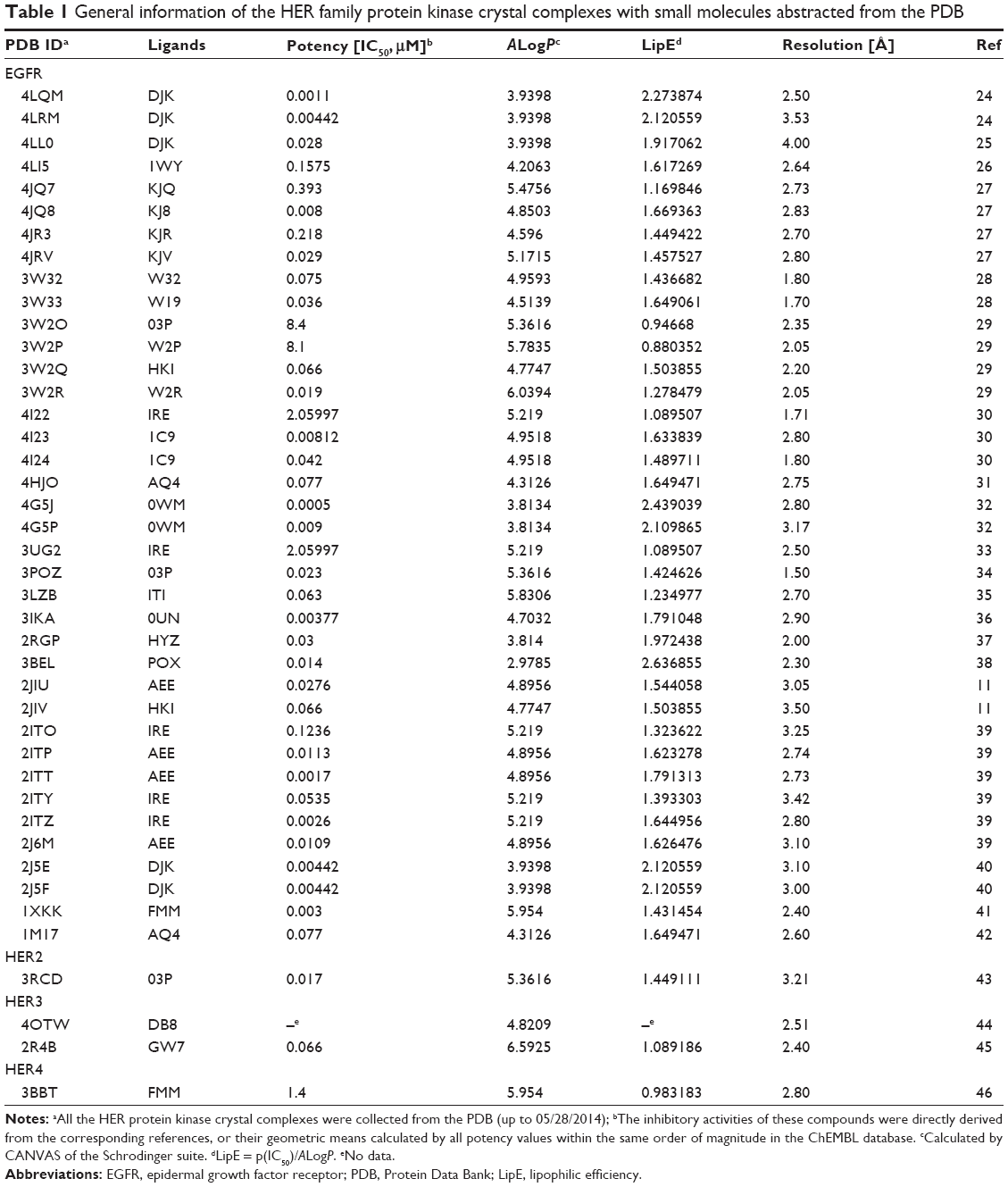 | Table 1 General information of the HER family protein kinase crystal complexes with small molecules abstracted from the PDB |
As to the protein conformation, there are three kinds of catalytic conformation (DFG-in, DFG-out, and DFG-out-like) among the protein crystals of kinase domains. Moreover, the distance between the αC carbon atoms of DxDFG.81 and EαC.24 could be used for the classification. According to the KLIFS database,48 one cluster that contained structures with distances 4–7.2 Å was marked as αC-in; the second main cluster, marked αC-out, contained structures with distances of 9.3 Å to >14 Å; the third that presented structures with distances of 7.2–9.3 Å, was marked as αC-out-like. However, Table 2 reveals that the range of distances should be revised as follows: 7.394–9.460 Å for αC-in, and 10.997–13.635 Å for αC-out. Considering the pose of the DFG motif and the position of the αC-helix, we extracted the binding modes in the catalytic cleft of all the 42 EGFR protein crystal complexes (Figures S1 and S2). In addition, we also presented all the mutations in the kinase domains of the 42 EGFR proteins according to sequence alignment based on the 1M17 wild-type protein (Table 2), in which there were 12 entries containing T790M gatekeeper mutation.
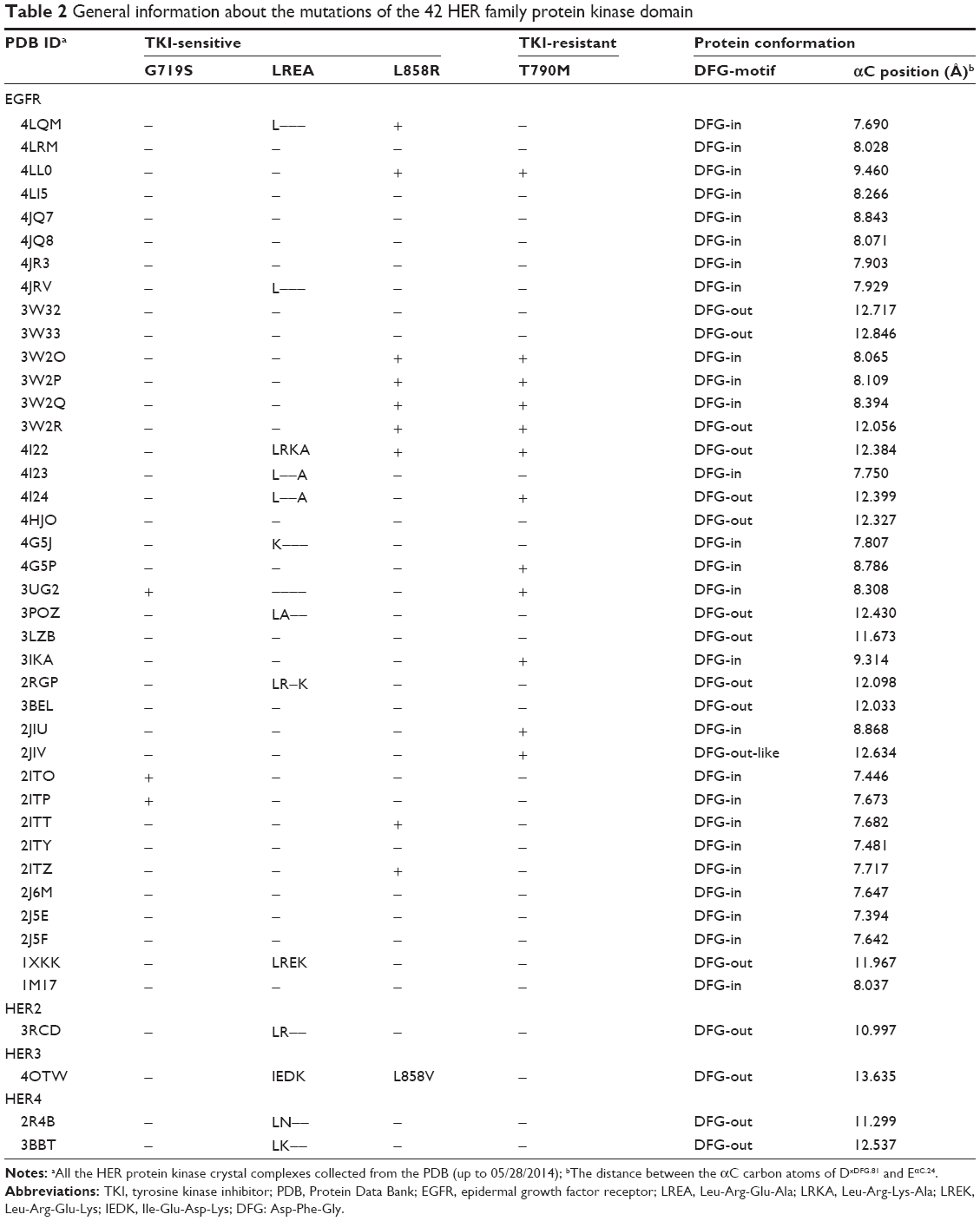 | Table 2 General information about the mutations of the 42 HER family protein kinase domain |
In order to investigate the binding pockets of EGFR proteins, all the 42 EGFR protein–ligand complexes were superposed together based on the 1M17 protein template (Figure 3A). Correspondingly, 24 small-molecule ligands that could be divided into type I and II inhibitors were also put together in the same coordinate space. Accordingly, important structural features of the kinase domain were labeled as in Figure 3B: β-sheets I–VIII, G-rich loop (gl), bl = loop connecting αC-helix to IV, gatekeeper, linker, catalytic loop (cl), DFG-motif, activation loop (al), and the αC-helix in the N-terminal domain. The 85 conserved amino acids mentioned in the KLIFS database48 were obtained through the sequence alignment of 42 EGFR proteins (Figures S1 and S4). Even though there were HER2/3/4 proteins among these protein complexes, the 85 conserved amino acids still seemed highly similar (Figure 3C).
The protein–ligand interaction space could provide new insights into the structural requirements of kinase binding that will be useful in ligand discovery and design studies. Therefore, in the context of a systematic analysis of the EGFR binding pocket space, we attempted to design new EGFR inhibitors through portraying the 24 superposed ligands carefully in the further step. We first decomposed the 24 crystal ligands into 25 different fragments with two aromatic rings (Figure 4), and then put them into the corresponding subregions of the binding pocket (Table S1). Interestingly, based on the analysis of these 75 fragments covering 25 different scaffolds, it was notable that the same scaffold derived from different inhibitors (even though a different type of inhibitor) was located in the same subregions or pocket context. Subsequently, this experience led to the hypothesis that we could complete the drug design by employing the different moieties from different subregions in the binding pocket. For validating the reasonableness of drug design, we finally synthesized the targeted compound and evaluated its biological activity (Figure 4).
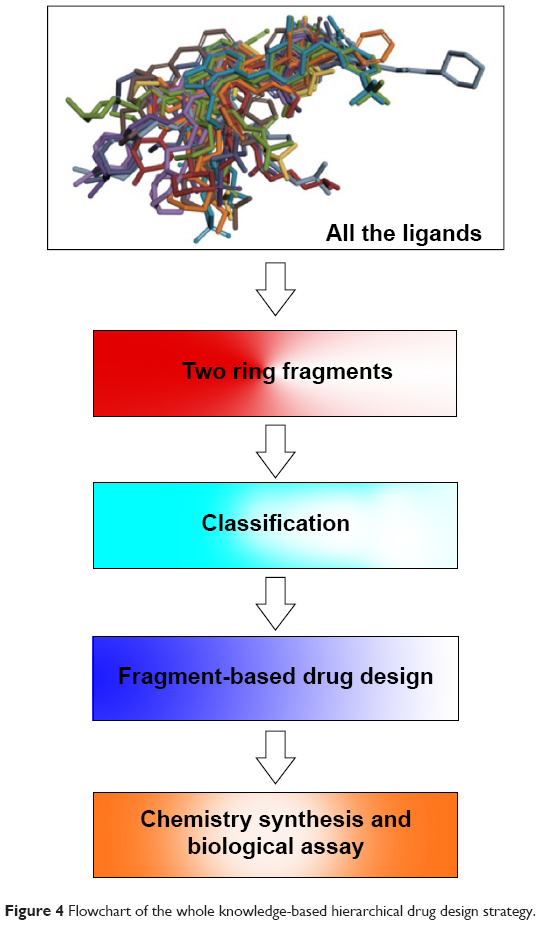 | Figure 4 Flowchart of the whole knowledge-based hierarchical drug design strategy. |
Design EGFR TKIs based on the study of the binding pockets
Because fragments with two aromatic rings (MW: 200–300 Da) were superior to fragments with one or three aromatic rings in FBDD, we collected all the fragments with two aromatic rings from the 42 crystal ligands and annotated the corresponding source ligand. According to the values of LipE of each protein item, we reckoned the matched values of fragment lipophilicity efficiency (formula in Table S1) of each fragment. Furthermore, in order to develop new EGFR TKIs for overcoming the T790M mutation, we meanwhile marked these important fragments retrieved from the protein crystal complexes with T790M mutations as the preference scaffolds when designing small molecules (Table S1).
Putting these fragments together (Figure 5A), it was clearly seen that the 4-anilinoquinazoline moiety (Fragment 1-11) was more effective than others such as pyrrolo[2,3-d]pyrimidin (Fragment 1-9), benzylfuro[2,3-d]pyrimidin (Fragment 1-10), and phenylthieno[3,2-d]pyrimidin (Fragment 1-14) when focused on the adenine binding and K subpockets (A/K regions). Additionally, fragments 1-3 and 1-16 located in the back pocket performed better than the others, especially 1-16, which can be selected as the preferential scaffold for targeting EGFR kinase with T790M mutation. Thus, we simply provided a rough procedure for producing new templates of EGFR inhibitors by combining the effective fragments located in the A/K subpockets and in the back pockets (Figure 5B) and paying attention to the fragments in the A/R subpockets (1-5, and 1-19~1-22). In this study, for producing highly effective EGFR inhibitors, we adopted the technique of combining fragments 1-11 and 1-6. As seen in Figure 5C, considering the values of 1-6, which contained seven detailed fragments extracted from seven unique protein kinase crystal complexes (Table S1, 1-6-1: 3W2O; 1-6-2: 3W2P; 1-6-3: 3W2R; 1-6-4: 3W32; 1-6-5: 3W33; 1-6-6: 3POZ; 1-6-7: 3RCD), we employed the fragments in the back pockets of 3W32 and 3W2R as the other part of new templates (A series and B series). The next step was to finish the process of drug design based on these two templates.
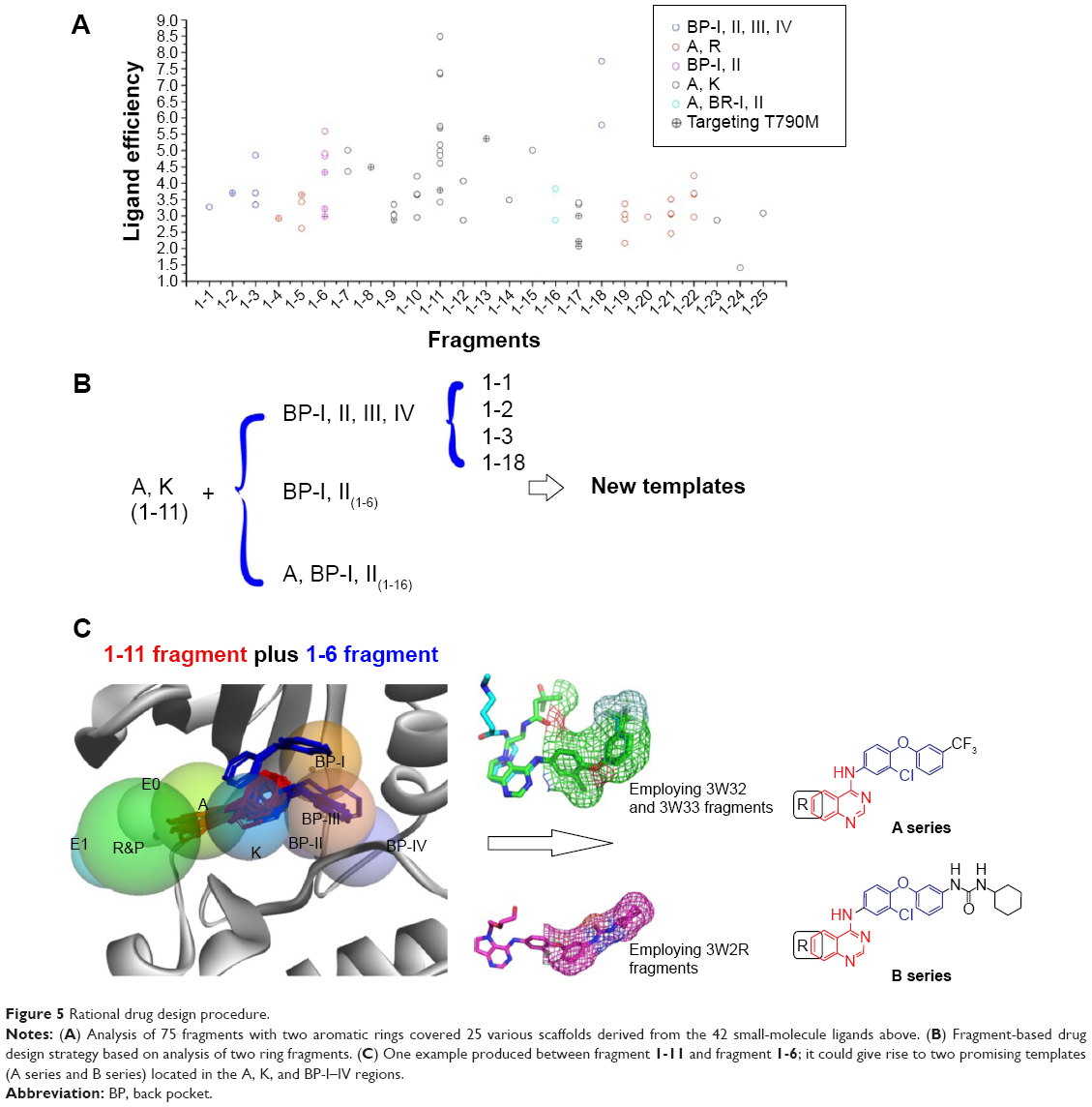 | Figure 5 Rational drug design procedure. |
As mentioned above, the modifications of the 6,7-positions of the new templates produced 20 new EGFR inhibitors (Figure 6, A-1–B-10). Based on a detailed investigation of the ligand-related references, these composed moieties located in the R/P subregions were mainly extracted from ten kinds of crystal ligands as follows: DJK, KJ8, KJV, 03P, HKI, IRE, AQ4, 0WM, FMM, and 1C9 (chemical structure in Figure S2).
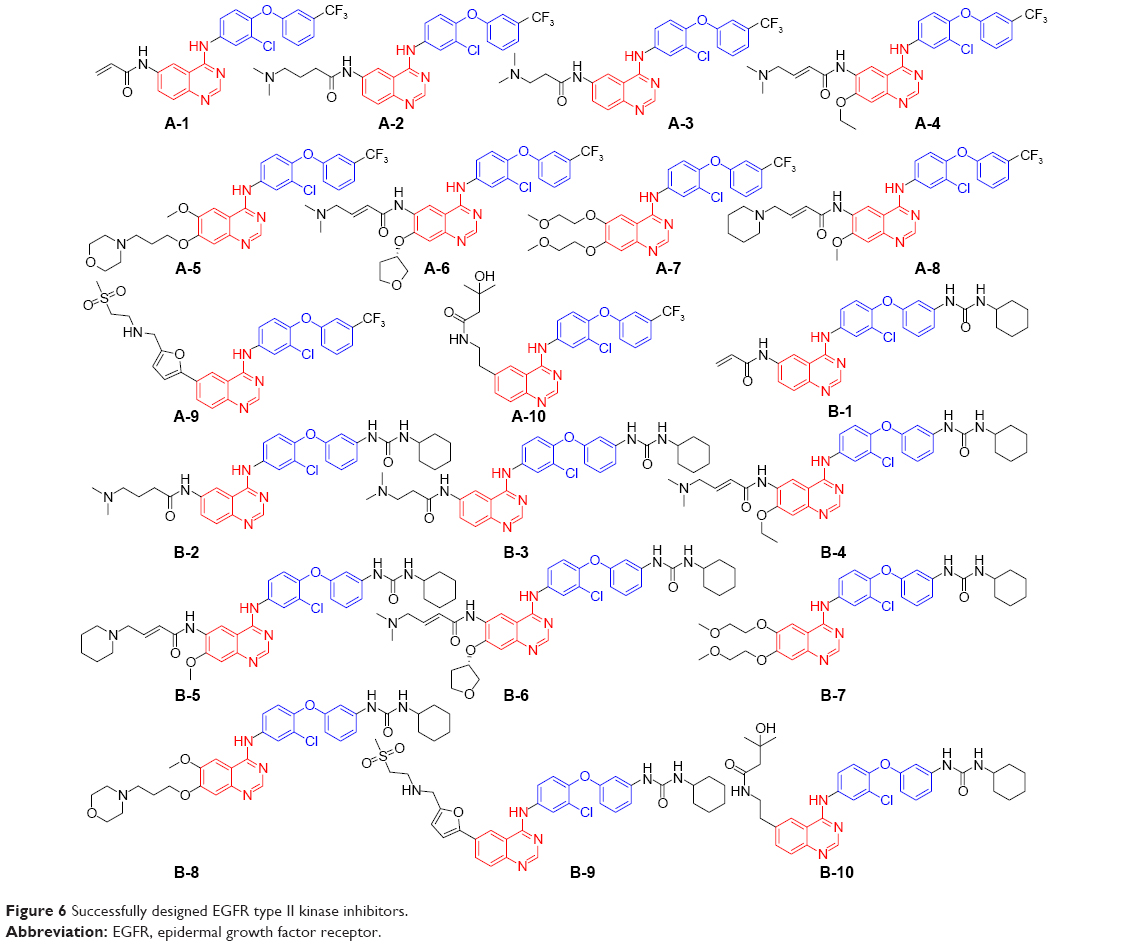 | Figure 6 Successfully designed EGFR type II kinase inhibitors. |
For these 20 new molecules, we evaluated their performance by molecular docking study based on the wild-type EGFR protein (PDB code: 3W33) and the dual T790M/L858R mutant protein (PDB code: 3W2R). With the aim of obtaining more precise outcome, the three docking protocols, CDOCKER docking, Glide sp docking, and Glide xp docking, were adopted. The detailed results are shown in Table S2. For most of the compounds in the CDOCKER docking study, this kind of algorithm could not search enough refined poses, possibly because this protocol could not perform well when small molecules possessed more rotatable bonds, particular compounds B-1–B-10. In addition, it was clearly noted that, in comparison with the control W2R and W19, compound A-10 performed better in a series of molecular docking studies, no matter whether 3W33 or 3W2R protein was used. Therefore, based on this kind of screening result, compound A-10 can be regarded as important candidate for chemical synthesis and biology evaluation.
Validation of availability of the design strategy
The detailed synthesis of compound A-10 is described in the “Methods” section. Figure 7 shows that 2-chloro-1-fluoro-4-nitrobenzene (1) as the starting material can directly react with 3-(trifluoromethyl)phenol (2) to give 2-chloro-4-nitro-1-(3-(trifluoromethyl)phenoxy)benzene (3), the subsequent reduction of which by Fe can produce 3-chloro-4-(3-(trifluoromethyl)phenoxy)aniline (4). On the other hand, cyclization of quinazoline mediated by dimethylformamide dimethyl acetal, together with compound 4, finally produced 6-chloro-N-(3-chloro-4-(3-(trifluoromethyl)phenoxy)phenyl)quinazolin-4-amine (7). Replacement of the halogen atom at the 6-position of compound 7 with boric acid moiety and subsequent amidation of the intermediate could generate the target compound A-10.
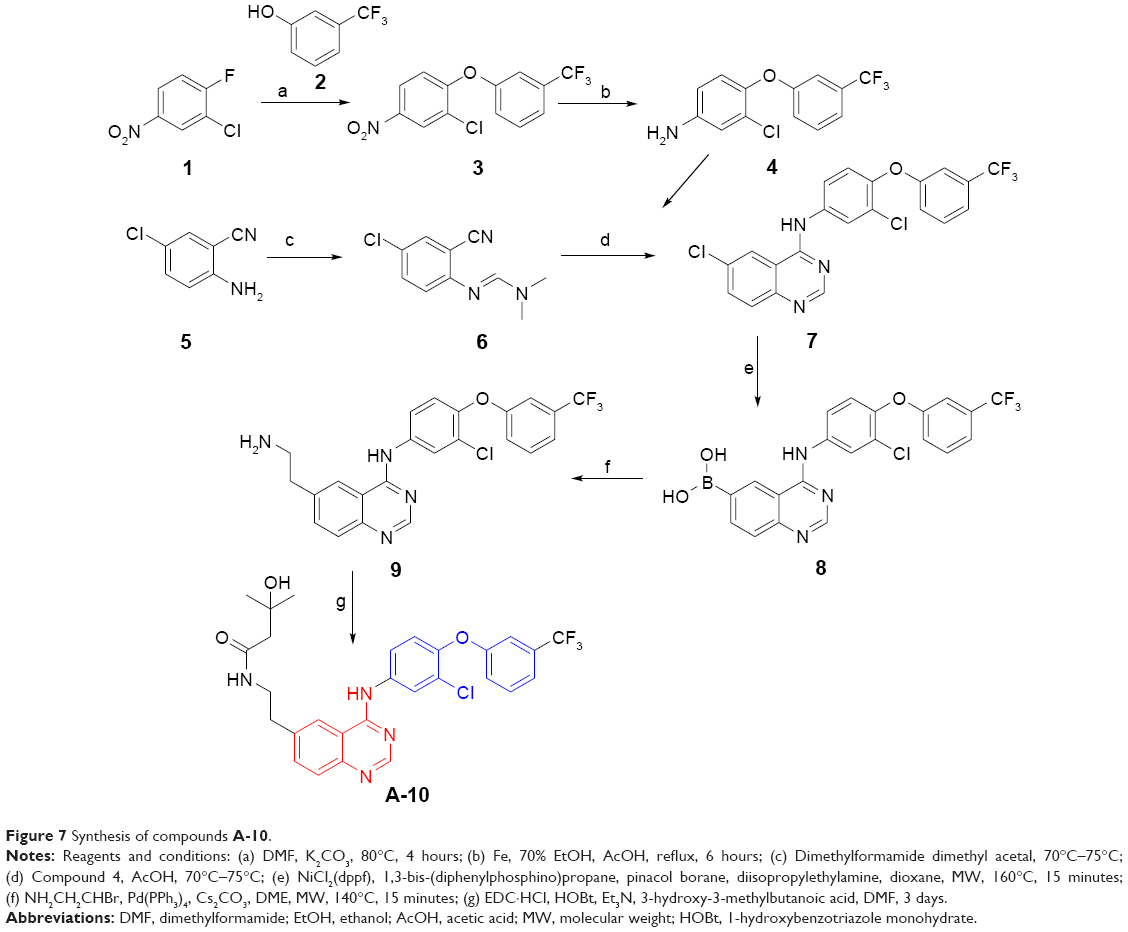 | Figure 7 Synthesis of compounds A-10. |
As to compound A-10, we examined the inhibitory activity of the compound against EGFR protein tyrosine kinase (Table 3), whose half maximal inhibitory concentration (IC50) values could reach up to 9.8 nM, 33 nM, and 19 nM for EGFRwild-type, EGFRT790M/L858R, and HER2, respectively, which are obviously superior to that of the two control molecules (gefitinib and erlotinib). Although this compound seemed to be less comparable to gefitinib or erlotinib in its inhibition toward EGFRwild-type, it showed at least 30-fold more potency against EGFR T790M than the two control molecules erlotinib and gefitinib in vitro. Moreover, it is worth noting that A-10 exhibited more potent activity against HER2 protein.
 | Table 3 Enzyme activities of compound A-10 against human EGFR and HER2 kinases |
In addition, the antiproliferative activities of compound A-10 are shown in Table 4. In accordance with kinase inhibition of A-10, it could meanwhile disturb the proliferative activities of three different cancer cells (A431: overexpression of wild-type EGFR; H1975: overexpression of dual mutation EGFR; MCF-7: overexpression of HER2), and performed better than the two EGFR drugs. Therefore, based on preliminary biological assay, it can be regarded as an important clinical candidate in the development of the third-generation EGFR inhibitors.
 | Table 4 In vitro cellular activities of A-10 |
To understand better the potency and relative selectivity for EGFR T790M, we predicted the binding modes of compound A-10 at the binding site of EGFR protein by molecular docking (Figure 8). It turned out that irrespective of whether the docking study was performed on 3W2R (Figure 8B–D) or 3W33 protein (Figure 8E–H), compound A-10 could bind the cleft tightly, in which the moiety (1-chloro-2-(3-(trifluoromethyl)phenoxy)benzene) played a key role in the kinase selectivity. For example, comparing 3W2R with 3W33, the orientation of this fragment seemed reversed, probably due to the fact that the gatekeeper mutation (T790M) could change the volume of the pocket context (BP-I subpocket). There is one more point to note: no matter whether the docking study was used by CDOCKER or Glide, the binding mode of A-10 in the active pocket remained unchanged.
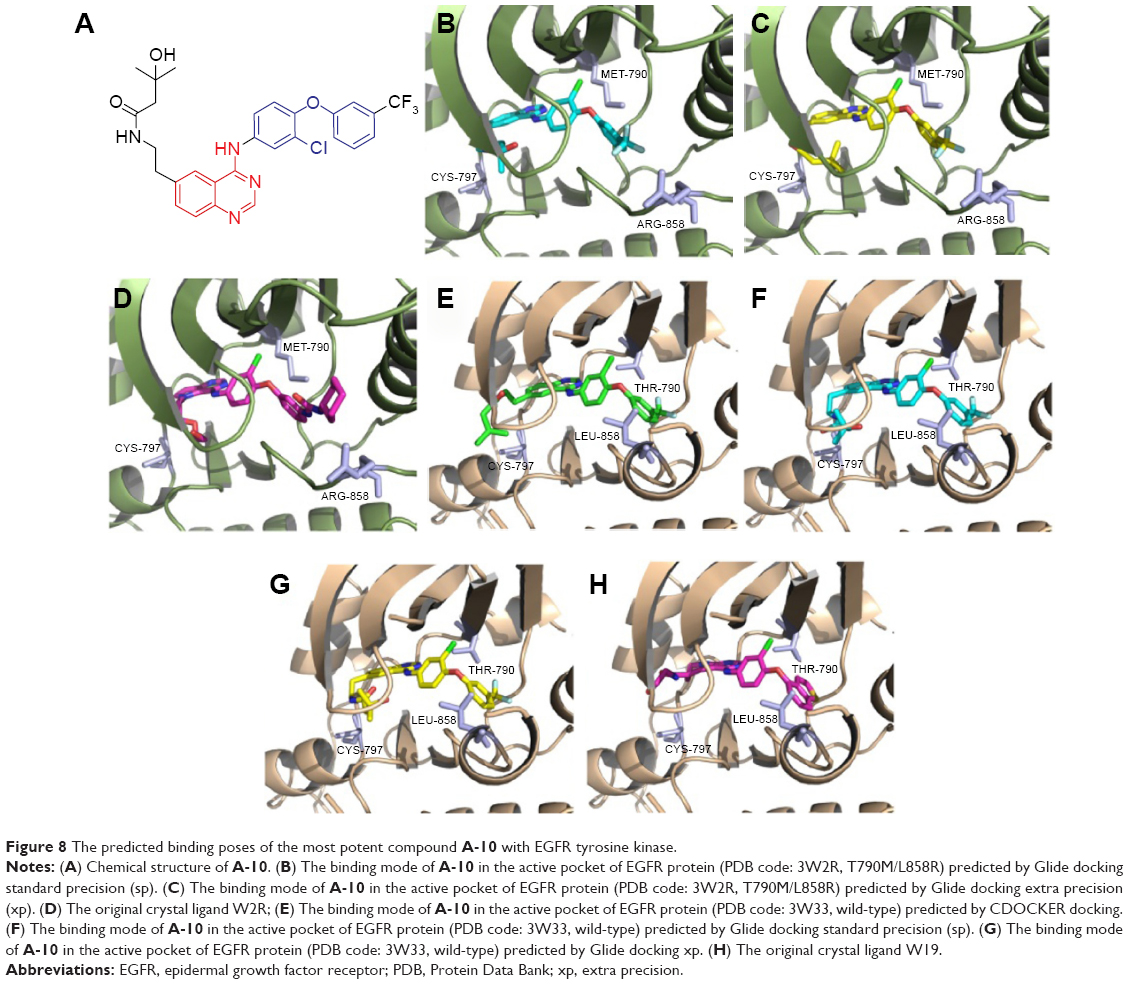 | Figure 8 The predicted binding poses of the most potent compound A-10 with EGFR tyrosine kinase. |
Conclusion
T790M mutation is a common mechanism of drug resistance to EGFR kinase inhibitors. A mixture of various approaches based on FBDD and structure-based drug design can be applied to identify novel agents specifically against drug resistance or oncogenic mutations implicated in human cancers. However, most of these approaches seemed a bit rough, so that the optimized compounds could not go further in the complicated biological assay. In this paper, based on the analysis of the binding pocket space of the EGFR family protein tyrosine kinase and the rationale of FBDD, we provided a unique strategy for developing a new EGFR inhibitor and identified a novel EGFR kinase inhibitor (A-10) that was effective in vitro against EGFR with dual T790M/L858R mutation. Thus, compared to the common protocol in medicinal chemistry, this kind of protocol in the field of drug design seems to be more precise. Besides, further studies are needed to determine whether this EGFR inhibitor will be clinically effective in in vivo models harboring the EGFR T790M mutation.
Acknowledgments
This work was financed by six grants below. It includes Health Department of Heilongjiang Province of China (grant number: 2013086), National Natural Science Foundation of China (grant number: 30772540, 81172214 and 81301991), Natural Science Foundation of Heilongjiang Province, China (grant number: LC2012C08), and Harbin Medical University Cancer Hospital (JJMS2014-04).
Disclosure
The authors declare no conflicts of interest in this work.
References
Olayioye MA, Neve RM, Lane HA, Hynes NE. The ErbB signaling network: receptor heterodimerization in development and cancer. EMBO J. 2000;19:3159–3167. | ||
Burgess AW, Cho HS, Eigenbrot C, et al. An open-and-shut case? Recent insights into the activation of EGF/ErbB receptors. Mol Cell. 2003;12:541–552. | ||
Zhang J, Yang PL, Gray NS. Targeting cancer with small molecule kinase inhibitors. Nat Rev Cancer. 2009;9:28–39. | ||
Pao W, Miller V, Zakowski M, et al. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci U S A. 2004;101:13306–13311. | ||
Raymond E, Faivre S, Armand JP. Epidermal growth factor receptor tyrosine kinase as a target for anticancer therapy. Drugs. 2000;60(suppl 1):15–23. [discussion 12–41]. | ||
Higa GM, Abraham J. Lapatinib in the treatment of breast cancer. Expert Rev Anticancer Ther. 2007;7:1183–1192. | ||
Lin NU, Winer EP, Wheatley D, et al. A phase II study of afatinib (BIBW 2992), an irreversible ErbB family blocker, in patients with HER2-positive metastatic breast cancer progressing after trastuzumab. Breast Cancer Res Treat. 2012;133:1057–1065. | ||
Zhang Q, Dai HH, Dong HY, Sun CT, Yang Z, Han JQ. EGFR mutations and clinical outcomes of chemotherapy for advanced non-small cell lung cancer: a meta-analysis. Lung Cancer. 2014;85:339–345. | ||
Shigematsu H, Lin L, Takahashi T, et al. Clinical and biological features associated with epidermal growth factor receptor gene mutations in lung cancers. J Natl Cancer Inst. 2005;97:339–346. | ||
Sequist LV, Bell DW, Lynch TJ, Haber DA. Molecular predictors of response to epidermal growth factor receptor antagonists in non-small-cell lung cancer. J Clin Oncol. 2007;25:587–595. | ||
Yun CH, Mengwasser KE, Toms AV, et al. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc Natl Acad Sci U S A. 2008;105:2070–2075. | ||
Kobayashi S, Boggon TJ, Dayaram T, et al. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. J Med. 2005;352:786–792. | ||
Wu JY, Wu SG, Yang CH, et al. Lung cancer with epidermal growth factor receptor Exon 20 mutations is associated with poor gefitinib treatment response. Clin Cancer Res. 2008;14:4877–4882. | ||
Costa DB, Schumer ST, Tenen DG, Kobayashi S. Differential responses to erlotinib in epidermal growth factor receptor (EGFR)-mutated lung cancers with acquired resistance to gefitinib carrying the L747S or T790M secondary mutations. J Clin Oncol. 2008;26:1182–1184. [author reply 1184–1186]. | ||
Balak MN, Gong Y, Riely GJ, et al. Novel D761Y and common secondary T790M mutations in epidermal growth factor receptor-mutant lung adenocarcinomas with acquired resistance to kinase inhibitors. Clin Cancer Res. 2006;12:6494–6501. | ||
Bean J, Riely GJ, Balak M, et al. Acquired resistance to EGFR kinase inhibitors associated with a novel T854A mutation in a patient with EGFR-mutant lung adenocarcinoma. Clin Cancer Res. 2008;14:7519–7525. | ||
Zhou W, Liu X, Tu Z, et al. Discovery of Pteridin-7(8H)-one-based irreversible inhibitors targeting the epidermal growth factor receptor (EGFR) kinase T790M/L858R mutant. J Med Chem. 2013;56:7821–7837. | ||
Xu S, Xu T, Zhang L, et al. Design, synthesis, and biological evaluation of 2-Oxo-3,4-dihydropyrimido[4,5-d]pyrimidinyl derivatives as new irreversible epidermal growth factor receptor inhibitors with improved pharmacokinetic properties. J Med Chem. 2013;56:8803–8813. | ||
Yang J, Wang LJ, Liu JJ, et al. Structural optimization and structure – activity relationships of N2-(4-(4-Methylpiperazin-1-yl)phenyl)-N8-phenyl-9H-purine-2,8-diamine derivatives, a new class of reversible kinase inhibitors targeting both EGFR-activating and resistance mutations. J Med Chem. 2012;55:10685–10699. | ||
Domarkas J, Dudouit F, Williams C, et al. The combi-targeting concept: synthesis of stable nitrosoureas designed to inhibit the epidermal growth factor receptor (EGFR). J Med Chem. 2006;49:3544–3552. | ||
Morales GA, Garlich JR, Su J, et al. Synthesis and cancer Stem cell-based activity of substituted 5-Morpholino-7H-thieno[3,2-b]pyran-7-ones designed as next generation PI3K inhibitors. J Med Chem. 2013;56:1922–1939. | ||
Wu G, Robertson DH, Brooks CL 3rd, Vieth M. Detailed analysis of grid-based molecular docking: a case study of CDOCKER – a CHARMm-based MD docking algorithm. J Comput Chem. 2003;24: 1549–1562. | ||
Friesner RA, Banks JL, Murphy RB, et al. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem. 2004;47:1739–1749. | ||
Yasuda H, Park E, Yun CH, et al. Structural, biochemical, and clinical characterization of epidermal growth factor receptor (EGFR) Exon 20 insertion mutations in lung cancer. Sci Transl Med. 2013;5:216ra177. | ||
Red Brewer M, Yun CH, Lai D, Lemmon MA, Eck MJ, Pao W. Mechanism for activation of mutated epidermal growth factor receptors in lung cancer. Proc Natl Acad Sci U S A. 2013;110:E3595–E3604. | ||
Ward RA, Anderton MJ, Ashton S, et al. Structure- and reactivity-based development of covalent inhibitors of the activating and gatekeeper mutant forms of the epidermal growth factor receptor (EGFR). J Med Chem. 2013;56:7025–7048. | ||
Peng YH, Shiao HY, Tu CH, et al. Protein kinase inhibitor design by targeting the Asp-Phe-Gly (DFG) Motif: the role of the DFG Motif in the design of epidermal growth factor receptor inhibitors. J Med Chem. 2013;56:3889–3903. | ||
Kawakita Y, Seto M, Ohashi T, et al. Design and synthesis of novel pyrimido[4,5-b]azepine derivatives as HER2/EGFR dual inhibitors. Bioorg Med Chem. 2013;21:2250–2261. | ||
Sogabe S, Kawakita Y, Igaki S, et al. Structure-based approach for the discovery of Pyrrolo[3,2-d]pyrimidine-based EGFR T790M/L858R mutant inhibitors. ACS Med Chem Lett. 2013;4:201–205. | ||
Gajiwala KS, Feng J, Ferre R, et al. Crystal structure of the wild-type EGFR kinase domain in com dacomitinib (soaked). Structure. 2013;21:209–219. | ||
Park JH, Liu Y, Lemmon MA, Radhakrishnan R. Erlotinib binds both inactive and active conformations of the EGFR tyrosine kinase domain. Biochem J. 2012;448:417–423. | ||
Solca F, Dahl G, Zoephel A, et al. Target binding properties and cellular activity of afatinib (BIBW 2992), an irreversible ErbB family blocker. J Pharmacol Exp Ther. 2012;343:342–350. | ||
Yoshikawa S, Kukimoto-Niino M, Parker L, et al. Structural basis for the altered drug sensitivities of non-small cell lung cancer-associated mutants of human epidermal growth factor receptor. Oncogene. 2013;32:27–38. | ||
Aertgeerts K, Skene R, Yano J, et al. Structural analysis of the mechanism of inhibition and allosteric activation of the kinase domain of HER2 protein. J Biol Chem. 2011;286:18756–18765. | ||
Fidanze SD, Erickson SA, Wang GT, et al. Imidazo[2,1-b]thiazoles: multitargeted inhibitors of both the insulin-like growth factor receptor and members of the epidermal growth factor family of receptor tyrosine kinases. Bioorg Med Chem Lett. 2010;20:2452–2455. | ||
Zhou W, Ercan D, Chen L, et al. Novel mutant-selective EGFR kinase inhibitors against EGFR T790M. Nature. 2009;462:1070–1074. | ||
Xu G, Abad MC, Connolly PJ, et al. 4-Amino-6-arylamino-pyrimidine-5-carbaldehyde hydrazones as potent ErbB-2/EGFR dual kinase inhibitors. Bioorg Med Chem Lett. 2008;18:4615–4619. | ||
Xu G, Searle LL, Hughes TV, et al. Discovery of novel 4-amino-6-arylaminopyrimidine-5-carbaldehyde oximes as dual inhibitors of EGFR and ErbB-2 protein tyrosine kinases. Bioorg Med Chem Lett. 2008;18:3495–3499. | ||
Yun CH, Boggon TJ, Li Y, et al. Structures of lung cancer-derived EGFR mutants and inhibitor complexes: mechanism of activation and insights into differential inhibitor sensitivity. Cancer Cell. 2007;11:217–227. | ||
Blair JA, Rauh D, Kung C, et al. Structure-guided development of affinity probes for tyrosine kinases using chemical genetics. Nat Chem Biol. 2007;3:229–238. | ||
Wood ER, Truesdale AT, McDonald OB, et al. A unique structure for epidermal growth factor receptor bound to GW572016 (Lapatinib): relationships among protein conformation, inhibitor off-rate, and receptor activity in tumor cells. Cancer Res. 2004;64:6652–6659. | ||
Stamos J, Sliwkowski MX, Eigenbrot C. Structure of the epidermal growth factor receptor kinase domain alone and in complex with a 4-anilinoquinazoline inhibitor. J Biol Chem. 2002;277:46265–46272. | ||
Ishikawa T, Seto M, Banno H, et al. Design and synthesis of novel human epidermal growth factor receptor 2 (HER2)/epidermal growth factor receptor (EGFR) dual inhibitors bearing a Pyrrolo[3,2-d]pyrimidine Scaffold. J Med Chem. 2011;54:8030–8050. | ||
Littlefield P, Moasser MM, Jura N. An ATP-competitive inhibitor modulates the allosteric function of the HER3 pseudokinase. Chem Biol. 2014;21:453–458. | ||
Wood ER, Shewchuk LM, Ellis B, et al. 6-Ethynylthieno[3,2-d]- and 6-ethynylthieno[2,3-d]pyrimidin-4-anilines as tunable covalent modifiers of ErbB kinases. Proc Natl Acad Sci U S A. 2008;105:2773–2778. | ||
Qiu C, Tarrant MK, Choi SH, et al. Mechanism of activation and inhibition of the HER4/ErbB4 kinase. Structure. 2008;16:460–467. | ||
Cui JJ, McTigue M, Nambu M, et al. Discovery of a novel class of exquisitely selective mesenchymal-epithelial transition factor (c-MET) protein kinase inhibitors and identification of the clinical candidate 2-(4-(1-(quinolin-6-ylmethyl)-1H-[1,2,3]triazolo[4,5-b]pyrazin-6-yl)-1H-pyrazol-1-yl)ethanol (PF-04217903) for the treatment of cancer. J Med Chem. 2012;55:8091–8109. | ||
van Linden OP, Kooistra AJ, Leurs R, de Esch IJ, de Graaf C. KLIFS: a knowledge-based structural database to navigate kinase-ligand interaction space. J Med Chem. 2014;57:249–277. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.