")
Back to Journals » Drug Design, Development and Therapy » Volume 8
N-n-butyl haloperidol iodide inhibits H2O2- induced Na+/Ca2+-exchanger activation via the Na+/H+ exchanger in rat ventricular myocytes
Authors Huang Y, gao F, Wang B, Zheng F, Zhang Y, Chen Y, Huang Z, Zheng Y, Zhong S, Shi G
Received 26 February 2014
Accepted for publication 31 May 2014
Published 9 September 2014 Volume 2014:8 Pages 1257—1267
DOI https://doi.org/10.2147/DDDT.S63163
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Yong-Pan Huang,1,* Fen-Fei Gao,1,* Bin Wang,1 Fu-Chun Zheng,2 Yan-Mei Zhang,1 Yi-Cun Chen,1 Zhan-Qin Huang,1 Yan-Shan Zheng,1 Shu-Ping Zhong,3 Gang-Gang Shi1,4
1Department of Pharmacology, 2Department of Pharmacy, First Affiliated Hospital, Shantou University Medical College, Shantou, Guangdong, People's Republic of China; 3Department of Biochemistry and Molecular Biology, University of Southern California, Los Angeles, CA, USA; 4Department of Cardiovascular Diseases, First Affiliated Hospital, Shantou University Medical College, Shantou, Guangdong, People's Republic of China
*These authors contributed equally to this work
Abstract: N-n-butyl haloperidol iodide (F2), a novel compound, has shown palliative effects in myocardial ischemia/reperfusion (I/R) injury. In this study, we investigated the effects of F2 on the extracellular signal-regulated kinase kinase (MEK)/extracellular signal-regulated kinase (ERK)/Na+/H+ exchanger (NHE)/Na+/Ca2+ exchanger (NCX) signal-transduction pathway involved in H2O2-induced Ca2+ overload, in order to probe the underlying molecular mechanism by which F2 antagonizes myocardial I/R injury. Acute exposure of rat cardiac myocytes to 100 µM H2O2 increased both NHE and NCX activities, as well as levels of phosphorylated MEK and ERK. The H2O2-induced increase in NCX current (INCX) was nearly completely inhibited by the MEK inhibitor U0126 (1,4-diamino-2,3-dicyano-1,4-bis[o-aminophenylmercapto]butadiene), but only partly by the NHE inhibitor 5-(N,N-dimethyl)-amiloride (DMA), indicating the INCX increase was primarily mediated by the MEK/mitogen-activated protein kinase (MAPK) pathway, and partially through activation of NHE. F2 attenuated the H2O2-induced INCX increase in a concentration-dependent manner. To determine whether pathway inhibition was H2O2-specific, we examined the ability of F2 to inhibit MEK/ERK activation by epidermal growth factor (EGF), and NHE activation by angiotensin II. F2 not only inhibited H2O2-induced and EGF-induced MEK/ERK activation, but also completely blocked both H2O2-induced and angiotensin II-induced increases in NHE activity, suggesting that F2 directly inhibits MEK/ERK and NHE activation. These results show that F2 exerts multiple inhibitions on the signal-transduction pathway involved in H2O2-induced INCX increase, providing an additional mechanism for F2 alleviating intracellular Ca2+ overload to protect against myocardial I/R injury.
Keywords: N-n-butyl haloperidol, hydrogen peroxide, Na+/Ca2+ exchanger, Na+/H+ exchanger
Introduction
Reperfusion of an ischemic myocardium leads to heart dysfunction and cardiomyocyte injury. Such myocardial ischemia/reperfusion (I/R) injury is characterized by impaired blood flow, metabolic dysfunction, contractile dysfunction, dysrhythmias, cellular necrosis, and apoptosis.1 I/R injury is a complex process involving numerous mechanisms, including cytosolic and mitochondrial Ca2+ overload, release of reactive oxygen species (ROS), acute inflammatory response, and shift in substrate use.2
ROS, produced as by-products of oxidative metabolism, are easily managed under normal conditions by reactive oxygen scavengers.3,4 Several forms of ROS are generated during I/R, including superoxide (O2−), H2O2, and the highly reactive hydroxyl radical (·OH), which cause lipid peroxidation and myocardial injury and trigger the contractile dysfunction observed during reperfusion.5,6 It has also been suggested that the burst in ROS upon reperfusion may contribute to Ca2+ overload in cardiomyocytes.7,8 Rothstein et al9 and Sabri et al10 found that low doses of H2O2 (50 μM, similar to those generated during I/R) cause Ca2+ overload in cultured neonatal rat ventricular myocytes, which is associated with activation of the Na+/H+ exchanger (NHE) in part through extracellular signal-regulated kinase (ERK)-1/2-mediated phosphorylation of NHE-1, the only NHE isoform in the myocardium. A link between H2O2 and diastolic Ca2+ overload in neonatal rat ventricular myocytes was proposed. Exposure to H2O2 results in the alteration of signaling proteins involved in the mitogen-activated protein-kinase (MAPK) pathway, ultimately leading to extracellular signal-regulated kinase kinase (MEK) activation, which then phosphorylates and activates ERK1/2. Activated ERK1/2 subsequently phosphorylates the COOH tail of NHE-1, increasing its exchanger activity to elevate intracellular Na+ concentrations. The resulting rise in intracellular Na+ decreases the activity of the Na+/Ca2+ exchanger (NCX), leading to an increase in diastolic Ca2+ levels.9 Therefore, pharmacological approaches to decrease MAPK, NHE-1, and/or NCX activity may ameliorate the alterations in Ca2+ homeostasis that contribute to myocardial tissue injury following I/R.
N-n-butyl haloperidol iodide (F2), a novel quaternary ammonium salt derivative of haloperidol synthesized in our laboratory, can maintain the effects of coronary artery relaxation without adverse extrapyramidal reactions.11 Our previous studies show that F2 can attenuate myocardial I/R injury, as evidenced by amelioration of hemodynamics and myocardial enzyme activity, reduction in myocardial infarction size, prevention of ventricular arrhythmias, and decreases in myocardial inflammation.11–14 The cardioprotective mechanism of F2 was thought to be associated with calcium-homeostasis maintenance against intracellular Ca2+ overload by inhibiting cardiocyte L-type Ca2+ channels.12,13 However, intracellular Ca2+ overload during I/R results primarily from the functional coupling of NHE and NCX. Ischemic hearts develop intracellular acidosis, which activates NHE to extrude H+ in exchange for an influx of Na+. Upon reperfusion, loss of extracellular H+ causes further extrusion of H+ in exchange for Na+. The subsequent elevation in intracellular Na+ promotes an increase Ca2+ influx into the cytosol via the reverse mode of NCX, resulting in Ca2+ overload.15 It is suggested that the mechanism of F2 antagonizing myocardial I/R injury might not be only related to suppression of the L-type Ca2+ channel. In this study, we used rat ventricular myocytes to investigate the effects of F2 on the MEK/ERK/NHE/NCX signal-transduction pathway involved in H2O2-induced Ca2+ overload in order to probe the underlying molecular mechanism by which F2 maintains intracellular calcium homeostasis and antagonizes myocardial I/R injury.
Materials and methods
Materials
F2 (synthesized by our lab and identified by the Shanghai Organic Chemistry Institute of the Chinese Academy of Sciences; purity greater than 98%) was prepared as a 0.1 M stock solution in dimethyl sulfoxide and diluted to the desired concentration with extracellular solution before each experiment. HEPES (4,[2-hydroxyethyl]-1-piperazine-ethanesulphonic acid]), CsCl, 1,2-bis(2-aminophenoxy)-ethane-N,N,N′,N′-tetraacetic acid (BAPTA), ouabain, nifedipine, ryanodine, epidermal growth factor (EGF), angiotensin (Ang) II and 5-(N,N-dimethyl)-amiloride (DMA) were purchased from Sigma-Aldrich Co. (St Louis, MO, USA). U0126 (1,4-diamino- 2,3-dicyano-1,4-bis[o-aminophenylmercapto]butadiene) was from Merck Millipore (Billerica, MA, USA), 2,7-bis(2-carboxyethyl)-5(6)-carboxyfluorescein-acetoxymethyl ester (BCECF-AM), and Pluronic® F127 were from Thermo Fisher Scientific (Waltham, MA, USA). Anti-MEK, anti-phosphorylated (p)-MEK, anti-ERK, and anti-p-ERK antibodies were from Cell Signaling Technology (Danvers, MA, USA), anti-NCX from Santa Cruz Biotechnology Inc., (Dallas, TX, USA), anti-NHE-1 antibody from Abcam (Cambridge, UK), anti-β-actin from Sigma-Aldrich, and secondary antibody (horseradish peroxidase-conjugated goat antirabbit immunoglobulin (Ig)G) from BosterBio (Pleasanton, CA, USA).
Isolation of ventricular myocytes
Adult male Sprague Dawley rats (180–250 g) were obtained from the Laboratory Animal Breeding and Research Center (Shantou, People’s Republic of China). All experiments were conducted in strict accordance with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (publication 85-23, revised 1996).16 The protocol was approved by the Medical Animal Care and Welfare Committee of Shantou University Medical College (permit SUMC2010-093). All surgery was performed under sodium pentobarbital anesthesia, and all efforts were made to minimize suffering. Single ventricular myocytes were isolated by an enzymatic dissociation method described previously.17,18 Single ventricular myocytes were harvested after filtration through a nylon mesh (pore size 200 mm).
INCX recording
Myocytes were perfused with extracellular solution (140 mM NaCl, 1 mM CaCl2, 1 mM MgCl2, 0.33 mM NaH2PO4, 10 mM glucose, 10 mM HEPES, 0.02 mM ouabain, 0.01 mM nifedipine, 2 mM CsCl, and 0.01 mM ryanodine, pH 7.2) at a rate of 1 mL/minute in a recording chamber. Patch pipettes were forged from 1.5 mm diameter glass capillaries with a two-stage microelectrode puller (pp-830; Narishige, Tokyo, Japan). The pipette resistance was 2–3 MΩ when filled with the pipette solution (20 mM NaCl, 20 mM BAPTA, 10 mM CaCl2 [free Ca2+ concentration of 226 nM], 120 mM CsOH, 3 mM MgCl2, 50 mM aspartic acid, 5 mM Mg-adenosine triphosphate, and 10 mM HEPES, pH 7.2).19 NCX current (INCX) was recorded by a tight-seal whole-cell voltage clamp with the use of an Axopatch™ 200B amplifier (Molecular Devices, Sunnyvale, CA, USA) with low-pass filtering at 2 kHz, digitized with a DigiData 1322A interface, and processed by pCLAMP® 8.2 software (Molecular Devices, Sunnyvale, CA, USA). The electrode capacitance was maximally compensated by use of the amplifier. No compensation was made for membrane capacitance or series resistance.
For recording INCX, the extracellular solution contained ouabain, nifedipine, Cs+, and ryanodine to block Na+/K+ pump current, ICa, IK, and Ca2+ release channels of the sarcoplasmic reticulum, respectively. INCX was induced by ramp-voltage pulses from a holding potential of −60 mV to +60 mV, and then hyperpolarizing to −150 mV before ramping back to the holding potential at a rate of 600 mV/second. The descending limb (from +60 to −150 mV) was plotted as the current–voltage (I–V) relationship without capacitance compensation.20 INCX was measured as the Ni2+-sensitive current that could be selectively inhibited by 5 mM NiCl2.
Measurement of intracellular pH and NHE activity
Intracellular pH (pHi) was measured by monitoring the fluorescence of the pH-sensitive dye BCECF.9,10 Myocytes placed in a petri dish were loaded with BCECF by incubation for 15 minutes in the dark at room temperature with the acetoxymethyl ester form (BCECF-AM, 2 μM) in modified Krebs solution (135 mM NaCl, 5.9 mM KCl, 1.5 mM CaCl2, 1.2 mM MgCl2, 11.5 mM glucose, 11.6 mM HEPES, pH 7.4) supplemented with 0.1% bovine serum albumin and 0.02% Pluronic F127. The cells were then washed three times and incubated for an additional 45 minutes in fresh Krebs solution in the presence or absence of the MEK inhibitor U0126 (5 μM). BCECF fluorescence was recorded using confocal microscopy (FluoView FV1000; Olympus, Tokyo, Japan). A ratio of fluorescence emitted at 515 nm from excitation at 490 nm to that at 440 nm was converted to intracellular pHi using the nigericin high-K+ protocol of Thomas et al.21
NHE activity was measured by monitoring the recovery rate from rapid acidification using the NH4Cl prepulse technique.21,22 After determination of basal pHi, cells were exposed to Krebs solution containing 25 mM NH4Cl for 5 minutes to cause rapid alkalinization as NH3 diffused into the cells and titrated intracellular H+. Then, perfusion with Na+-free Krebs solution (Na+ isosmotically replaced with N-methylglucamine) removed NH4+ from the external medium to cause a rapid decrease in pHi. There was no recovery from this acid load in the absence of Na+. pHi recovered when the perfusate was switched to an Na+-containing Krebs solution. This Na+-dependent recovery was operationally defined as NHE activity. To quantify the rate of pHi recovery, the slope of a straight line fitted to the initial 60 seconds after the onset of recovery was measured.10
Western blotting
Total protein extracts were prepared from cells using cell-lysis buffer containing a protease-inhibitor cocktail (aprotinin, leupeptin, pepstatin A, and phenylmethylsulfonyl fluoride). The protein concentration was determined by a Bradford protein-assay kit (Bio-Rad Laboratories Inc., Hercules, CA, USA). Equal amounts of total protein (40 μg) were subjected to sodium dodecyl sulfate polyacrylamide gel electrophoresis (10%), followed by electrophoretic transfer to nitrocellulose membranes (GE Healthcare UK Ltd., Little Chalfont, UK). The blots were incubated with primary antibody (rabbit antirat) at 4°C overnight, followed by secondary antibody (horseradish peroxidase-conjugate goat antirabbit IgG) for 2 hours at room temperature. The bound antibodies were detected by the use of a SuperSignal Western blotting kit (Thermo Fisher Scientific ). Densitometric analysis of protein bands was performed with Quantity One® software (version 4.5.2; Bio-Rad Laboratories Inc.,).
Statistical analysis
All values are presented as means ± standard error of the mean. Statistical analysis was carried out using paired Students’s t-tests or one-way analysis of variance followed by the Student–Newman–Keuls test, with P<0.05 considered statistically significant.
Results
F2 inhibits the H2O2-induced increase of INCX
Currents were recorded when myocytes were perfused in sequence with the control extracellular solution, and solutions containing H2O2 (100 μM), H2O2 + F2 (0.1, 1.0, or 10 μM), and NiCl2 (5 mM) for 10 minutes, respectively. Bidirectional outward and inward INCX were induced by 1 mM Ca2+ and 140 mM Na+ in the external solution, and 20 mM Na+ and 226 nM free Ca2+ in the pipette solution. Under these ionic conditions, the reversal potential of INCX with a 3Na+:1Ca2+ stoichiometry was calculated to be −65 mV at room temperature according to the equation ENa/Ca=3ENa − 2ECa.23,24 Figure 1A illustrates the I–V relation of control myocytes (a), and myocytes exposed to H2O2 (b), H2O2 + 0.1, 1.0, or 10 μM F2 (c–e), and NiCl2 (f). The net Ni2+-sensitive currents all crossed the voltage axis at about −65 mV (Figure 1B), confirming that the Ni2+-sensitive currents were INCX. Both outward and inward INCX increased after perfusion with 100 μM H2O2. F2 diminished the increase of INCX in a concentration-dependent manner, with reverse-mode NCX being greater than forward-mode inhibition (Figure 1C).
 | Figure 1 Effect of F2 on the H2O2-induced increase in INCX. |
U0126 and DMA inhibit H2O2- induced INCX increases
To confirm the involvement of the MAPK pathway and the NHE in H2O2-induced NCX activation, we tested the effects of U0126, a highly selective inhibitor of MEK, and DMA, an NHE inhibitor, on the H2O2-induced increase in INCX. We initially determined the minimal effective concentrations that completely blocked H2O2-induced MEK activation and NHE-1 activity, and used those concentrations to examine the roles of MEK and NHE in F2-mediated inhibition of H2O2-mediated induction of INCX activity. Results showed that perfusion of 5 μM U0126 for 10 minutes, which alone did not affect INCX,25 significantly inhibited the H2O2-induced increase in INCX at 60 mV by 81.13%±3.63% and at −150 mV by 93.64%±4.52% (n=5) (Figure 2A and B). In contrast, perfusion of 20 μM DMA for 10 minutes only inhibited the H2O2-induced increase by 39.98%±3.00% at 60 mV, and by 32.42%±1.78% at −150 mV (n=5) (Figure 2C and D). This result indicates that the H2O2-induced increase in INCX was primarily mediated by the MEK/MAPK pathway, and partially through activation of NHE-1.
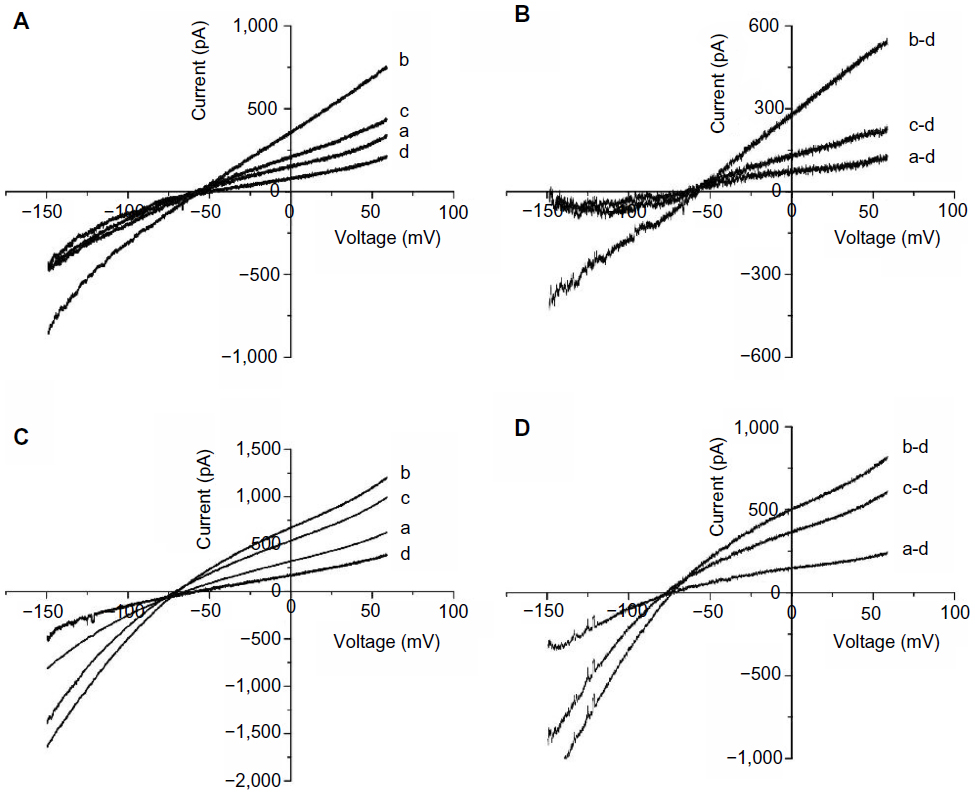 | Figure 2 Effects of U0126 and DMA on the H2O2-induced INCX increase. |
F2 inhibits H2O2-induced MEK/ERK activation and EGF-induced INCX increases
To investigate whether F2 modulates MEK activity, we examined the effect of F2 on H2O2-induced and EGF-induced MEK/ERK activation. As shown in Figure 3A, H2O2 (100 μM) and EGF (50 ng/mL) led to a significant increase in the level of phosphorylated MEK and ERK, and 1 μM F2 inhibited both H2O2-induced and EGF-induced MEK and ERK activation. We then observed the effect of F2 on the INCX increase induced by EGF. INCX was increased by EGF, and treatment with 1 μM F2 resulted in a significant reduction in EGF-induced INCX rise at 60 mV by 72.88%±5.76% and at −150mV by 71.14%±3.19% (n=8) (Figure 3B).
 | Figure 3 Effects of F2 on H2O2-induced MEK/ERK activation and EGF-induced INCX increase. |
F2 inhibits H2O2-induced and Ang II- induced NHE activity
To investigate the effects of F2 on NHE activity, we examined its effects on H2O2-induced and Ang II-induced, Na+-dependent recovery from acid load in rat ventricular myocytes. The mean resting pH of ventricular myocytes in bicarbonate-free Krebs solution at room temperature was 7.48±0.13 (n=10). The addition and removal of NH4Cl from the external medium caused a rapid rise and decrease in pHi. Cells were unable to recover from this acid load in Na+-free medium. Reintroduction of Na+ led to a rapid recovery of pHi that approached resting values (Figure 4A). This Na+-dependent recovery was completely blocked by DMA (25 μM) (Figure 4B). Exposure to 100 μM H2O2 caused an increase in Na+-dependent recovery of pHi from acid load (4.8±0.6×10−3 ΔpH/second [n=5] versus 2.5±0.3×10−3 ΔpH/minute in controls [n=5], P<0.05) that was again completely blocked by DMA, indicating that H2O2-mediated enhancement of recovery from acid load is mediated by the NHE (Figure 4C and D). Similar to DMA, pretreatment with the MEK inhibitor U0126 abolished H2O2-induced NHE activity (2.4±0.4×10−3 ΔpH/minute [n=4], P<0.05 versus control) (Figure 4E). Perfusion with 1 μM F2 completely blocked both Na+-dependent recovery in the presence of H2O2 (Figure 4F) and NHE activity in the presence of 1 nM Ang II (Figure 4G). These results suggest that F2 exerted its cardioprotective effects by blocking NHE activity.
 | Figure 4 Effects of F2 on H2O2-induced and Ang II-induced NHE activity. |
F2 inhibits Ang II-induced INCX increases
Ang II at a low concentration stimulates NHE-1 activity to elevate intracellular Na+ levels,26,27 which reverses NCX activity and leads to INCX increases. We observed that 1 nM Ang II increased outward INCX at 60 mV by 26.92%±4.40% and inward INCX at −150 mV by 14.26%±2.95% (n=5), consistent with a prior report.28 Addition of 1 μM F2 resulted in a significant reduction in the Ang II-induced INCX rise at 60 mV by 62.27%±3.42% and at −150 mV by 46.19%±3.36% (n=5) (Figure 5), consistent with a role for F2 in blocking NHE activation.
 | Figure 5 Effect of F2 on Ang II-induced INCX increase. |
Effects of F2 on the protein expression of NHE and NCX
Exchanger activity is regulated by changes in protein expression and by phosphorylation of existing exchangers or a closely associated modulatory protein.29–33 Therefore, we examined the effects of F2 on the protein expression of NHE and NCX. The results showed that the total protein expression of NHE and NCX did not change after myocytes were treated with H2O2, EGF, and Ang II for 30 minutes, and that F2 had no significant effect on the total protein expression of either NHE or NCX (Figure 6).
 | Figure 6 Effect of F2 on NHE and NCX protein expression. |
Discussion
The present study describes the effects of F2 on the H2O2-induced signal-transduction pathway for INCX increase in rat ventricular myocytes. F2 can inhibit the signal-transduction pathway involved in H2O2-induced INCX increase at multiple sites.
Excess ROS production and intracellular Ca2+ overload play a prominent role in I/R injury. Moreover, there is a reciprocal interaction between excess ROS production and accumulation of cytosolic and mitochondrial Ca2+ due to the cross talk between ROS and Ca2+.34–36 Ca2+ can enhance ROS generation.37 ROS can activate MAPKs (ERK, Jun N-terminal kinase [JNK], p38),38 which are activated during ischemia, and to a greater extent on reperfusion.39,40 Activated ERK1/2 leads to phosphorylation and activation of NHE-1,9,41 and this may contribute to a feed-forward activation loop (Ca2+ → ROS → ERK → more Na+ → more Ca2+), enhancing Ca2+overload in I/R injury.37 The ability to disrupt this vicious cycle will exert beneficial effects on recovery from I/R injury. In this study, we demonstrated that F2 can inhibit H2O2-induced increase in NCX activity through inhibiting both MEK/ERK activation and NHE activity, blocking intracellular Ca2+ overload to protect against myocardial I/R injury.
Our results show that acute exposure of cardiac myocytes to 100 μM H2O2 causes the INCX to increase, along with a rapid activation of MEK and an increase in NHE activity. The H2O2-induced INCX increase was blocked almost completely by the MEK inhibitor U0126, but only partly by the NHE inhibitor DMA (Figure 2), indicating the INCX increase was primarily mediated by the MEK MAPK pathway and partially through activation of NHE, consistent with prior reports.9,25 Furthermore, the H2O2-induced increase in NHE activity was abolished by pretreatment with the MEK inhibitor U0126 (Figure 4E), suggesting that MAPKs act upstream of NHE in H2O2-induced INCX increase. The present study shows that F2 blocks MEK activation-induced by not only H2O2 but also EGF (Figure 3A), suggesting that F2 directly inhibits MEK activation.
Dyck et al found an increase in steady-state levels of NHE-1 messenger ribonucleic acid in chronic ischemia in rat myocardium, suggesting that increased activity is due to an increase in protein expression.31 However, in our experiments, acute exposure to H2O2 caused a rapid activation of NHE and NCX activity in the absence of changes in total NHE and NCX. The most likely explanation is that the exposure to H2O2 in our experiment was too short for changes in protein expression, indicating that posttranslational modification rather than gene expression played the major role in the rapid time course for regulation of exchanger activity. Unfortunately, we could not detect phosphorylation of NHE-1 and NCX due to the absence of antibodies for phospho-NHE-1 and phospho-NCX, which was a limitation of this study.
NHE activation increases INCX through increasing intracellular Na+ concentration. NCX is one of the major mechanisms for regulating intracellular Ca2+ concentration in cardiac myocytes. Under physiological conditions, the Na+/Ca2+ exchanger operates in forward mode, extruding Ca2+ from the cell to maintain intracellular Ca2+ homeostasis. Conversely, during I/R, a large burst of ROS contributes to Ca2+ loading via activation of the NCX Ca2+-influx mode, which accelerates intracellular Ca2+ overload.42 H2O2 increases NCX activity, leading to Ca2+ overload via activation of the MEK/ERK/NHE pathway.4,9,10,25,43 Our previous studies demonstrate that F2 blocks L-type Ca2+ channels and protects the activity of sarco/endoplasmic reticulum Ca2+-adenosine triphosphatases to attenuate Ca2+ overload against I/R injury in cardiac myocytes.12,14,18,44 We now show an additional mechanism for F2 in the regulation of calcium homeostasis, demonstrating that F2 inhibits both MEK activation and NHE activity to diminish H2O2-induced INCX increase, but we do not rule out inhibition by F2 on NCX activity. Figure 7 illustrates the possible signaling pathways from H2O2 to NCX and the target of F2 action.
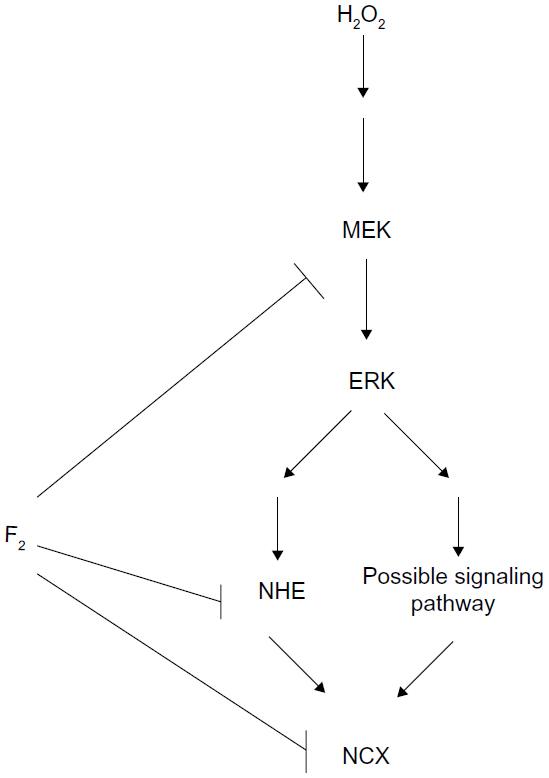 | Figure 7 Scheme of possible signaling pathways for NCX activation by H2O2 and the target of F2 action. |
In conclusion, we demonstrate an additional mechanism by which F2 can alleviate intracellular Ca2+ overload, and thus protect against myocardial I/R injury. F2, a novel quaternary ammonium salt derivative of haloperidol with a different chemical structure from classical Ca2+-channel antagonists, seems like an undesirable drug due to its broad, nonspecific effects, but the study of its structure–function relationship may help to develop new drugs for the treatment of ischemic heart disease.
Acknowledgments
We are grateful for the support from the Center for Neuroscience, Shantou University Medical College for the utilization of the laser confocal microscopy. This work was supported by National Natural Science Foundation of China–Guangdong Joint Funds (U0932005), the National Natural Science Foundation of China (81173048 and 81072633), central government special funds supporting the development of local colleges and universities.
Disclosure
The authors report no conflicts of interest in this work.
References
Zhao ZQ, Vinten-Johansen J. Postconditioning: reduction of reperfusion-induced injury. Cardiovasc Res. 2006;70(2):200–211. | |
Dirksen MT, Laarman GJ, Simoons ML, Duncker DJ. Reperfusion injury in humans: a review of clinical trials on reperfusion injury inhibitory strategies. Cardiovasc Res. 2007;74(3):343–355. | |
Gumina RJ, Daemmgen J, Gross GJ. Inhibition of the Na(+)/H(+) exchanger attenuates phase 1b ischemic arrhythmias and reperfusion-induced ventricular fibrillation. Eur J Pharmacol. 2000;396(2–3):119–124. | |
Moor AN, Gan XT, Karmazyn M, Fliegel L. Activation of Na+/H+ exchanger-directed protein kinases in the ischemic and ischemic-reperfused rat myocardium. J Biol Chem. 2001;276(19):16113–16122. | |
Farber NE, Pieper GM, Thomas JP, Gross GJ. Beneficial effects of iloprost in the stunned canine myocardium. Circ Res. 1988;62(2):204–215. | |
Droge W. Free radicals in the physiological control of cell function. Physiol Rev. 2002;82(1):47–95. | |
Goldhaber JI, Weiss JN. Oxygen free radicals and cardiac reperfusion abnormalities. Hypertension. 1992;20(1):118–127. | |
Goldhaber JI. Free radicals enhance Na+/Ca2+ exchange in ventricular myocytes. Am J Physiol. 1996;271(3 Pt 2):H823–H833. | |
Rothstein EC, Byron KL, Reed RE, Fliegel L, Lucchesi PA. H2O2-induced Ca2+ overload in NRVM involves ERK1/2 MAP kinases: role for an NHE-1-dependent pathway. Am J Physiol Heart Circ Physiol. 2002;283(2):H598–H605. | |
Sabri A, Byron KL, Samarel AM, Bell J, Lucchesi PA. Hydrogen peroxide activates mitogen-activated protein kinases and Na+-H+ exchange in neonatal rat cardiac myocytes. Circ Res. 1998;82(10):1053–1062. | |
Gao FF, Shi GG, Zheng JH, Liu B. Protective effects of N-n-butyl haloperidol iodide on myocardial ischemia-reperfusion injury in rabbits. Chin J Physiol. 2004;47(2):61–66. | |
Huang ZQ, Shi GG, Zheng JH, Liu B. Effects of N-n-butyl haloperidol iodide on rat myocardial ischemia and reperfusion injury and L-type calcium current. Acta Pharmacol Sin. 2003;24(8):757–763. | |
Huang Z, Shi G, Gao F, et al. Effects of N-n-butyl haloperidol iodide on L-type calcium channels and intracellular free calcium in rat ventricular myocytes. Biochem Cell Biol. 2007;85(2):182–188. | |
Gao FF, Hao SY, Huang ZQ, et al. Cardiac electrophysiological and antiarrhythmic effects of N-n-butyl haloperidol iodide. Cell Physiol Biochem. 2010;25(4–5):433–442. | |
Lee C, Dhalla NS, Hryshko LV. Therapeutic potential of novel Na+-Ca2+ exchange inhibitors in attenuating ischemia-reperfusion injury. Can J Cardiol. 2005;21(6):509–516. | |
Bayne K. Revised Guide for the Care and Use of Laboratory Animals available. American Physiological Society. Physiologist. 1996;39(4):199, 208–211. | |
Cheng H, Lederer WJ, Cannell MB. Calcium sparks: elementary events underlying excitation-contraction coupling in heart muscle. Science. 1993;262(5134):740–744. | |
Xiao JF, Wang CY, Huang YP, et al. N-n-butyl haloperidol iodide preserves cardiomyocyte calcium homeostasis during hypoxia/ischemia. Cell Physiol Biochem. 2011;27(5):433–442. | |
Watanabe Y, Kimura J. Inhibitory effect of azimilide on Na+/Ca2+ exchange current in guinea-pig cardiac myocytes. J Pharmacol Sci. 2010;114(1):111–114. | |
Brittain MK, Brustovetsky T, Brittain JM, Khanna R, Cummins TR, Brustovetsky N. Ifenprodil, a NR2B-selective antagonist of NMDA receptor, inhibits reverse Na+/Ca2+ exchanger in neurons. Neuropharmacology. 2012;63(6):974–982. | |
Thomas JA, Buchsbaum RN, Zimniak A, Racker E. Intracellular pH measurements in Ehrlich ascites tumor cells utilizing spectroscopic probes generated in situ. Biochemistry. 1979;18(11):2210–2218. | |
Roos A, Boron WF. Intracellular pH. Physiol Rev. 1981;61(2):296–434. | |
Kimura J, Miyamae S, Noma A. Identification of sodium-calcium exchange current in single ventricular cells of guinea-pig. J Physiol. 1987;384:199–222. | |
Blaustein MP, Lederer WJ. Sodium/calcium exchange: its physiological implications. Physiol Rev. 1999;79(3):763–854. | |
Hinata M, Matsuoka I, Iwamoto T, Watanabe Y, Kimura J. Mechanism of Na+/Ca2+ exchanger activation by hydrogen peroxide in guinea-pig ventricular myocytes. J Pharmacol Sci. 2007;103(3):283–292. | |
Eguti DM, Thieme K, Leung GP, Mello-Aires M, Oliveira-Souza M. Regulation of Na+/H+ exchanger isoform 1 (NHE1) by calmodulin-binding sites: role of angiotensin II. Cell Physiol Biochem. 2010;26(4–5):541–552. | |
Oliveira-Souza M, De Mello-Aires M. Interaction of angiotensin II and atrial natriuretic peptide on pH(i) regulation in MDCK cells. Am J Physiol Renal Physiol. 2000;279(5):F944–F953. | |
Aiello EA, Villa-Abrille MC, Cingolani HE. Autocrine stimulation of cardiac Na(+)-Ca(2+) exchanger currents by endogenous endothelin released by angiotensin II. Circ Res. 2002;90(4):374–376. | |
Sardet C, Counillon L, Franchi A, Pouyssegur J. Growth factors induce phosphorylation of the Na+/H+ antiporter, glycoprotein of 110 kD. Science. 1990;247(4943):723–726. | |
Sardet C, Fafournoux P, Pouyssegur J. Alpha-thrombin, epidermal growth factor, and okadaic acid activate the Na+/H+ exchanger, NHE-1, by phosphorylating a set of common sites. J Biol Chem. 1991;266(29):19166–19171. | |
Dyck JR, Maddaford TG, Pierce GN, Fliegel L. Induction of expression of the sodium-hydrogen exchanger in rat myocardium. Cardiovasc Res. 1995;29(2):203–208. | |
Wakabayashi S, Bertrand B, Ikeda T, Pouyssegur J, Shigekawa M. Mutation of calmodulin-binding site renders the Na+/H+ exchanger (NHE1) highly H(+)-sensitive and Ca2+ regulation-defective. J Biol Chem. 1994;269(18):13710–13715. | |
Wakabayashi S, Bertrand B, Shigekawa M, Fafournoux P, Pouyssegur J. Growth factor activation and “H(+)-sensing” of the Na+/H+ exchanger isoform 1 (NHE1). Evidence for an additional mechanism not requiring direct phosphorylation. J Biol Chem. 1994;269(8):5583–5588. | |
Yan Y, Wei CL, Zhang WR, Cheng HP, Liu J. Cross-talk between calcium and reactive oxygen species signaling. Acta Pharmacol Sin. 2006;27(7):821–826. | |
Sedlic F, Sepac A, Pravdic D, et al. Mitochondrial depolarization underlies delay in permeability transition by preconditioning with isoflurane: roles of ROS and Ca2+. Am J Physiol Cell Physiol. 2010;299(2):C506–C515. | |
Feissner RF, Skalska J, Gaum WE, Sheu SS. Crosstalk signaling between mitochondrial Ca2+ and ROS. Front Biosci (Landmark Ed). 2009;14:1197–1218. | |
Brookes PS, Yoon Y, Robotham JL, Anders MW, Sheu SS. Calcium, ATP, and ROS: a mitochondrial love-hate triangle. Am J Physiol Cell Physiol. 2004;287(4):C817–C833. | |
Levonen AL, Patel RP, Brookes P, et al. Mechanisms of cell signaling by nitric oxide and peroxynitrite: from mitochondria to MAP kinases. Antioxid Redox Signal. 2001;3(2):215–229. | |
Naito Z, Kudo M, Xu G, et al. Immunohistochemical localization of mitogen-activated protein kinase (MAPK) family and morphological changes in rat heart after ischemia-reperfusion injury. Med Electron Microsc. 2000;33(2):74–81. | |
Yue TL, Wang C, Gu JL, et al. Inhibition of extracellular signal-regulated kinase enhances ischemia/reoxygenation-induced apoptosis in cultured cardiac myocytes and exaggerates reperfusion injury in isolated perfused heart. Circ Res. 2000;86(6):692–699. | |
Snabaitis AK, Hearse DJ, Avkiran M. Regulation of sarcolemmal Na(+)/H(+) exchange by hydrogen peroxide in adult rat ventricular myocytes. Cardiovasc Res. 2002;53(2):470–480. | |
Soliman D, Hamming KS, Matemisz LC, Light PE. Reactive oxygen species directly modify sodium-calcium exchanger activity in a splice variant-dependent manner. J Mol Cell Cardiol. 2009;47(5):595–602. | |
Wang H, Silva NL, Lucchesi PA, et al. Phosphorylation and regulation of the Na+/H+ exchanger through mitogen-activated protein kinase. Biochemistry. 1997;36(30):9151–9158. | |
Huang ZQ, Shi GG, Gao FF, et al. Effects of N-n-butyl haloperidol iodide on L-type calcium channels and intracellular free calcium in rat ventricular myocytes. Biochem Cell Biol. 2007;85(2):182–188. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.