")
Back to Journals » Drug Design, Development and Therapy » Volume 8
Multipotent cholinesterase/monoamine oxidase inhibitors for the treatment of Alzheimer’s disease: design, synthesis, biochemical evaluation, ADMET, molecular modeling, and QSAR analysis of novel donepezil-pyridyl hybrids
Authors Bautista-Aguilera O, Esteban G, Chioua M, Nikolic K, Agbaba D, Moraleda I, Iriepa I, Soriano E, Samadi A, Unzeta M, Marco-Contelles J
Received 18 June 2014
Accepted for publication 15 July 2014
Published 13 October 2014 Volume 2014:8 Pages 1893—1910
DOI https://doi.org/10.2147/DDDT.S69258
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Oscar M Bautista-Aguilera,1,* Gerard Esteban,2,* Mourad Chioua,1 Katarina Nikolic,3 Danica Agbaba,3 Ignacio Moraleda,4 Isabel Iriepa,4 Elena Soriano,5 Abdelouahid Samadi,1 Mercedes Unzeta,2 José Marco-Contelles1
1Laboratory of Medicinal Chemistry (Institute of General Organic Chemistry [IQOG], National Research Council [CSIC]), Madrid, Spain; 2Department of Biochemistry and Molecular Biology, Institute of Neurosciences, Autonomous Barcelona University, Barcelona, Spain; 3Institute of Pharmaceutical Chemistry, Faculty of Pharmacy, University of Belgrade, Belgrade, Serbia; 4Department of Organic Chemistry, Faculty of Pharmacy, University of Alcalá, Ctra Barcelona, Alcalá de Henares, Spain; 5Synthesis, and Structure of Organic Compounds (SEPCO) (IQOG, CSIC), Madrid, Spain
*These authors have equally contributed to this work
Abstract: The design, synthesis, and biochemical evaluation of donepezil-pyridyl hybrids (DPHs) as multipotent cholinesterase (ChE) and monoamine oxidase (MAO) inhibitors for the potential treatment of Alzheimer’s disease (AD) is reported. The 3D-quantitative structure-activity relationship study was used to define 3D-pharmacophores for inhibition of MAO A/B, acetylcholinesterase (AChE), and butyrylcholinesterase (BuChE) enzymes and to design DPHs as novel multi-target drug candidates with potential impact in the therapy of AD. DPH14 (Electrophorus electricus AChE [EeAChE]: half maximal inhibitory concentration [IC50] =1.1±0.3 nM; equine butyrylcholinesterase [eqBuChE]: IC50 =600±80 nM) was 318-fold more potent for the inhibition of AChE, and 1.3-fold less potent for the inhibition of BuChE than the reference compound ASS234. DPH14 is a potent human recombinant BuChE (hBuChE) inhibitor, in the same range as DPH12 or DPH16, but 13.1-fold less potent than DPH15 for the inhibition of human recombinant AChE (hAChE). Compared with donepezil, DPH14 is almost equipotent for the inhibition of hAChE, and 8.8-fold more potent for hBuChE. Concerning human monoamine oxidase (hMAO) A inhibition, only DPH9 and 5 proved active, compound DPH9 being the most potent (IC50 [MAO A] =5,700±2,100 nM). For hMAO B, only DPHs 13 and 14 were moderate inhibitors, and compound DPH14 was the most potent (IC50 [MAO B] =3,950±940 nM). Molecular modeling of inhibitor DPH14 within EeAChE showed a binding mode with an extended conformation, interacting simultaneously with both catalytic and peripheral sites of EeAChE thanks to a linker of appropriate length. Absortion, distribution, metabolism, excretion and toxicity analysis showed that structures lacking phenyl-substituent show better druglikeness profiles; in particular, DPHs13–15 showed the most suitable absortion, distribution, metabolism, excretion and toxicity properties. Novel donepezil-pyridyl hybrid DPH14 is a potent, moderately selective hAChE and selective irreversible hMAO B inhibitor which might be considered as a promising compound for further development for the treatment of AD.
Keywords: donepezil-pyridyl hybrids, ChE, MAO, 3D-QSAR, molecular modeling, ADMET
Introduction
Alzheimer’s disease (AD) is a neurodegenerative disorder characterized by dementia and other cognitive impairments.1 Previous biochemical studies indicated that amyloid-β (Aβ) deposits,2 hyperphosphorylated τ-protein aggregation,3 and oxidative stress4 play crucial roles in the pathophysiology of the disease. AD is characterized by a selective loss of cholinergic neurons as a consequence of decreasing levels of acetylcholine (ACh) in specific brain regions that mediate memory and learning functions.5 Inhibitors of acetylcholinesterase (AChE; EC 1.1.1.7) prevent the hydrolysis of ACh and thus increase ACh concentration in the synaptic cleft. Donepezil,6 galantamine,7 and rivastigmine8 are US Food and Drug Administration (FDA)-approved drugs that improve AD symptoms by inhibiting AChE. Also, some symptoms of AD are related to the alterations in the dopaminergic9 and serotoninergic neurotransmitter systems.10
Monoamine oxidase (MAO; EC 1.4.3.4) is the enzyme that catalyzes the oxidative deamination of various biogenic amines,11 rendering the corresponding aldehyde, ammonia, and hydrogen peroxide as metabolic products. Thus, MAO inhibitors might increase amine neurotransmission and exert valuable biochemical effects in the treatment of AD.11 Increased levels of MAO B due to enhanced astrogliosis in the brain of AD patients has been reported,12 indicating that dual inhibition of MAO A/B may be valuable AD therapy. Beneficial properties of MAO inhibitors are also related to the reduction of the formation of the reactive oxygen species, which may influence the increased neuronal damage.13,14 Finally, recent biochemical and clinical studies indicated that the propargylamine group of the MAO B inhibitors, such as rasagiline and ladostigil, is responsible for neuroprotective, antiapoptotic activities,15–17 and inhibitory effect on the Aβ aggregation of these compounds.18
Because of the multifactorial nature of AD and diverse cerebral mechanisms implicated in the control of AD,1,11 multi-target-directed ligands have been extensively examined as novel drug candidates with beneficial effects in therapy of AD.7,15,19,20
In this context, in previous communications from our laboratory, we have identified the pyridine structural and functional motif affording molecules endowed with cholinesterase inhibition potential.21
Our recently synthesized hybrids22 exerted appreciable MAO A/B, AChE, and butyrylcholinesterase (BuChE) inhibitory activity. These molecules, exemplified by the most attractive compound ASS234 (Figure 1), are hybrids of donepezil, a potent AChE inhibitor currently used to treat AD, and PF9601N, an inhibitor of MAO B,23 connected through an appropriate linker to facilitate the binding of both N-benzylpiperidine and indole-propargylamine moieties at catalytic anionic site (CAS) and peripheral anionic site (PAS), respectively, in AChE.
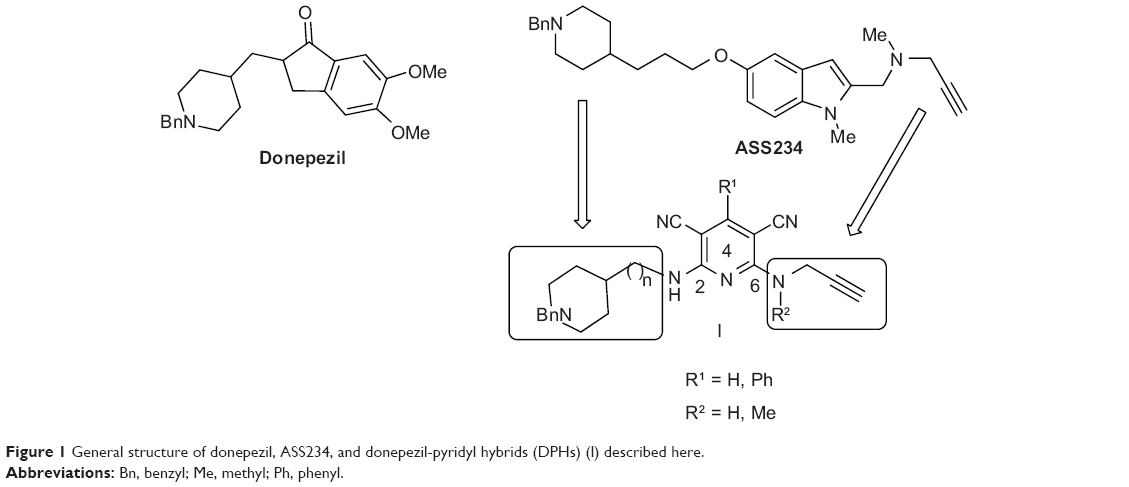 | Figure 1 General structure of donepezil, ASS234, and donepezil-pyridyl hybrids (DPHs) (l) described here. |
With these ideas in mind, we have designed, synthesized, and submitted to biochemical evaluation donepezil-pyridyl hybrids (DPHs) of type I (Figure 1), as multipotent ChE and MAO inhibitors.
In a recent communication we have reported preliminary results on DPHs1–8 (Figure 2).24 These compounds are based on donepezil and ASS234 by fixing the N-benzylpiperidine motif linked by a methylene carbon chain (n=0 to 3) attached through an amino group to C2 of a pyridine substituted with a phenyl group or a hydrogen at C4, used now as the central heterocyclic core instead of the indole motif, while retaining the N(H,Me)-propargylamine moiety at C6. We have observed24 that most of these DPHs showed no MAO inhibition activity, and that the only active compound (DPH5) was a very weak MAO A inhibitor (Table 1).24 Consequently, more work was needed in order to improve in power and selectivity the multipotent profile of our molecules. As a result, we have synthesized new DPHs9–16 (Table 1), and report here the biochemical evaluation for the inhibition of MAO A/B, AChE, and BuChE, as well as a 3D-quantitative structure-activity relationship (QSAR) study used to define specific molecular determinants for MAO A/B, AChE, and BuChE inhibition and to create QSAR models for evaluation of MAO A/B, AChE, and BuChE activity of the designed multipotent inhibitors. From this work, we have identified the novel donepezil-pyridyl hybrid DPH14 as a moderately potent, selective AChE and MAO B inhibitor, whose molecular modeling has also been investigated.
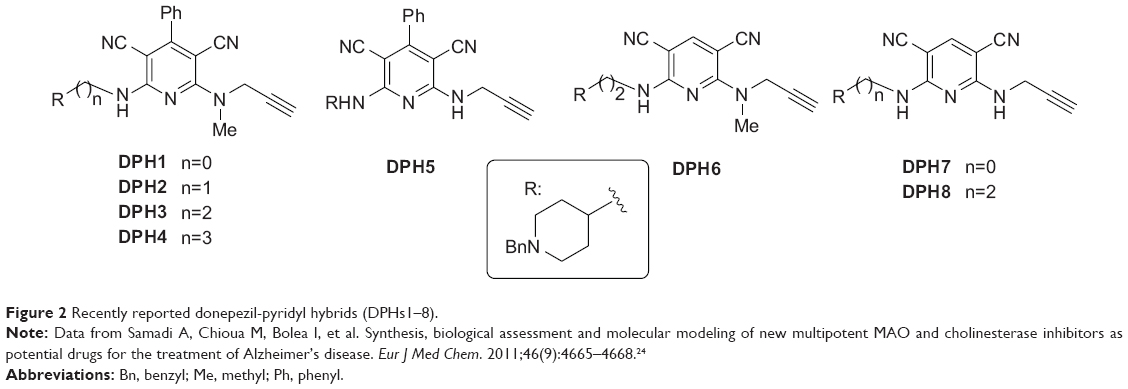 | Figure 2 Recently reported donepezil-pyridyl hybrids (DPHs1–8). |
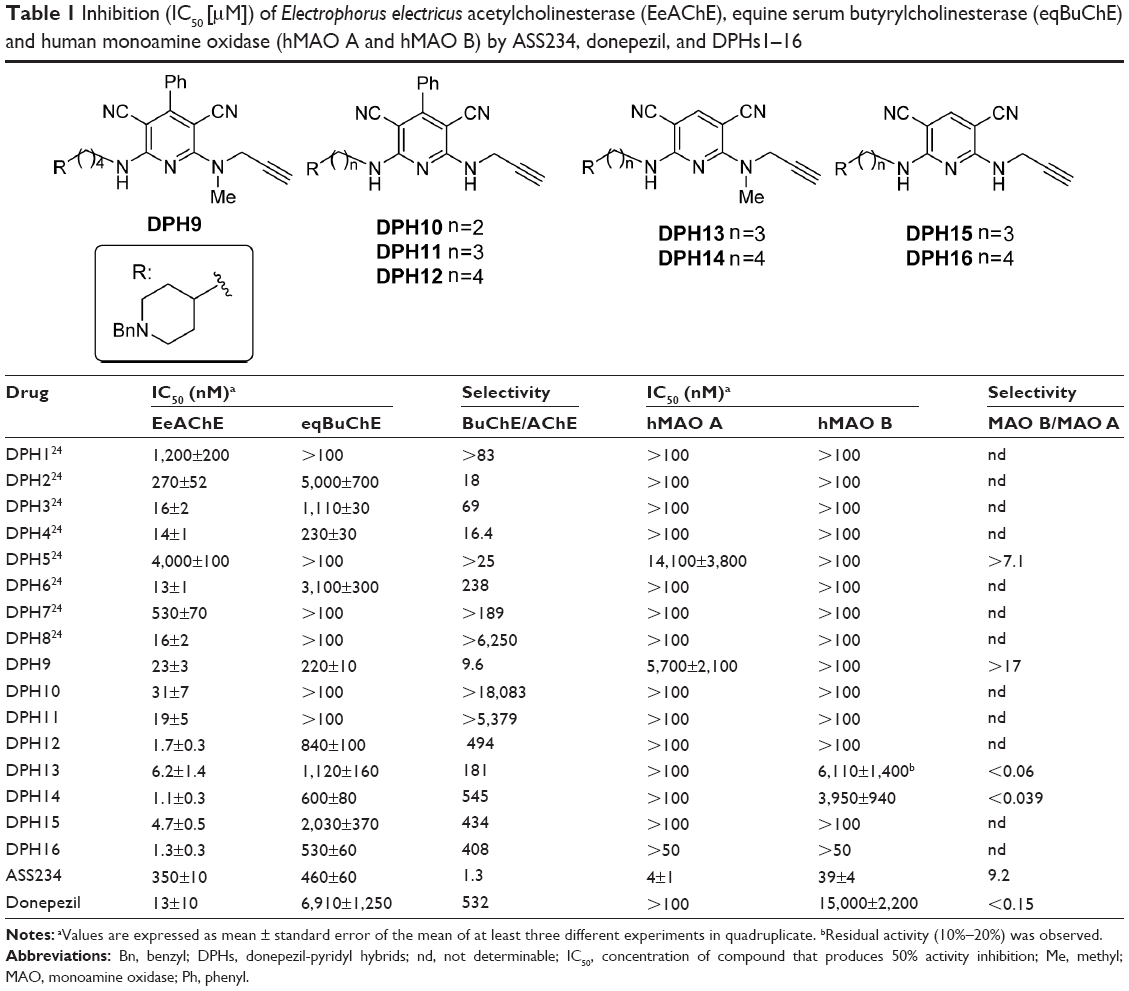 | Table 1 Inhibition (IC50 [μM]) of Electrophorus electricus acetylcholinesterase (EeAChE), equine serum butyrylcholinesterase (eqBuChE) and human monoamine oxidase (hMAO A and hMAO B) by ASS234, donepezil, and DPHs1–16 |
Materials and methods
General
Melting points were determined on a Koffler apparatus, and are uncorrected. Infrared spectra were recorded in a Perkin-Elmer apparatus (“Spectrum One”; PerkinElmer Inc., Waltham, MA, USA). 1H nuclear magnetic resonance (NMR) and 13C NMR spectra were, recorded in deuterated chloroform (CDCl3) or deuterated dimethylsulfoxide (DMSO-d6) at 300, 400, or 500 MHz and at 75, 100, or 125 MHz, respectively, in a Brucker Avance III HD (Brucker Corporation, Billerica, MA, USA) apparatus, using solvent peaks [CDCl3: 7.27 (D), 77.2 (C) ppm] as internal reference. The assignment of chemical shifts was based on standard NMR experiments (1H, 13C, DEPT [Distortionless Enhancement by Polarization Transfer], COSY, gHSQC, gHMBC [2D NMR correlation spectroscopy experiments]). High-resolution mass spectrometry spectra were recorded in a Hewlett-Packard (Palo Alto, CA, USA) HP-5973 mass selective detector apparatus. Mass spectra were recorded on a gas chromatography-mass spectrometry spectrometer with an electrospray ionization source in a Hewlett-Packard HP-1100 MSD apparatus. Elemental analyses were performed at Centro Quimica Organica (Consejo Superior de Investigaciones Cientificas, Madrid, Spain) in a Carlo Erba (Milan, Italy) instrument (CHNS/O EA1108). Thin-layer chromatography were performed on silica F254 and detection by ultraviolet light at 254 nm or by charring with either ninhydrin, anisaldehyde, or phosphomolybdic-H2SO4 dyeing reagents. Anhydrous solvents were used in all experiments. Column chromatography was performed on silica gel 60 (230 mesh). All known compounds have been synthesized as reported.
Chemistry
The synthesis of the target DPH molecules was carried out as shown in schemes 1 and 2 (Supplementary material http://www.iqog.csic.es/iqog/sites/default/files/public/User/Jos%C3%A9%20Luis%20Marco%20Contelles/Supplementary%20material.pdf).
Target DPHs py(C6)N(Me)propargylamines 9, 13, and 14 have been synthesized starting from known intermediates 2,6-dichloro-4-phenylpyridine-3,5-dicarbonitrile (17)25 and 2,6-dichloropyridine-3,5-dicarbonitrile (18),26 easily prepared from readily available precursors 2-amino-6-chloro-4-phenylpyridine-3,5-dicarbonitrile27 and 2-amino-6-chloropyridine-3,5-dicarbonitrile,28 respectively. The reaction of pyridines 17 and 18 with N-methylprop-2-yn-1-amine afforded pyridines 1924 and 20,24 respectively, is shown in Figure 3. In the synthesis of compound 20, minor amounts of N,N′-di(C2,6)alkylated product were isolated and characterized (Supplementary material http://www.iqog.csic.es/iqog/sites/default/files/public/User/Jos%C3%A9%20Luis%20Marco%20Contelles/Supplementary%20material.pdf). Next, the reaction of compound 19 with 4-(1-benzylpiperidin-4-yl)butan-1-amine (22)29 gave the desired target compound DPH9 in good yield (Figure 3). Similarly, the reaction of 20 with amines 3-(1-benzylpiperidin-4-yl)propan-1-amine (21),30 and 2229 gave the desired target compounds DPHs13 and 14 in good yields (Figure 3).
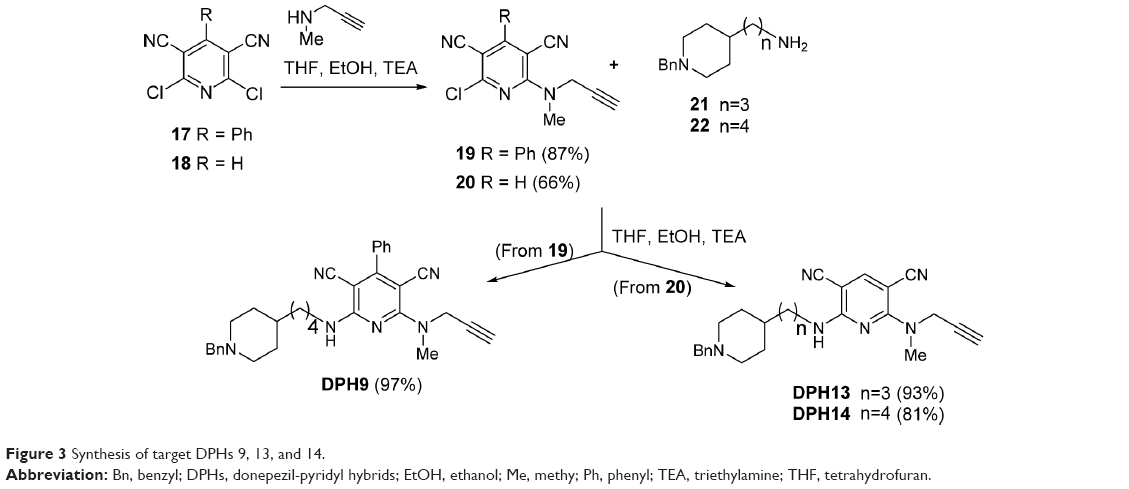 | Figure 3 Synthesis of target DPHs 9, 13, and 14. |
Next, we attacked the synthesis of target DPHs py(C6)N(H)propargylamines 10–12, 15, and 16. Thus, the reaction of 2,6-dichloropyridine 1725 with propargylamine gave 2-chloro-4-phenyl-6-(prop-2-yn-1-ylamino)pyridine-3,5-dicarbonitrile (23) (Figure 4) that after coupling with amines 2-(1-benzylpiperidin-4-yl)ethanamine (24),31 21,30 and 2229 cleanly afforded the desired target molecules DPHs10–12 in good overall yield (Figure 4). Finally, the synthesis of DPHs15 and 16 was carried out from the key intermediate 25 (prepared as usual from precursor 18)26 and reaction with amines 21 and 22, respectively (Figure 4).
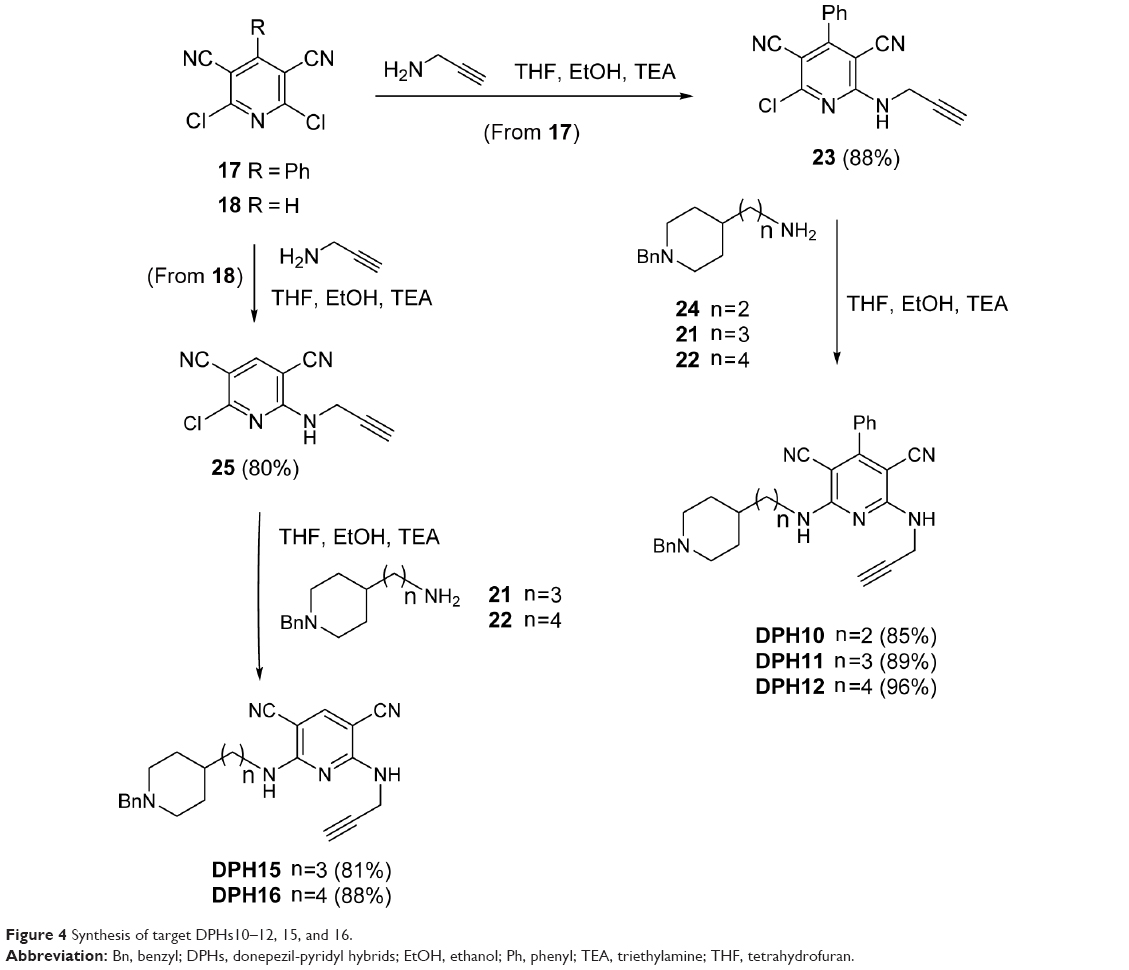 | Figure 4 Synthesis of target DPHs10–12, 15, and 16. |
Biochemical methods
Inhibition experiments of AChE and BuChE
To assess the inhibition of the activities of AChE from Electrophorus electricus (type V-S), human recombinant AChE (hAChE) or BuChE from equine serum (lyophilized powder) and human recombinant BuChE (hBuChE) (Sigma-Aldrich Co., St Louis, MO, USA), the spectrophotometric method of Ellman was followed.32 The reactions took place in a final volume of 300 μL in a phosphate-buffered solution (0.1 M) at pH 8, containing 116.7 U/L of AChE or 166.7 U/L of BuChE and 0.35 mM of 5,5′-dithiobis-2-nitrobenzoic acid (DTNB; Sigma-Aldrich Co.). Inhibition curves were made by pre-incubating this mixture with at least nine concentrations of each compound for 20 minutes. A sample with no compound was always present to determine the 100% of the enzyme activity. After this pre-incubation period, 0.35 mM acetylthiocholine iodide or 0.5 mM butyrylthiocholine iodide (Sigma-Aldrich Co.) were added, allowing the enzymatic reaction for 5 minutes with AChE and 30 minutes with BuChE while the DTNB produces the yellow anion 5-thio-2-nitrobenzoic acid along with the enzymatic degradation of the substrates. Changes in absorbance were detected at 405 nm in a spectrophotometric plate reader (FluoStar OPTIMA; BMG Labtech, Ortenberg, Germany). Compounds inhibiting AChE or BuChE activity would reduce the color generation, thus the half maximal inhibitory concentration (IC50) values were calculated as the concentration of compound that produces 50% activity inhibition. Data are expressed as means ± standard error of the mean (SEM) of at least three different experiments in quadruplicate.
Inhibition experiments of MAO A/B
MAO activities from recombinant human MAO A/B (Sigma-Aldrich Co.) were performed using a fluorometric method.33 Tyramine hydrochloride was used as substrate for both enzymes in a 96-well black opaque microplate (OptiPlate-96F, PerkinElmer Inc.) in a final volume of 200 μL. Serial dilutions of each inhibitor were pre-incubated for 30 minutes at 37°C with 360 U/L human monoamine oxidase (hMAO) A or 67.5 U/L hMAO B. Following the pre-incubations, enzymatic reactions were started by adding 100 μL of a mixture containing 1 mM tyramine, 40 U/L horseradish peroxidase, and 25 μM Amplex UltraRed (Life Technologies, Eugene, OR, USA) reagent in 0.25 mM sodium phosphate pH 7.4 as final concentrations. The fluorescence production associated with peroxidase-coupled production of resorufin from Amplex UltraRed was constantly measured for at least 1 hour at 530 nm in a spectrophotometric plate reader (FluoStar OPTIMA, BMG Labtech). Control experiments were carried out simultaneously by replacing the inhibitors with distilled water. In addition, the possible capacity of compounds to modify the fluorescence generated in the reaction mixture due to nonenzymatic inhibition was determined by adding these compounds to solutions containing only the Amplex UltraRed reagent in a sodium phosphate buffer. Samples with no substrate were used as blanks.
Determination of IC50 values
IC50 values were determined from dose–response curves, plotted by using the GraphPad “PRISM” software (version 3.0; GraphPad Software, Inc., La Jolla, CA, USA), as the inhibitor concentration producing 50% of activity inhibition. Data are expressed as mean ± SEM of at least three different experiments performed in triplicate.
Test of reversibility inhibition of human recombinant MAO B by DPH14
Reversibility of MAO B inhibition by DPH14 was determined by studying the recovery of the enzymatic activity after a large dilution of the complex. MAO B concentration of 100-fold over the concentration required for the activity assay was used with 50 μM DPH14 and 0.5 μM R-deprenyl (as standard irreversible MAO B inhibitor), concentrations equivalent to 10-fold their IC50 values previously determined. After 30 minutes pre-incubation at 37°C, the mixture was rapidly diluted 100-fold into reaction buffer containing 40 U/L horseradish peroxidase, 25 μM Amplex UltraRed reagent, and 1 mM p-tyramine in order to initiate enzymatic reaction. Curves of each sample were plotted and compared to that of enzyme samples incubated and diluted in absence of inhibitor. After enzyme dilutions, enzyme concentrations were equal to that used in previous dose-curve experiments, but MAO B concentration changing from 10× IC50 to 0.1× IC50 upon dilution.
The resulting progress curves were plotted and followed for at least 1 hour reaction. Activity rates were calculated and final data were expressed as percentage of remaining enzyme activity as mean ± SEM of at least three different experiments.
Results and discussion
Biochemical evaluation
The in vitro activity of DPHs9–16 derivatives inhibiting E. electricus AChE (EeAChE) and equine BuChE (eqBuChE) was determined using the Ellman’s method.32 Donepezil and ASS234 were also assayed for comparative purposes. In order to test their potential multipotent profile, DPHs9–16 were also evaluated as human recombinant MAO A/B inhibitors using a fluorometric assay,33 giving the values shown in Table 1, where we have incorporated the corresponding IC50 values of compound DPHs1–8 for comparative purposes.24
Regarding ChEs inhibition, DPHs were very active and selective EeAChE versus (vs) eqBuChE inhibitors. Particularly, DPH12, 14, and 16 were potent, in the low nanomolar range, AChE inhibitors, DPH14 being the most active (AChE: IC50 =1.1±0.3 nM; BuChE: IC50 =600±80 nM), exhibiting one of the most active ChE inhibitory potencies ever determined by us.34
DPH14 bears a hydrogen at C4, four methylene groups in the chain linker, and an NMe-propargylamine moiety at C6, three structural motifs that seem relevant for a strong AChE inhibition power in this family of compounds, although structural changes in these groups still afford potent acetylcholinesterase inhibitors (AChEI) (Table 1). As shown in Table 1, and compared with the reference compound ASS234,22 DPH14 was 318-fold more potent for the inhibition of AChE, but 1.3-fold less active for the inhibition of BuChE, while, by comparison with donepezil, DPH14 was 11.8- and 11.5-fold more active at inhibiting AChE and BuChE, respectively.
As shown in Table 1, on-going from DPH1 to DPH4 and DPH9, by increasing the linker length (n) from 0 to 4, while retaining a phenyl group at C4 and an NMe-propargylamine at C6, the most potent AChEI was DPH4 (n=3) (AChE: IC50 =14±1 nM), 1.6-fold more potent than DPH9 (n=4), and in the same range than DPH3 (n=2). Concerning the selectivity (BuChE/AChE), DPH1 and DPH3 showed the highest values, while DPH9 was the most potent BuChE inhibitor (IC50 =220±10 nM), similar to DPH4.
Next, in DPHs5, 10–12 compared to DPHs1, 3, 4, and 9, the NMe-propargylamine at C6 was changed by an NH-propargylamine. In this group, the most potent AChEI was DPH12 (n=4) (AChE: IC50=1.7±0.3 nM), being the only active BuChE inhibitor (IC50 =840±100 nM). DPH10 and 11 were significantly less active than DPH12. It is also remarkable that for the same value of n (0, 2, and 3), by comparing DPH1 with DPH5 (n=0), DPH3 with DPH10 (n=2), and DPH4 with DPH11 (n=3), the most potent was always the DPH bearing the NMe-propargylamine moiety, while for n=4, by comparing DPH9 with DPH12, the most potent was the DPH bearing the NH-propargylamine moiety.
In compounds DPHs6–8, and 13–16 (Table 1), the same type of modifications was explored, but instead of a phenyl group at C4, a proton was incorporated at the same position.
Concerning DPHs6, 13, and 14, bearing the NMe-propargylamine moiety at C6, the power and selectivity increased from n=2 to 4, DPH14 being the most active (AChE: IC50 =1.1±0.3 nM; BuChE: IC50 =600±80 nM). However, compared with DPH9, both bearing the same value of n (4) and an NMe-propargylamine moiety at C6, DPH14 was 20-fold more potent at inhibiting AChE, but 2.7-fold less active at inhibiting BuChE.
Regarding hybrids DPHs7, 8, 15, and 16, bearing an NH-propargylamine moiety at C6, the inhibitory potency and selectivity also increased from n=0, 2 to 4, DPH16 being the most active inhibitor (AChE: IC50 =1.3±0.3 nM; BuChE: IC50 =530±60 nM). DPH12 and DPH16, both bearing the same n value (4) and an NH-propargylamine moiety at C6, were almost equipotent at inhibiting AChE and BuChE, respectively, but highly selective for the inhibition of AChE. Concerning AChE inhibition, it was also noticed that for the same n values (n=2 or 4), DPH6 and DPH8, DPH14 and DPH16, were equipotent, whereas comparing DPH13 with DPH15 (n=3), the most potent was the DPH15 bearing the NH-propargylamine moiety. However, in the BuChE inhibition, for the same n value, in the case of DPH6 and 8 (n=2) and DPH13 and DPH15 (n=3), the most potent was the DPH bearing the NMe-propargylamine moiety, while DPH14 and 16 (n=4)] were equipotent.
Based on these results, the activity of DPHs9, 12–16, possessing potent EeAChE/eqBuChE inhibitory profiles, was also determined as hAChE and hBuChE inhibitors.
As shown in Table 2, all selected compounds exhibited similar trends as previously observed in nonhuman ChEs, revealing hAChE-selectivity and displaying IC50 values in low nanomolar range. DPH15 (IC50 =1±0.4 nM) and DPH12 (IC50 =770±140 nM) were the most potent hAChE and hBuChE inhibitors, respectively. DPH16 showed the most satisfactory balance in terms of inhibitory activity power for both enzymes [hAChE (IC50 =2.9±0.8 nM); hBuChE (IC50 =790±110 nM)], but DPH15 was the most selective hAChE inhibitor (selectivity index: 1920). Very interestingly, DPH14 is still a potent hBuChE inhibitor, in the same range as DPH12 or DPH16, but 13.1-fold less potent than DPH15 for the inhibition of hAChE. Finally, note that DPH16 was 3.1- and 9.2-fold more active than donepezil for the inhibition of hAChE and hBuChE, respectively.
 | Table 2 Inhibition (IC50 [μM]) of human recombinant acetylcholinesterase (hAChE) and human recombinant butyrylcholinesterase (hBuChE) by DPHs9, 12–16, ASS234, and donepezil |
Concerning MAO inhibition by DPHs (Table 1), some of them (DPHs10–12, 15, 16) were inactive, or moderate inhibitors, such as DPHs9, 5, 13, and 14. Among those active, DPH9 [IC50 (MAO A) =5,700±2,100 nM] and DPH14 [IC50 (MAO B) =3,950±940 nM] exhibited the most interesting profile, as moderately selective MAO A/B inhibitors, respectively, in the low μM range.
Regarding our multi-target approach, DPH14 showed a very potent and slightly selective EeAChE inhibition, and a moderately selective hMAO B inhibition activity. DPH5 showed a potent and selective hAChE inhibition as well as a moderate, but selective hMAO A inhibition activity.
Once IC50 values were determined as a preliminary evaluation, DPH14 was selected as the most relevant MAO B inhibitor to next investigate its mechanism of inhibition.
Thus, the study of reversibility of hMAO B inhibition by DPH14 was assessed by pre-incubating for 30 minutes 100-fold enzyme concentration used in the experiments previously described with inhibitor concentrations equivalent to 10-fold their IC50 values: 50 μM DPH14 and 0.5 μM R-deprenyl, as standard irreversible MAO B inhibitor. After pre-incubations, the mixture was diluted 100-fold into a buffered solution containing 1 mM p-tyramine to recover previous enzyme activity levels. Figure 5 shows that initial MAO B inhibition by DPH14 (55%) was not recovered after a 100-fold dilution in saturated-substrate concentration, suggesting an irreversible MAO B inhibition by DPH14. Control tests were carried out by pre-incubating and diluting in the absence of inhibitors.
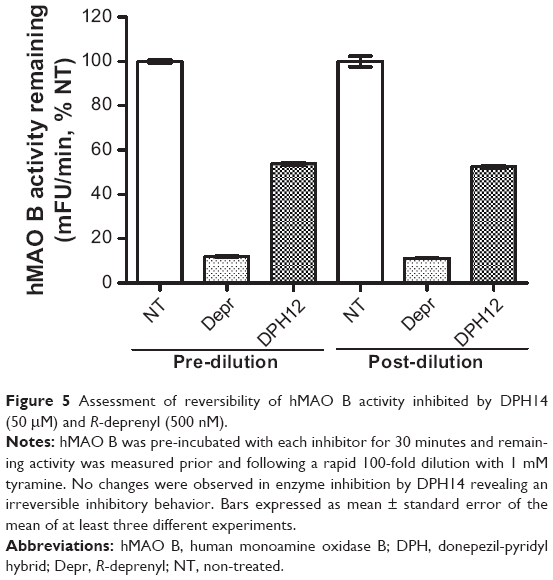 | Figure 5 Assessment of reversibility of hMAO B activity inhibited by DPH14 (50 μM) and R-deprenyl (500 nM). |
In order to further characterize the mechanism of hMAO B inhibition by DPH14, dose–response curves (IC50) were obtained by varying pre-incubation times (15–60 minutes). A time-dependent inhibition was clearly observed since IC50 values were found to decrease in a pre-incubation time-dependent manner (Table 3). These findings agree with the previously determined irreversible behavior.
 | Table 3 Time-dependent inhibition (IC50) of human recombinant MAO B by DPH14 following different pre-incubation times |
3D-QSAR study of novel DPHs
The 3D-QSAR study has been carried out on these hybrids with main goal to define structural requirements for high inhibitory activity on the enzymes involved, following the usual methods (Supplementary material http://www.iqog.csic.es/iqog/sites/default/files/public/User/Jos%C3%A9%20Luis%20Marco%20Contelles/Supplementary%20material.pdf).
The IC50 inhibition values of MAO A/B and AChE/BuChE by donepezil-pyridine hybrids (Chart S1, Supplementary material http://www.iqog.csic.es/iqog/sites/default/files/public/User/Jos%C3%A9%20Luis%20Marco%20Contelles/Supplementary%20material.pdf), and 37 donepezil-indole (Charts S2 and S3, Supplementary material http://www.iqog.csic.es/iqog/sites/default/files/public/User/Jos%C3%A9%20Luis%20Marco%20Contelles/Supplementary%20material.pdf)22–24 derivatives were used for the 3D-QSAR study. In the examined data set, the pIC50 (MAO A) interval spanned 6 log units (3.07–9.10), the pIC50 (MAO B) interval was 6.6 log units (4.00–10.60), the pIC50 (AChE) activity was 4.2 log units (4.00–8.17), and pIC50 (BuChE) activity was 2.6 log units (4.00–6.64). Relatively wide activity interval of the training set provides extensive applicability domain for the formed 3D-QSAR models.
Hydrogen bond acceptor (HBA) properties of O-bridge create crucial favorable interactions (v892: N1-TIP) (Figure S1, Supplementary material http://www.iqog.csic.es/iqog/sites/default/files/public/User/Jos%C3%A9%20Luis%20Marco%20Contelles/Supplementary%20material.pdf) for MAO A inhibiting activity with steric properties of the propargylamine moiety (Donz-D4/Donz-D9, Figure S2A/B, Supplementary material http://www.iqog.csic.es/iqog/sites/default/files/public/User/Jos%C3%A9%20Luis%20Marco%20Contelles/Supplementary%20material.pdf). The unsaturated bond of the propargylamine moiety also forms a favorable interaction (v287: TIP-TIP, v344: TIP-TIP) with the nearest terminal hydrogen (Donz-D4/Donz-D9, Figure S2A/B, Supplementary material http://www.iqog.csic.es/iqog/sites/default/files/public/User/Jos%C3%A9%20Luis%20Marco%20Contelles/Supplementary%20material.pdf). The 3D-QSAR study confirmed previous experimental findings,22–23 which indicated the essential role of the propargylamine moiety for the MAO A inhibiting activity. Substitution of the terminal hydrogen of propargylamine moiety (Donz-D4/Donz-D9, Figure S2A/B, Supplementary material http://www.iqog.csic.es/iqog/sites/default/files/public/User/Jos%C3%A9%20Luis%20Marco%20Contelles/Supplementary%20material.pdf) with bulky groups could enhance MAO A inhibiting activity of the examined compounds by facilitating the favorable interactions (v892: N1-TIP, v287: TIP-TIP, v344: TIP-TIP).
Also, HBA O-bridge forms very specific unfavorable interactions (v854: N1-TIP and v868: N1-TIP) with N-Me group of the indole moiety (Donz-D9, Figure S2B, Supplementary material http://www.iqog.csic.es/iqog/sites/default/files/public/User/Jos%C3%A9%20Luis%20Marco%20Contelles/Supplementary%20material.pdf). Furthermore, the N-Me group of the indole moiety forms unfavorable interactions (v301: TIP-TIP, v325: TIP-TIP, and v332: TIP-TIP) with meta/para positions of the benzyl moiety (Donz-D4/Donz-D9, Figure S2A/B, Supplementary material http://www.iqog.csic.es/iqog/sites/default/files/public/User/Jos%C3%A9%20Luis%20Marco%20Contelles/Supplementary%20material.pdf).
Finally, the HBD properties of the N-atom in the piperidinyl or piperazinyl moiety create very specific unfavorable interactions (v783: O-TIP) with meta positions of the benzyl moiety (Donz-D4/Donz-D9, Figure S2A/B, Supplementary material http://www.iqog.csic.es/iqog/sites/default/files/public/User/Jos%C3%A9%20Luis%20Marco%20Contelles/Supplementary%20material.pdf).
Therefore, substitution of the methyl group with hydrogen in the N-Me group of the indole could enhance MAO A inhibiting activity, while bulky groups in meta/para positions of the benzyl moiety could decrease MAO A inhibiting activity.
The HBA group of compounds does not have propargylamine moiety to form essential pharmacophores (v287: TIP-TIP and v892: N1-TIP) for MAO A inhibiting activity.
Predictive potential of the developed 3D-QSAR (MAO A) model was tested by use of leave-one-out cross validation of the training set (Q2: 0.87, R2Observed vs Predicted: 0.977, and root mean square error of estimation [RMSEE]: 0.229) and verification set (R2Observed vs Predicted: 0.804 and root mean square error of prediction [RMSEP]: 0.578) (Table S1, Supplementary material http://www.iqog.csic.es/iqog/sites/default/files/public/User/Jos%C3%A9%20Luis%20Marco%20Contelles/Supplementary%20material.pdf). The obtained statistical parameters indicated that the created 3D-QSAR (MAO A) model has good prognostic capacity for MAO A inhibiting activity.
Hydrogen bond donor (HBD) properties of amino groups of propargylamine and indole moieties create very specific favorable interactions (v765: O-TIP, v775: O-TIP) (Figure S3, Supplementary material http://www.iqog.csic.es/iqog/sites/default/files/public/User/Jos%C3%A9%20Luis%20Marco%20Contelles/Supplementary%20material.pdf) for MAO B inhibiting activity with unsaturated back chain (Donz-D3/Donz-D5, Figure S4A/B, Supplementary material http://www.iqog.csic.es/iqog/sites/default/files/public/User/Jos%C3%A9%20Luis%20Marco%20Contelles/Supplementary%20material.pdf). Thus, new electron-donating groups in propargylamine or indole moiety could increase electron densities of the groups and therefore selectively enhance MAO B activity of the compounds. The propargylamine group creates favorable interaction (v295: TIP-TIP, v314: TIP-TIP) with N-CH3 group of the indole moiety, and also forms favorable interaction with meta/para positions of the benzyl moiety (Donz-D3/Donz-D5, Figure S4A/B, Supplementary material http://www.iqog.csic.es/iqog/sites/default/files/public/User/Jos%C3%A9%20Luis%20Marco%20Contelles/Supplementary%20material.pdf). Therefore, substitution of the methyl substituent on N-atom of the indole moiety and meta/para positions of the benzyl moiety with ethyl or other bulkier group could selectively increase MAO B activity of the compounds. The 3D-QSAR study confirmed previous experimental findings15–17 that propargylamine moiety has essential positive influence on the MAO B inhibiting activity.
Also, the hydrophobic part of the propargylamine moiety forms unfavorable interactions (v477: N1-DRY, v527: N1-DRY) with HBA ortho-C-atom of the benzyl moiety (Donz-D3, Figure S4A, Supplementary material http://www.iqog.csic.es/iqog/sites/default/files/public/User/Jos%C3%A9%20Luis%20Marco%20Contelles/Supplementary%20material.pdf). Therefore, electron withdrawing groups at meta/para positions of the benzyl moiety could enhance MAO B inhibiting activity of the examined compounds.
The HBA group of compounds does not have propargylamine moiety to form essential pharmacophores (v765: O-TIP, v775: O-TIP, v295: TIP-TIP, and v314: TIP-TIP) for MAO B inhibiting activity.
Predictive potential of the developed 3D-QSAR (MAO B) model was tested by use of leave-one-out cross validation of the training set (Q2: 0.82, R2Observed vs Predicted: 0.956, and RMSEE: 0.314) and verification set (R2Observed vs Predicted: 0.924 and RMSEP: 0.410) (Table S2, Supplementary material http://www.iqog.csic.es/iqog/sites/default/files/public/User/Jos%C3%A9%20Luis%20Marco%20Contelles/Supplementary%20material.pdf). The obtained statistical parameters indicated that the created 3D-QSAR (MAO B) model has good prognostic capacity for MAO B inhibiting activity.
HBD properties of N-atom in the piperidine ring and secondary N-atom in the propargylamine group create very specific unfavorable interactions (v133: O-O) (Figure S5, Supplementary material http://www.iqog.csic.es/iqog/sites/default/files/public/User/Jos%C3%A9%20Luis%20Marco%20Contelles/Supplementary%20material.pdf) for AChE inhibition (DPH8, Figure S6A, Supplementary material http://www.iqog.csic.es/iqog/sites/default/files/public/User/Jos%C3%A9%20Luis%20Marco%20Contelles/Supplementary%20material.pdf). Thus, extension of the N-alkyl bridge could selectively enhance AChE inhibiting activity of the pyridine derivatives.
HBD feature of N-atom in piperidinyl moiety creates very specific unfavorable interaction (v678: O-N1) with the cyano group (DPH4, Figure S6B, Supplementary material http://www.iqog.csic.es/iqog/sites/default/files/public/User/Jos%C3%A9%20Luis%20Marco%20Contelles/Supplementary%20material.pdf). The propargylamine group creates unfavorable interactions (v334: TIP-TIP) with meta/para position of benzyl moiety (DPH8 and 4, Figure S6A/B, Supplementary material http://www.iqog.csic.es/iqog/sites/default/files/public/User/Jos%C3%A9%20Luis%20Marco%20Contelles/Supplementary%20material.pdf). All these results indicated that extension of the N-alkyl bridge could selectively decrease three very specific unfavorable interactions, v133: O-O, v334: TIP-TIP, and v678: O-N1, and therefore significantly enhance AChE inhibiting activity of the pyridine derivatives.
HBA feature of N-atom in the pyridine ring creates favorable interactions with meta/para position of benzyl moiety (v895: N1-TIP) for AChE inhibition (DPH8 and 4, Figure S6A/B, Supplementary material http://www.iqog.csic.es/iqog/sites/default/files/public/User/Jos%C3%A9%20Luis%20Marco%20Contelles/Supplementary%20material.pdf). The propargylamine moiety creates crucial favorable interaction with hydrophobic phenyl substituent of the pyridine moiety (v803: DRY-TIP) (DPH4, Figure S6B, Supplementary material http://www.iqog.csic.es/iqog/sites/default/files/public/User/Jos%C3%A9%20Luis%20Marco%20Contelles/Supplementary%20material.pdf). Therefore, phenyl substituent in the pyridine ring is able to selectively enhance AChE inhibiting activity. Since meta position of the phenyl substituent of the pyridine moiety forms one unfavorable interaction with N-atom of the pyridine ring (v909: N1-TIP), substitution of the meta position of the phenyl substituent could result in decrease of AChE inhibiting activity (DPH4, Figure S6B, Supplementary material http://www.iqog.csic.es/iqog/sites/default/files/public/User/Jos%C3%A9%20Luis%20Marco%20Contelles/Supplementary%20material.pdf).
HBD properties of N-bridge forms a very specific favorable interaction (v785: O-TIP) with the propargylamine group (DPH8 and 4, Figure S6A/B, Supplementary material http://www.iqog.csic.es/iqog/sites/default/files/public/User/Jos%C3%A9%20Luis%20Marco%20Contelles/Supplementary%20material.pdf).
Predictive potential of the developed 3D-QSAR (AChE) model was tested by use of leave-one-out cross validation of the training set (Q2: 0.87, R2Observed vs Predicted: 0.979, and RMSEE: 0.203) and verification set (RMSEP: 0.574). The obtained statistical parameters indicated that the created 3D-QSAR (AChE) model has good prognostic capacity for AChE inhibiting activity.
HBD feature of N-atom in piperidinyl moiety creates crucial favorable interactions (v690: O-N1) (Figure S7, Supplementary material http://www.iqog.csic.es/iqog/sites/default/files/public/User/Jos%C3%A9%20Luis%20Marco%20Contelles/Supplementary%20material.pdf) for BuChE inhibition with the HBA properties of N-atom of the pyridine ring (DPH3 and 4, Figure S8A/B, Supplementary material http://www.iqog.csic.es/iqog/sites/default/files/public/User/Jos%C3%A9%20Luis%20Marco%20Contelles/Supplementary%20material.pdf).
HBA feature of N-atom in the pyridine ring also creates a crucial favorable interaction (v984: N1-TIP) with meta/para position of benzyl moiety (DPH3 and 4, Figure S8A/B, Supplementary material http://www.iqog.csic.es/iqog/sites/default/files/public/User/Jos%C3%A9%20Luis%20Marco%20Contelles/Supplementary%20material.pdf).
The propargylamine group creates two favorable interactions (v334: TIP-TIP, v351: TIP-TIP) with meta/para position of benzyl moiety (DPH3 and 4, Figure S8A/B, Supplementary material http://www.iqog.csic.es/iqog/sites/default/files/public/User/Jos%C3%A9%20Luis%20Marco%20Contelles/Supplementary%20material.pdf).
The propargylamine (DPH3, Figure S8A, Supplementary material http://www.iqog.csic.es/iqog/sites/default/files/public/User/Jos%C3%A9%20Luis%20Marco%20Contelles/Supplementary%20material.pdf) or benzyl (DPH4, Figure S8B, Supplementary material http://www.iqog.csic.es/iqog/sites/default/files/public/User/Jos%C3%A9%20Luis%20Marco%20Contelles/Supplementary%20material.pdf) moieties create crucial favorable interactions (v317: TIP-TIP) with phenyl substituent of the pyridine ring. Therefore, the donepezil-pyridine hybrids with phenyl substituent in the pyridine ring are good leads for further design of novel multipotent ligands. Since meta position of the phenyl substituent of the pyridine moiety forms one unfavorable interaction with N-atom of the pyridine ring (v884: N1-TIP), substitution of the meta position of the phenyl substituent could result in decrease of BuChE inhibiting activity (DPH3 and 4, Figure S8B, Supplementary material http://www.iqog.csic.es/iqog/sites/default/files/public/User/Jos%C3%A9%20Luis%20Marco%20Contelles/Supplementary%20material.pdf).
HBA properties of cyano group of compound DPH3 forms a crucial unfavorable interaction (v907: N1-TIP) with meta position of benzyl moiety, while the compounds with longer N-alkyl chain (such as compound DHH4) do not create the v907: N1-TIP unfavorable interaction for BuChE inhibiting activity (DPH3 and 4, Figure S8A/B, Supplementary material http://www.iqog.csic.es/iqog/sites/default/files/public/User/Jos%C3%A9%20Luis%20Marco%20Contelles/Supplementary%20material.pdf).
These results indicated that extension of the N-alkyl bridge could decrease unfavorable interaction, v907: N1-TIP, and therefore influence a modest increase of BuChE inhibiting activity.
Predictive potential of the developed 3D-QSAR (BuChE) model was tested by use of leave-one-out cross validation of the training set (Q2: 0.78, R2Observed vs Predicted: 0.931, and RMSEE: 0.254) and verification set (R2Observed vs Predicted: 0.619 and RMSEP: 0.550). The obtained statistical parameters indicated that the created 3D-QSAR (BuChE) model has good prognostic capacity for BuChE inhibiting activity.
The created 3D-QSAR models were further applied for design of novel multipotent inhibitors, prediction of their inhibitory activity on the enzymes, and for selection of the most promising ligands. Results of the 3D-QSAR (AChE) study were mainly applied for design of novel multipotent MAO/ChE ligands with strong AChE inhibiting activity (Figure 2). Since the QSAR (AChE) study indicated that the longer N-butyl bridge and phenyl substituent in the pyridine ring are crucial for enhancement of AChE inhibiting activity of the donepezil-pyridine derivatives, several new donepezil-pyridine hybrids have been designed, examined for MAO/ChE inhibiting activity. Since the designed compounds contain unique N-butyl bridge between two pharmacophores, while the QSAR-training set ligands contain O-methyl, O-ethyl, O-propyl, N-methyl, N-ethyl, and N-propyl, bridge, relatively good agreement was obtained between QSAR-predicted and observed MAO/ChE inhibiting activity for the novel compounds. The designed compounds with 3D-QSAR predicted pIC50 (AChE) >6.0 were selected for synthesis (Table 1) and examined in vitro (Tables 4 and 5).
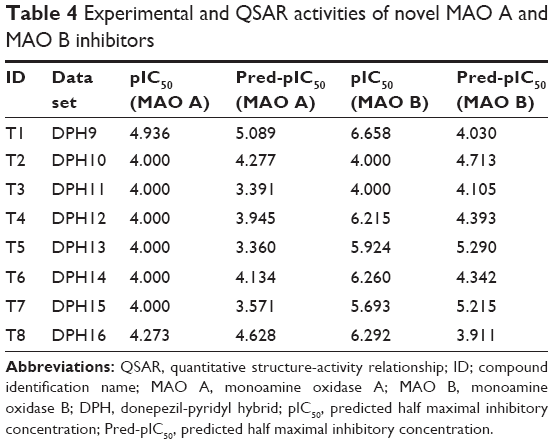 | Table 4 Experimental and QSAR activities of novel MAO A and MAO B inhibitors |
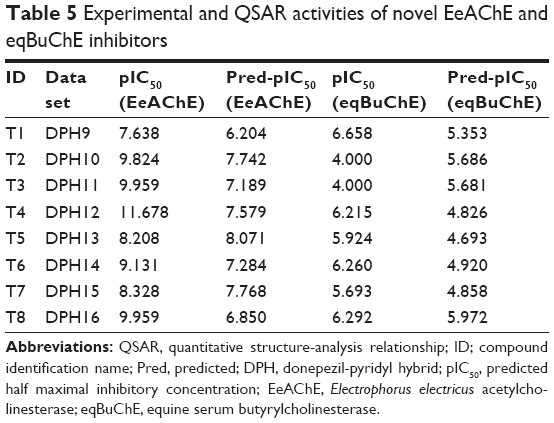 | Table 5 Experimental and QSAR activities of novel EeAChE and eqBuChE inhibitors |
Next, we have investigated the molecular modeling of compound DPH14 following the usual methods (Supplementary material http://www.iqog.csic.es/iqog/sites/default/files/public/User/Jos%C3%A9%20Luis%20Marco%20Contelles/Supplementary%20material.pdf).
Molecular modeling of compound DPH14
Inhibition of AChE and BuChE
The ligand-enzyme binding interactions of the potent ChE inhibitor DPH14, (EeAChE IC50 =0.0011±0.0003 μM; eqBuChE IC50 =0.60±0.08 μM) were investigated by molecular modeling studies. We have used the 3D structure of the enzyme species (EeAChE and eqBuChE), which was also used for the biochemical studies.
Docking simulations of EeAChE (Protein Data Bank [PDB]: 1C2B) and compound DPH14 were performed using Autodock Vina program (Molecular Graphics Laboratory, Scripps Research Institute, La Jolla, CA, USA).35 The binding site for docking was designed so that the entire receptor molecule was included within the selection grid. Autodock Vina introduces side chain flexibility into the target macromolecule. Eight side chains, Trp286, Tyr124, Tyr337, Tyr72, Asp74, Thr75, Trp86, and Tyr341, are allowed to change their conformations at the same time as the ligand that is being docked.
The docking study of inhibitor DPH14 within EeAChE indicates that two major binding modes at the enzyme-binding site can be proposed. In Figure 6, the two most favored binding modes are presented along with the first shell of residues surrounding DPH14. In both modes, compound DPH14 showed a binding mode with an extended conformation and interacted simultaneously with both catalytic and peripheral site of EeAChE thanks to a linker of appropriate length.
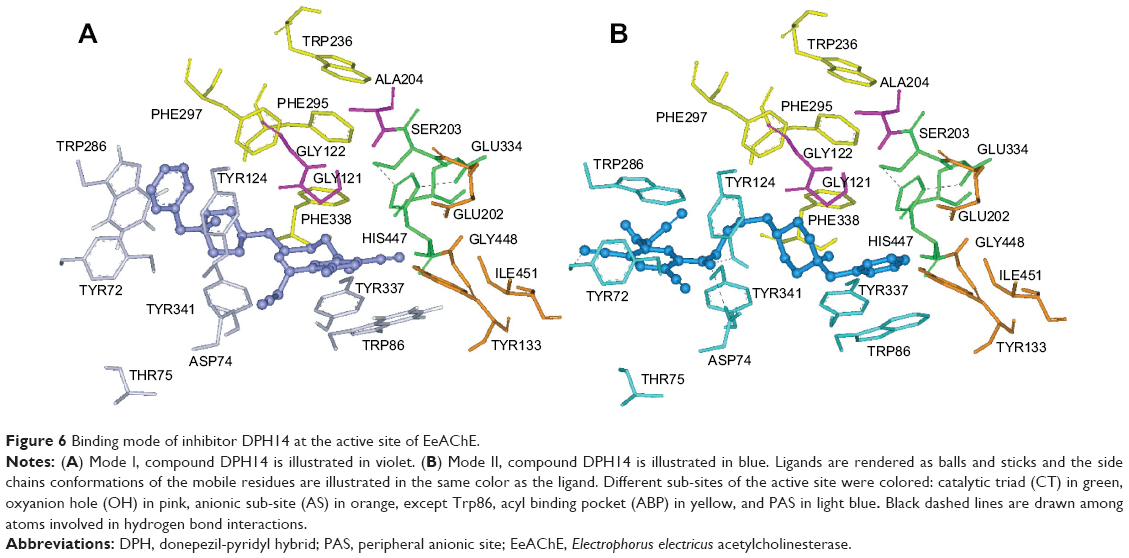 | Figure 6 Binding mode of inhibitor DPH14 at the active site of EeAChE. |
In Mode I (Figure 6A; binding energy: −13.1 kcal/mol), the N-benzylpiperidine structural motif was oriented toward the PAS, where the benzene ring was stacked over Trp286 and Tyr124. The pyridine ring was positioned at the bottom of the active site and it was stacked between Trp86 and a tyrosine pocket comprising Tyr124, Tyr337, and Tyr341. This ligand also showed hydrophobic interactions with His447 at the choline-binding site.
In Mode II (Figure 6B; binding energy: −11.9 kcal/mol), the ligand is oriented the other way around than in Mode I. As can be seen in Figure 6B, compound DPH14 had several interactions along the active site of EeAChE. Near the bottom of the gorge, the phenyl ring stacked against the Trp86 indole ring. At the top of the gorge, the pyridine ring and Trp286 indole ring formed a favorable face-to-face π–π interaction. The nitrogen of the cyano group was undergoing hydrogen bonding with the hydroxyl group of Tyr72. The amino-alkyl linker was positioned midway through the active site gorge and the NH group was undergoing hydrogen bonding with the hydroxyl group of Tyr124 and the carboxylate group of Asp74.
On the other hand, the docking study of DPH14 within the active site of eqBuChE was also carried out. In the absence of X-ray structure of eqBuChE, a homology model was used. SWISS-MODE,36–38 accesible via the ExPASy web server (swissmodel.expasy.org/), a fully automated protein structure homology-modeling server was used to design the receptor. A putative 3D structure of eqBuChE has been created based on the crystal structure of hBuChE (pdb: 2PM8) as these two enzymes exhibited 89% sequence identity.
In order to simulate the binding of compound DPH14 to eqBuChE, docking experiments were performed as blind dockings following the same computational protocol used for EeAChE. As depicted in Figure 7, compound DPH14 was well accommodated inside the active site gorge and two major binding modes (Modes I and II) can be proposed. In both modes, compound DPH14 showed a binding mode with a U-shaped conformation. Modes I and II (Figure 7a and b) placed the substituted-pyridine moiety into the binding pocket interacting with the residues involved in catalysis. In these orientations, the phenyl moieties interact with Trp82 allowing π–π stacking interactions.
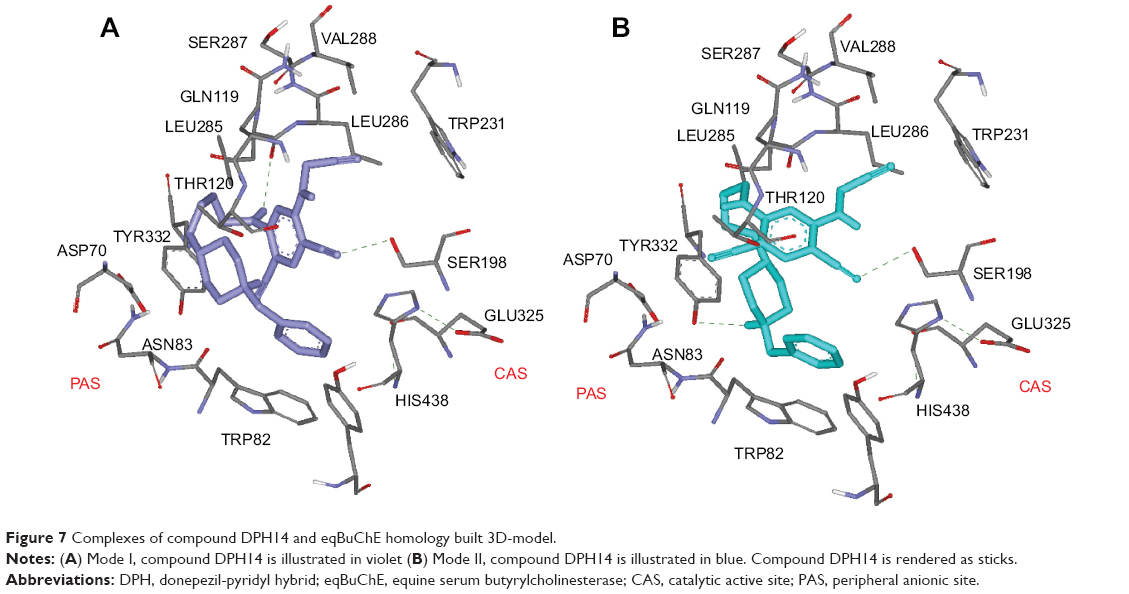 | Figure 7 Complexes of compound DPH14 and eqBuChE homology built 3D-model. |
In Mode I (Figure 7A; binding energy: −9.3 kcal/mol), a close examination of the first shell of residues surrounding DPH14 revealed that the cyano group formed a hydrogen bond with the hydroxyl of Ser198. The NH group of the linker formed a hydrogen bond with residue Leu286. The bound ligand is also stabilized by hydrophobic interactions with the catalytic triad residue His438.
In Mode II (Figure 7B; binding energy: −9.8 kcal/mol), two hydrogen bonds were observable. Compound DPH14 preserved the hydrogen bond already observed in Mode I, and it is established between the cyano group and the hydroxyl group of Ser198. The NH group of the protonated piperidine ring was also undergoing hydrogen-bonding interaction with the hydroxyl group of Tyr332.
Analysis of the intermolecular interactions indicated key residues responsible for ligand binding. The cyano group is likely to be an important feature for these derivatives to exhibit both AChE- and BuChE-inhibitory activities.
It is significant to note that the linear conformation allows DPH14 (Modes I and II) to span both CAS and PAS that contributes to its superior binding toward EeAChE.
Inhibition of MAO A and MAO B
In order to explore the nature of the ligand–receptor interactions, the ligand was docked to the active site of both MAO A and MAO B isoforms using the program Autodock Vina.35 We have focused on the compound DPH14, which showed the best hMAO B (IC50 =3.95±0.94 μM) inhibitory activity, with very significant EeAChE inhibitory potency.
Since inhibition data were determined on human MAOs, docking simulations were run on the human model of the MAO A and MAO B isoforms. The 3D structures for hMAOs were retrieved from the PDB (PDB ID: 2Z5X for hMAO A and PDB ID: 2V5Z for hMAO B). Following the protocol previously described for eqBuChE, we have theoretically investigated the recognition process between compound DPH14 (chosen as reference compound) and hMAOs.
Results from several studies have shown that it must be the neutral amine that reaches the active site of MAO A and MAO B that allows the chemistry.39–42 The docking simulations were done with compound DPH14 as neutral species despite that at physiological pH, most of the piperidine rings would be in the protonated, positively charged form.
As reported in our previous paper,43 a number of six structural water molecules were explicitly considered in the docking simulations. These water molecules are labeled as w72, w193, w11, w23, w15, and w53 in accordance with the numbering reported for the hMAO B crystallographic structure (PDB ID: 1S3E) and they are located near the flavin adenine dinucleotide (FAD) cofactor.
Figure 8 illustrates the binding mode of DPH14 into the hMAO A binding cavity (binding energy: −6.5 kcal/mol). Visual inspection of the pose of compound DPH14 into the MAO A binding site revealed that the phenyl ring placed in the “aromatic cage” framed by Tyr407, Tyr444 side chains, as well as the isoalloxazine FAD ring and the aromatic ring is oriented to establish π–π stacking interactions with the carbonyl group of Gln215 residue. The pyridine moiety is located in a hydrophobic core delimited by residues Val93, Leu97, and Ala111. Moreover, the amino group of the propargylamine moiety is also able to form a hydrogen bond with Ser209 side chain.
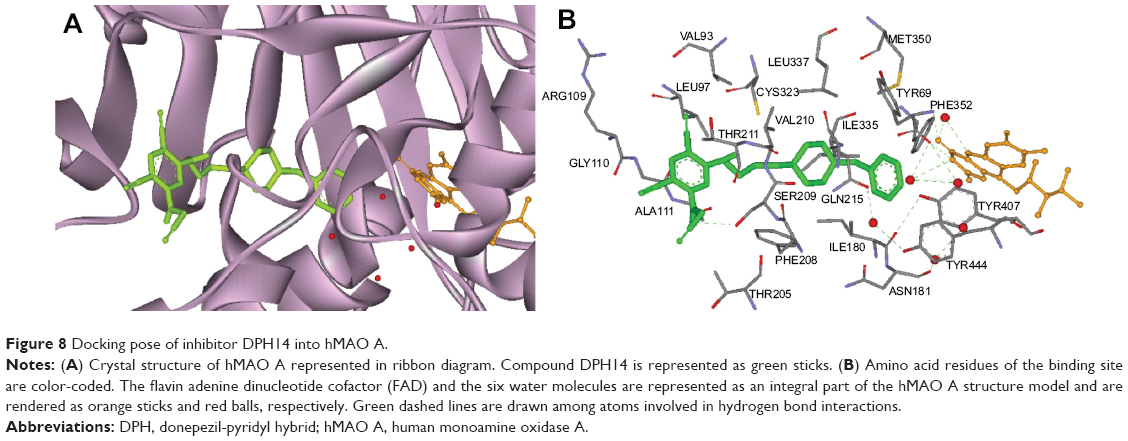 | Figure 8 Docking pose of inhibitor DPH14 into hMAO A. |
To rationalize the selectivity of MAO A/B, blind docking studies of compound DPH14 into the MAO B were done. The six structural water molecules selected for hMAO A were also included in the study.
Visual inspection of the pose of compound DPH14 into the MAO B binding site revealed that this inhibitor also crosses both cavities, presenting the piperidine nucleus located between the “entrance” and “catalytic” cavities, separated by the residues Ile199 and Tyr326 (Figure 9; binding energy: −8.3 kcal/mol). The substituted-pyridine ring is oriented toward the bottom of the substrate cavity, interacting with the FAD cofactor as well as Tyr398, Tyr435, and Gln206 through van der Waals, hydrophobic interactions and π–π interactions. In addition, the predicted orientation of the substituted-pyridine moiety allowed the interaction of the NH hydrogen of the propargylamine moiety with Cys172, showing a favorable geometry for the formation of hydrogen bond between the ligand and the receptor. Moreover, the cyano group in ortho-position with respect to the propargylamine group formed a hydrogen bond with a water molecule. Finally, the phenyl ring is oriented to an entrance cavity, a hydrophobic sub-pocket, which is defined by Pro102, Thr201, Thr314, and Ile316.
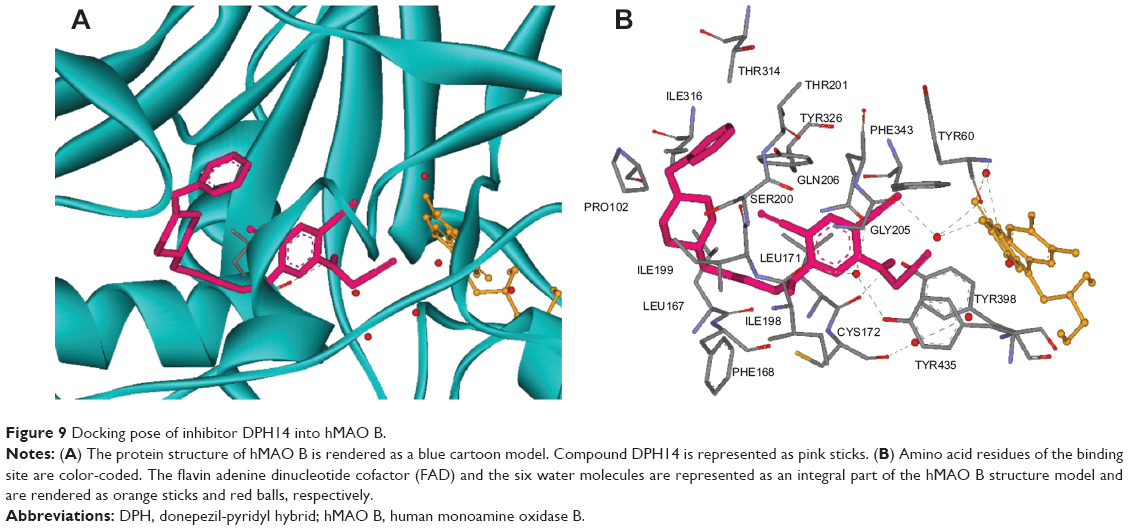 | Figure 9 Docking pose of inhibitor DPH14 into hMAO B. |
The study confirmed the selectivity of compound DPH14 for MAO B isoform. Thus, selectivity is likely due to the orientation of the pyridine and phenyl moieties of DPH14 in MAO A and in MAO B. For MAO A, the pyridine system was hosted in the entrance cavity and for MAO B this system occupied the substrate cavity. Compound DPH14 established more interactions with the MAO B active site than with the MAO A active site, indicating that DPH14 might interact more tightly with MAO B.
Absortion, distribution, metabolism, excretion and toxicity analysis
Absortion, distribution, metabolism, excretion and toxicity (ADMET; ADMET Predictor, v.6.5, Simulations Plus, Inc., Lancaster, CA, USA) analysis has been carried out following the usual methods (Supplementary material http://www.iqog.csic.es/iqog/sites/default/files/public/User/Jos%C3%A9%20Luis%20Marco%20Contelles/Supplementary%20material.pdf).44
Drugs that penetrate the central nervous system (CNS) should have lower polar surface areas than other kinds of molecules,45 namely, in the range 60–90 Å2. In our study, all the compounds present appropriate values.
The lipophilicity increases with the hydrocarbon tether chain, in such a way that DPH4, 9, and the unmethylated structures DPH11 and DPH12 show logP >5 (and/or MlogP >4.1).46 Similarly, the molecular weight of DPHs4, 9, and 12 fall outside the recommended range (molecular weight <500). Therefore, these structures violate the Lipinski’s rule of five.47 Moreover, a more rigid rule for CNS drug-like characteristics48 (molecular weight ≤450, hydrogen bond donor ≤3, hydrogen bond acceptors ≤7, logP ≤5, polar surface area ≤90, and number of rotatable bonds ≤8) is only satisfied for DPH5 and 6. DPHs13–16 also show good values despite being too flexible.
The blood–brain barrier (BBB) is a separation of circulating blood and cerebrospinal fluid in the CNS. Predicting BBB penetration means predicting whether compounds pass across the BBB. This is crucial in drug design because CNS-active compounds must pass across it and CNS-inactive compounds must not pass across it in order to avoid CNS side effects. According to the computed values, one of the models predicts that DPHs4, 6, 9–11, 13–16 should be good candidates. In particular, DPHs13–15 show a brain penetration sufficient for CNS activity.49,50
Peff quantifies permeability across the intestinal membrane and characterizes absorption whether by passive diffusion, active transport, facilitated diffusion, paracellular diffusion, or any other mechanism. According to the predictions, all the structures show an adequate permeability to be good candidates (Peff >0.1),51 and should be well absorbed compounds (% human intestinal absorption).52 Moreover, a middle Caco-2 cell permeability is suggested.53
The human Ether-à-go-go Related Gene (hERG) encodes potassium channels, which are responsible for the normal repolarization of the cardiac action potential. Blockage or any other impairment of these channels in the heart cells can lead to fatal cardiac problems. Therefore, drug-induced blockage of potassium channels has been a major concern for the pharmaceutical industry. DPHs4, 6, 9, 14 and 16 show hERG liability.54 On other hand, DPHs1–5, 9–12 could induce carcinogenicity, whereas none of the molecules is predicted to present hepatotoxicity.55
To sum up, the structures lacking phenyl substituent show better druglikeness profiles; in particular, DPHs13–15 present the more suitable ADMET properties of the DPH compounds studied here (Table S3, Supplementary material http://www.iqog.csic.es/iqog/sites/default/files/public/User/Jos%C3%A9%20Luis%20Marco%20Contelles/Supplementary%20material.pdf).
Conclusion
In this paper, the synthesis and biochemical evaluation of the designed novel DPHs is reported. Compound DPH9 was identified as a very potent hAChE inhibitor (IC50 =25±3 nM) and moderate hBuChE inhibitor (IC50 =1,190±310 nM) with low selectivity toward hMAO B. However, DPH14 was revealed to be an active hAChE (IC50 =13.1±2.1 nM) and hBuChE (IC50 =835±139 nM) inhibitor and a selective moderate irreversible hMAO B inhibitor (IC50 =3,950±940 nM). As suggested by one of the reviewers, the increased potency on BuChE inhibition might potentiate emetogenic side effects certainly, but as it is very well-known, in old AD patients, AChE levels are decreased, and BuChE activity is elevated,56 suggesting that ACh hydrolysis in AD may largely occur via BuChE catalysis.57 Consequently, increased and/or specific inhibition of BuChE is important in raising ACh levels and improving cognition.58
Taking these initial results into account, and considering that in AD brain MAO B isoform is overexpressed and contributing consequently to the oxidative stress,59 DPH14 was selected as the most promising candidate to be further studied.
The 3D-QSAR study of 37 donepezil-indolyl and DPHs was used to define 3D-pharmacophores for inhibition of MAO A/B, AChE, and BuChE enzymes and to design DPHs as novel multipotent ligands (Supplementary material http://www.iqog.csic.es/iqog/sites/default/files/public/User/Jos%C3%A9%20Luis%20Marco%20Contelles/Supplementary%20material.pdf). The 3D-QSAR study has selected structural modifications of the examined hybrids that could selectively enhance inhibiting activity on MAO A/B, AChE, and BuChE. Substitution of the terminal hydrogen of propargylamine moiety with bulky groups and replacement of the methyl substituent with hydrogen in the N-Me group of the indole moiety could selectively increase MAO A inhibiting activity, while bulky groups in meta/para positions of the benzyl moiety are able to decrease MAO A inhibiting activity of the examined hybrids. New electron-donating substituents in propargylamine or indole moiety and also bulky substituents on N-atom of the indole moiety could selectively enhance MAO B activity. Furthermore, an electron withdrawing bulky groups at meta/para positions of the benzyl moiety are able to selectively increase MAO B inhibiting activity of the examined hybrids. Extension of the N-alkyl bridge could selectively and strongly enhance AChE inhibiting activity, while BuChE inhibiting activity could be slightly increased too. Phenyl substituent in the pyridine ring is able to selectively enhance AChE inhibiting activity, while substitution in meta position of the phenyl group could result in decrease of AChE inhibiting activity.
In addition, we have found that the structures lacking phenyl substituent show better druglikeness profiles; in particular, DPH14 presents the more suitable ADMET properties of the series.
Molecular modeling of inhibitor DPH14 within EeAChE showed a binding mode with an extended conformation, interacting simultaneously with both catalytic and peripheral sites of EeAChE thanks to a linker of appropriate length. It is clear that the linear conformation allows DPH14 (Modes I and II) to span both CAS and PAS which contributes to its superior binding toward EeAChE. Similarly, visual inspection of the pose of compound DPH14 into the MAO B binding site revealed that this inhibitor also crosses both cavities, presenting the piperidine nucleus located between the entrance and catalytic cavities, separated by the residues Ile199 and Tyr326. The observed MAO A/B selectivity is likely due to the orientation of the pyridine and phenyl moieties of DPH14 in MAO A and in MAO B. For MAO A, the pyridine system was hosted in the entrance cavity and for MAO B this system occupied the substrate cavity. Compound DPH14 established more interactions with the MAO B active site compared to the MAO A active site, which may indicate that DPH14 interacts more tightly with MAO B.
To sum up, donepezil-pyridyl hybrid DPH14 is a promising new multipotent molecule for the potential prevention and treatment of AD.
Acknowledgments
This work was supported by the European Cooperation in Science and Technology (COST) action CM1103. KN and DA acknowledge project supported by the Ministry of Education and Science of the Republic of Serbia, Contract 172033. JMC and MU thank Ministerio de Economía y Competitividad (MINECO) (Spain) for support (SAF2009-07271; SAF2012-33304). OMB-A thanks MINECO (Spain) for an Formación del Personal Investigador (FPI) fellowship.
Disclosure
The authors report no conflicts of interest in this work.
References
Goedert M, Spillantini MG. A Century of Alzheimer’s disease. Science. 2006;314(5800):777-781. | ||
Kung HF. The β-amyloid hypothesis in Alzheimer’s disease: seeing is believing. ACS Med Chem Lett. 2012;3(4):265-267. | ||
Ballatore C, Lee VM, Trojanowski JQ. Tau-mediated neurodegeneration in Alzheimer’s disease and related disorders. Nat Rev Neurosci. 2007; 8(9):663–672. | ||
Gella A, Durany N. Oxidative stress in Alzheimer disease. Cell Adh Migr. 2009;3(1):88-93. | ||
Jakob-Roetne R, Jacobsen H. Alzheimer’s disease: from pathology to therapeutic approaches. Angew Chem Int Ed Engl. 2009;48(17): 3030–3059. | ||
Sugimoto H, Yamanishi Y, Iimura Y, Kawakami Y. Donepezil hydrochloride (E2020) and other acetylcholinesterase inhibitors. Curr Med Chem. 2000;7(3):303–339. | ||
Marco-Contelles J, do Carmo Carreiras M, Rodríguez C, Villarroya M, Garcia AG. Synthesis and pharmacology of galantamine. Chem Rev. 2006;106(1):116–133. | ||
Anand P, Singh B. A review on cholinesterase inhibitors for Alzheimer’s disease. Arch Pharm Res. 2013;36(4):375–399. | ||
Barbanti P, Fabbrini G, Ricci A, et al. Reduced density of dopamine D2-like receptors on peripheral blood lymphocytes in Alzheimer’s disease. Mech Ageing Dev. 2000;120(1–3):65–75. | ||
Thompson AJ, Lummis SC. The 5-HT3 receptor as a therapeutic target. Expert Opin Ther Targets. 2007;11(4):527–540. | ||
Shih JC, Chen K, Ridd MJ. Monoamine oxidase: from genes to behavior. Annu Rev Neurosci. 1999;22:197–217. | ||
Riederer P, Danielczyk W, Grünblatt E. Monoamine oxidase-B inhibition in Alzheimer’s disease. Neurotoxicology. 2004;25(1–2):271–277. | ||
Pizzinat N, Copin N, Vindis C, Parini A, Cambon C. Reactive oxygen species production by monoamine oxidases in intact cells. Naunyn Schmiedebergs Arch Pharmacol. 1999;359(5):428-431. | ||
Kristal BS, Conway AD, Brown AM, et al. Selective dopaminergic vulnerability: 3,4-dihydroxyphenylacetaldehyde targets mitochondria. Free Radical Biol Med. 2001;30(8):924-931. | ||
Sterling J, Herzig Y, Goren T, et al. Novel dual inhibitors of AChE and MAO derived from hydroxy aminoindan and phenethylamine as potential treatment for Alzheimer’s disease. J Med Chem. 2002;45(24): 5260–5279. | ||
Yogev-Falach M, Bar-Am O, Amit T, Weinreb O, Youdim MB. A multifunctional, neuroprotective drug, ladostigil (TV3326), regulates holo-APP translation and processing. FASEB J. 2006;20(12):2177-2179. | ||
Youdim MB, Weinstock M. Molecular basis of neuroprotective activities of rasagiline and the anti-Alzheimer drug TV3326 [lpar;N-Propargyl-(3R) aminoindan-5-YL-ethyl methyl carbamate]. Cell Mol Neurobiol. 2002;21(6):555-573. | ||
Bar-Am O, Amit T, Weinreb O, Youdim MB, Mandel S. Propargylamine containing Compounds as modulators of proteolytic cleavage of amyloid protein precursor: involvement of MAPK and PKC activation. J Alzheimer’s Dis. 2010;21(2):361-371. | ||
Bruehlmann C, Ooms F, Carrupt PA, et al. Coumarins derivatives as dual inhibitors of acetylcholinesterase and monoamine oxidase. J Med Chem. 2001;44(19):3195–3198. | ||
Zheng H, Fridkin M, Youdim MB. Novel chelators targeting cell cycle arrest, acetylcholinesterase, and monoamine oxidase for Alzheimer’s therapy.Curr Drug Targets. 2012;13(8):1096–1113. | ||
Samadi A, Marco-Contelles J, Soriano E, et al. Multipotent drugs with cholinergic and neuroprotective properties for the treatment of Alzheimer and neuronal vascular diseases. I. Synthesis, biological assessment, and molecular modeling of simple and readily available 2-aminopyridine-, and 2-chloropyridine-3,5-dicarbonitriles. Bioorg Med Chem. 2010;18(16):5861–5872. | ||
Bolea I, Juárez-Jiménez J, de los Ríos C, et al. Synthesis, biological evaluation, and molecular modeling of donepezil and N-[(5-(benzyloxy)-1-methyl-1H-indol-2-yl)methyl]-N-methylprop-2-yn-1-amine hybrids as new multipotent cholinesterase/monoamine oxidase inhibitors for the treatment of Alzheimer’s disease. J Med Chem. 2011;54(24): 8251–8270. | ||
Pérez V, Marco JL, Fernández-Álvarez E, Unzeta M. Relevance of benzyloxy group in 2-indolyl methylamines in the selective MAO-B inhibition. Brit J Pharmacol. 1999;127(4):869–876. | ||
Samadi A, Chioua M, Bolea I, et al. Synthesis, biological assessment and molecular modeling of new multipotent MAO and cholinesterase inhibitors as potential drugs for the treatment of Alzheimer’s disease. Eur J Med Chem. 2011;46(9):4665–4668. | ||
Peinador C, Veiga MC, Vilar J, Quintela. A synthesis of heterocyclic ring systems. Pyrido[3′,2′:4,5]thieno[2,3-b]pyrrolizine and pyrido[6′,5′:4,5][3′,2′:4,5]dithieno[2,3-b‘:2,3-b]dipyrrolizine. Heterocycles. 1994;38(6):1299–1305. | ||
Vilarelle DV, Peinador C, Quintela JM. Synthesis of pyrido and pyrazinodithienodipyrimidine-4,8(3H,9H)-dione derivatives by the aza-Wittig methodology. Tetrahedron. 2004;60(2):275–283. | ||
Murray TJ, Zimmerman SC, Kolotuchin SV. Synthesis of heterocyclic compounds containing three contiguous hydrogen bonding sites in all possible arrangements. Tetrahedron. 1995;51(2):635–648. | ||
Piper JR, McCaleb GS, Montgomery JA, Kisliuk RL, Gaumont Y, Sirotnak FM. Syntheses and antifolate activity of 5-methyl-5-deaza analogs of aminopterin, methotrexate, folic acid, and N10-methylfolic acid. J Med Chem. 1986;29(6):1080–1087. | ||
Samadi A, Estrada M, Pérez C, et al. Pyridonepezils, new dual AChE inhibitors as potential drugs for the treatment of Alzheimer’s disease: Synthesis, biological assessment, and molecular modelling. Eur J Med Chem. 2012;57:296–301. | ||
Atsushi K, Mitsuru O, Hisashi T; inventors. Fujisawa Pharmaceutical Co.; applicant. Oxadiazole derivatives having acetylcholinesterase-inhibitory and muscarinic agonist activity. Patent publication number WO9313083A1. 1993 Jul 8. | ||
Contreras JM, Rival YM, Chayer S, Bourguignon JJ, Wermuth CG. Aminopyridazines as acetylcholinesterase inhibitors. J Med Chem. 1999;42(4):730–741. | ||
Ellman GL, Courtney KD, Andres V Jr, Feather-stone RM. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem Pharmacol. 1961;7(2):88–95. | ||
Zhou M, Panchuk-Valashina N. A one-step fluorometric method for the continuous measurement of monoamine oxidase activity. Anal Biochem. 1997;253(2):169–174. | ||
León R, Marco-Contelles J. A step further towards multitarget drugs for Alzheimer and neuronal vascular diseases: targeting the cholinergic system, amyloid-β aggregation and Ca2+ dyshomeostasis.Curr Med Chem. 2011;18(4):552–576. | ||
Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31(2):455–461. | ||
Arnold K, Bordoli L, Kopp J, Schwede T. The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics. 2006;22(2):195–201. | ||
Kiefer F, Arnold K, Künzli M, Bordoli L, Schwede T. The SWISS-MODEL Repository and associated resources. Nucl Acids Res. 2009; 37(Database issue):D387–D392. | ||
Guex N, Peitsch MC, Schwede T. Automated comparative protein structure modeling with SWISS-MODEL and Swiss-PdbViewer: a historical perspective. Electrophoresis. 2009;30(Suppl 1):S162–S173. | ||
Edmonson DE, Mattevi A, Binda C, Li M, Hubalek F. Structure and mechanism of Monoamine Oxidase. Curr Med Chem. 2004;11(15): 1983–1993. | ||
Jones TZ, Balsa D, Unzeta M, Ramsay RR. Variations in activity and inhibition with pH: the protonated amine is the substrate for monoamine oxidase, but uncharged inhibitors bind better. J Neural Transm. 2007; 114(6):707–712. | ||
Wang J, Edmonson DE. 2H kinetic isotope effects and pH dependence of catalysis as mechanistic probes of rat Monoamine Oxidase A: comparisons with the human enzyme. Biochemistry. 2011;50(35):7710–7717. | ||
Scerrer RA, Leo AJ. Multi-pH QSAR: a method to differentiate the activity of neutral and ionized species and obtain true correlations when both species are involved. Molecular Informatics. 2010;29(10): 687–693. | ||
Samadi A, de los Ríos C, Bolea I, et al. Multipotent MAO and cholinesterase inhibitors for the treatment of Alzheimer’s disease: synthesis, pharmacological analysis and molecular modeling of heterocyclic substituted alkyl and cycloalkyl propargyl amine. Eur J Med Chem. 2012;52:251–262. | ||
ACD/Percepta 14.0.0, Advanced Chemistry Development, 2013. | ||
Kragh-Hansen U. Molecular aspects of ligand binding to serum albumin. Pharmacol Rev. 1981;33:17–53. | ||
Moriguchi I, Hirono S, Liu Q, Nakagome I, Matsushita Y. Simple method of calculating octanol/water partition coefficient. Chem Pharm Bull. 1992;40(1):127–130. | ||
Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 2001; 46(1–3):3–26. | ||
Pajouhesh H, Lenz GR. Medicinal chemical properties of successful central nervous system drugs. NeuroRx. 2005;2(4):541–553. | ||
Ma XL, Chen C, Yang Y. Predictive model of blood-brain barrier penetration of organic compounds. Acta Pharmacol Sin. 2005;26(4): 500–512. | ||
Cheng F, Li W, Zhou Y, et al. admetSAR: a comprehensive source and free tool for assessment of chemical ADMET properties. J Chem Inf Model. 2012;52(11):3099–3105. | ||
Lennernas H. Human intestinal permeability. J Pharm Sci. 1998;87(4): 403–410. | ||
Yee S. In vitro permeability across caco-2 cells (Colonic) can predict in vivo (small intestinal) absorption in man – Fact or myth. Pharm Res. 1997;14(6):763–766. | ||
Yamashita S, Furubayashi T, Kataoka M, Sakane T, Sezaki H, Tokuda H. Optimized conditions for prediction of intestinal drug permeability using Caco-2 cells. Eur J Pharm Sci. 2000;10(3):195–204. | ||
Gold LS, Manley NB, Slone TH, Rohrbach L. Supplement to the Carcinogenic Potency Database (CPDB): results of animal bioassays published in the general literature in 1993 to 1994 and by the National Toxicology Program in 1995 to 1996. Enviro Health Perspect. 1999; 107(Suppl 4):527–600. | ||
Matthews EJ, Kruhlak NL, Benz RD, Contrera JF. Assessment of the health effects of chemicals in humans: I. QSAR estimation of the maximum recommended therapeutic dose (MRTD) and no effect level (NOEL) of organic chemicals based on clinical trial aata1. Curr Drug Discov Technol. 2004;1(1):61–76. | ||
Arendt T, Bruckner MK, Lange M, Bigl V. Changes in acetylcholinesterase and butyrylcholinesterase in Alzheimer’s disease resemble embryonic development. A study of molecular forms. Neurochem Int. 1992;21(3):381–396. | ||
Darvesh S, Hopkins DA, Geula C. Neurobiology of butyrylcholinesterase. Nat Rev Neurosci. 2003;4(2):131–138. | ||
Greig NH, Utsuki T, Ingram DK, et al. Selective butyrylcholinesterase inhibition elevates brain acetylcholine, augments learning and lowers Alzheimer β-amyloid peptide in rodent. Proc Natl Acad Sci U S A. 2005;102(47):17213–17218. | ||
Saura J, Bleuel Z, Ultrich J, et al. Molecular neuroanatomy of human monoamine oxidases A and B revealed by quantitative enzyme radioautography and in situ hybridization histochemistry. Neuroscience. 1996;70(3):755–774. |
Supplementary material
Available from: http://www.iqog.csic.es/iqog/sites/default/files/public/User/Jos%C3%A9%20Luis%20Marco%20Contelles/Supplementary%20material.pdf
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.