")
Back to Archived Journals » Antibody Technology Journal » Volume 4
Monoclonal antibodies for the prevention of rabies: theory and clinical practice
Authors Nagarajan T, Marissen W, Rupprecht C
Received 20 August 2013
Accepted for publication 1 October 2013
Published 20 January 2014 Volume 2014:4 Pages 1—12
DOI https://doi.org/10.2147/ANTI.S33533
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Thirumeni Nagarajan,1 Wilfred E Marissen,2 Charles E Rupprecht3,4
1Biological E. Limited, Shameerpet, Hyderabad, Andhra Pradesh, India; 2Crucell Holland BV, Leiden, the Netherlands; 3Ross University School of Veterinary Medicine, Biomedical Sciences, Basseterre, St. Kitts, West Indies; 4The Global Alliance For Rabies Control, Manhattan, KS, USA
Abstract: Monoclonal antibodies (MAbs) have become a unique and attractive class of biologics, possessing several desirable characteristics for use in human medicine. Anti-infective MAbs for several medically important viral agents, including rabies virus (RABV), have been developed and are currently at different stages of clinical development. Rabies is a vaccine-preventable but neglected zoonosis. After severe bite exposures, prompt administration of a combination of potent rabies vaccine and rabies immunoglobulin (RIG) is recommended. Due in part to cost, equine RIG has been largely used instead of human RIG, especially in the developing world. With an estimated 10 million RABV exposures annually, the use of MAbs has emerged in concept as a potential alternative to polyclonal RIG for future prophylaxis needs. Murine MAbs, although efficacious, are less attractive because of immunogenicity. However, human MAbs seem to have the potential to replace polyclonal RIG because they possess all the desirable characteristics for an intended biologic. The exquisite specificity of the MAbs for a single epitope is generally believed to result in narrow spectrum of RABV neutralization and perceived generation of escape mutants. These issues can be mitigated by formulating a cocktail of candidate MAbs that are directed against distinct, nonoverlapping epitopes. Expression of recombinant human MAbs in mammalian cell lines, such as Chinese hamster ovary and human retinal, is central to the economical production at an industrial scale. Thus far, human MAbs developed by two companies have successfully passed through Phase I or II clinical trials in countries such as the US and India.
Keywords: rabies, postexposure prophylaxis, polyclonal RIG, monoclonal antibody, rabies immunoglobulin, clinical trials
Introduction
Monoclonal antibodies (MAbs) are versatile molecules with an undisputed usefulness for biomedical applications. Besides their utility in diagnostic applications, MAbs are also attractive as therapeutic candidates because of their stability, tolerance, functionality, and amenability for engineering to enhance various desirable characteristics, such as reduced immunogenicity, longer half-lives, higher affinity, and better effector functions. Over the past decade, MAbs have become a thriving class of biologically active molecules for therapeutics.1 Nearly one in five biotherapeutic molecules belongs to this category. Thus far, more than 25 antibodies have been licensed for human use (Table 1).2–12 Nearly 200 other candidates are in different stages of development (Table 2). Though MAbs are indicated mainly for noninfectious diseases such as cancer, inflammatory conditions, and autoimmune disorders, they have also been investigated for their potential as anti-infective agents, although with limited success to date. For example, of 46 anti-infective MAbs tested clinically, only one, palivizumab, was approved by the US Food and Drug Administration as a prophylaxis for respiratory syncytial virus infection in high-risk pediatric patients. Of course, ample medical needs exist for the development of safe and efficacious targeted anti-infective MAbs that would complement the current arsenal of vaccines and anti-infective drugs.1 Moreover, there is a renewed interest in the development of antiviral MAbs because of potential safety issues and supply limitations associated with the use of polyclonal antibodies derived from human plasma. For the purpose of this review, our discussion is concentrated mainly upon the topic of antiviral MAbs intended for the prevention of rabies in humans.
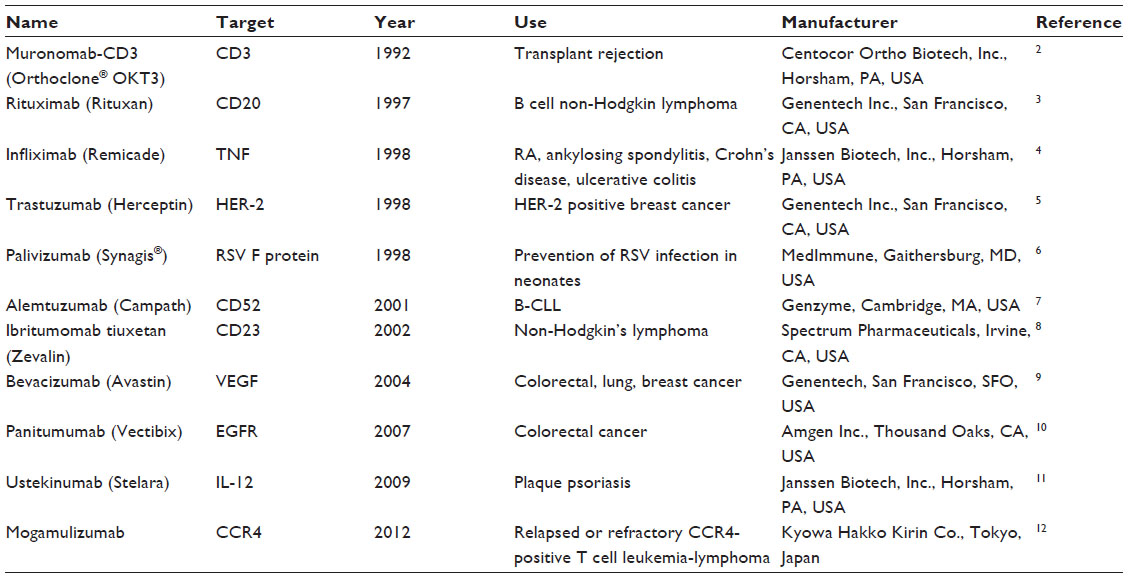 | Table 1 Selected examples of licensed therapeutic monoclonal antibodies |
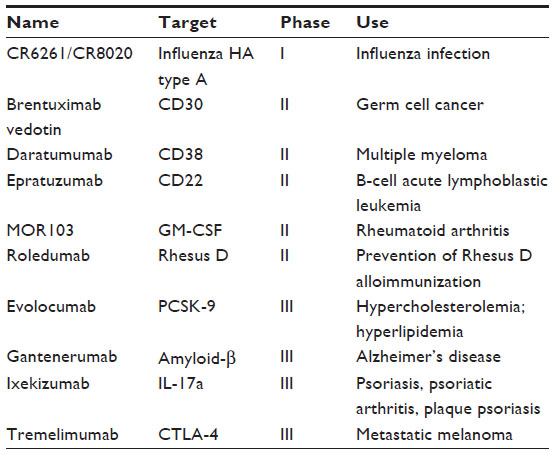 | Table 2 Selected examples of therapeutic monoclonal antibodies in clinical development |
Postexposure prophylaxis in the prevention of human rabies
Rabies is one of the most feared zoonotic diseases because it has the highest human case–fatality proportion of all conventional infectious diseases.13 This neglected disease is caused by several ribonucleic acid viruses in the family Rhabdoviridae, genus Lyssavirus. Although all lyssaviruses cause rabies, the most significant member of the genus is rabies virus (RABV). Only a few cases of survival have been documented, and this acute progressive encephalitis is considered incurable.14,15 Globally, rabies occurs in more than 150 countries and territories. More than 3 billion people live in areas in which the disease is enzootic (Figure 1).16 Worldwide, millions of exposures are registered, resulting in tens of thousands of human deaths, with most occurring in Asia and Africa, despite the availability of safe and efficacious biologics. Of this burden, 30%–60% of the victims of dog bites are children under the age of 15 years.17 Historically, vaccination forms the cornerstone of a multipronged approach to rabies prevention. Interestingly, rabies differs from most other vaccine-preventable infectious diseases, because modern intervention permits prevention both before exposure (pre-exposure prophylaxis) and after exposure (postexposure prophylaxis [PEP]). Vaccination involves the use of inactivated rabies vaccines derived either from cultured cells (human diploid cells, primary chick embryo cells, and Vero cells) or avian embryo cells (duck). Annually, more than 15 million people worldwide are estimated to receive PEP to prevent the disease, which is estimated to prevent hundreds of thousands of rabies deaths.18 Based on the types of interaction with suspected rapid animals, exposure is broadly classified into three categories viz. I, II, and III (Table 3).19 Used promptly and appropriately, modern rabies vaccines are highly effective in the rapid induction of virus-neutralizing antibodies. Understandably, vaccination alone is inadequate to protect rabies victims from all animal bite contacts, especially associated with multiple and severe bites.20 Based upon viral dose and route, even accelerated vaccination schedules may be inadequate after severe exposures.21 Today, most people die of rabies because of a lack of proper education about the disease and access to affordable, lifesaving biologics. Occasional PEP failures may occur, in part due to lack of timely administration of RIG, in concert with vaccination.22,23 When PEP is administered in a timely and appropriate manner, RABV may be cleared before a productive infection of the central nervous system manifests.24 Although protective immunity against lyssaviruses is deemed to be complex and due to a suite of factors, humoral immunity directed against the outer viral G is felt to be paramount. The mode of protection is likely to be a combination of local virus neutralization by antibodies or via antibody-mediated clearance of virus-infected cells.25 Because the bite of a rabid or suspected rabid animal is presumed to have virus excreted in the saliva, PEP includes immediate local treatment of all bite wounds and scratches with thorough washing and disinfection, local wound infiltration with RIG, and vaccination. This combination of active and passive immunization is considered the status quo for PEP, except for those persons who have been previously immunized with a rabies vaccine via a recognized schedule or a documented adequate RABV antibody titre.26 The aim of passive immunization using RIG is to confer short-lived immunity characterized by rapid onset and lack of immunological memory, while the goal of active immunization using rabies vaccine is to elicit durable immunity characterized by delayed onset and immunological memory.27 Understandably, the antibodies administered passively compensate for the time necessary for vaccine-induced antibodies to appear. Hence, the RIG should be given usually through the seventh day after the first dose of vaccine is administered. However, beyond the seventh day, RIG is not indicated, as an active antibody response to cell culture rabies vaccine is presumed to have occurred.28 Although the combination of rabies vaccine and RIG is nearly 100% effective in prevention before illness, attempts to use rabies vaccine or RIG after the onset of symptomatic rabies have not been proven beneficial.29
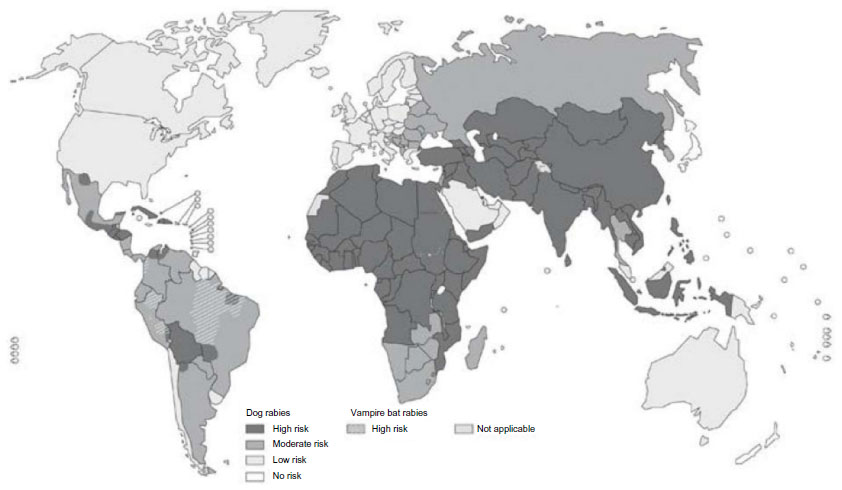 | Figure 1 Global prevalence of rabies. Four categories of countries or areas, from those at no risk to those at low, moderate and high risk. |
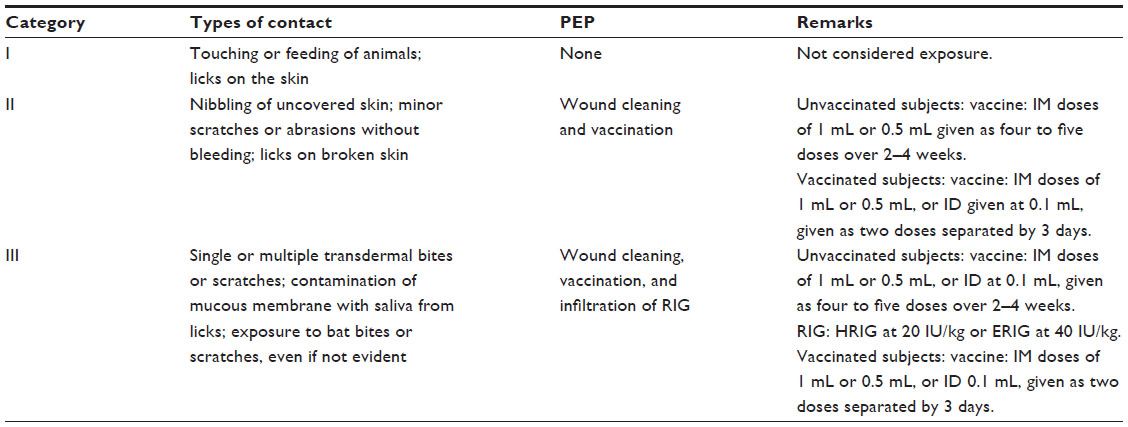 | Table 3 Categories of rabies contacts for consideration of prophylaxis |
Rabies immunoglobulin – polyclonal
Historically, polyclonal RIG has been used for PEP of human rabies since the mid-20th century. The polyclonal RIG is relatively easy to produce, possesses high potency, a broad spectrum of virus-neutralizing activity, polyspecificity that prevents the selection of neutralization escape mutants, and multiple effector functions, mediated by several isotypes, due to its heterogeneous nature.30 Typically, manufacture involves purification of immunoglobulin (Ig)G from the plasma of immune human or animal donors, such as horses (equine rabies immunoglobulin [ERIG]). Both ERIG and human rabies immunoglobulin (HRIG) are currently licensed for use in humans and widely used in developing and developed countries, respectively. The ERIG is a heterologous molecule and highly immunogenic in humans, resulting in induction of human antiequine antibody responses, leading to rapid clearance of ERIG, necessitating higher dosing and culminating in induction of severe type III hypersensitivity reactions and serum sickness, which is sometimes fatal.31 Consequently, often, physicians are hesitant to use ERIG, thus providing incomplete PEP, which may result in failures.32 These adverse events are essentially due to the Fc region of the Ig, and hence ERIG devoid of the Fc region: ie, F(ab’)2, which is obtained by pepsin digestion of IgG, is a format currently in use.33 The F(ab’)2 of ERIG per se retains effectiveness because of its ability to bind to the target mediated by Fab region, which is a prerequisite for neutralization and elimination of the pathogen. Nevertheless, the ERIG is known to pose some risks to humans, which may be mitigated to some extent by improved purification processes.33 Medical concerns may be managed by performing skin tests or by premedication with antihistamine and corticosteroids.34,35 Compared with HRIG, the less expensive nature of ERIG seems to be the primary reason for its continued use in developing countries. All licensed RIGs are expected to neutralize all known RABV variants, but no available product will neutralize all described lyssaviruses.36
The quest for an improved alternative to heterologous animal serum products resulted in the initial development of HRIG, which is its homologous equivalent. Modern HRIG is safe, nonimmunogenic, and well tolerated by humans. The incidence of anaphylaxis or serum sickness is virtually unknown.37 However, as a human plasma product, it has the potential for transmission of blood-borne infectious agents, which can be mitigated by treatment with solvents or detergents38 or heat treatment.39 Such processes are expensive and not generally affordable in developing countries, where canine rabies remains the primary public health problem.40 The worldwide inaccessibility of HRIG and its high cost of production place it out of reach of most patients in the developing world.31 Nevertheless, HRIG has largely replaced ERIG in several countries, and future animal welfare concerns may further limit the availability of ERIG. With the idea of exploiting certain desirable qualities of polyclonal RIG and to overcome the limitations of existing ERIG and HRIG in parallel, RIG production has been reported originating from other species, including chickens,41 rabbits,42 and sheep.43 It remains to be seen whether polyclonal RIG from these species would be viable for clinical use. Regardless of the species of origin, polyclonal RIG in general has certain constraints, including the need for donor recruitment and immunization; multiple inoculations and bleeding procedures; donor retention; ethical problems; lower specific activity, which may necessitate the use of more protein, which may lead to higher viscosity and adverse events; variable batch-to-batch consistency; supply limitations from competing use of plasma products; and the potential risk of transmission of infectious agents. For such economic, supply, and safety reasons, replacement of HRIG and ERIG is advocated, and the World Health Organization strongly encourages development of alternative products.44
Alternatives to polyclonal immunoglobulins
The invention of the concept of MAb technology by Kohler and Milstein45 in 1975 revolutionized biomedicine and has become a billion-dollar industry. For the first time, researchers and clinicians were able to replicate and harness the therapeutic power of single antibodies created by the immune system.46 Clearly, MAbs have contributed immensely to the fields of basic research and disease diagnosis. The versatility of MAbs for in vitro applications prompted scientists to explore their usefulness for therapeutic applications, which resulted in the launch of the first US Food and Drug Administration-approved mouse MAb (Orthomab OKT3) for treating acute organ transplantation complications.47 The use of hybridomas in MAb production enables a sustained production of antibody and is not dependent on the life of the donor host as with polyclonal antibody production.48 The rationale behind using MAbs for therapy is that they provide a more potent product with better activity than their polyclonal counterparts.49 Additionally, they do not seem to have the inherent variability with regards to epitope and isotype,30 and are homogeneous in nature and hence exhibit relatively low lot-to-lot variability. Significantly, the duration of action of MAbs is predictable and likely to be related to the biological half-life.50 High specific activity of MAbs essentially makes administration of a low amount of protein and volume possible, which per se avoids several adverse events, including the concern for compartment syndrome. Although MAbs have significant promise as therapeutic agents, they are not without limitations: eg, 1) they may be expensive; 2) by definition, they target a single epitope and hence provide one type of effector function corresponding to their isotype; 3) although the specificity of MAbs is a strength, a pathogen that possesses rapid antigenic variation poses a significant hurdle for broader MAb development; and 4) they may select for neutralization escape variants as a result of microbial mutation or microevolution.51 The use of MAbs that target conserved areas of viral particles, or a cocktail of MAbs that target various epitopes, can obviate this concern.30,52 In fact, the World Health Organization has advocated the use of antirabies MAb cocktails for rabies PEP and does not recommend the use of single MAbs, due to the potential of viral escape.44 However, this approach of using a cocktail of MAbs would also have the drawback of increasing the cost of production and the complexity of regulatory issues involving their efficacy and safety (Figure 2).53
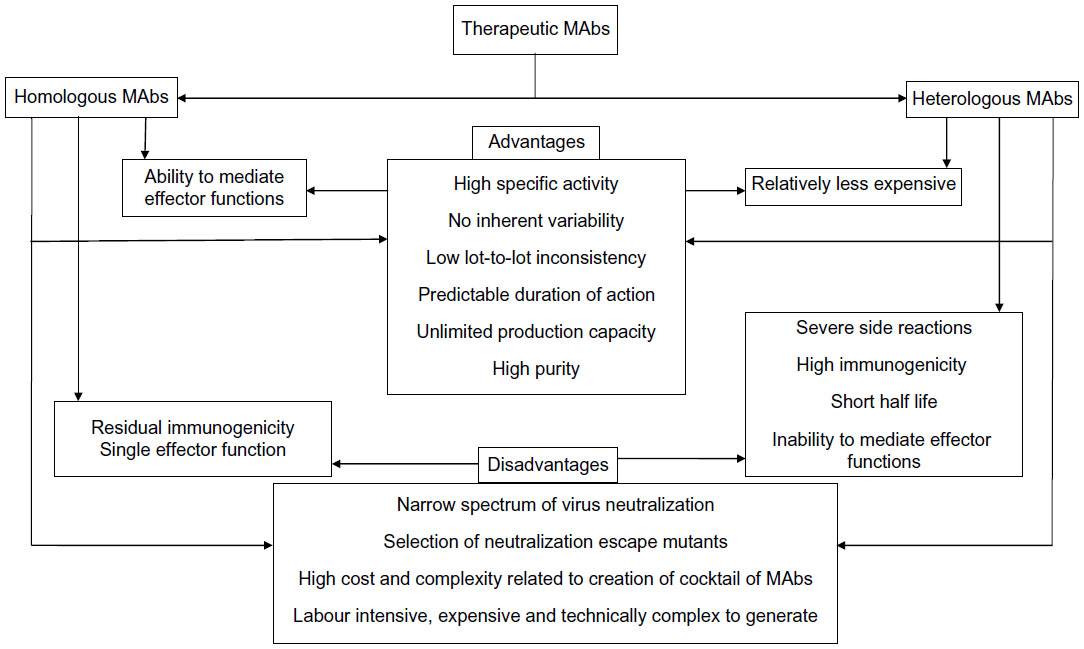 | Figure 2 Advantages and disadvantages of therapeutic monoclonal antibodies (MAbs). |
Native – mouse MAbs – full length
During the last 3 decades, numerous murine MAbs against the RABV G that neutralize RABV and other lyssaviruses, both in vitro and in vivo, have been developed and extensively characterized by several groups worldwide.54–62 They are specific to one of five distinct antigenic sites on the RABV G (antigenic sites I, II, III, IV, and minor site a),56,61,63,64 with the vast majority of them recognizing either antigenic site II or III (Figure 3).65 Antigenic site II is discontinuous and conformation dependent,59 whereas antigenic site III is predicted to be continuous and conformation dependent.61 No single MAb will neutralize all known RABV variants,67 so MAbs can be broadly neutralizing only when used in combination,60,68,69 which is not the case when used alone, because the G is prone to a high level of diversity in nature. Besides neutralization activity in cell culture, MAbs directed against the RABV G have also been shown to protect Syrian hamsters from lethal challenge.32,56,60 Cocktails of mouse MAbs have been envisioned to be less expensive alternatives to polyclonal RIG for PEP to prevent rabies in humans, because they performed as well as HRIG in animal models (Table 4).70,71 Initially, the first generation of mouse-derived MAbs suffered side effects due to an unwanted immune reaction in humans, referred to as a “human antimouse antibody” response.72,73 This response, characterized by fever, chills, arthralgia, and life-threatening anaphylaxis, was similar to the serum sickness observed several decades earlier with animal antisera.46 Instability of some murine hybridomas, immunogenicity, potential short half-life, and contamination with potential pathogen of murine MAbs pose both scalability and safety risks that essentially constrain their use in human therapy. The full promise of murine MAbs could be realized by engineering to “humanize” murine antibodies to make them less immunogenic and safer.46 To this end, a chimeric mouse–human version of MAb 62-71-3 was expressed in tobacco leaves and found to be an appropriate candidate MAb in the making of a novel antibody cocktail.74
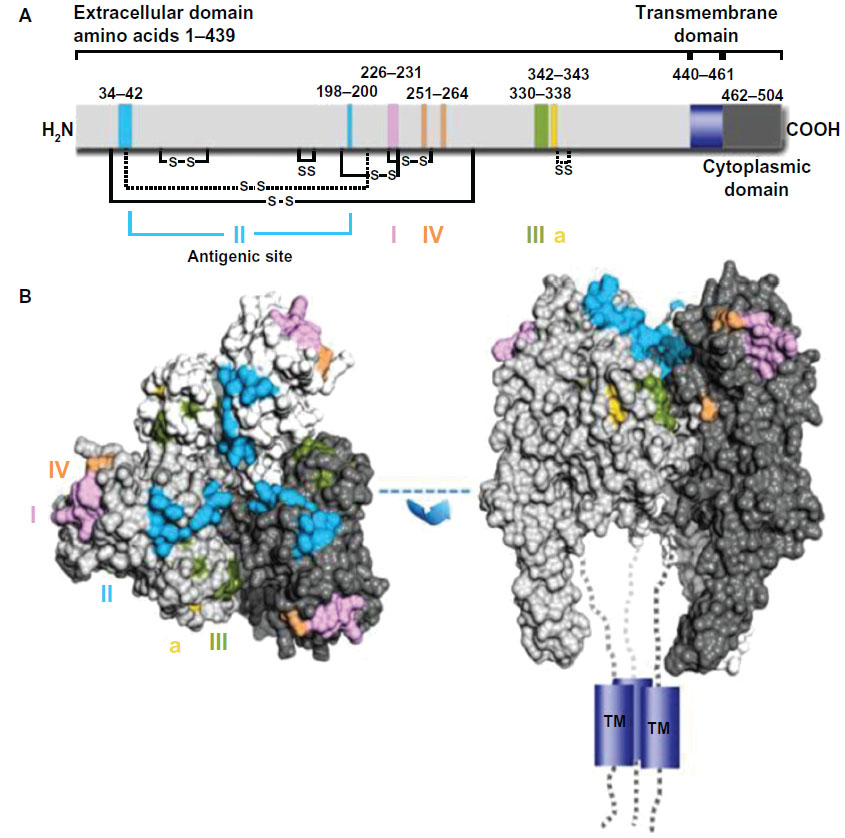 | Figure 3 Representation of antigenic sites on the mature rabies virus glycoprotein G. |
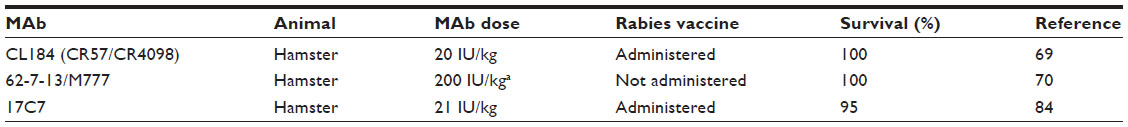 | Table 4 In vivo efficacy data of antirabies monoclonal antibodies (MAbs) in simulated postexposure prophylaxis models |
Native – human MAbs – full length
Human MAbs are nonimmunogenic molecules that essentially retain all the desirable qualities of murine MAbs. They are ideally suited for prophylaxis because they undergo affinity maturation in vivo and represent the natural Ig repertoire.75 The development of human MAbs by hybridoma technology was difficult at first due to poor accessibility to primed human B cells and a lack of ideal myeloma fusion partners.76 However, human B-cell immortalization could be accomplished by employing myeloma or heteromyeloma fusion partners and Epstein–Barr virus (EBV), although with varying levels of efficiency and clonal instability.77 Interestingly transgenic mice expressing a human antibody repertoire allowed the creation of human MAbs through the development of murine hybridomas in a relatively simpler manner.78 Prior work showed that EBV-transformed cell lines often secrete low amounts of antibodies that are of the IgM class, and that human MAbs so derived may not be suitable for biomedical applications due to the presence of EBV antigens.79 This problem could be overcome by stable and high-efficiency expression of recombinant human MAbs in heterologous or homologous systems such as Chinese hamster ovary (CHO) and human retinal (PER.C6) cell lines, respectively.80,81
Several RABV-neutralizing human MAbs were established by using the human x mouse heterohybridoma method,64,79,82 EBV transformation,65,80 phage display technology,83 and transgenic mouse technology.84 Two human MAbs viz CR57 (antigenic site I) and CR4098 (antigenic site III) recognize nonoverlapping, noncompeting epitopes, with broader neutralization of street RABV isolates when used as a combination, displaying in vitro neutralization of all RABV tested so far and demonstrating an efficacy profile similar to HRIG in animal studies (Table 4).69,83,85 A single antigenic site III specific human MAb RAB1 derived from transgenic mouse (Medarex, Inc, Princeton, NJ, USA) has been shown to efficiently neutralize many currently identified RABV isolates except a fixed RABV: ie, CVS-11, and a bat RABV from Peru in vitro,67 and protect Syrian hamsters from lethal challenge (Table 4).84 Similarly, an antigenic site II specific human MAb No 254 created through EBV transformation of human B cells exhibited a broad spectrum of RABV neutralization and has been shown to be as effective as ERIG in an experimental animal model.80 A human MAb will be of value to the majority of people only if it can be produced in large amounts for a cost comparable with the cost of current ERIG but lower than HRIG products. Initial industrial-level scale-up of heterohybridoma cell lines may not be cost-effective because of instability and low levels of antibody production.79
Homologous products (recombinant) – fully human antibody fragments
An early focus of antibody engineering was on reducing the immunogenicity of rodent antibodies via chimerization and humanization. This focus later expanded to include engineering for enhancing several other desirable traits.86 Phage and ribosomal display technologies for discovery and selection of antibodies in vitro are less time-consuming and highly suitable. The antibodies are usually displayed as Fab (VH–CH, VL–CL)87 and scFv (VH–VL) fragments.88 There are several reports on isolation of human scFvs specific for RABV G by phage display89 and ribosome display,90 and Fabs by phage display91 and conversion of scFv.92 The scFvs are relatively less stable and low in half-life than the Fab, due to their lower molecular weight.93 A Fab displayed better tissue penetration and efficacy sufficient to replace HRIG when administered with nanoparticles (Fab094–CPNPs).91 Nevertheless, antibody fragments may hold promise as a potential alternative to full-length human MAbs due to their simpler production process and lower production costs.94 However, they are not as ideal as HRIG when it comes to mediation of viral clearance through antibody-dependent cellular cytotoxicity and will have much lower half-life compared with full antibodies. Presumably, the antibody fragments may be suitable for PEP if they are appropriately engineered. To this end, an interesting derivative of scFv, an scFv–Fc (scFv–hinge–CH2–CH3) fusion protein expressed in yeast, was found to mediate RABV neutralization95 and effector functions.96
Homologous products (recombinant) – fully human intact IgG
Important parameters that drive selection of the most optimal systems for expression of antibodies include basic structure, protein folding and glycosylation,97 productivity, ease of purification,98 product quality and quantity, safety issues, time to clinic and market, and economic considerations such as cost of goods and regulatory issues.99 In general, eukaryotic systems such as mammalian cells,99,100 insect cells,97 yeast,101 plants,74,102 green algae,103 and transgenic animals104 hold the greatest promise for expressing recombinant human MAbs. However, mammalian cells are predominantly used because they can correctly fold, assemble, glycosylate, and secrete antibodies.105 It is possible to achieve yields in excess of 3 g/L using optimized cell culture processes. However, development of a stable and high-producing cell line is quite challenging and is a critical step in the development of biotherapeutics.106
Several mammalian cells, namely CHO, mouse myeloma (NSO, Sp2/0), baby hamster kidney (BHK-21), human embryonic kidney (HEK-293), and PER.C6 cells, have gained regulatory approval for production of recombinant proteins. The CHO cell line, in particular, has become the workhorse for industrial manufacture, and has even surpassed some microbial systems in productivity.107 However, it has certain limitations, such as the need for gene amplification and the selection pressure that is considered responsible for instability of expression.108 Interestingly, a recombinant rhabdovirus-based vector suitable for rapid and cost-effective industrial-scale production of antibody, which utilizes the CHO cell line as a substrate, has been reported.109,110 A human origin cell line PER.C6 has been developed as an alternative to the CHO cell line for large-scale manufacturing of recombinant human MAbs.100 It can be adapted to grow under different conditions, produces a high level of recombinant proteins in a stable manner, does not require gene amplification, and does not add nonhuman glycan structures to proteins.81 The choice for a certain cell expression system will have to take into account the production cost in relation to the target population, which for many neglected diseases is disproportionately the poor people living in developing countries.111 Hence, in some instances, it may be beneficial to explore inexpensive expression systems such as yeast112 and plants.113
Clinical trials
Understandably, human clinical trials for new types of rabies biologics are not easy to accomplish today. In general, a clinical study involves applied research using human volunteers that is intended to add to medical knowledge related to the treatment, diagnosis, or prevention of diseases or conditions, in this case a fatal viral zoonosis. Historically, there are two main types of such studies: 1) actual clinical trials and 2) observational studies, such as occurred with the introduction of RIG and the human diploid cell vaccine. In a clinical trial, human volunteers receive specific interventions according to a protocol created by the investigators. These interventions may be medical products such as drugs or devices. The investigators try to determine the safety and efficacy of the intervention by measuring certain outcomes in the participants. Such clinical trials in drug development are described by various phases.114 These include Phase 0 (exploratory study), Phase I (safety), Phase II (effectiveness), Phase III (safety and effectiveness), and Phase IV (postmarketing safety, efficacy, and optimal use).115 To date, one anti-RABV human MAb (SII Rmab) derived from an engineered CHO cell line has successfully passed through a Phase I clinical trial116 (Table 5), and a Phase II clinical trial has been initiated very recently.117 Similarly, a combination of two anti-RABV human MAbs, called CL184, produced using the innovative MAbstract technology and PER.C6 cell line has successfully progressed through Phase I and Phase II clinical trials (Table 5).18 Phase III clinical trials are expected to be conducted in the near future. For the first time in history, these products may be launched globally and eventually reach the clinic sooner rather than later. It is tempting to speculate that human MAbs will slowly replace the polyclonal RIGs that are currently in use and over the next several years slowly dominate the market place. Considering the slow evolution of rabies biologics over the past century, a period of more than 3 decades of such research for such a major paradigm shift to occur for effective PEP of humans with MAbs seems well worth the wait.
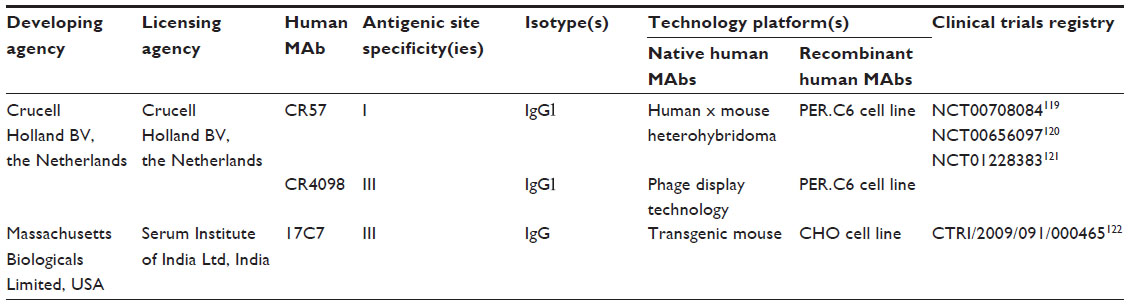 | Table 5 Human anti-rabies virus MAbs that have passed through Phase I/II clinical trials |
Disclosure
The authors report no conflicts of interest in this work.
References
Reichert JM, Dewitz MC. Anti-infective monoclonal antibodies: perils and promise of development. Nat Rev Drug Dis. 2006;5(3):191–195. | |
Cohen DJ, Benvenistry AI, Cianci J, Hardy MA. OKT3 prophylaxis in cadaveric kidney transplant recipients with delayed graft rejection. Am J Kidney Dis. 1989;14(5 Suppl 2):19–27. | |
Maloney DG, Grillo-Lopez AJ, White CA, et al. IDEC-C2B8 (rituximab) anti-CD20 monoclonal antibody therapy in patients with relapsed low-grade non-Hodgkin’s lymphoma. Blood. 1997;90(6):2188–2195. | |
Onrust SV, Lamb HM. Infliximab: a review of its use in Crohn’s disease and rheumatoid arthritis. Bio Drugs. 1998;10(5):397–422. | |
Albanell J, Baselga J. Trastuzumab, a humanized anti-HER2 monoclonal antibody, for the treatment of breast cancer. Drugs Today (Barc). 1999;35(12):935–946. | |
Storch GA. Humanized monoclonal antibody for prevention of respiratory syncytial virus infection. Pediatrics. 1998;102(3 Pt 1):648–651. | |
Ferrajoli A, O’Brien S, Keating MJ. Alemtuzumab: a novel monoclonal antibody. Expert Opin Biol Ther. 2001;1(6):1059–1065. | |
Krasner C, Joyce RM. Zevalin: 90yttrium labeled anti-CD20 (ibritumomab tiuxetan), a new treatment for non-Hodgkin’s lymphoma. Curr Pharm Biotechnol. 2001;2(4):341–349. | |
Kerr DJ. Targeting angiogenesis in cancer: clinical development of bevacizumab. Nat Clin Pract Oncol. 2004;1(1):39–43. | |
Ohenuram M, Saif MW. Panitumumab the first fully human monoclonal antibody: from the bench to the clinic. Anticancer Drugs. 2007; 18(1):7–15. | |
Cingoz O. Ustekinumab. mAbs. 2009;1(3):216–221. | |
Subramaniam JM, Whiteside G, McKeage K, Croxtall JC. Mogamulizumab. Drugs. 2012;72(9):1293–1298. | |
Rupprecht CE, Hanlon CA, Hemachudha T. Rabies re-examined. Lancet Infect Dis. 2002;2(6):327–343. | |
Jackson AC, Warrell MJ, Rupprecht CE, et al. Management of rabies in humans. Clin Infect Dis. 2003;36(1):60–63. | |
Wilde H, Hemachudha T, Jackson AC. View point: management of human rabies. Trans R Soc Trop Med Hyg. 2008;102(10):979–982. | |
World Health Organization. Rabies vaccines. WHO position paper. Wkly Epidemiol Rec. 2007;82:425–436. | |
World Health Organization. Rabies Information System of the WHO Collaboration Centre for Rabies Surveillance and Research. What is rabies? Available on: http://www.who-rabies-bulletin.org/about_rabies/What_is_rabies.aspx. Accessed October 2, 2013. | |
World Health Organization. Recommendations on Rabies Post-Exposure Treatment and the Correct Technique of Intradermal immunizationagainst Rabies. World Health Organization; 1996. Available from: http://www.who.int. Accessed on July 21, 2013. | |
World Health Organization. WHO recommendations on rabies post-exposure treatment and the correct technique of intradermal immunization. WHO, Geneva. 1996. Available from: http://whqlibdoc.who.int/hq/1996/WHO_EMC_ZOO_96.6.pdf. Accessed October 2, 2013. | |
World Health Organization. State of the art of new vaccine research and development. Section 7. Zoonotic infections. Chapter 7.5. Rabies. Pages 88–90. WHO, Geneva. 2006. Available from: http://whqlibdoc.who.int/hq/2006/WHO_IVB_06.01_eng.pdf. Accessed October 2, 2013. | |
Wilde H, Khawplod P, Hemachudha T, Sitprija V. Postexposure treatment of rabies infection: can it be done without immunoglobulin? Clin Infect Dis. 2002;34(4):477–480. | |
Gacouin A, Bourhy H, Renaud JC, Camus C, Suprin E, Thomas R. Human rabies despite post-exposure vaccination. Eur J Clin Microbiol Infect Dis. 1999;18(3):233–235. | |
Hemachudha T, Mitrabhakdi E, Wilde H, Vejabhuti A, Siripataravanit S, Kingnate D. Additional reports of failure to respond to treatment after rabies exposure in Thailand. Clin Infect Dis. 1999;28(1):143–144. | |
Dietzschold B, Kao M, Zheng YM, et al. Delineation of putative mechanisms involved in antibody-mediated clearance of rabies virus from the central nervous system. Proc Natl Acad Sci U S A. 1992;89(15):7252–7256. | |
Dietzschold B. Antibody-mediated clearance of viruses from the mammalian central nervous system. Trends Microbiol. 1993;1(2):63–66. | |
World Health Organization. Rabies fact sheet N 99. WHO, Geneva. Available from: http://www.who.int/mediacentre/factsheets/fs099/en/print.html. Cited May 18, 2007. Accessed October 2, 2013. | |
Goldsby RA, Kindt TJ, Osborne BA, Kuby J. Immunology. 5th ed. New York: W.H. Freeman and Company; 1996. | |
Khawplod P, Wilde H, Chomckey P, et al. What is an acceptable delay in rabies immune globulin administration when vaccine alone had been given previously? Vaccine. 1996;14(5):389–391. | |
Both L, Banyard AC, van Dolleweerd C, Horton DL, Ma JK, Fooks AR. Passive immunity in the prevention of rabies. Lancet Infect Dis. 2012;12(5):397–407. | |
Saylor C, Dadachova E, Casadevall A. Monoclonal antibody-based therapies for microbial diseases. Review. Vaccine. 2009;27(6):G38–G46. | |
Wilde H, Chomchey P, Prakongsri S, Puyaratabandhu P, Chutivongse S. Adverse effects of equine rabies immune globulin. Vaccine. 1989;7(1):10–11. | |
Muhamuda K, Madhusudana SN, Ravi V. Use of neutralizing murine monoclonal antibodies to rabies glycoprotein in passive immunotherapy against rabies. Hum Vaccin. 2007;3(5):192–195. | |
Fernandes A, Kaundinya JO, Daftary G, Saxena L, Banerjee S, Pattnaik P. Chromatographic purification of equine immunoglobulin G F(ab’)2 from plasma. J Chromatogr B Analyt Technol Biomed Life Sci. 2008;876(1):109–115. | |
Cupo P, de Azevedo-Marques MM, Sarti W, Hering SE. Proposal of abolition of the skin sensitivity test before equine rabies immune globulin application. Rev Inst Med Trop Sao Paulo. 2001;43(1):51–53. | |
Hanlon CA, DeMattos CA, DeMatto CC, et al. Experimental utility of rabies virus-neutralizing human monoclonal antibodies in postexposure prophylaxis. Vaccine. 2001;19(28–29):3834–3842. | |
Keller MA, Stiehm ER. Passive immunity in prevention and treatment of infectious diseases. Clin Microbiol Rev. 2000;13(4):602–614. | |
Helmick CG, Johnstone C, Summer J, Winkler WG, Fager S. A clinical study of Merieux human rabies immune globulin. J Biol Stand. 1982;10(4):357–367. | |
Grifols Therapeutics Inc. Rabies immune globulin (human): HyperRAB® S/D. Available from: http://www.talecris-pi.info/inserts/hyperrab.pdf. Cited Sep 2012. Accessed October 2, 2012. | |
Sanofi Pasteur SA. Rabies immune globulin (human) USP, heat treated. Imogam® Rabies – HT. Available from: https://www.vaccineshoppe.com/image.cfm?doc_id=10902&image_type=product_pdf. Cited Dec 2005. Accessed October 2, 2012. | |
Wilde H, Chomchey P, Prakongsri S, Punyaratabandhu P. Safety of equine rabies immune globulin. Lancet. 1987;2(8570):1275. | |
Motoi Y, Sato K, Hatta H, Morimoto K, Inoue S, Yamada A. Production of rabies neutralizing antibody in hen’s egg using a part of the G protein expressed in Escherichia coli. Vaccine. 2005;23(23):3026–3032. | |
Liu X, Liu Q, Feng X, et al. Rabbit anti-rabies immunoglobulins production and evaluation. Trop Biomed. 2011;28(1):138–148. | |
Redwan el RM, Fahmy A, El Hanafy A, Abd El-Baky N, Sallam SM. Ovine anti-rabies antibody production and evaluation. Comp Immunol Microbiol Infect Dis. 2009;32(1):9–19. | |
World Health Organization. WHO consultation on a monoclonal antibody cocktail for rabies post exposure treatment. WHO, Geneva. May 23–24, 2002. | |
Kohler G, Milstein C. Continuous cultures of fused cells secreting antibodies of predetermined specificity. Nature. 1975;256: 495–497. | |
Buelow R, van Schooten W. Ernst Schering Foundation symposium proceedings, Berlin, Germany: Springer-Verlag; 2007. | |
Goldstein G; Ortho Multicenter Transplant Study Group. A randomized clinical trial of OKT3 monoclonal antibody for acute rejection of cadaveric renal transplants. Ortho multicenter transplant study group. N Engl J Med. 1985;313(6):337–342. | |
Boenisch T. Antibodies. Chapter 1. Immunochemical Staining Methods. 5th ed. Carpinteria, CA: Dako North America; 2009. | |
Marasco WA, Sui J. The growth and potential of human antiviral monoclonal antibody therapeutics. Nat Biotechnol. 2007;25(12):1421–1434. | |
Peterson E, Owens SM, Henry RL. Monoclonal antibody form and function: manufacturing the right antibodies for treating drug abuse. AAPS J. 2006;8(2):E383–E390. | |
Zharikova D, Mozdzanowska K, Feng J, Zhang M, Gerhard W. Influenza type A virus escape mutants emerge in vivo in the presence of antibodies to the ectodomain of matrix protein 2. J Virol. 2005;79(11):6644–6654. | |
Law M, Hangartner L. Antibodies against viruses: passive and active immunization. Curr Opin Immunol. 2008;20(4):486–492. | |
Casadevall A, Dadachova E, Pirofski LA. Passive antibody therapy for infectious diseases. Nat Rev Microbiol. 2004;2(9):695–703. | |
Flamand A, Wiktor TJ, Koprowski H. Use of hybridoma monoclonal antibodies in the detection of antigenic differences between rabies and rabies-related virus proteins. II. The glycoprotein. J Gen Virol. 1980;48(1):105–109. | |
Lafon M, Ideler J, Wunner WH. Investigation of the antigenic structure of rabies virus glycoprotein by monoclonal antibodies. Dev Biol Stand. 1984;57:219–225. | |
Lafon M, Wiktor TJ, Macfarlan RI. Antigenic sites on the CVS rabies virus glycoprotein: analysis with monoclonal antibodies. J Gen Virol. 1983;64(Pt 4):843–851. | |
Nagarajan T, Reddy GS, Mohanasubramanian B, et al. A simple immuno-capture ELISA to estimate rabies viral glycoprotein antigen in vaccine manufacture. Biologicals. 2006;34(1):21–27. | |
Ni Y, Tominaga Y, Honda Y, Morimoto K, Sakamoto S, Kawai A. Mapping and characterization of a sequential epitope on the rabies virus glycoprotein which is recognized by a neutralizing monoclonal antibody, RG719. Microbiol Immunol. 1995;39(9):693–702. | |
Prehaud C, Coulon P, Lafay F, Thiers C, Flamand A. Antigenic site II of the rabies virus glycoprotein: structure and role in viral virulence. J Virol. 1988;62(1):1–7. | |
Schumacher CL, Dietzschold B, Ertl HCJ, Hong-Shun N, Rupprecht CE, Koprowski H. Use of mouse anti-rabies monoclonal antibodies in postexposure treatment of rabies. J Clin Invest. 1989;84(3):971–975. | |
Seif I, Coulon P, Rollin PE, Flamand A. Rabies virulence: effect on pathogenicity and sequence characterization of rabies virus mutations affecting antigenic site III of the glycoprotein. J Virol. 1985;53(3):926–934. | |
Wiktor TJ, Koprowski H. Monoclonal antibodies against rabies virus produced by somatic cell hybridization: detection of antigenic variants. Proc Natl Acad Sci U S A. 1978;75(8):3938–3942. | |
Benmansour A, Leblois H, Coulon P, et al. Antigenicity of rabies virus glycoprotein. J Virol. 1991;65(8):4198–4203. | |
Dietzschold B, Gore M, Casali P, et al. Biological characterization of human monoclonal antibodies to rabies virus. J Virol. 1990;64(6):3087–3090. | |
Lafon M, Edelman L, Bouvet JP, et al. Human monoclonal antibodies specific for the rabies virus glycoprotein and N protein. J Gen Virol. 1990;71(8):1689–1696. | |
Walker PJ, Kongsuwan K. Deduced structural model for animal rhabdovirus glycoproteins. J Gen Virol. 1999;80:1211–1220. | |
Kuzmina NA, Kuzmin IV, Ellison JA, Rupprecht CE. Conservation of binding epitopes for monoclonal antibodies on the rabies virus glycoprotein. J Antivir Antiretrovir. 2013;5(2):37–43. | |
Montano-Hirose JA, Lafage M, Weber P, Badrane H, Tordo N, Lafon M. Protective activity of a murine monoclonal antibody against European bat lyssavirus 1 (EBL1) infection in mice. Vaccine. 1993;11(12):1259–1266. | |
Goudsmit J, Marissen WE, Weldon WC, et al. Comparison of an anti-rabies human monoclonal antibody combination with human polyclonal anti-rabies immune globulin. J Infect Dis. 2006;193(6):796–801. | |
Muller T, Dietzschold B, Ertl H, et al. Development of a mouse monoclonal antibody cocktail for post-exposure rabies prophylaxis in humans. PLoS Negl Trop Dis. 2009;3(11):1–10. | |
Wang Y, Rowley KJ, Booth BJ, Sloan SE, Ambrosino DM, Babcock GJ. G glycoprotein amino acid residues required for human monoclonal antibody RAB1 neutralization are conserved in rabies virus street isolates. Antiviral Res. 2011;91(2):187–194. | |
Badger CC, Anasetti C, Davis J, Bernstein ID. Treatment of malignancy with unmodified antibody. Pathol Immunopathol Res. 1987;6(5–6):419–434. | |
Khazaeli MB, Conry RM, LoBuglio AF. Human immune response to monoclonal antibodies. J Immunother Emphasis Tumor Immunol. 1994;15(1):42–52. | |
Both L, van Dolleweerd C, Wright E, et al. Production, characterization, antigenic specificity of recombinant 62-71-3, a candidate monoclonal antibody for rabies prophylaxis in humans. FASEB J. 2013;27(5):2055–2065. | |
Harding FA, Stickler MM, Razo J, DuBridge RB. The immunogenicity of humanized and fully human antibodies. mAbs. 2010;2(3):256–265. | |
Winter G, Milstein C. Man-made antibodies. Nature. 1991;349(6307):293–299. | |
Sugimoto M, Furuichi Y, Ide T, Goto M. Incorrect use of “immortalization” for B-lymphoblastoid cell lines transformed by Epstein-Barr virus. J Virol. 1999;73(11):9690–9691. | |
Jakobovits A, Amado RG, Yang X, Roskos L, Schwab G. From XenoMouse technology to panitumumab, the first fully human antibody product from transgenic mice. Nat Biotechnol. 2007;25(10):1134–1143. | |
Nagarajan T, Rupprecht CE, Dessain SK, Rangarajan PN, Thiagarajan D, Srinivasan VA. Human monoclonal antibody and vaccine approaches to prevent human rabies. Curr Top Microbiol Immunol. 2008; 317: 67–101. | |
Matsumoto T, Yamada K, Noguchi K, et al. Isolation and characterization of novel human monoclonal antibodies possessing neutralizing ability against rabies virus. Microbiol Immunol. 2010;54(11):673–683. | |
Jones DH, Kross N, Anema R, et al. High-level expression of recombinant IgG in the human cell line PER.C6. Biotechnol Prog. 2003;19(1):163–168. | |
Champion JM, Kean RB, Rupprecht CE, et al. The development of monoclonal human rabies virus-neutralizing antibodies as a substitute for pooled human immune globulin in the prophylactic treatment of rabies virus exposure. J Immunol Methods. 2000;235(1–2):81–90. | |
Bakker AB, Marissen WE, Kramer RA, et al. Novel human monoclonal antibody combination effectively neutralizing natural rabies virus variants and individual in vitro escape mutants. J Virol. 2005;79(14):9062–9068. | |
Sloan SE, Hanlon C, Weldon W, et al. Identification and characterization of a human monoclonal antibody that potently neutralizes a broad panel of rabies virus isolates. Vaccine. 2007;25(15):2800–2810. | |
de Kruif J, Bakker AB, Marissen WE, et al. A human monoclonal antibody cocktail as a novel component of rabies postexposure prophylaxis. Annu Rev Med. 2007;58:359–368. | |
Presta LG. Molecular engineering and design of therapeutic antibodies. Curr Opin Immunol. 2008;20(4):460–470. | |
McCafferty J, Griffiths AD, Winter G, Chiswell DJ. Phage antibodies: filamentous phage displaying antibody variable domains. Nature. 1990;348(6301):552–554. | |
Huston JS, Levinson D, Mudgett Hunter M, et al. Protein engineering of antibody binding sites: recovery of specific activity in an anti-digoxin single-chain Fv analogue produced in Escherichia coli. Proc Natl Acad Sci U S A. 1988;85(16):5879–5883. | |
Zhao XL, Yin J, Chen WQ, Jiang M, Yang G, Yang ZH. Generation and characterization of human monoclonal antibodies to G5, a linear neutralization epitope on glycoprotein of rabies virus, by phage display technology. Microbiol Immunol. 2008;52(2):89–93. | |
Zhao XL, Chen WQ, Yang ZH, Li JM, Zhang SJ, Tian LF. Selection and affinity maturation of human antibodies against rabies virus from a scFv gene library using ribosome display. J Biotechnol. 2009;144(4):253–258. | |
Liu X, Lin H, Tang Q, et al. Characterization of human antibody fragment Fab and its calcium phosphate nanoparticles that inhibit rabies virus infection with vaccine. PLoS One. 2011;6(5):1–8. | |
Li C, Zhang F, Lin H, et al. Generation and characterization of the human neutralizing antibody fragment Fab019 against rabies virus. Acta Pharmacol Sin. 2011;32(3):329–337. | |
Raag R, Whitlow M. Single chain Fvs. FASEB J. 1995;9(1):73–80. | |
Duan Y, Gu TJ, Jiang CL, et al. A novel disulfide-stabilized single-chain variable antibody fragment against rabies virus G protein with enhanced in vivo neutralizing potency. Mol Immunol. 2012;51(2):188–196. | |
Wang DD, Su MM, Sun Y, Huang SL, Wang J, Yan WQ. Expression, purification and characterization of a human single-chain Fv antibody fragment fused with the Fc of an IgG1 targeting a rabies antigen in Pichia pastoris. Protein Expr Purif. 2012;86(1):75–81. | |
Repp R, Kellner C, Muskulus A, et al. Combined Fc-protein- and Fc-glyco-engineering of scFv-Fc fusion proteins synergistically enhances CD16a binding but does not further enhance NK-cell mediated ADCC. J Immunol Methods. 2011;373(1–2):67–78. | |
Palmberger D, Rendic D, Tauber P, Krammer F, Wilson IBH, Grabherr R. Insect cells for antibody production: evaluation of an efficient alternative. J Biotechnol. 2011;153(3–4):160–166. | |
Chadd HE, Chamow SM. Therapeutic antibody expression technology. Curr Opin Biotechnol. 2001;12(2):188–194. | |
de Kruif J, Kramer A, Nijhuis R, et al. Generation of stable cell clones expressing mixtures of human antibodies. Biotechnol Bioeng. 2010;106(5):741–750. | |
Jones DH, van Berkel PHC, Logtenberg T, Bout A. PER.C6 cell-line for human antibody production. Genetic Eng News. 2002;22(10):50. | |
Barnard GC, Kull AR, Sharkey NS, et al. High-throughput screening and selection of yeast cell lines expressing monoclonal antibodies. J Ind Microbiol Biotechnol. 2010;37(9):961–971. | |
Ko K, Tekoah, Y, Rudd PM, et al. Function and glycosylation of plant-derived antiviral monoclonal antibody. Proc Natl Acad Sci U S A. 2003;100(13):8013–8018. | |
Tran M, Zhou B, Pettersson PL, Gonzalez MJ, Mayfield SP. Synthesis and assembly of a full length human monoclonal antibody in algal chloroplasts. Biotechnol Bioeng. 2009;104(4):663–673. | |
Pollock DP, Kutzko JP, Birck-Wilson E, et al. Transgenic milk as a method for the production of recombinant antibodies. J Immunol Methods. 1999;231(1–2):147–157. | |
Andersen DC, Krummen L. Recombinant protein expression for therapeutic applications. Curr Opin Biotechnol. 2002;13(2):117–123. | |
Sabourin M, Huang Y, Dhulipala P, et al. Increasing antibody yield and modulating final product quality using the Freedom™ CHO-S™ production platform. BMC Proc. 2011;5(8):P102. | |
Wurm F, Hacker D. First CHO genome. Nat Biotechnol. 2011;29(8):718–720. | |
Bailey LA, Hatton D, Field R, Dickson AJ. Determination of Chinese hamster ovary cell line stability and recombinant antibody expression during long-term culture. Biotechnol Bioeng. 2012;109(8):2093–2103. | |
Morimoto K, Schnell MJ, Pulmanausahakul R, et al. High level expression of a human rabies virus-neutralizing monoclonal antibody by a rhabdovirus-based vector. J Immunol Methods. 2001;252(1–2):199–206. | |
Prosniak M, Faber M, Hanlon CA, Rupprecht CE, Hooper DC, Dietzschold B. Development of a cocktail of recombinant-expressed human rabies virus neutralizing monoclonal antibodies for post exposure prophylaxis of rabies. J Infect Dis. 2003;187:53–56. | |
Paul M, Dolleweerd CV, Drake PMW, et al. Molecular pharming: future targets and aspirations. Human Vaccin. 2011;7(3):375–382. | |
Fan JY, Shen Z, Wang G, et al. Secretory expression of human scFv against keratin in Pichia pastoris and its effects on cultured keratinocytes. Arch Dermatol Res. 2009;301(5):367–372. | |
Rainer F, Jurgen D, Neil E, et al. Towards molecular farming in the future: Pichia pastoris-based production of single-chain antibody fragments. Biotechnol Appl Biochem. 1999;30(Pt 2):117–120. | |
US National Institutes of Health. Learn about clinical studies. Available from: http://clinicaltrials.gov/ct2/about-studies/learn. Cited Aug 2012. Accessed October 2, 2013. | |
US National Institutes of Health. Glossary definition. Available from: http://clinicaltrials.gov/ct2/help/glossary/phase. Accessed October 2, 2013. | |
Gogtay N, Thatte U, Kshirsagar N, et al. Safety and pharmacokinetics of a human monoclonal antibody to rabies virus: a randomized, dose-escalation phase 1 study in adults. Vaccine. 2012;30(50):7315–7320. | |
Shelton ML. Clinical trial for rabies monoclonal antibody. Available from: http://www.eurekalert.org/pub_releases/2012–2008/uomm-ctf080712.php. Cited August 7, 2012. Accessed October 2, 2013. | |
Bakker AB, Python C, Kissling CJ, et al. First administration to humans of a monoclonal antibody cocktail against rabies virus: safety, tolerability and neutralizing activity. Vaccine. 2008;26(47):5922–5927. | |
Crucell Holland BV. Randomized Phase II Trial on Safety and Neutralizing Activity of CL184 and Rabies Vaccine Versus Human Rabies Immune Globulin (HRIG) and Rabies Vaccine in Children and Adolescents. Available from: http://clinicaltrials.gov/show/NCT00708084. Accessed October 21, 2013. | |
Crucell Holland BV. A Randomized Phase II Trial to Compare the Safety and Neutralizing Activity of CL184 in Combination With Rabies Vaccine vs. HRIG or Placebo in Combination With Rabies Vaccine in Healthy Adult Subjects. Available from: http://clinicaltrials.gov/show/NCT00656097. Accessed October 21, 2013. | |
Crucell Holland BV. Rabies Virus Neutralizing Activity and Safety of CL184, a Monoclonal Antibody Cocktail, in Simulated Rabies Post-Exposure Prophylaxis in Healthy Adults. Available from: http://clinicaltrials.gov/show/NCT01228383. Accessed October 21, 2013. | |
Serum Institute of India Ltd. A clinical trial to assess the safety and rabies antibody levels in blood following a new Human Monoclonal Antibody to Rabies (SII RMab) in comparison with Human Rabies Immune Globulin, when given together with Rabies Vaccine (RABIVAX®) in healthy adults. Available from: http://www.ctri.nic.in/Clinicaltrials/pmaindet2.php?trialid=680. Accessed October 21, 2013. | |
Marissen WE, Kramer A, Rice A, et al. Novel Rabies Virus-Neutralizing Epitope Recognized by Human Monoclonal Antibody: Fine Mapping and Escape Mutant Analysis. J. Virol. 2005;79(8):4672–4678. | |
World Health Organization. WHO Expert Consultation on Rabies, Second report. WHO Techinical Report Series 982. Available from: http://apps.who.int/iris/bitstream/10665/85346/1/9789241209823_eng.pdf. Accessed November 28, 2013. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.