")
Back to Journals » Drug Design, Development and Therapy » Volume 14
Naringenin Prevents Propofol Induced Neurodegeneration in Neonatal Mice Brain and Long-Term Neurocognitive Impacts on Adults
Authors Zou L, Ning M, Wang W, Zheng Y, Ma L, Lv J
Received 9 September 2020
Accepted for publication 28 October 2020
Published 10 December 2020 Volume 2020:14 Pages 5469—5482
DOI https://doi.org/10.2147/DDDT.S280443
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Tuo Deng
Lili Zou,1,* Mingliang Ning,2,* Wenjuan Wang,1 Yuemei Zheng,1 Liping Ma,1 Jing Lv3
1Department of Anesthesiology, General Hospital of NingXia Medical University, Yinchuan, NingXia 750000, People’s Republic of China; 2Department of Oncological Surgery, General Hospital of NingXia Medical University, Yinchuan, NingXia 750000, People’s Republic of China; 3Department of Anesthesiology, Union Hospital of Tongji Medical College, Huazhong University of Science and Technology, Wuhan, Hubei 430000, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Jing Lv
Department of Anesthesiology, Union Hospital of Tongji Medical College, Huazhong University of Science and Technology, Wuhan, Hubei 430000, People’s Republic of China
Tel/Fax +86-27-85351618
Email [email protected]
Background: Natural products have shown neuroprotective effects in neurodegenerative conditions. Naringenin is a natural flavonoid with various pharmacological activities especially antioxidant, anti-inflammatory and neuroprotective properties. We investigated the effects of naringenin on anesthetic propofol-induced impacts on neonatal mouse brain development and consequent long-term neurocognitive impacts during adulthood.
Materials and Methods: Female C57Bl/6 and male CD-1 mice and postnatal day 7 (P7) pups were exposed to propofol (2.5 mg/kg) and propofol with naringenin (50 mg/kg). Mice pups were allowed to grow until week 10 (adulthood), and memory and learning were assessed.
Results: Propofol caused neurodegeneration by inducing apoptosis in the neonatal mouse brains while naringenin administration prevented neuronal cell loss by preventing neuronal apoptosis in neonatal mouse brains. Propofol caused degenerative alterations in metabolic factors pH, PO2, glucose and lactate, which were subsequently restored by naringenin treatment. Propofol-exposed mice, when developed into adults, showed long-term neuronal deficits, impaired neurocognitive functions, and memory and learning restrictions.
Conclusion: Administration of naringenin to propofol-exposed mice resulted in significant neuroprotective effects by restoring long-term neurocognitive functions. The molecular mechanism behind the effects of naringenin was mediated by suppressing apoptosis and preventing cellular inflammation. These findings suggest that propofol administration requires careful consideration and that naringenin may prevent neurodegeneration and neurocognitive functions.
Keywords: naringenin, propofol, neonatal, neurodegeneration, apoptosis, cognitive functions
Background
Neonatal surgical procedures have evolved into advantageous pediatric surgeries that require increased use of anesthetics. Anesthetic agents have long been used in clinical and preclinical experimental studies.1,2 Early exposure of anesthetics prior to the completion of synaptogenesis in neonates has resulted in extensive neurodegradation characterized by neuronal apoptosis and late learning disability. Synaptogenesis is an early developmental stage in the nervous system and a rapid brain growth stage characterized by the formation of synapses between neurons. Anesthetics and sedative agents have shown contradictory responses in neuronal systems.3 However, anesthetics have shown neurodegenerative responses in neonates and impaired neurocognitive functions upon entering adulthood, especially when administered during synaptogenesis. Studies conducted on animal models of neuronal disease have demonstrated that administration of anesthetics and sedative agents at synaptogenesis may induce severe neurodegenerative responses, including neuronal apoptosis and morphological degenerations in neurons. These detrimental changes could further lead to learning and behavioral abnormalities during adulthood.3,4 Notably, the commonly used anesthetics and sedative agents in clinical practices act by enhancing GABAA receptors and by blocking N-methyl-d-aspartate (NMDA) receptors, or both. However, contrary to the mature brain, the neonatal brain exhibits neurodegenerative responses when exposed to the transient pharmacological blockage of NMDA receptors, as characterized by intense apoptotic cell death in neurons.4 However, another volatile anesthetic agent, sevoflurane, is particularly useful in infants and children, with rapid induction and recovery.5 Sevoflurane acts by enhancing GABAA receptors and by blocking NMDA receptors. In vivo and in vitro experimental studies have reported that sevoflurane may affect cell survival by inducing neuronal apoptosis in the hippocampus of the brain.6
Propofol (Pentothal), sodium thiopental in chemical nature, is a commonly used anesthetic agent for the induction of anesthesia, because recovery from propofol is more rapid than other anesthetics.7 Propofol induces neuronal apoptosis by modulating the activities of NMDA and GABAA receptors, as well as by blocking Na+ channels. Isoflurane causes prevalent neurodegeneration and neuronal death when administered during the rapid brain growth period (synaptogenesis) in monkey neonates. It further causes long-term behavioral abnormalities and deficiencies in learning and memory during adulthood in animals.8 Nonetheless, preventive stimulations attenuate the propofol-induced apoptosis of neurons and protect brain development in neonatal rodents.9 Propofol was shown to causes neuronal degeneration in neonatal mice and subsequent long-term neurocognitive impairments in mice at adulthood.10 Propofol administration was also associated with a rare but serious side effect due to prolonged infusion of propofol (usually more than 4mg/kg per h for more than 24 hours). This is denoted as the propofol infusion syndrome which presents physiological characteristics of metabolic acidosis, hyperkalemia, hyperlipidemia, and rhabdomyolysis, renal dysfunction and even cardiac failure.11 The pharmacokinetic studies of propofol administration in the range of 3–8 mg/kg showed a significant impact on preterm infants.12 Thus, it is a prerequisite to evaluate the effects of anesthetics on neuronal growth, especially the impact on neuronal apoptosis and associated histopathological changes. Thus, the demand for clinically safe anesthetics continues to increase as well as other protective measures to reduce the neurodegenerative burden.
Several natural products have shown neuroprotective properties in mature as well as neonatal models. Flavonoids are natural dietary polyphenols associated with the prevention of several diseases especially oxidative stress induced pathophysiological conditions.13 Naringenin (4ʹ,5,7-trihydroxyflavanone) is a flavonoid that is colorless and bitter in taste and is predominantly found in a variety of grapes, berries and citrus fruits.14 Naringenin exerts a plethora of pharmacological activities in various test systems, such as antioxidant,15 anti-inflammatory,16 immunomodulatory,17 anticancer and neuroprotective properties.13,18 Naringenin has been reported to have a number of biological effects, such as neuroprotective and memory enhancing activity19 and as monoamine oxidase (MOX) inhibitor20 and anti-inflammatory as well as antioxidant.21 Naringenin mediates its effects mainly by modulating nuclear factor-kappa B (NF-κB) and peroxisome proliferator-activated receptor (PPAR).18 The free radical scavenging activities of naringenin may prevent neurodegenerative responses by modulating cell growth and other cellular signals.13,15,18 Naringenin exerts its neuroprotective effects by inhibiting monoamine oxidase and anti-inflammatory, as well as antioxidant and memory enhancing, activities.20 The neuroprotective effects of naringenin in experimental systems are mediated mainly through NF-κB-mediated neuroinflammation and the prevention of neurodegeneration by preventing apoptosis.18 Neuroprotective effects of naringenin were shown in experimental model of stroke through suppression of NF-κB-mediated neuroinflammation.18 Naringenin was also reported as a neuroprotective agent in neurodegenerative diseases like Parkinson’s disease, amnesia, Alzheimer’s disease, as well as it prevented oxidative injury in several pathophysiological disorders affecting the brain.22–24
Despite of a number of beneficial effects of naringenin reported by several studies still the neuroprotective effects in the brain were needed to elaborate. Also to assess the contradictory neurotoxic and cytotoxic effects of anesthetics were necessitated. Thus, this study aimed to assess the effects of propofol on the neonatal mouse brain followed by preventive management using naringenin. Neuronal apoptosis was assessed in the neonatal brain and long-term neurocognitive impacts on adult mice were evaluated. The current study emphasizes that naringenin protects against the neurodegenerative impacts of propofol on mouse brains, followed by recovery of impairments in learning and memory behavior during adulthood. The novelty of this study relies towards the exploration of the deleterious effects of propofol in neonates and long-term behavioral effects, as well as the neuroprotective effects of naringenin on the neonatal mouse brain during the developmental stage (synaptogenesis).
Materials and Methods
Experimental Animals and Drug Treatment
All the animal experimental procedures were performed in accordance with approval from the Institutional Animal Ethical Committee of Huazhong University of Science and Technology (Ref#2018/01/AEC19) following the Ministry of Science and Technology (MOST), Govt. of China, issued Regulations for the Administration of Affairs Concerning Experimental Animals. The study required female C57Bl/6 and male CD-1 mice. Animals were housed in a climate-controlled room at a temperature of 25±2°C. Animals were given standard chow and water ad libitum. The lighting conditions were regulated for a 12-h cycle (dark and light). Animals were allowed to mate to produce offspring. The offspring were randomly inducted in the study at P7 (postnatal day 7). P7 offspring mice were further randomly divided into three different treatment groups (n=15 per group): Group-I: control treated with normal saline (NS) as a vehicle control; Group-II: propofol treatment at 2.5 mg/kg; and Group-III: propofol (2.5 mg/kg) followed by naringenin (50 mg/kg). Drug agents and vehicle were administered intraperitoneally. Earlier studies have suggested that use of propofol in the range of 2 to 5 mg/kg body weight of neonatal mouse could cause neuronal degeneration.10–12
Respiratory and Metabolic Factors Analysis
Five mouse pups in each treatment group were subjected to respiratory and metabolic factors analysis at 6 and 12 h following NS or drug exposure. The temporal profile of physiological variables was established by collecting arterial blood from mouse pups at different time intervals. Blood was collected from the right carotid artery by aspiration. Physiological parameters, such as pH, O2, CO2, and glucose and lactate concentrations, were measured using the SMART analyzer system (i-Stat Corp., East Windsor, NJ, USA).
Histological Examination of the Mouse Brain
At 12 h after drug or vehicle administration, five mice from each treatment group were sacrificed and brains were collected. The brains were processed for hematoxylin and eosin (H&E) staining according to a standard method. Briefly, mouse brain hippocampal sections were dissected and fixed in 0.1% methanol. Dissected tissue sections were dehydrated and embedded in paraffin wax. Paraffin wax-embedded mouse brains were sectioned to 5-μm thickness using an automated microtome (Leica Biosystems Nussloch GmbH, Germany) and stained with H&E. Stained slides were visualized under a light microscope (Olympus Magnus, PA, USA) at 40x magnification. Morphological changes in the CA1 and CA3 regions of the hippocampus were examined and evaluated.
Immunohistochemistry (IHC) for Apoptosis
At 12 h after drug or vehicle administration, five mice from each treatment group were sacrificed and brains were collected. Brains were processed for the apoptosis assay by using the caspase-3 active (C-3A) antiserum assay kit (Cell Signaling Technology, Danvers, MA, USA) according to the manufacturer’s instructions. Briefly, dissected mouse brain tissues were fixed in 4% paraformaldehyde through a left ventricular cardiotomy. Brain sections were then cut using an automated microtone at 40-µm thickness. The sections were blocked on glass slides using 3% H2O2. Glass slide-blocked sections were then incubated with C-3A at a 1:500 dilution overnight at 4°C. Antibody-treated sections were subjected to treatment with a horseradish peroxidase-labeled anti-rabbit secondary antibody for 4 h at room temperature. Sections were visualized using the DAB chromogen system (Sigma-Aldrich, MO, USA) followed by counterstaining with H&E. Sections were then visualized under a light microscope (Olympus Magnus, PA, USA) at 40x magnification. The documentation of caspase-3-positive regions in different tissue sections was performed, and the scoring of C-3A-positive cells is presented per mm2 brain section. Counting and scoring were performed by blinded investigators. The total count of C-3A-positive cells is presented in the form of apoptotic cell percentage relative to the vehicle control.
TUNEL Assay for Apoptosis
At 12 h after drug or vehicle administration, five mice from each treatment group were sacrificed and brains were collected. The TUNEL (Terminal deoxynucleotidyl transferase dUTP Nick End Labeling) assay was performed to estimate indications of cellular apoptosis using the In Situ Cell Death Detection Kit, Fluorescein (Roche Diagnostics, Risch-Rotkreuz, Switzerland). Briefly, mouse brain sections were fixed and permeated according to a standard procedure. Sections were then end-labeled with digoxigenin-11-deoxyuridine triphosphate by terminal deoxynucleotidyl transferase enzyme. Sections were incubated and treated with stop-washing buffer. Nuclei were stained with DAPI (4ʹ-6-diamidino-2-phenylindole) and TUNEL-positive regions were observed under a light microscope at 40x magnification. The visualized sections were scored for TUNEL-positive cells per mm2 brain section by blinded investigators. The total count of TUNEL-positive cells is presented in the form of stained cell percentage relative to the vehicle control.
RNA Isolation and Quantitative Real-Time PCR (qPCR)
Mouse brains collected at 12 h after drug or vehicle administration were sectioned for hippocampal slices. Three hippocampal slices (n=3 per group) were used for RNA isolation using Trizol reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. Isolated RNA was quantified and reverse transcription was performed for cDNA synthesis using the Reverse Transcriptase SuperScript III Kit (Invitrogen, USA). An equivalent amount of RNA (1 μg per sample group) was reverse-transcribed into cDNA. An equivalent amount of cDNA (2 µL per sample group) was used for qPCR on an ABI 7500 Real Time PCR System (Applied Biosystems, Foster City, CA, USA). The specific primer sets for caspase-3, PARP, Bcl-xL, NF-κB, iNOS and Cox-2 genes were used for qPCR. Relative quantification of mRNAs was performed by normalizing their levels to that of 18s RNA. Changes in gene expression are presented as fold change in the treatment groups relative to the control (one-fold).
Analysis of Long-Term Cellular and Neurocognitive Impacts
The long-term cellular and neurocognitive impacts of propofol and naringenin-treated P7 mouse pups were analyzed when mice were grown to adulthood at P28. For this purpose, five mice from each treatment group were placed in separate chambers and grown until P28. At this stage of growth, mice were separated by gender and then placed into different chambers to grow until week 10 (adulthood). Neurocognitive tests were performed in male and female mouse pairs. After completion of neurocognitive tests, mice were euthanized, and brains were excised and processed for neuronal nuclei (NeuN) staining and other assays.
NeuN Immunohistochemistry
NeuN protein is specific to neurons, where it localizes in the nuclei of postmitotic neurons and perikarya. NeuN IHC staining was performed in adult mouse brains (n=3 per group) after perfusion with chilled 0.9% NS and 4% paraformaldehyde. Briefly, brains were embedded in paraffin sections of 40-µm thickness and rehydrated for antigen retrieval with 100 mM citrate-phosphate buffer (pH 6.30). Sections were then washed with Tris-buffered saline containingTween-20 (TBST) and submerged in primary NeuN antibody (Chemicon International Inc., Temecula, CA, USA) at 1:100 dilution overnight. Slides were rinsed in TBST and incubated with donkey anti-mouse IgG secondary antibody (Cell Signaling Technology) for 3 h. Slides were rinsed again in TBST, coverslipped, and mounted. The density of NeuN-stained neurons was determined using laser scanning confocal microscopy (Nikon, Tokyo, Japan) as described previously.25–27 Brain sections containing NeuN-stained neurons were examined in two regions: the CA3 pyramidal cell layer of the hippocampus and retrosplenial cortical regions adjacent to the brain midline. Data are quantitatively expressed as raw neuronal density counts relative to the control.
Behavioral Observations and Neurocognitive Testing
The Morris water maze (MWM) test is performed to analyze spatial learning and memory in behavioral neurosciences, as described previously.28 The offspring were grouped and housed with a corresponding dam up to P28. Male and female offspring pairs of each dam were used for the MWM test between the hours of 10 a.m. and 5 p.m. in a homogenously illuminated room. A pool (100 cm diameter x 50 cm latitude) with black internal coating was filled with water (27°C) and a camera was placed 2.5 m above the pool. The pool containing spatial cues was divided into four quadrants: NW, northwest; NE, northeast; SW southwest; and SE, southeast. Spatial learning was examined on a 10 cm (diameter) escape platform placed in the second quadrant (2 cm submerged; 25 cm from the edge). Mice could find the location of the platform by training four times per day during four successive days. Starting positions for each trial were set to be SE, SW, NW, and NE. The latency time represents spatial learning ability, which was scored for finding the hidden platform from the start position (SE). A probe trial was performed for the spatial memory test beginning from the start position (SE) on day 5 in a platform-less pool. The time of first platform crossing and the frequency of platform crossing were recorded.
Statistical Analysis
Data are presented as the mean ± standard deviation from at least three experimental repeats. Data were compared by one-way analysis of variance (ANOVA) with Tukey’s post-hoc tests for multiple comparisons. The MWM test data were analyzed by two-way ANOVA. The relative expression of genes by qPCR was compared by Student’s t-test. P values <0.05 were considered statistically significant.
Results
Effect of Naringenin on Propofol-Induced Morphological Changes
Brain sections of P7 neonatal mice were stained with H&E and evaluated for changes following exposure to propofol (2.5 mg/kg, body weight [b.w.]) and subsequent treatment with naringenin (50 mg/kg, b.w.). The histological staining images of neonatal mouse brain slices are presented in Figure 1. First, the effects of propofol were analyzed and compared with vehicle-treated control mice. Propofol exposure in neonates caused swelling of pyramidal neurons in the CA1 region of the hippocampus. Other prominent structural changes included cellular clumping and degenerative patches in brain sections treated with propofol. These patches were prominent, with darker and shrunken nuclei in propofol-treated brain sections compared to the control. These observations indicate that propofol treatment to neonatal mice causes cellular damage and degenerative structural changes in the brain. Anesthetic and sedative agents, such as lidocaine and propofol, show similar degenerative changes in neonatal mice as well as apoptotic neurodegeneration in the developing mouse brain.29 The effect of propofol on brain morphology was analyzed and compared to treatment with naringenin in the developmental stage of the mouse brain. Treatment of propofol-exposed mice with naringenin repaired the morphological changes in mouse brains compared to propofol treatment alone and in reference to the control (Figure 1A and B). These observations suggest that naringenin exerts a preventive impact on the mouse brain undergoing degenerative structural changes.
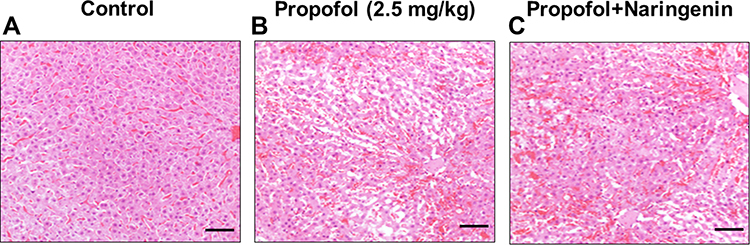 |
Figure 1 Histological assessment of neonatal mice brain cortical sections. (A) control group; (B) propofol treatment group; (C) propofol+naringenin. Magnification 40x, Scale bar 100 μM. |
Naringenin Prevents Propofol-Induced Alterations in Respiratory and Metabolic Factors
Respiratory and metabolic activities regulate the physiological acid-base balance. Acidosis is a primary increase in the partial pressure of carbon dioxide (PCO2) with or without a compensatory bicarbonate (HCO3−) level. Thus, pH usually maintains at a low but near to normal level. The reduction in respiratory rate and/or volume (hypoventilation) is typically caused by alterations in the central nervous system or pulmonary or iatrogenic conditions.30 Respiratory acidosis can be acute or chronic, and the chronic form is asymptomatic and may characterize some pathophysiological conditions. Thus, alterations in respiratory and metabolic factors suggest that the pathophysiological conditions arise from morphological changes in neonatal mouse brains. The measurement of serum electrolytes and other metabolites, as well as physiological parameters, may aid in clinical diagnosis. We analyzed the effect of propofol treatment on neonatal mice by measuring respiratory and metabolic functions. Arterial blood was collected from the right carotid artery by aspiration from experimental mice in different treatment groups (n=5 per group). The values for pH, PCO2 (mm Hg), PO2 (mm Hg), glucose (mg/dL), and lactate (mg/L) are presented in Table 1. Exposing mouse neonates to propofol (2.5 mg/kg) caused respiratory and metabolic imbalances, showing critical effects on brain development. Exposure to propofol reduced the pH in a time-dependent manner. After 12 h of propofol exposure, the pH was reduced to 7.02±0.4 (P<0.05 vs control). Treatment of propofol-exposed neonatal mice with naringenin (50 mg/kg) restored the pH value (7.32±0.3) to that of the control group; however, the pH value was significantly lower than that of the propofol group (P<0.05). Upon analyzing the ratio and level of gaseous exchange, it was observed that PCO2 was slightly reduced by exposure to propofol and was restored by treatment with naringenin; however, these changes were not statistically significant. However, the PO2 level was reduced in propofol-treated mice in a time-dependent manner. The PO2 level was significantly reduced to 126 and 122 mm Hg compared to the control (average 143 mm Hg) (P<0.05). Treatment of propofol-exposed neonatal mice with naringenin (at 12 h) restored the PO2 level to 136 mmHg, which was significantly reduced compared to the propofol group (P<0.05). Glucose and lactate levels are indicators of biochemical metabolic activities that may be affected by chronic conditions. The glucose level was reduced in propofol-treated mice in a time-dependent manner. The glucose level was significantly reduced to 98 and 92 mg/dL compared to the control (P<0.05). Treatment of propofol-exposed neonatal mice with naringenin caused an increase in glucose levels to 105 and 119 mg/dL at 6 and 12 h, respectively, which was significantly higher than the propofol group (P<0.05). Likewise, the lactate level was significantly elevated to 4.8 and 6.5 mM compared to the control (P<0.05). Treatment of propofol-exposed neonatal mice with naringenin caused a reduction in the level of lactate to 5.1 and 4.2 mM at 6 and 12 h, respectively, which was significantly lower than the propofol group at 12 h (P<0.05). These observations indicate that exposure of neonatal mice to propofol causes metabolic acidosis, as demonstrated by the lower pH and alterations in blood glucose and lactate, and that these changes are subsequently restored by naringenin administration.
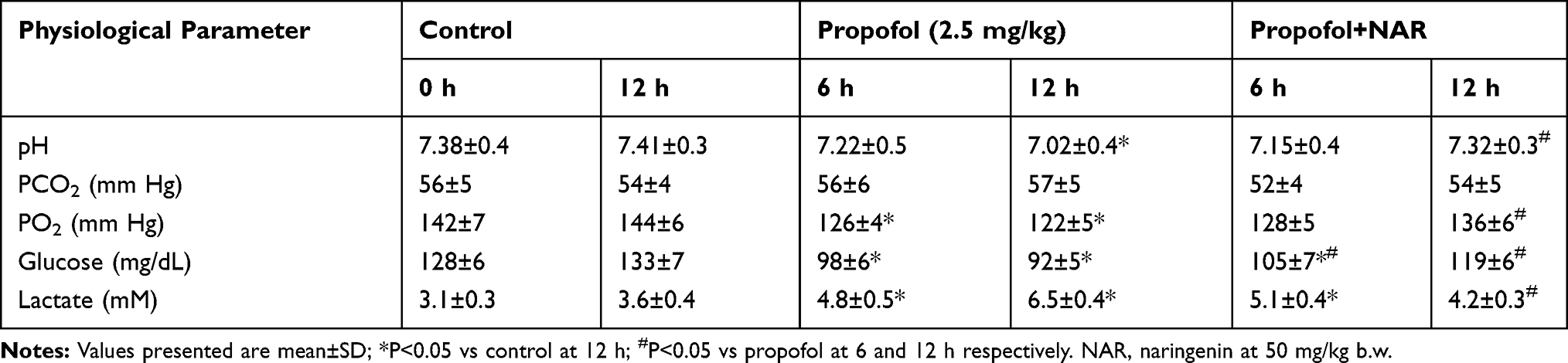 |
Table 1 Physiological Factors Assessment in Neonatal P7 Mice Exposed to Different Treatment Conditions |
Naringenin Prevents Propofol-Induced Apoptotic Neurodegeneration
Neurodegenerative changes at the cellular level were measured by assaying apoptosis in the brains of mouse neonates exposed to propofol followed by naringenin administration. Apoptosis was evaluated by C-3A IHC and the TUNEL assay. Exposure of neonatal mice to propofol induced cellular degeneration in the neonatal mouse brain, as assessed by IHC staining of activated caspase-3 in cortical sections (Figure 2). IHC staining also revealed that the hippocampal sections of control mouse brains showed negligible C-3A. Neonatal mouse brain sections exposed to propofol showed drastically increased staining for C-3A. Furthermore, treatment of propofol-exposed mice with naringenin showed reduced expression of C-3A compared to propofol-treated mice. Caspase-3 staining revealed that propofol treatment induced neuronal apoptosis by 47±6% compared to the control (1%). Subsequent treatment of propofol-exposed mice with naringenin caused a reduction in C-3A IHC expression to the level of 14±3% (P<0.05 vs propofol). C-3A IHC staining results were supported by the TUNEL staining of cortical sections of neonatal mouse brains (Figure 3). TUNEL staining revealed that propofol treatment in mouse neonates caused an increase in apoptotic staining in the hippocampal (CA1 and CA3) regions in propofol-treated mice compared to control mice. Furthermore, treatment of propofol-exposed mice with naringenin caused a dramatic reduction in TUNEL-positive staining of the hippocampal (CA1 and CA3) regions. The quantification of TUNEL-positive staining showed that propofol induced neuronal apoptosis by 48±7% compared to the control (6%). The percentage of TUNEL-positive cells was 23±5% following naringenin treatment in propofol-exposed mice, which was significantly lower than the propofol treatment group (P<0.05). These results indicate that propofol induces neuronal cell death in neonatal mouse brains, which can be prevented by naringenin administration.
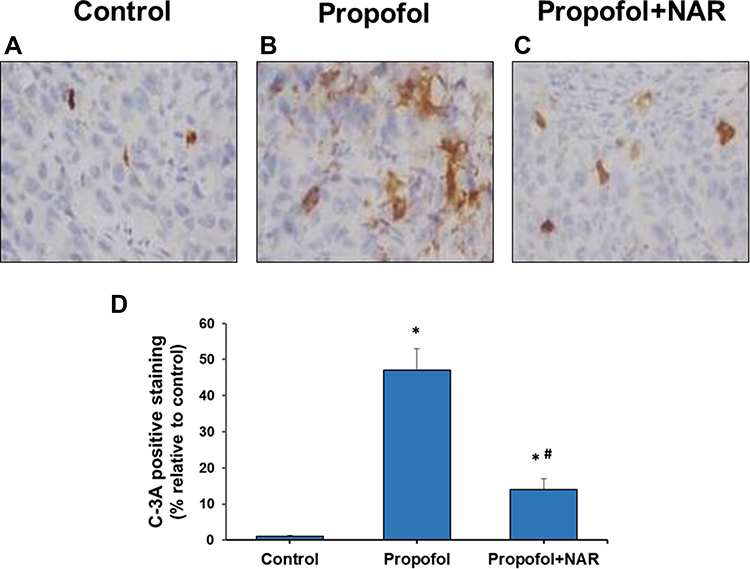 |
Figure 2 Activated caspase-3 (C-3A) IHC staining in brain cortex slices from neonatal mice. (A) control group; (B) propofol treatment group; (C) propofol+naringenin. (D) Quantitative expression of C-3A positive staining. *P<0.05 values versus control group; #P<0.05 values versus propofol group. |
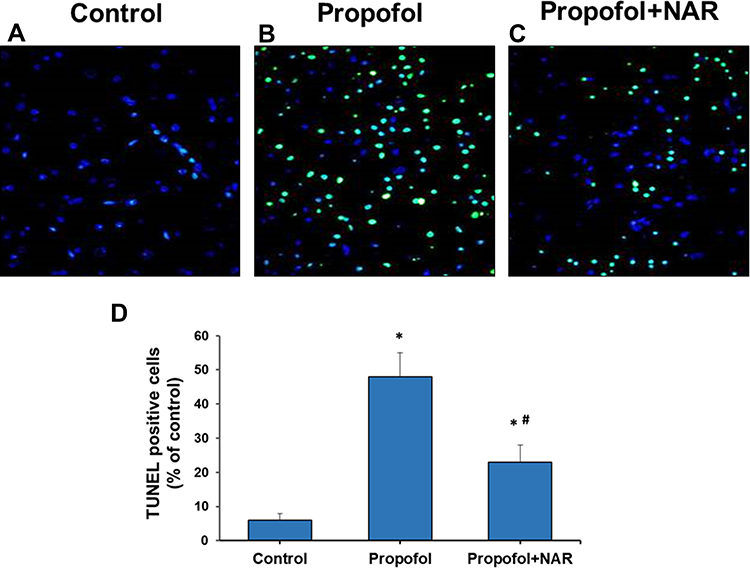 |
Figure 3 Apoptosis measurement by TUNEL staining of brain cortex slices from neonatal mice. (A) control group; (B) propofol treatment group; (C) propofol+naringenin. (D) Quantitative expression of TUNEL positive staining. *P<0.05 versus control group; #P<0.05 versus propofol group. |
Naringenin Prevents Propofol-Induced Apoptosis and Inflammation
Propofol-induced apoptosis in neonatal mouse brains was confirmed by C-3A staining and the TUNEL assay. Molecular signatures for apoptosis induction were confirmed by analyzing the expression of genes involved in the apoptotic pathway. Total cellular RNA was isolated from hippocampal tissues, and cDNA was synthesized and further used for qPCR analysis of caspase-3, PARP and Bcl-xL (Figure 4A). Gene expression was normalized to the control gene and is presented as the fold change relative to the control. Propofol exposure caused a significant increase in the expression of caspase-3, PARP and Bcl-xL by 2.8-, 1.8- and 2.2-fold, respectively, compared to the control (onefold) (P<0.05). Subsequent naringenin treatment significantly reduced gene expression by 1.6-, 1.4- and 1.5-fold, respectively, compared to the propofol treatment group (P<0.05), which was similar to the control group. These results collectively indicate that propofol induces apoptosis in neonatal mouse brains by activating the apoptosis pathway, which can be prevented by naringenin administration.
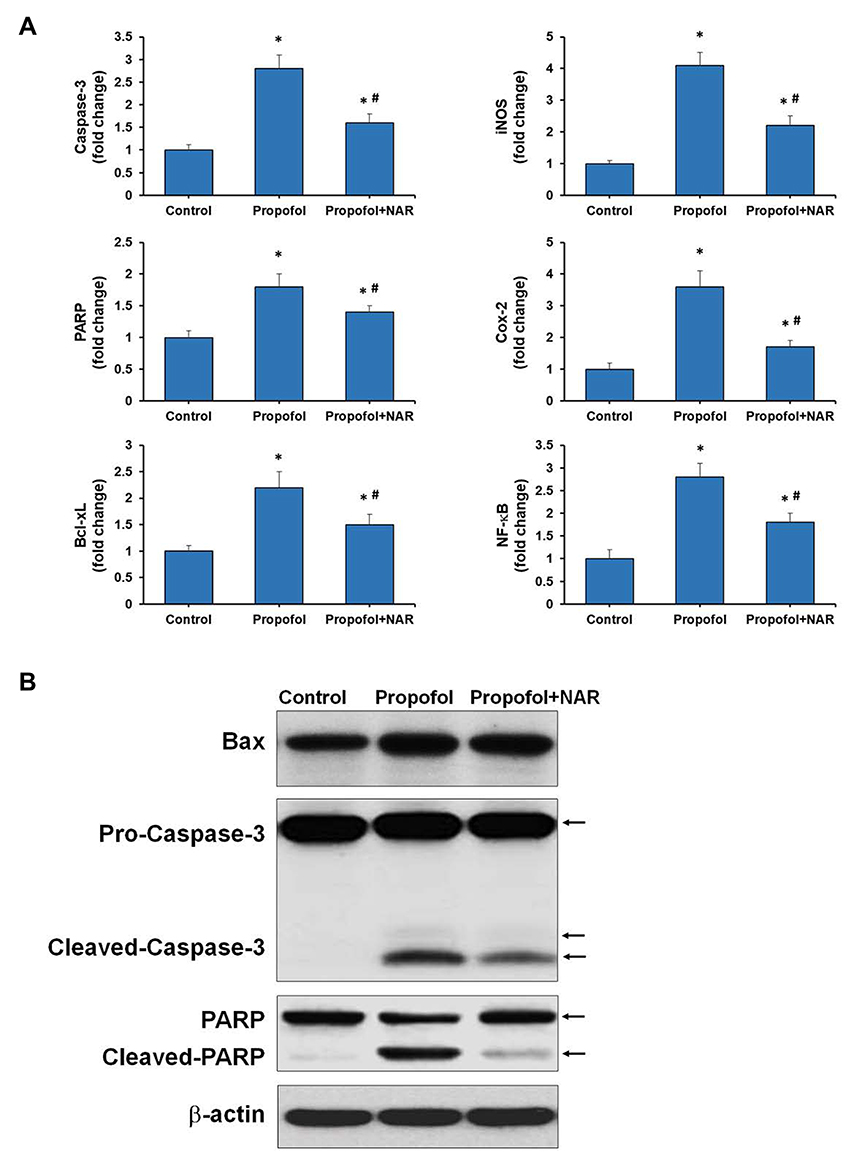 |
Figure 4 Gene and protein expression analysis for apoptosis and inflammation related genes. (A) qPCR was performed for Caspase-3, PARP, Bcl-xL, iNOS, COX-2, and NF-κB genes. Expression of genes was presented as fold change relative to control (one-fold). *P<0.05 versus control group; #P<0.05 versus propofol group. (B) Western blotting was performed for Bax, Caspase-3, PARP, and β-actin. NAR, naringenin. The arrows corresponding to the molecular weight of proteins are mentioned respectively. |
Next, we analyzed the expression of genes in inflammation-related pathways, such as iNOS, COX-2, TNF-α, and NF-κB (Figure 4B). Propofol exposure caused a drastic increase in the expression of iNOS and COX-2 (4.1- and 3.6-fold) relative to the control (onefold). Subsequent naringenin treatment significantly reduced the expression of iNOS and COX-2 by 2.2- and 1.7-fold, respectively, relative to the propofol group (P<0.05). Propofol exposure caused overexpression of TNF-α and NF-κB by 2.4- and 2.8-fold, respectively, which were subsequently suppressed by naringenin administration to 1.8- and 1.5-fold, respectively. Increases in the expression of iNOS, COX-2, TNF-α, and NF-κB are directly associated with the inflammatory response in the brains of neonatal mice exposed to propofol. And suppression of these increments by naringenin administration may represent one of the molecular mechanisms of its preventive effects. These results collectively indicate that propofol induces apoptosis in neonatal mouse brain sections by activating apoptosis and pro-inflammatory signaling pathways, which can be prevented by naringenin administration.
Naringenin Improves Propofol-Induced Neuronal Impairments and Neurocognitive Functions
The long-term cellular and neurocognitive functions in neonatal mice exposed to propofol followed by naringenin treatment were evaluated by IHC staining for NeuN. IHC for NeuN antigen showed that propofol-induced degenerative changes in mouse brain sections. Neonatal mice exposed to propofol showed neuronal deficits and reduced neuronal density upon reaching 10 weeks of age. IHC NeuN staining is intended to analyze the neuropathological changes, highlighting the physiological state of neuronal cells in brain sections. NeuN antigen expression is observed specifically in post-mitotic neurons and acts as a primary indicator of neuronal nucleus development and function.31 Mouse brains were analyzed for NeuN expression at the hippocampal CA2/3 and cortical regions (Figure 5). Control mouse brains showed strong NeuN expression, demonstrating healthy neuronal development and function. However, rigorous degenerative cellular loss was observed in adult mice exposed to propofol at the neonatal stage. Mouse brain sections showed labeling cell bodies, axons and dendrites in NeuN IHC staining. The analysis of NeuN positivity in postembryonic life propofol-exposed mice (at neonatal stage) showed degenerative signs of neuronal differentiation. In mice, propofol-induced changes in brain tissue correlate with alterations in the functional and morphological development of the brain during adulthood. Furthermore, when propofol-exposed mice were treated with naringenin, NeuN expression improved and the degenerative structural signs were reduced. Naringenin restored neuronal development in propofol-exposed mice. The quantification of NeuN expression was performed and compared between groups with reference to the control group (100%). Propofol treatment caused a drastic reduction in NeuN expression by 47±5% (P<0.05 vs control). Treatment of propofol-exposed mice with naringenin caused a restoration of NeuN expression by 72±9% compared to the control. Although the improvement in NeuN expression was significantly lower than the control (P<0.03), the significance with respect to the propofol group was highly notable (P<0.001). These results demonstrate that propofol exposure in neonatal mice may cause altered neuronal development and neurodegeneration that can be visualized at adulthood. Subsequent treatment of propofol-exposed mice with naringenin was significantly preventive, as it enhanced NeuN expression.
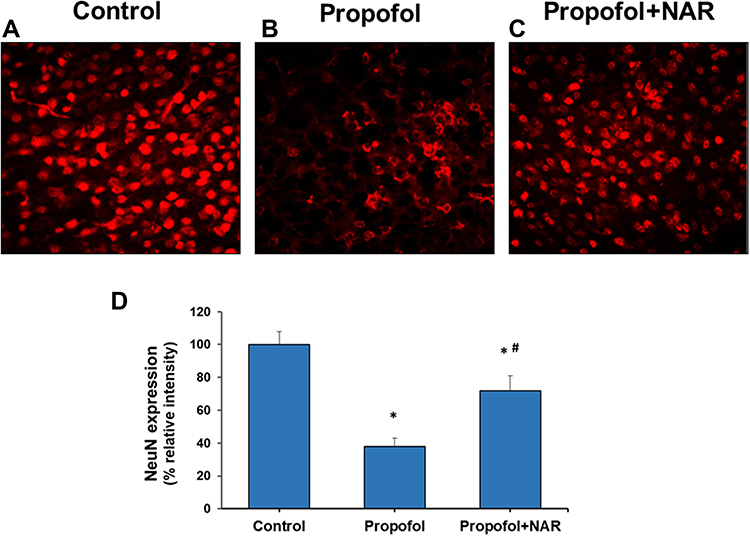 |
Figure 5 Neuronal nucleus (NeuN) expression analysis in cortical brain section of adult mice. (A) control group; (B) propofol treatment group; (C) propofol+naringenin. (D) Quantitative expression of NeuN expression. *P<0.05 versus control group; #P<0.05 versus propofol group. |
The neurocognitive functions of mice after propofol exposure and naringenin administration were assessed at adulthood by assessing latency time (Figure 6). The latency time for the control group exhibited a decreasing trend from days 1 to 5 by 100.8, 96.2, 88.6, 82.8 and 76.2 s, respectively. The latency time was slightly reduced in propofol-treated mice yet significantly higher than the control group. Propofol-treated mice showed a latency time of 101.2, 98.8, 98.6, 94.6 and 88.3 s at 1 to 5 days, respectively. The propofol group data were significantly reduced relative to the control on days 3 to 5 (P<0.05). Furthermore, administration of naringenin to propofol-exposed mice caused a notable improvement in latency time. The naringenin-treated group showed a latency time of 102.7, 96.4, 92.4, 86.4 and 81.2 s at 1 to 5 days, respectively. The naringenin group latency time data were significantly reduced relative to the propofol-treated group on days 3 to 5 (P<0.05) and these values remained close to the control (P<0.05). An increasing trend in latency time is an indicator of impaired learning and behavior in mice, as caused by propofol exposure. Subsequent treatment of propofol-treated mice with naringenin improved the latency time, indicating that naringenin mediates the increased learning response.
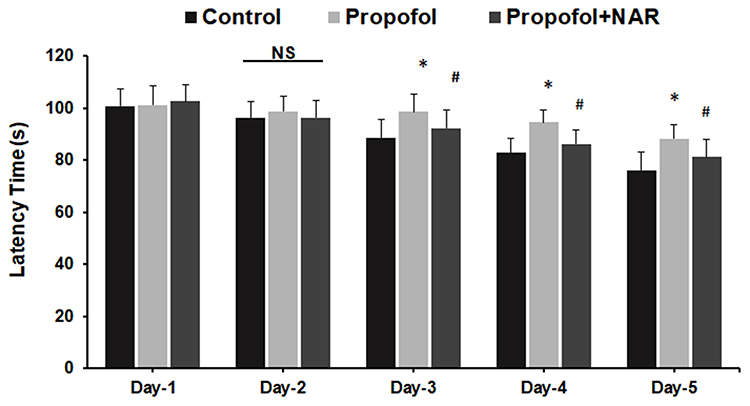 |
Figure 6 Assessment of learning behavior by measuring latency time. Latency time (s) was measured by calculating mice to find the hidden platform during day 1 to day 5 in the MWM test. *P<0.05 versus control group on test day; #P<0.05 versus propofol group on test day. Abbreviation: NS, not significant. |
Further, the time and frequency of first platform crossing in the MWM test were evaluated following exposure to propofol and naringenin treatment (Table 2). The comparative analysis revealed that control group mice took 48.4±6.8 s to cross the first platform, with a frequency of2.4±0.4. The propofol-treated group took longer to cross the first platform (68.7±8.5 s) and showed a reduced frequency of platform crossing (1.2±0.2). These values were significantly altered compared to the control group (P<0.05). Subsequent treatment of propofol-exposed mice with naringenin improved the time it took to cross the first platform (54.3±5.3 s) and the frequency of platform crossing (1.9±0.3). These values were significantly altered compared to the propofol group (P<0.05) and similar to the control group (P>0.05). The probe trial demonstrated that mouse in propofol group showed mean time 19.3±1.5 s which reduced in mouse group treated with propofol by mean time 13.6±2.0 s and the data was statistically significant (P = 0.006). While naringenin administration to propofol-treated mice showed mean time 16.7±1.5 s which was statistically not significant to propofol group (P > 0.05) yet marginally significant as compared to control (P = 0.047). Thus, the data shows that time spent by propofol group in the quadrant with the removed platform was reduced and the same was improved by naringenin administration yet statistically not significant (Supplementary Figure 1).
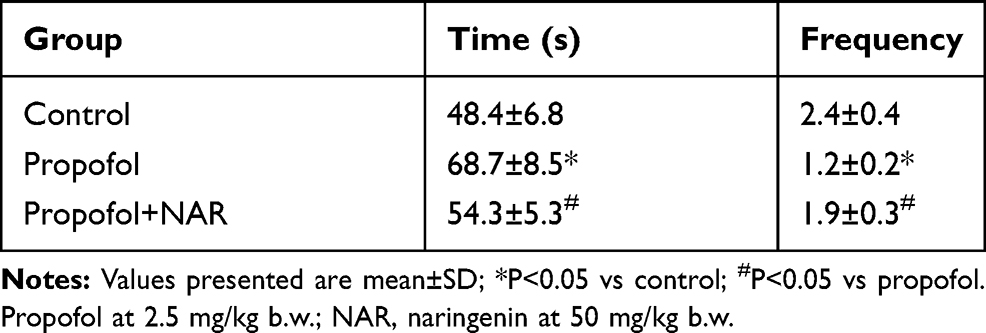 |
Table 2 Effect of Naringenin on Propofol Induced Behavioral Changes in Adult Mice |
Discussion
This study evaluated the effects of the anesthetic agent propofol on neonatal mice and explored the effects on neuronal growth, as well as brain development and learning and behavioral functions at adulthood. Furthermore, the prevention of neurodegeneration was assessed by using naringenin. The observations from this study demonstrate that propofol induced neurodegeneration in mouse brains and neuronal cell death. It also caused degenerative signs in mature neurons at adulthood, causing learning, memory and behavioral impairments. Propofol-exposed mice treated with naringenin exhibited reduced neuronal apoptosis in neonatal mouse brains and improved neurocognitive functions in adult mice, indicating the preventive effects of naringenin. Thus, the findings from this study suggest that naringenin may be a potent preventive natural remedy against propofol-induced neurodegeneration.
Anesthetic agents have shown various clinical and physiological indications, with contradictory signs of neuronal development in neonates and neurocognitive functions in adults. Anesthetic and sedative agents, when administered in rodents during synaptogenesis, have shown prevalent neuronal apoptosis and neuronal degeneration, thus impairing learning and behavioral abnormalities at adulthood.3–5 Anesthetics agents have shown variable long-term effects on the developing brain and subsequent cognitive functions. Anesthetics agents like ketamine, midazolam, isoflurane and propofol have been reported to cause neurodegenerative changes in the developing brain and subsequent cognitive dysfunction in several animal models.9,32–35 The occurrence of such long-term neurocognitive impacts by anesthetic agents has raised concerns about their use, especially in neonates. Propofol is, chemically, sodium thiopental (Pentothal), an intravenously administered short-acting hypnotic/amnestic agent that has been used for the induction and maintenance of general anesthesia and sedation. Propofol induces neuronal apoptosis by acting as NMDA and GABAA agonists and as a Na+ channel blocker.3,34 Additionally, propofol exerts protective functions in hypoxic brains by reducing arterial blood flow and intracranial pressure, as well as by maintaining metabolism.36,37 Therefore, investigating the role of neurotransmitter receptors such as GABAA and NMDA remains an important challenge when assessing the effects of anesthetic and sedative agents for their use in neonates.
This study demonstrated that propofol caused cellular degeneration in mouse brains (Figure 1) and modulated physiological parameters, such as blood pH, PO2, and glucose and lactate levels (Table 1). Propofol caused impairments to cell proliferation and inhibited neurogenesis in the brains of mice infants possibly by inducing cognitive dysfunction.38 Naringenin treatment in propofol-exposed mice showed corrective measures: it suppressed the level of neuronal apoptosis and maintained physiological and biochemical parameters. The propofol-induced reduction in blood glucose may represent a key biochemical mechanism regulating cellular degeneration in the brain, which was maintained by naringenin administration. Propofol-induced neuronal apoptosis in neonatal mouse brains was subsequently suppressed by naringenin administration. The detrimental effects of propofol exposure on neonatal mice were long-term neuronal deficits at adulthood, which were assessed by NeuN staining. Naringenin restored NeuN staining and functions that were suppressed by propofol. Propofol eventually impaired neurocognitive deficits and had long-term impacts on memory and learning in adults, as assessed by the MWM test for spatial learning and memory. Notably, naringenin administration to propofol-exposed mice was significantly effective at repairing cognitive issues, and improved learning and memory functions in adult mice. Propofol exposure in mouse neonates caused degenerative signatures in brain regions, including swelling of pyramidal neurons in the CA1 region of the hippocampus. This result indicates that propofol causes cellular damage and degenerative structural modifications in the developing brain. Treatment of propofol-exposed mice with naringenin showed drastic improvements in degenerative morphology in brain sections. Propofol showed to cause swelling of the pyramidal neurons in the CA1 region of the hippocampus in a dose-dependent manner in an infant mice model.10 The measurement of apoptosis by TUNEL staining and caspase-3 IHC suggest that propofol causes significant apoptotic changes in the CA1 and CA3 hippocampal regions of the brain. Furthermore, naringenin may inhibit these changes and suppress the apoptotic response. The activation of apoptosis in mouse brains was also confirmed by qPCR analysis of apoptosis-related genes (caspase-3, PARP and Bcl-xL), which revealed that propofol caused an apparent increase in the expression of these genes. The expression of these genes directly correlates with apoptosis induction. In addition, naringenin treatment reversed the effects of propofol and prevented apoptotic neuronal cell death. Propofol was shown to significantly increase the staining of hippocampal CA1 and CA3 regions at 5 mg/Kg dose in a time-dependent manner and increased apoptosis.10
Since the impact of propofol exposure on neonates may exert long-term consequences when the animals develop to adulthood, the impact was analyzed for long-term cellular and neurocognitive functions. NeuN IHC analysis was applied to emphasize the physiological status of neurons in the brain. Propofol caused rigorous cell loss in adults compared to the control group, which was subsequently restored by naringenin treatment. NeuN immunoreactivity declines significantly when exposed to severe injuries, such as cerebral hypoxia/ischemia and fetal distress or perinatal asphyxia.31 Thus, NeuN expression is a direct marker of propofol-induced neuronal damage and loss of neuronal morphology, which leads to long-term neurodegeneration in mouse brains. These results were accompanied by observations from behavior and learning studies. Propofol increased the latency time, which was subsequently reduced by treating propofol-exposed mice with naringenin. The high latency time in the propofol-exposed group is an indicator of poor learning and behavioral response in mice, which were restored when mice were treated with naringenin. Propofol-exposed mice took a significantly longer time to cross the first platform, with a reduced frequency, which were restored by naringenin administration, suggesting that naringenin may act as a potent preventive agent against propofol-induced poor learning behavior and neurocognitive responses in mice at adulthood. Propofol causes neurodegeneration by inhibiting the biosynthesis of amino acid neurotransmitters such as aspartate, glutamate, glycine, and GABAA.39,40 Although the exact mechanisms behind anesthetics induced neurotoxicity and neuronal damage remain complicated and unclear. However, modulation of apoptotic signalling is identified as one of the major mechanisms for the cellular responses of anesthetic agents which further correlates with the activation of many deleterious signalling events in cell death signalling, cleavage of cytoskeletal proteins, signaling kinases and DNA repair enzymes in neuronal and nonneuronal cells.3,34–38,41,42 Reversal of the propofol-altered biochemical and cellular events may represent a plausible mechanism that is involved in naringenin-mediated neuroprotection which can be utilized in preventive and therapeutic neuroprotection.
Conclusion
The results of the current study indicate that propofol exposure causes neuronal degeneration in mice at the neonatal stage, leading to learning and behavioral impairments at adulthood. Propofol exposure caused cellular degenerative changes in the mouse brain and modulated the levels of physiological and biochemical parameters (pH, PCO2, PO2, glucose and lactate). Propofol exposure ultimately induced neuronal apoptosis in mouse brains, which could cause long-term neuronal deficits in adults. Propofol-exposed mice showed impaired neurocognitive functions and poor spatial learning and reference memory. In addition, administration of naringenin to propofol-exposed mice exerted preventive effects by suppressing degenerative effects, possibly restoring the physiological and biochemical alterations. Naringenin prevented neuronal apoptosis in mouse brains and improved the learning memory response. This study emphasizes that naringenin may exert preventive effects against propofol exposure and prevent neurodegenerative processes in neonatal mouse brains. This study also suggests that careful consideration should be taken when using anesthetic agents for neonates, and preventive measures, such as natural flavonoids, may be utilized.
Disclosure
The authors declare they have no conflicts of interest.
References
1. Yan J, Jiang H. Dual effects of ketamine: neurotoxicity versus neuroprotection in anesthesia for the developing brain. J Neurosurg Anesthesiol. 2014;26(2):155–160. doi:10.1097/ANA.0000000000000027
2. Yu D, Li L, Yuan W. Neonatal anesthetic neurotoxicity: insight into the molecular mechanisms of long-term neurocognitive deficits. Biomed Pharmacother. 2017;87:196–199.
3. Jevtovic-Todorovic V, Hartman RE, Izumi Y, et al. Early exposure to common anesthetic agents causes widespread neurodegeneration in the developing rat brain and persistent learning deficits. J Neurosci. 2003;23(3):876–882. doi:10.1523/JNEUROSCI.23-03-00876.2003
4. Loepke AW, Istaphanous GK, McAuliffe JJ
5. Fredriksson A, Archer T, Alm H, Gordh T, Eriksson P. Neurofunctional deficits and potentiated apoptosis by neonatal NMDA antagonist administration. Behav Brain Res. 2004;153(2):367–376. doi:10.1016/j.bbr.2003.12.026
6. Nishikawa K, Harrison NL. The actions of sevoflurane and desflurane on the gamma-aminobutyric acid receptor type A: effects of TM2 mutations in the alpha and beta subunits. Anesthesiology. 2003;99(3):678–684. doi:10.1097/00000542-200309000-00024
7. Walsh CT. Propofol: milk of amnesia. Cell. 2018;175(1):10–13. doi:10.1016/j.cell.2018.08.031
8. Brambrink AM, Evers AS, Avidan MS, et al. Isoflurane-induced neuroapoptosis in the neonatal rhesus macaque brain. Anesthesiology. 2010;112(4):834–841. doi:10.1097/ALN.0b013e3181d049cd
9. Liu JR, Liu Q, Li J, et al. Noxious stimulation attenuates ketamine-induced neuroapoptosis in the developing rat brain. Anesthesiology. 2012;117(1):64–71. doi:10.1097/ALN.0b013e31825ae693
10. Li P-X, Ru F, Lu Y-M, Zhang L. Propofol causes neuronal degeneration in neonatal mice and long-term neurocognitive consequences in adult mice. Trop J Pharm Res. 2016;15(9):1889. doi:10.4314/tjpr.v15i9.11
11. Michel-Macías C, Morales-Barquet DA, Reyes-Palomino AM, Machuca-Vaca JA, Orozco-Guillén A. Single dose of propofol causing propofol infusion syndrome in a newborn. Oxford Med Case Rep. 2018;2018(6):omy023. doi:10.1093/omcr/omy023
12. Allegaert K, Peeters MY, Verbesselt R, et al. Inter-individual variability in propofol pharmacokinetics in preterm and term neonates. Br J Anaesth. 2007;99(6):864–870. doi:10.1093/bja/aem294
13. Sarkar A, Angeline MS, Anand K, Ambasta RK, Kumar P. Naringenin and quercetin reverse the effect of hypobaric hypoxia and elicit neuroprotective response in the murine model. Brain Res. 2012;1481:59–70. doi:10.1016/j.brainres.2012.08.036
14. Williams RJ, Spencer JP, Rice-Evans C. Flavonoids: antioxidants or signalling molecules? Free Radic Biol Med. 2004;36(7):838–849. doi:10.1016/j.freeradbiomed.2004.01.001
15. Mershiba SD, Dassprakash MV, Saraswathy SD. Protective effect of naringenin on hepatic and renal dysfunction and oxidative stress in arsenic intoxicated rats. Mol Biol Rep. 2013;40(5):3681–3691. doi:10.1007/s11033-012-2444-8
16. Jayaraman J, Jesudoss VA, Menon VP, Namasivayam N. Anti-inflammatory role of naringenin in rats with ethanol induced liver injury. Toxicol Mech Methods. 2012;22(7):568–576. doi:10.3109/15376516.2012.707255
17. Wang HK, Yeh CH, Iwamoto T, Satsu H, Shimizu M, Totsuka M. Dietary flavonoid naringenin induces regulatory T cells via an aryl hydrocarbon receptor mediated pathway. J Agric Food Chem. 2012;60(9):2171–2178. doi:10.1021/jf204625y
18. Raza SS, Khan MM, Ahmad A, et al. Neuroprotective effect of naringenin is mediated through suppression of NF-kappaB signaling pathway in experimental stroke. Neuroscience. 2013;230:157–171. doi:10.1016/j.neuroscience.2012.10.041
19. Zbarsky V, Datla KP, Parkar S, Rai DK, Aruoma OI, Dexter DT. Neuroprotective properties of the natural phenolic antioxidants curcumin and naringenin but not quercetin and fisetin in a 6-OHDA model of Parkinson’s disease. Free Radic Res. 2005;39(10):1119–1125. doi:10.1080/10715760500233113
20. Olsen HT, Stafford GI, van Staden J, Christensen SB, Jäger AK. Isolation of the MAO-inhibitor naringenin from Mentha aquatica L. J Ethnopharmacol. 2008;117(3):500–502. doi:10.1016/j.jep.2008.02.015
21. Shi Y, Dai J, Liu H, et al. Naringenin inhibits allergen-induced airway inflammation and airway responsiveness and inhibits NF-kappaB activity in a murine model of asthma. Can J Physiol Pharmacol. 2009;87(9):729–735. doi:10.1139/Y09-065
22. Baluchnejadmojarad T, Roghani M. Effect of naringenin on intracerebroventricular streptozotocin-induced cognitive deficits in rat: a behavioral analysis. Pharmacology. 2006;78(4):193–197. doi:10.1159/000096585
23. Heo HJ, Kim MJ, Lee JM, et al. Naringenin from citrus junos has an inhibitory effect on acetylcholinesterase and a mitigating effect on amnesia. Dement Geriatr Cogn Disord. 2004;17(3):151–157. doi:10.1159/000076349
24. Mercer LD, Kelly BL, Horne MK, Beart PM. Dietary polyphenols protect dopamine neurons from oxidative insults and apoptosis: investigations in primary rat mesencephalic cultures. Biochem Pharmacol. 2005;69(2):339–345. doi:10.1016/j.bcp.2004.09.018
25. Williams RW, Rakic P. Three-dimensional counting: an accurate and direct method to estimate numbers of cells in sectioned material. J Comp Neurol. 1988;278(3):344–352. doi:10.1002/cne.902780305
26. Peterson DA. Quantitative histology using confocal microscopy: implementation of unbiased stereology procedures. Methods. 1999;18(4):493–507. doi:10.1006/meth.1999.0818
27. Howell K, Hopkins N, McLoughlin P. Combined confocal microscopy and stereology: a highly efficient and unbiased approach to quantitative structural measurement in tissues. Exp Physiol. 2002;87(6):747–756. doi:10.1113/eph8702477
28. Vorhees CV, Williams MT. Morris water maze: procedures for assessing spatial and related forms of learning and memory. Nat Protoc. 2006;1(2):848–858. doi:10.1038/nprot.2006.116
29. Lee JH, Park YH, Song HG, et al. The effect of lidocaine on apoptotic neurodegeneration in the developing mouse brain. Korean J Anesthesiol. 2014;67(5):334–341. doi:10.4097/kjae.2014.67.5.334
30. Engelking LR. Chapter 90 - respiratory acidosis. In: Engelking LR, editor. Textbook of Veterinary Physiological Chemistry.
31. Lavezzi AM, Corna MF, Matturri L. Neuronal nuclear antigen (NeuN): a useful marker of neuronal immaturity in sudden unexplained perinatal death. J Neurol Sci. 2013;329(1–2):45–50. doi:10.1016/j.jns.2013.03.012
32. Young C, Jevtovic-Todorovic V, Qin Y-Q, et al. Potential of ketamine and midazolam, individually or in combination, to induce apoptotic neurodegeneration in the infant mouse brain. Br J Pharmacol. 2005;146(2):189–197. doi:10.1038/sj.bjp.0706301
33. Stratmann G, Sall JW, May LD, et al. Isoflurane differentially affects neurogenesis and long-term neurocognitive function in 60-day-old and 7-day-old rats. Anesthesiology. 2009;110(4):834–848. doi:10.1097/ALN.0b013e31819c463d
34. Bercker S, Bert B, Bittigau P, et al. Neurodegeneration in newborn rats following propofol and sevoflurane anesthesia. Neurotox Res. 2009;16(2):140–147.
35. Bittigau P, Sifringer M, Genz K, et al. Antiepileptic drugs and apoptotic neurodegeneration in the developing brain. Proc Natl Acad Sci U S A. 2002;99(23):15089–15094. doi:10.1073/pnas.222550499
36. Hara M, Kai Y, Ikemoto Y. Propofol activates GABAA receptor-chloride ionophore complex in dissociated hippocampal pyramidal neurons of the rat. Anesthesiology. 1993;79(4):781–788. doi:10.1097/00000542-199310000-00021
37. Kochs E, Hoffman WE, Werner C, Thomas C, Albrecht RF, Schulte Am Esch J. The effects of propofol on brain electrical activity, neurologic outcome, and neuronal damage following incomplete ischemia in rats. Anesthesiology. 1992;76(2):245–252. doi:10.1097/00000542-199202000-00014
38. Huang WY, Wu H, Li DJ, et al. Protective effects of blueberry anthocyanins against H2O2-induced oxidative injuries in human retinal pigment epithelial cells. J Agric Food Chem. 2018;66(7):1638–1648. doi:10.1021/acs.jafc.7b06135
39. Hirose K, Chan PH. Blockade of glutamate excitotoxicity and its clinical applications. Neurochem Res. 1993;18(4):479–483. doi:10.1007/BF00967252
40. Kollegger H, McBean GJ, Tipton KF. Reduction of striatal N-methyl-D-aspartate toxicity by inhibition of nitric oxide synthase. Biochem Pharmacol. 1993;45(1):260–264. doi:10.1016/0006-2952(93)90401-H
41. Mishra SK, Kang JH, Lee CW, et al. Midazolam induces cellular apoptosis in human cancer cells and inhibits tumor growth in xenograft mice. Mol Cells. 2013;36(3):219–226. doi:10.1007/s10059-013-0050-9
42. Yang CS, Landau JM, Huang MT, Newmark HL. Inhibition of carcinogenesis by dietary polyphenolic compounds. Annu Rev Nutr. 2001;21(1):381–406. doi:10.1146/annurev.nutr.21.1.381
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.