")
Back to Journals » Drug Design, Development and Therapy » Volume 9
Modeling, molecular dynamics, and docking assessment of transcription factor rho: a potential drug target in Brucella melitensis 16M
Authors Pradeepkiran JA, Kumar KK, Kumar YN, Bhaskar M
Received 4 November 2014
Accepted for publication 9 December 2014
Published 31 March 2015 Volume 2015:9 Pages 1897—1912
DOI https://doi.org/10.2147/DDDT.S77020
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 6
Editor who approved publication: Professor Shu-Feng Zhou
Jangampalli Adi Pradeepkiran,1 Konidala Kranthi Kumar,1 Yellapu Nanda Kumar,2 Matcha Bhaskar1
1Division of Animal Biotechnology, Department of Zoology, Sri Venkateswara University, Tirupati, 2Biomedical Informatics Centre, Vector Control Research Centre, Indian Council of Medical Research, Pondicherry, India
Abstract: The zoonotic disease brucellosis, a chronic condition in humans affecting renal and cardiac systems and causing osteoarthritis, is caused by Brucella, a genus of Gram-negative, facultative, intracellular pathogens. The mode of transmission and the virulence of the pathogens are still enigmatic. Transcription regulatory elements, such as rho proteins, play an important role in the termination of transcription and/or the selection of genes in Brucella. Adverse effects of the transcription inhibitors play a key role in the non-successive transcription challenges faced by the pathogens. In the investigation presented here, we computationally predicted the transcription termination factor rho (TtFRho) inhibitors against Brucella melitensis 16M via a structure-based method. In view the unknown nature of its crystal structure, we constructed a robust three-dimensional homology model of TtFRho’s structure by comparative modeling with the crystal structure of the Escherichia coli TtFRho (Protein Data Bank ID: 1PVO) as a template in MODELLER (v 9.10). The modeled structure was optimized by applying a molecular dynamics simulation for 2 ns with the CHARMM (Chemistry at HARvard Macromolecular Mechanics) 27 force field in NAMD (NAnoscale Molecular Dynamics program; v 2.9) and then evaluated by calculating the stereochemical quality of the protein. The flexible docking for the interaction phenomenon of the template consists of ligand-related inhibitor molecules from the ZINC (ZINC Is Not Commercial) database using a structure-based virtual screening strategy against minimized TtFRho. Docking simulations revealed two inhibitors compounds – ZINC24934545 and ZINC72319544 – that showed high binding affinity among 2,829 drug analogs that bind with key active-site residues; these residues are considered for protein-ligand binding and unbinding pathways via steered molecular dynamics simulations. Arg215 in the model plays an important role in the stability of the protein-ligand complex via a hydrogen bonding interaction by aromatic-π contacts, and the ADMET (absorption, distribution, metabolism, and excretion) analysis of best leads indicate nontoxic in nature with good potential for drug development.
Keywords: brucellosis, rho proteins, transcription inhibitors, SMD simulations, ADMET analysis, therapeutics
Introduction
“Rho proteins” are molecular switches in systemic transcription regulation, and the resulting translated products assist in bacterial survival. “Rho” is a hexameric protein that causes transcription termination and it binds to the nascent RNAs. By analogy with quite similar sequences of Escherichia coli, Mycobacterium tuberculosis, and Rickettsia prowazekii Rho factors, the binding should include up to four ionic (largely basic) and hydrophobic motifs, effecting release of nascent messenger RNA (mRNA). Rho proteins contain distinct binding domains – that is, adenosine triphosphate (ATP) and guanosine-5′-triphosphate (GTP) binding regions – which regulate the bacterial transcription by binding the corresponding ATP or GTP analogs.1 In prokaryotes, two commonly used types of transcription termination are rho dependent and rho independent. Rho plays a regulatory role in bacterial transcription. Rho terminates the synthesis of mRNA for a significant number of operons. Rho dissociates RNA polymerase from the DNA template to release RNA, deriving energy by hydrolyzing ATP through its RNA-dependent ATPase activity.2
As a prerequisite of rho-dependent termination by cis-acting and trans-acting elements, the cis-acting elements are the main regulators of the rho protein when observed in C-rich transcribed RNA genomes where the Brucella facilitates rho-dependent termination.3 The ring-shaped hexameric protein rho possesses the helicase activities where C-rich coding sequences are good candidates for binding. A rho protein plays a successive role in the regeneration of bacteria through the mitosis of cells. The three-dimensional (3D) crystal structure of the rho protein contains the hexamer, which inhibits elongation during transcription. The rho protein contains the two domains of activities proteins with ATPase and helicase, which are required for additional factors such as N utilization substance protein A (nusA), which has been reported as a potential drug target in Brucella melitensis 16M with an important role in the release of the terminated transcripts.4,5 The rho GTPase activity most likely allows bacteria to invade epithelial cells that also become resistant to apoptosis and thus provide a safe spot, inaccessible to the host defenses.6
This translational antagonism of bacterial operons can be suppressed by rho or by its inhibitors. Transcription terminator rho proteins are among the best novel therapeutic drug targets and play an important role in the inhibition of bacterial pathogens, and the rho-dependent targeting analogs that mislead their successive operons misguide the proteins involved in invading the pathogens in the host environment. These rho proteins, coupled with G proteins, act as molecular regulators for various metabolic pathways. Rho proteins are highly conserved, and essential for the regulation of various gene clusters in bacteria.7
Brucella is a genus of Gram-negative facultative intracellular pathogens that cause immediate miscarriage in pregnant women and also cause arthritis and cardiac problems, among other issues. The Brucella genome has a high guanine-cytosine content, which links to an elevated average content of acidic and neutral hydrophobic amino acids in the expressed proteins (including rho).
Rho across bacterial species appears to have several RNA binding sites, and shares at least two basic polynucleotide-binding, carrier-protein-binding, nuclear-translocation-helping (PCN) motifs that have at least three (and at least 50%) basic residues, such as K[R]AKR and RPKR. The transcription terminator rho has two nucleic-acid binding sites and one ATP binding site for ATP hydrolysis and the translation action of RNA.8 Functional-based drug discovery against rho protein is one promising approach to treating brucellosis. The present computational approach helps in drug design, which can helps in the identification process of new lead molecules, and provides the most effective target-based treatment against human brucellosis strains like B. melitensis 16M, Brucella suis, Brucella abortus S19, and Brucella canis. Due to the nonavailability of the crystal structure of the rho protein in Brucella, the investigation presented here aimed to proceed with homology modeling, a molecular dynamics (MD) simulation technique that reveals the optimal reliable modeled protein structure in the surrounding solvent environment, and docking studies were performed to predict the inhibitors to rho by computational methods and validate protein inhibition in the presence of lead molecules by MD simulations.
Materials and methods
Molecular characterization and crystallizability
The protein sequence of the B. melitensis 16M transcription termination factor rho (TtFRho) was obtained from the NCBI (National Center for Biotechnology Information) protein sequence database with the accession number NP_538921.1.9 Crystallizability and molecular features were predicted by the XtalPred server.10 This server provides several publicly available programs for the calculation and prediction of protein features, including physicochemical properties (molecular weight [MW], theoretical isoelectric point [pI], amino-acid length, instability index) and grand average of hydropathicity (GRAVY), expert pool crystallization classes, random forest crystallization classes by biochemical and biophysical features with corresponding probability distributions obtained from TargetDB,11 and other features such as coiled-coil regions, longest disorder regions, transmembrane helices, signal peptides, low-complexity regions, and secondary structure, from the COILS,12 DISOPRED2,13 TMHMM (v 2.0),14 RPSP,15 SEG,16 and PSIPRED (v 3.3)17 servers, respectively. The protein subcellular localization and secretory signal peptides and their cleavage sites were predicted by the Cell-PLoc package fusing gene-ontology information,18 PSORTb (v 3.0),19 and SignalP (v 4.0) servers, respectively.20
Homology search, modeling, refinement, and evaluation
The primary amino-acid sequence was used to search for a suitable template, which was carried out by a homology search via a position-specific iterative Basic Local Alignment Search Tool algorithm (PSI-BLAST)21 against the Protein Data Bank (PDB)22 to generate a 3D coordinate structure. The homology search showed the crystal structure coordinates of the E. coli TtFRho complex with a single-stranded RNA (ssRNA) substrate and phosphoaminophosphonic acid-adenylate ester (ANP) (PDB ID: 1PVO),23 which was found to be the best hit based on query coverage, identity, E-value, and high similarity with the “A” chain, so was therefore considered as the template for homology modeling.
The initial alignment of target and template was generated using the ALIGN2D module, and then the 3D structure of the target protein was generated using a restrained-based approach in MODELLER (v 9.12) using a model-single module.24,25 Initially, we developed 100 3D structures; among those, the best ones were judged by low discrete optimized protein energy (DOPE) because this was a standard scoring function in MODELLER. The final model geometry was regularized by energy minimization – to satisfy the restraints on bond distances and dihedral angles of each amino acid – by changing the default optimization and refinement protocols – that is, variable target function method, optimization with conjugate gradients (CGs), and refinement using MD with simulated annealing.24
The final model was evaluated, before refinement and after, by calculating the stereochemical quality via PROCHECK,26 ERRAT (v 2.0),27 and Verify3D environment profile analysis methods28 by submitting to the SAVES (Structural Analysis and Verification Server). ProSA (Protein Structure Analysis) was also used to evaluate the final 3D structure for probable errors.29 The root mean square deviations (RMSDs) between the target model and template were calculated by structural superimposition with the Swiss-PdbViewer (v 4.10) and SuperPose (v 1.0) programs.30 Based on the results, the protein structure was refined to obtain reliable scores and low error values, and then the obtained structure was submitted to the Protein Model DataBase (PMDB).31
Pocket binding site and ligand predictions
Protein structure and function relevance were illuminating in regards to the ligand bindings to its active site and their specificity.32 We identified the protein-ligand binding domain by searching in the CASTp (Computed Atlas of the Surface Topography of Proteins) server33 and visualizing with PyMOL CASTp. Rho transcription termination factor (PDB ID: 1PVO) consist ligand, ie, phosphoaminophosphonic acid-adenylate ester (ANP) – was considered a template for the structure-based virtual screening study. A canonical simplified molecular-input line-entry system (SMILES) search for similar ligand molecules with 70% identity was performed, and the results were retrieved in the Mol2 format from the ZINC (ZINC Is Not Commercial) small-molecule database.34 These predicted structures were optimized in the Open Babel package (v 2.3.2) for refinement and repair of hydrogen and bonds through minimization by applying CGs with a united force field through 500 steps/cycles.35 The final optimized leads were prepared as Dataset A for docking studies.
Virtual screening and flexible docking approaches
The Dataset A molecules from the described approaches were used to virtually screen against the pocket site of protein with functional residues for molecular-docking simulations. AutoDockTools was used to establish the autogrid points via AutoGrid (v 4.0) by selecting active-site residues.36 The grid dimensions were predicted as per the domain area by the center x, y, and z coordinates. The Lamarckian geometric algorithm was used for the docking parameter for AutoDock Vina (v 2.0)37 in the PyRx (v 0.8) virtual screening software with 50-ligand exhaustiveness.38 The virtual screening results comprise the best molecules – those having a higher binding affinity with the receptor binding domain with functional residues than the positive control (ie, ANP) and separate them out. The top scoring 10% of molecules were selected for further flexible docking simulations by AutoDock (v 4.2).36 AutoDock was used to perform docking using flexible conformations of lead and protein molecules. A docking grid from the autogrid with a size of 25×25×25 Å was used. Center coordinates of the grid were obtained from the flexible atoms of the described virtual screening complex – 15 Å in each direction from the center x, y, and z coordinates. AutoDock shows the docking scores as the free energy of binding (kcal/mol). The best binding affinity leads were selected for toxic evaluation.
Protein-ligand interaction analysis
The hydrogen bond and hydrophobic interactions of protein-ligand complexes were analyzed by LigPlot+ (v 1.4.5)38 and PyMOL. “LigPlot+” is a graphical system that generates multiple two-dimensional (2D) diagrams of ligand–protein interactions from docked complexes. In the described investigation, the LigPlot+ program was used to discover the interacting residues, hydrogen bonds, and hydrophobic interactions of ligands in docked complexes. “PyMOL” is a widely used tool for biomolecular visualization. In the present study, the PyMOL program was used to analyze the protein-ligand interactions along with bond angle and bond distance, functional atoms involved in ligands, and protein functional amino-acid residues.
In silico toxicity and bioactivity prediction
In the drug-discovery process, prediction of drug toxicity is essential because unacceptable toxicity is still a major bottleneck.39 Hence, as an additional in silico investigation, for all the molecules, the rule of five, drug likeness, toxicity, and relationships with other proteins were investigated. The Lipinski rule, drug likeness, and molecular properties were calculated by the Molinspiration and MolSoft programs. These servers provide the ligand consists drug likeness properties like logP, MW, number of hydrogen-bond acceptors (HBA), number of hydrogen-bond donors (HBD) and other molecular properties such as molecular topological polar surface area (tPSA), number of rule of five violations, number of rotatable bonds, number of non-hydrogen atoms and molecular volume calculated by the method of contributions and correction factors were predicted for each lead molecule. The toxicity and drug-relevant properties of ligands were predicted by the OSIRIS Property Explorer (v 2.0).40 This program depicts the molecule toxicity (mutagenicity, tumorigenicity, irritating effect, and reproductive effect) for respective drawing ligands. The molecules selected from the described approaches, and all those that exhibited unfavorable features, were removed from the final set of ligands. The similarity ensemble approach (SEA) was applied to the ligands that passed the toxicity analysis to identify the relationships between the ligand-related proteins based on the chemical similarity of their specific ligands determined by searching with SEArch (SEA Search Tool; http://sea.bkslab.org/search/).41
Molecular dynamics simulations
Preliminary MD simulations for the modeled protein were performed using the program NAMD (NAnoscale Molecular Dynamics program; v 2.9),42 and all files were generated using visual molecular dynamics (VMD).43 The protein was solvated with a TIP3P water box with a 2.5 Å layer of water for each direction of the coordinate structure and a CHARMM (Chemistry at HARvard Macromolecular Mechanics) 22 parameter file for proteins and lipids; phi and psi cross-term map correction were used in the force field for proteins with similar chemical structures.44 For the minimization and equilibration of TtFRho in the water box, we assumed force-field parameters excluding scaling of 1.0 Å and a cutoff of Coulomb forces with a switching function starting at 12 Å, reaching zero at a distance of 10 Å, ending at 14 Å with a margin of 3.0 Å, and all atoms, including those of hydrogen, were illustrated explicitly. The hydrogen atom coordinates of TtFRho were generated using the VMD Tk-Console salvation command.42 Integrator parameters also included 2 fs/step for all rigid bonds and nonbonded frequencies were selected for 1 Å and full electrostatic evaluations for 2 Å were used, with ten steps for each cycle. The particle mesh Ewald method was used for electrostatic interactions of TtFRho system periodic boundary conditions with grid dimensions of 1.0 Å.45 The 3D structure of TtFRho contains 421 amino-acid residues and 8,828 water molecules; to eliminate bad water constraints, the protein preliminary energy was minimized via 1,000 steps of the Powell algorithm at constant temperature (310 K), followed by simulation of an additional 500,000 runs for 2 ns with Langevin dynamics to control the kinetic energy, temperature, and/or pressure of the system without a binding pocket, and the applying parameters do not change the protein conformation during the ligand unbinding.46 The method used Langevin dynamics via the following equation:
| (1) |
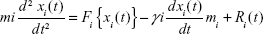
After the completion of primary simulations of the target protein, the protein-ligand complexes were thoroughly analyzed with selected inhibitors by MD simulations. The protein-ligand complexes containing flexible functional residues and ligand atoms were selected, and then parameterization of the protein and ligands was performed in Chimera (v 1.8.1)47 and SwissParam servers.48 The parameterized ligands were inserted into the protein and saved in the form of a protein-ligand complex by VMD with the binding pocket residues, and then the protein-ligand complex was immersed in the center of a 50 Å box of water molecules49,50 where all water molecule atoms (H-O-H) were closer than 1.5 Å. Finally, the solvated protein-ligand complex system was equilibrated with 1,000 minimization steps, 200,000 runs for 2 ns with velocity recycling every 2.0 ps, and each trajectory was saved at 100 ps for further analysis.
Steered molecular dynamics simulations
Ligand free energy calculations
Ligand binding free energies were calculated based on the Jarzynski equality.51 Steered molecular dynamics (SMD) simulations of the described complexes were conducted at constant velocity (cv) using the approach implemented in NAMD.52 For the initial simulated complexes, water was removed, and then the functional residue atom moving direction in the grid was measured; next, the atoms’ moving directions were fixed and saved as a reference file generated by VMD. These atoms were harmonically restrained to moving directions from the selected point with cv and constant forces (cf). The ligands were consequently pulled out of the protein. The external force applied to the ligands was calculated using the following equation:
| (2) |
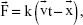
in which “![]() ” is the dissociation of the controlled atom with respect to its original position, “t” is time and “k” =4 kcal/mol Å2 for small fluctuations of applied force (x is due to thermal movement of the position of the controlled atom), where the term “small” is relative to the scale of
” is the dissociation of the controlled atom with respect to its original position, “t” is time and “k” =4 kcal/mol Å2 for small fluctuations of applied force (x is due to thermal movement of the position of the controlled atom), where the term “small” is relative to the scale of ![]() at this position; k was chosen to be large, such that the contraction of the ligands’ “spring” due to the development of the ligand and an unbinding path in excess of 1–2 Å resulted in an obvious drop in the force.53,54 The spring constant was 500 pN/Å, and pulling velocities of 1–10 Å/ns were used. The strength of the ligand binding to the binding pocket site of the protein was dependent on the pulling velocity.53,55 The ligand binding directions were calculated by VMD, and then the residue information was saved as a fixed reference file for one binding direction. In the parameter file, the SMD position was on without wrap water, and pulling velocities of 1–10 Å/ns were used (SMDVel 0.005); then, the simulations of binding pathways were run, with 1,000 minimization for 500,000 steps with 5 ns. From the calculations and output files, the simulations of unbinding pathways were run with 1,000 minimization for 200,000 steps with 2 ns. After the simulations, the results were analyzed in VMD by calculating the RMSD and root mean square fluctuation (RMSF) trajectories; the protein was found to contain overall translational and rotational motions removed by superimposing the backbone of the protein with all trajectories in each configuration using a least-squares fitting algorithm.56 The analyzed trajectories were plotted using Microsoft® Excel 2007.
at this position; k was chosen to be large, such that the contraction of the ligands’ “spring” due to the development of the ligand and an unbinding path in excess of 1–2 Å resulted in an obvious drop in the force.53,54 The spring constant was 500 pN/Å, and pulling velocities of 1–10 Å/ns were used. The strength of the ligand binding to the binding pocket site of the protein was dependent on the pulling velocity.53,55 The ligand binding directions were calculated by VMD, and then the residue information was saved as a fixed reference file for one binding direction. In the parameter file, the SMD position was on without wrap water, and pulling velocities of 1–10 Å/ns were used (SMDVel 0.005); then, the simulations of binding pathways were run, with 1,000 minimization for 500,000 steps with 5 ns. From the calculations and output files, the simulations of unbinding pathways were run with 1,000 minimization for 200,000 steps with 2 ns. After the simulations, the results were analyzed in VMD by calculating the RMSD and root mean square fluctuation (RMSF) trajectories; the protein was found to contain overall translational and rotational motions removed by superimposing the backbone of the protein with all trajectories in each configuration using a least-squares fitting algorithm.56 The analyzed trajectories were plotted using Microsoft® Excel 2007.
Results and discussion
Molecular features and crystallizability of target protein
The predicted physicochemical properties of TtFRho showed 421 amino acids length with a 46,970 kD MW. The pI was approximately 5.87 for those that were affected by the ionizing groups of the amino acid associated with electron withdrawal from the ligand (ATP) for several cellular processes, such as hydrolase, phosphoric ester, diester hydrolase, cyclic-nucleotide and 3′-5′-cyclic-nucleotide phosphodiesterase, catalytic activities. The GRAVY and the instability indexes was −0.21 and 42.75, respectively. The overall physicochemical properties revealed that the protein might be unstable and hydrophilic in nature and interact with ATP in cellular and biochemical processes. Crystallizability of the protein shows class-4 expert pool crystallization and random forest crystallization by molecular-surface features, such as surface entropy, surface hydrophobicity, and surface ruggedness, which were −1.23, −1.39, and 1.15, respectively. Thus, based on the crystallizability of the protein, there might be a suboptimal region comparable with crystallization structures from TargetDB. Furthermore, according to PSIPRED, the predicted secondary structure of the protein consists of 41 coil residues, 42 helix residues, and 17 strand residues. Hence, the mature protein might form a standard 3D structure with helix, coils, loops, and strands, and these are useful for prediction and stabilization of the protein by homology modeling. The other servers, such as COILS, DISOPRED2, TMHMM, RPSP, and SEG, depicted the other molecular features; among these predictions were, from COILS, that TtFRho does not form the leucine zipper domains in transcriptional regulators because of the absence of coiled-coil regions; and, from DISOPRED2, that the protein consists of 18 (5%) longest disorder regions. No transmembrane helices were identified from TMHMM, while eleven low-complexity regions were identified from SEG. Localization studies of TtFRho were predicted from the Cell-PLoc package by fusing the gene-ontology information prediction approach and PSORTb by SCL-BLAST. Both servers depicted the target protein significantly located in the cytoplasmic region of the cell with the score of 9.97, and the SignalP prediction showed that the protein did not have signal peptides. From the above investigation, the target TtFRho protein may form good and reasonable tertiary structure with substrate and ligand molecules. The class-4 crystallizability indicated that this target associated with previous crystal structures. Because of the fact that the target TtFRho protein consists of cytoplasmic localized ATP a nucleotide binding domain was vital for Brucella survival via several cellular processes. The results are depicted in Table 1.
 | Table 1 Molecular characterization and crystallizability predicted from the XtalPred server |
Three-dimensional model construction, optimization, and evaluation
To determine the probable function of B. melitensis 16M TtFRho, the sequence was subjected to comparative modeling using the target protein sequence as a query for PSI-BLAST. The crystal structure of the TtFRho complex with ssRNA substrate and ANP from E. coli (PDB ID: 1PVO) was selected as the template. The 1PVO crystal structure covers 98% of the query residues, 0.0 E-value, and 69% of the sequence identity with the target protein. The alignment with the ALIGN2D module in MODELLER showed the possible region for three target domains (172–177, 184–189, and 215 binding residues of ATP), and the residues showed 80% sequence identity with the template 1PVO residues. The target sequence showed the maximum conserved residue in the coverage of the nucleotide and ATP domains of the TtFRho chain. The PDB structure of 1PVO consists of ssRNA and ANP within the domain coordinates; hence, the supplementary modeling and analysis were processed in the ANP + ssRNA domain. The domain consists of conserved residues that were aligned appropriately with the functionally essential regions of the protein template. The 3D structure of the 1PVO chain A, at 3.0 Å, was used as the coordinate structure for homology modeling. The comparative modeling of the target Brucella’s TtFRho was performed by a restrained-based approach in MODELLER. A set of 100 initial models was constructed for the target protein. The resulting 3D models of TtFRho were sorted according to scores calculated from the DOPE scoring function. Thus, based on this model, number 38 (TtFRho.B99990038) contained lower DOPE energy other than the models, with a value of −46,435.96094 with 2,816.10596 molecular PDF energies. Therefore, model 38 was considered for further studies.
The final errors in the protein structure geometry were regularized via energy minimization to satisfy the special restraints on bond distances and dihedral angles of each amino acid; this was accomplished by applying forces via the MODELLER variable target function method by applying 500 steps of the steepest descent algorithm, optimized with 500 steps of the CG algorithm, and then refining the result using MD with simulated annealing for 1,000 steps. The final refined model was evaluated by stereochemical quality checking by submitting it to the SAVES. Before optimization, PROCHECK showed that 91.9% of residues were present in most favored regions (A, B, L), 6.8% of residues were located in additional allowed regions (a, b, l, p), and 1.4% of residues were located in generously allowed regions (~a, ~b, ~l, ~p); there were no residues present in disallowed regions, and the non-glycine and non-proline residues present in the protein were 27 and 27, respectively. ERRAT showed 80.535. Verify3D showed that 80.09% of the residues had average 3D–one-dimensional (1D) scores. After refinement of the model protein, PROCHECK showed that 90.5% of residues were present in most favored regions (A, B, L), 9.2% of residues were located in additional allowed regions (a, b, l, p), and 0.3% of residues were located in generously allowed regions (~a, ~b, ~l, ~p); there were no residues present in disallowed regions. ERRAT showed a 92.533 overall quality factor, and Verify3D showed a better environmental profile and that 86.06% of the residues had average 3D–1D scores (Figure 1). After optimization, overall quality factors were increased and the error values were reduced by satisfying the special restraints. The Verify3D plot shows a compatibility score above zero, indicating that the protein contains favored side-chain environments and good fold regions. The final optimized target protein secondary structure, and the values of ramachandran plot, Verify 3D plot profile, ERRAT was depicted in Figure 1A, B, C and D respectively.
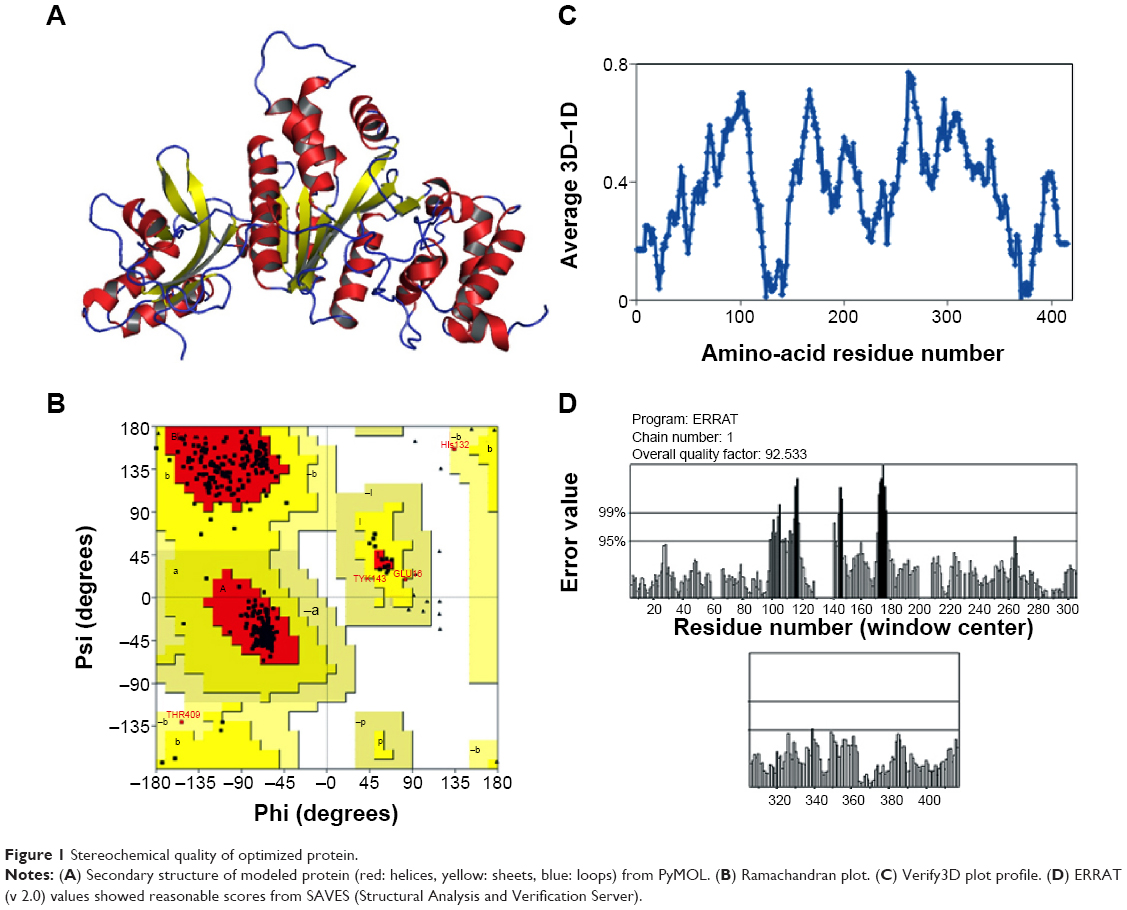 | Figure 1 Stereochemical quality of optimized protein. |
The final model that shared the lowest RMSD was traced out by the Swiss-PdbViewer (also known as “DeepView”) and SuperPose programs, and relative to the backbone and Cα atoms of the coordinate structure (1PVO), it showed 0.33 and 0.56 Å, respectively. The overall quality factors and RMSD results are illustrated in Table 2. Finally, the evaluated structure was submitted to the PMD; TtFRho B. melitensis 16M is available from the PMDB with the accession number PM0079545.
 | Table 2 Significant SAVES (Structure Analysis and Verification Server) results of modeled transcription termination factor Rho protein calculated before and after optimization |
Catalytic cleft analysis and lead optimization
The pocket binding site of the modeled TtFRho was predicted from the CASTp server because the function of an entire protein depends on the binding ability of small molecules or ligands within the binding pocket and interaction with active-site residues with a high degree of specificity.57 The highest volume of active-site residues within the binding pocket were selected for docking studies ie, pocket number 76. The CASTp server results revealed that binding-site 76 consists of 181 to 186 active-site residues involved in nucleotide binding with ATP ([KR]-x-G-K-[TS]-x); also, the 215th residue for ATP binding and 326 optional residues for RNA-binding 2 (by similarity with the ATP region) are also correlated with the template coordinate structure with 80% identity. Thus, the 76th pocket’s active-site residues were selected as grid dimensions. The template coordinate structure 1PVO’s ligand (ie, ANP) was selected as the coordinate template for the structure-based ligand prediction from the ZINC database. The ANP’s physicochemical properties and their structural information were retrieved from the PubChem database with their chemical structure, CID: 33113. Accordingly, the results revealed that ANP has a MW of 506.196266 (g/mol), molecular formula of C10H17N6O12P3, XLOGP: -6, eight HBD, and 17 HBA with standard SMILES. Based on the ANP properties with SMILES, we predict 2,821 potential drug-like compounds for docking studies with 70% structural identity with a nontoxic side-chain environment. The predicted compounds’ errors were resolved by lead optimization by applying the united force-field forces of the ligand bond angle and bond length. Then, the complete refined leads were prepared as datasets with the AutoDock ligand format.
Structure-based virtual screening and flexible docking approaches
The ANP-related structural analogs were used for structure-based virtual screening against the selected active site of the modeled TtFRho protein to understand their molecular interaction and inhibition mode by docking simulation. The grid parameter file was created by AutoGrid. The grid dimension is formed by the proper adjustment of nucleotide binding with ATP binding-site residues. The domain coordinates selected as grid dimensions – the x, y, and z center – and the total grid size values of the grid were −9.262, −23.686, 22.174 and 26.103, 27.756, 30.022, respectively. Subsequently, the TtFRho protein was converted into the AutoDock format, and the Lamarckian geometric algorithm was used for docking simulations. The docking was performed by AutoDock Vina by PyRx virtual screening software using the reference of the template substrate. The docking for 2,821 of the optimized ligands against TtFRho with 50 dock poses was calculated for each ligand. The best dock poses were selected and then refined and analyzed. Based on the virtual screening study, high-binding molecules were separated, and top scoring hits representing 10% of the molecules were selected for further standard flexible docking simulations by AutoDock.4
The residues were involved in the exact binding of incoming ligand molecules with Thr185, Gly186, Lys187, Thr188, and Arg215 within the predicted binding pockets. The PO group of substrates interacted with active-site residues of the N-terminal amino and CA groups of the target protein. Then, the top five ranking compounds were selected from the flexible docking. The docking scores of these compounds have ranges of −10.4 to −10.1 kcal/mol. In fact, among those, ZINC24934545 and ZINC72319544 have a docking score of −10.4 kcal/mol with twelve and ten hydrogen-bond contacts, respectively, with conserved residues involved in hydrogen-bonding interactions, including Arg180, Thr185, Gly186, Lys187, Thr188, Asp213, Arg215, Glu218, Phe235, and Phe358. Three other leads, ZINC49803082, ZINC58554413, and ZINC36093420, have docking scores of −10.3, −10.2, and −10.1, respectively. ZINC49803082 has seven hydrogen-bonding interactions with conserved residues; other pocket residues, such as Asn278, Thr279, and ZINC58554413, have eleven hydrogen-bonding interactions with conserved residues; and still other residues, such as Pro182, Glu218, Arg272, Thr326, and ZINC36093420, have ten hydrogen-bonding interactions with conserved residues only. ZINC24934545 forms two hydrogen bonds with Arg180 (the atoms CA-N interact with the PO group) and three hydrogen bonds with Thr188 (the residue atoms CB-OG1 interact with the PO of different positions). These conserved residues were involved in nucleotide binding with ATP-binding activity. ZINC72319544 formed three hydrogen bonds with Arg215, there are two bonds with CZ-NH2, and one bond with CA-N, interacting with the PO of the ligand. Arg215, an important conserved residue, is involved in ATP-binding activity. The two best binding leads were selected for further toxic evaluation. The predicted docking results of selected compound key features are illustrated in Table 3. Hydrogen bonds and hydrophobic interactions of protein-ligand complexes were analyzed by the LigPlot+ program (Figure 2). The molecular interaction features, such as bond angle, bond distance, the atoms involved in the protein, and lead molecules, were discovered using PyMOL software.
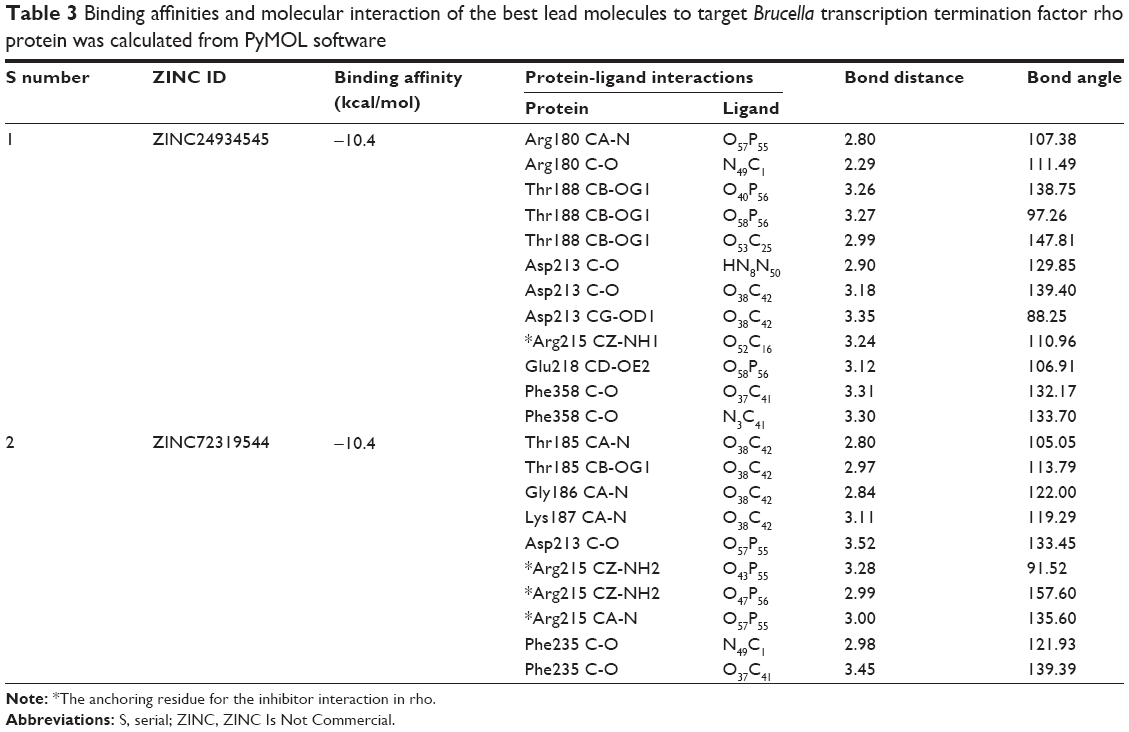 | Table 3 Binding affinities and molecular interaction of the best lead molecules to target Brucella transcription termination factor rho protein was calculated from PyMOL software |
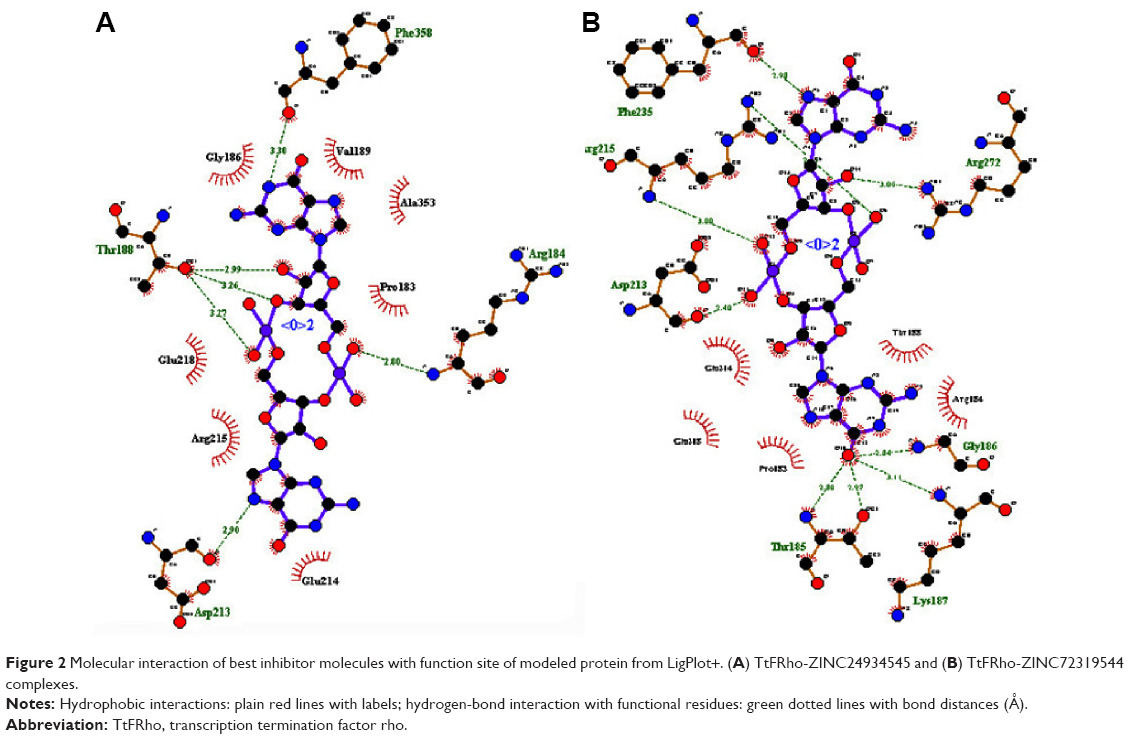 | Figure 2 Molecular interaction of best inhibitor molecules with function site of modeled protein from LigPlot+. (A) TtFRho-ZINC24934545 and (B) TtFRho-ZINC72319544 complexes. |
In silico toxicity and bioactivity prediction
The final set of best hit compounds was submitted to Molinspiration and MolSoft for the calculation of drug likeness and molecular properties. According to these criteria, the best molecules were selected for further studies. The selected compounds showed reasonable logP, MWs, HBA, HBD, molecular polar surface areas, rule of five violations, rotatable bonds, non-hydrogen atoms, and molecular volumes, as shown in Table 4. For the final set of lead candidates, we chose only the molecules with good adherence to the Lipinski rule and drug likeness, no toxicity risks (mutagenicity, tumorigenicity, irritability, and reproductive effectiveness), and the most favorable drug-score features via the OSIRIS server. Chemically related drugs frequently bind to naturally diverse protein targets, and protein receptors with similar sequences or structures do not constantly identify the similar ligands. The SEA tool judges the proteins via a chemo-centric analysis of the chemical similarity of the ligands relating them. Hence, ligand molecules that have similar molecules have similar biological functions and bind to similar protein targets. This classification is performed according to the significant and insignificant E-values (<1×10−10 and >1.0, respectively) of target ligands. The most prominent, interesting, and significant results for the selected leads were obtained for the best hits, ZINC24934545 and ZINC72319544, referenced via the enzyme-inhibition property with E-values of 3.62×10−15 and 1.62×10−10, respectively, as shown in Table 5. ZINC24934545 and ZINC72319544 were selected for complex simulation studies. For that reason, the ligands have high hydrogen-bonding interaction, the same Pfizer’s rule of five (xlogP =−5.07, MW =688.4, HBD =8, HBA =24) and similar bioactivity from Molinspiration (G protein-coupled receptor ligand =0.44, enzyme inhibitor =0.46), along with conserved residues with high binding affinity; reasonable bond angles and bond distances; and nontoxic and similar protein targets from the OSIRIS and SEA analyses. The overall predictions show that both ligands are reasonable drugs for the target protein, and they were analyzed thoroughly by MD simulations.
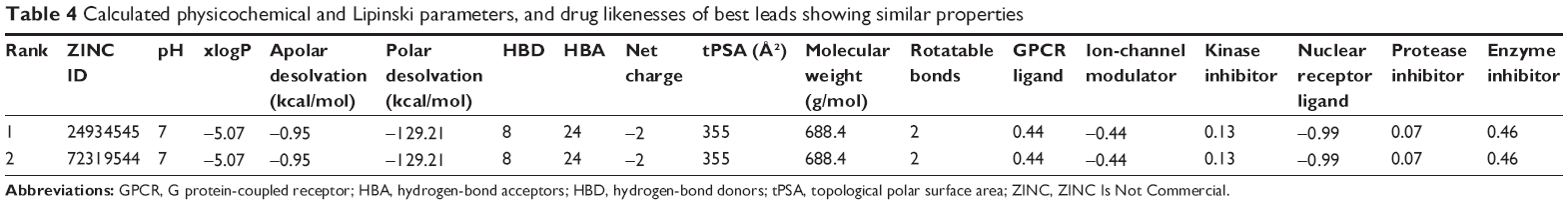 | Table 4 Calculated physicochemical and Lipinski parameters, and drug likenesses of best leads showing similar properties |
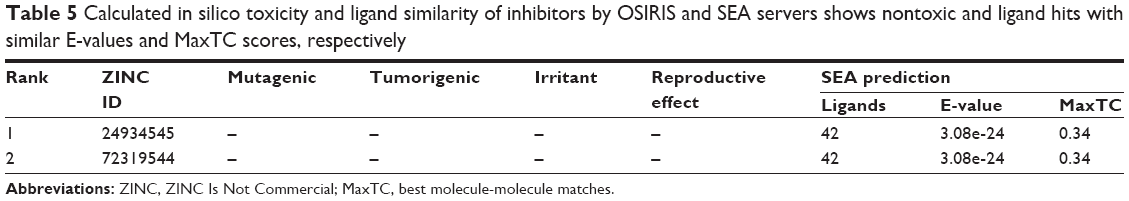 | Table 5 Calculated in silico toxicity and ligand similarity of inhibitors by OSIRIS and SEA servers shows nontoxic and ligand hits with similar E-values and MaxTC scores, respectively |
Protein-ligand molecular dynamics simulations
MD simulation is a well-known theoretical technique and is mainly used for evaluating the stability of any predicted 3D model. Therefore, the constructed 3D homology model of TtFRho was processed for primary MD simulation for a 5 ns timescale with Langevin dynamics to control the kinetic energy, temperature, and/or pressure of the system without binding pocket. It also does not change its conformation during the ligand unbinding in the explicit solvent condition in the NAMD 2.9 package. The results demonstrate the overall conformations of the protein contain RMSD values are normally below 3 Å; only the domain residues showed RMSD values between 3 and 2.5 Å. Hence, the protein water system appears to be equilibrated in a very short time after the start of the free MD simulations. After 2 ns of MD simulations, we prepared the protein inhibitor complex in VMD with parameterized ZINC24934545 and ZINC72319544 compounds. The protein-ligand complex systems were simulated for 2 ns with flexible residues involved in the binding pocket (Thr185, Gly186, Lys187, Thr188, and Arg215) in both alternative conformations. Completion of the total 2 ns free conventional simulation for both enzyme ligand complexes relaxed the functional residue atoms, and a reliable stable conformation was obtained. The RMSD values of both complexes contain alpha carbon atoms (Ca), and all atoms were calculated by taking their respective structures with reference conformation points. The RMSD values of all atoms of TtFRho-ZINC24934545 and TtFRho-ZINC72319544 were stabilized at 1.7 Å (Figure 3); accordingly, the results indicate that there was no change in hydrogen-bond distances (from 1.2 to 1.5 Å) from coordination bonds to the ligands in the simulations (Figure 4A), and average values of fluctuations were less than 1% from RMSF (Figure 5A). In addition, the conformations of the two complexes were stable.
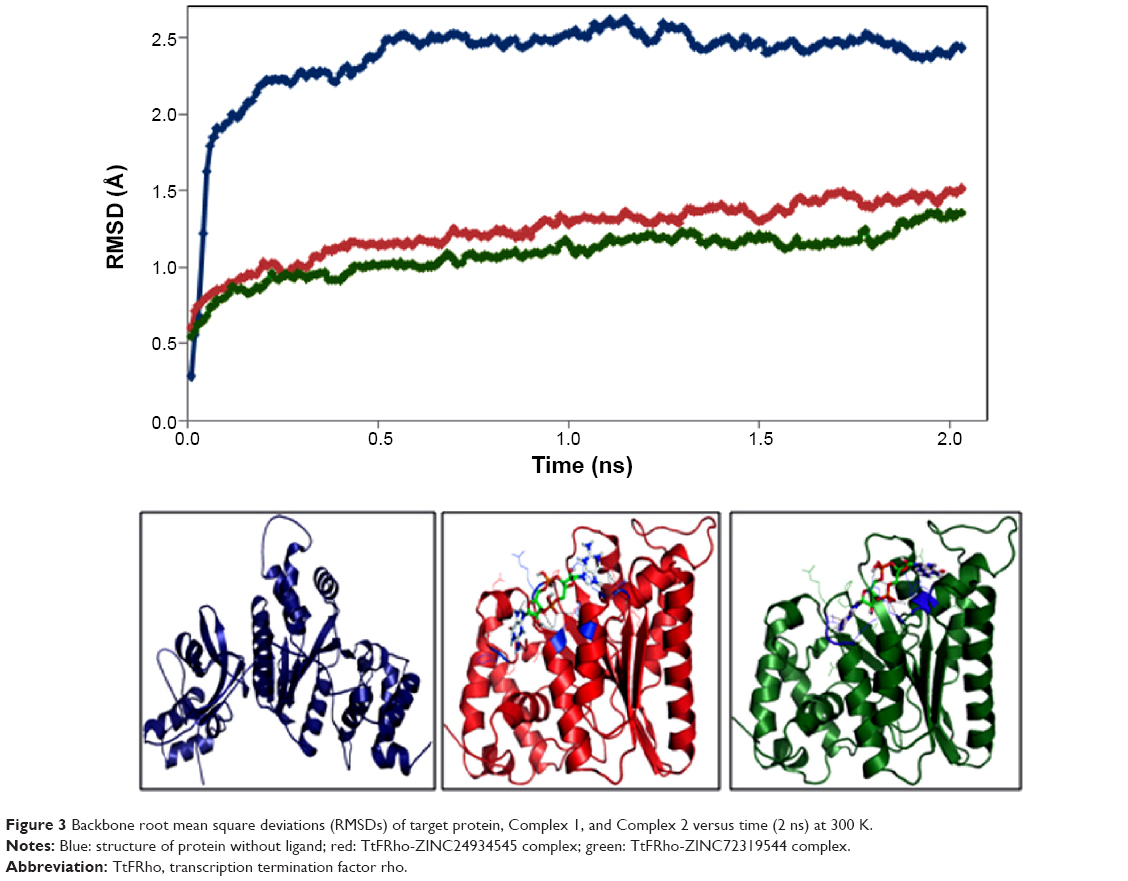 | Figure 3 Backbone root mean square deviations (RMSDs) of target protein, Complex 1, and Complex 2 versus time (2 ns) at 300 K. |
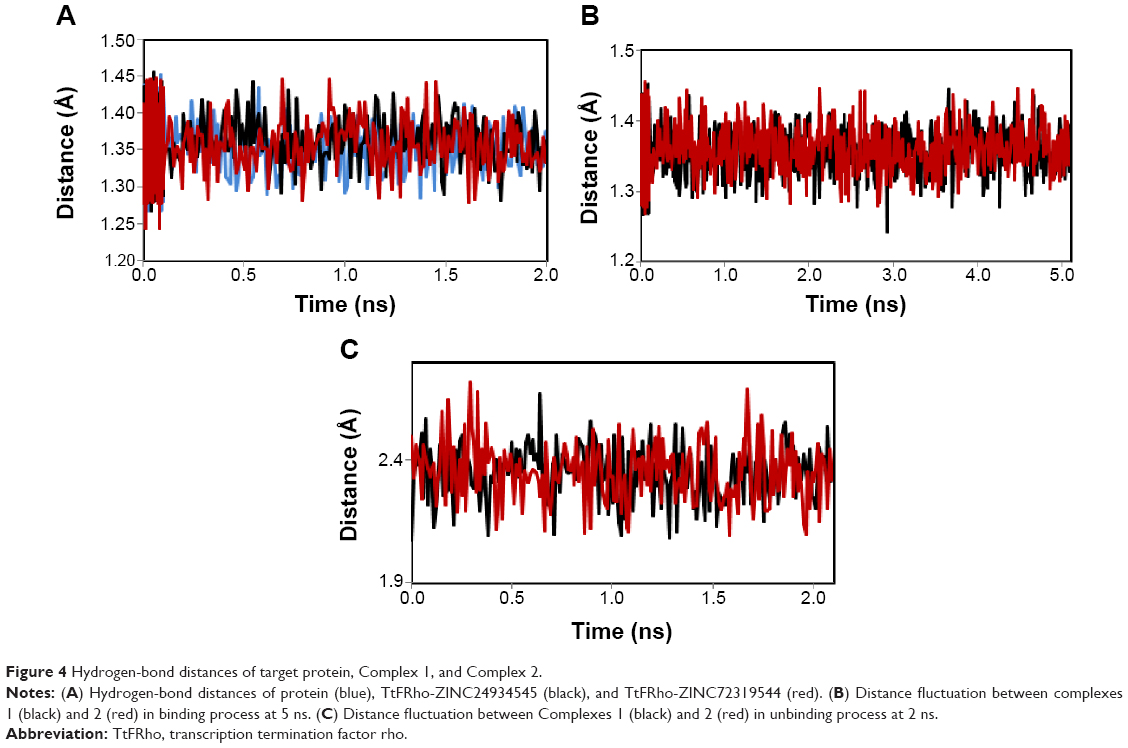 | Figure 4 Hydrogen-bond distances of target protein, Complex 1, and Complex 2. |
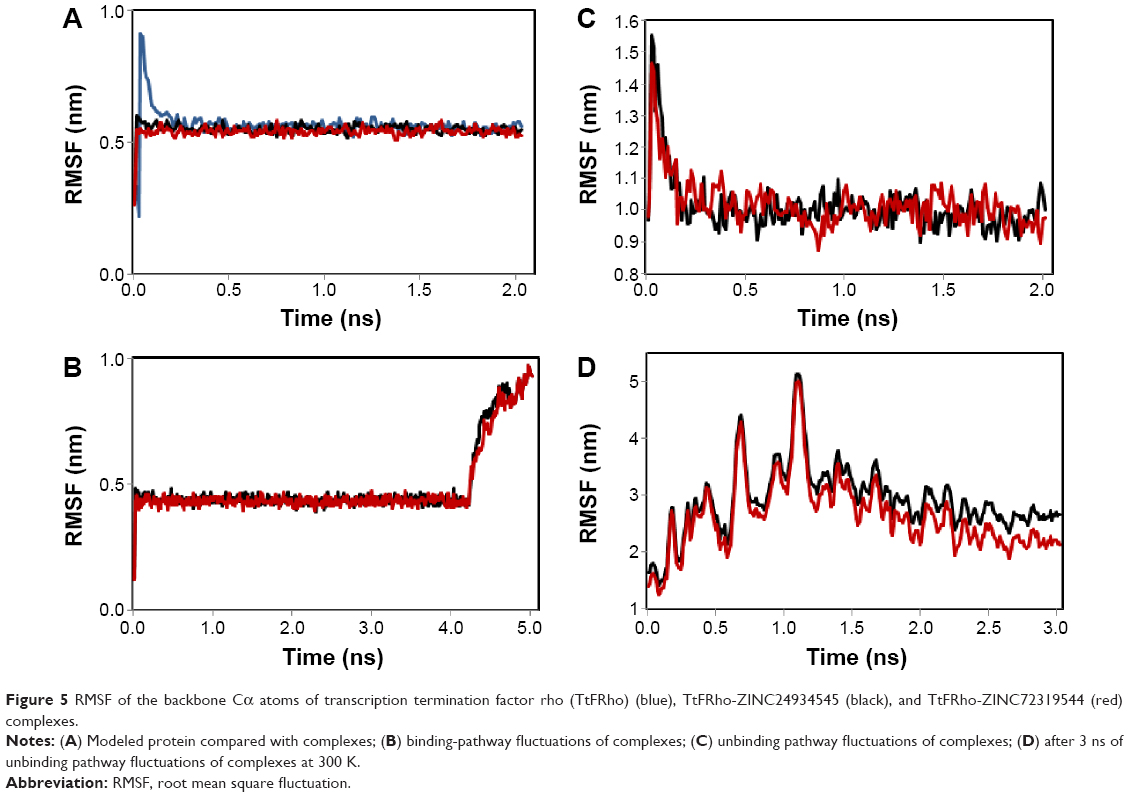 | Figure 5 RMSF of the backbone Cα atoms of transcription termination factor rho (TtFRho) (blue), TtFRho-ZINC24934545 (black), and TtFRho-ZINC72319544 (red) complexes. |
Steered molecular dynamics simulations
Ligand free energy calculations
The two potential binding pathways of ligands were calculated for 5 ns via the SMD simulations. The CHARMM forces applied to the ligands for functional residues were crucial to the specific directions. Here, cv and cf SMD simulations were performed for Ligands 1 (ZINC24934545) and 2 (ZINC72319544) by using fixed atomic binding directions. SMD simulations of binding directions were performed at a constant temperature of 300 K. The identified conformational changes of the protein path when binding to ligands and then stabilized at RMSD 1.5 were analyzed by VMD. The trajectory confirmations read 1–5 ns (Figure 6). A change in hydrogen-bond distances was observed from Complex 1 to Complex 2 – from 1.2 to 1.5 Å, respectively (Figure 4B). The average values of fluctuations were less than 1% from RMSF; hydrogen bonds stabilized between 0 and 4 ns, but increased bond stability was observed at 4 to 5 ns (1 ns timescale) (Figure 5B). The completion of 5 ns of binding-direction SMD simulations revealed that Complex 1 (ie, TtFRho-ZINC24934545) shows lower binding affinity than Complex 2, ie, TtFRho-ZINC72319544. Overall, hydrogen-bond distance and RMSFs in the PO group interaction with Arg218 in the protein very clearly indicate that ZINC72319544 has high binding affinity with low timescale when compared with ZINC24934545.
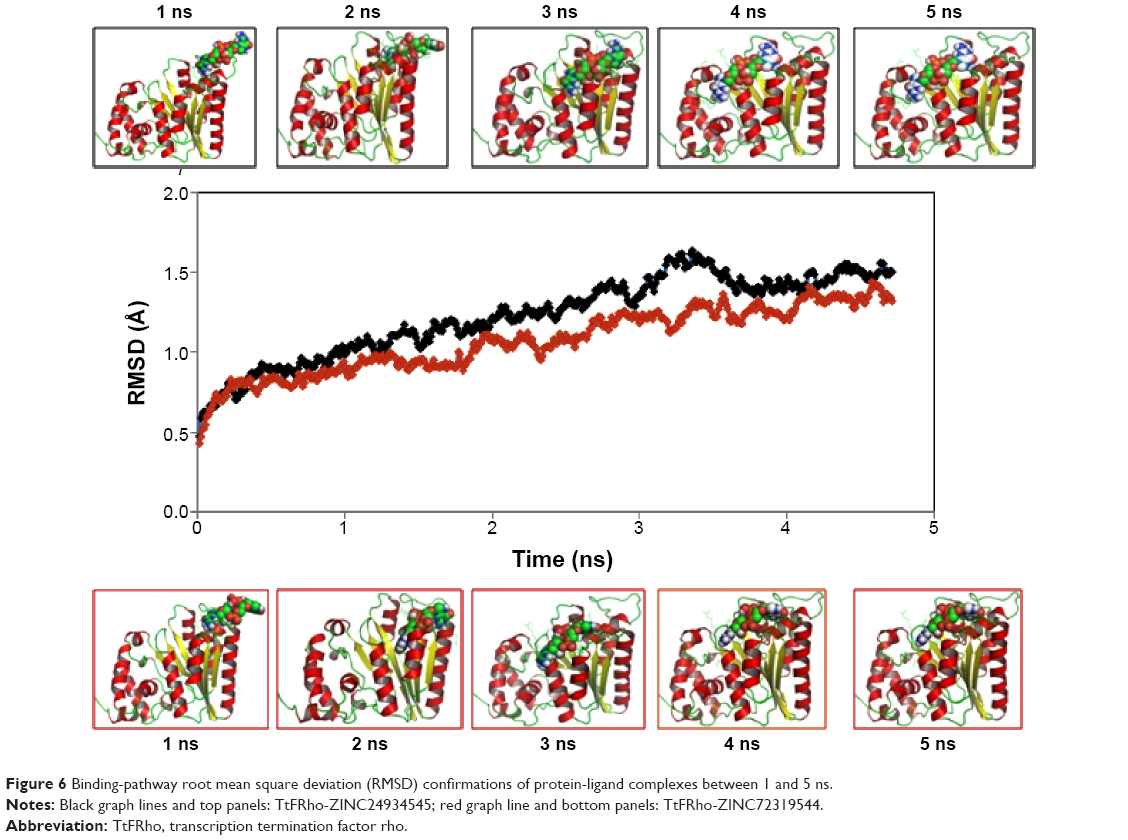 | Figure 6 Binding-pathway root mean square deviation (RMSD) confirmations of protein-ligand complexes between 1 and 5 ns. |
During the unbinding of ligands, forces were crucial for pulling the ligand out from the binding pocket of both protein-ligand complexes, along with possible pathways. Hence, cv and cf SMD simulations were performed to pull Ligands 1 (ZINC24934545) and 2 (ZINC72319544) out using fixed directions. The SMD simulations revealed that, due to divergent features of the inhibitor unbinding process, discovered after the careful examination of the ligand binding directions in both protein-ligand complexes, SMD simulations revealed that the two potential pathways were preferred to carry out the unbinding nature of the ligands. In TtFRho, the two potential pathway vectors are as follows: (1) Direction 1 is resolute by the vector from Arg215 CZ-NH1 to ZINC24934545 and Arg215 CZ-NH2 to ZINC24934545, and (2) Direction 2 is resolute by the vector from Arg215 CZ-NH1 to ZINC72319544 and Arg215 CZ-NH2 to ZINC72319544, in which Pathway 1 and 2 are quite similar but different from their binding atoms in that ZINC24934545 forms CO bonding with Arg215 but ZINC72319544 forms PO bonding with Arg215. Throughout the course of the 2 ns simulations, residues altered their interactions with the inhibitors and pulled out. During the simulations with the 2 ns timescale for the unbinding along Paths 1 and 2 of both complexes, an increase in the applied force was observed; the hydrogen bond made by the PO group of the inhibitor to one of the amino groups of Arg215 required less energy to break than the CO group of the inhibitor, while the broken bond mated to the other possible residue with Arg215, hence was weakened. This result was evident in that a sharp increase in the interaction energy of ZINC72319544 with Arg215, more so than with ZINC24934545; at this confirmation of the complexes, the pocket residues Arg180, Arg184, Thr185, Gly186, Lys187, and Thr188 formed the closed van der Waals interactions with the PO and CO groups of the ligands. The structural superimposition elucidated the RMSD value of 1.5 to 1.0 Å of Cα atoms between the two protein complexes, indicating that the cv SMD of Complex 2 was more stabilized than that of Complex 1. As the cf SMD of both complexes was within 1.5 Å of the RMSD, both are similar and correlated. For each direction in two complexes, four SMD simulations were carried out with the stiff spring constant. All the cv curves showed different changing behavior; cf curves showed similar correlated curves, which strictly suggested that the forces were common for both complexes, but velocity changes were judged by the functional group of the ligand in the protein. These results strongly suggest that the PO group of ZINC72319544 was a much stronger binder to the loop of Arg215 than ZINC24934545 (Figure 7). The change in hydrogen-bond distances was observed from Complex 1 to Complex 2 at a distance of 2.4 Å (Figure 4C). The average value of fluctuations from the RMSF was less than 10%, and hydrogen bonds between them stabilized at 0 to 2 ns with a distance of 1 Å (Figure 5B); however, a standard increased bond stability deviation at 1.5 to 3.0 ns was observed (1.5 ns timescale) (Figure 5D).Therefore, the suite of SMD simulations for binding and unbinding direction analysis revealed that the ZINC72319544 ligand has a higher binding energy and a lower unbinding energy than ZINC24934545, with good drug-like properties and a nontoxic nature.
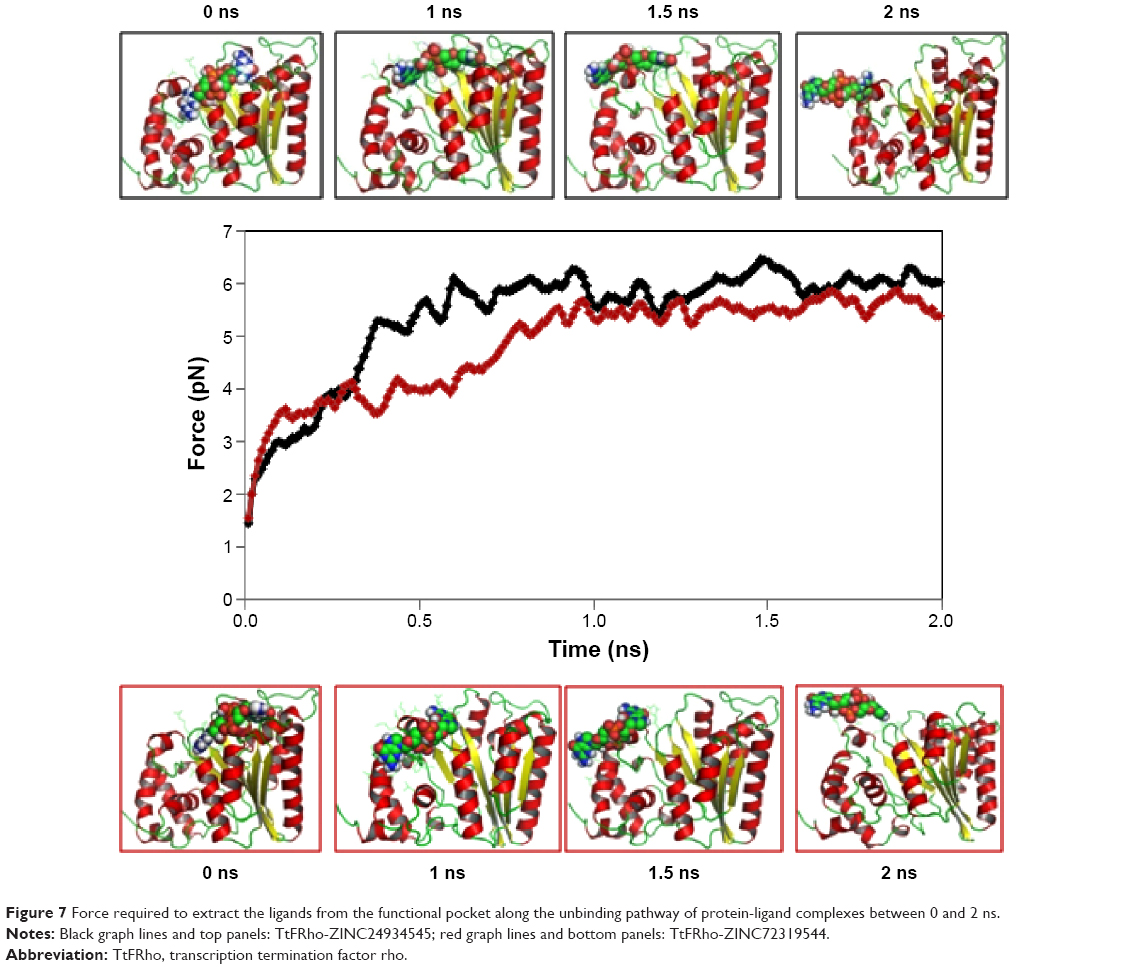 | Figure 7 Force required to extract the ligands from the functional pocket along the unbinding pathway of protein-ligand complexes between 0 and 2 ns. |
Overall this work reveals that both ligands can be used to target therapies for brucellosis by preclinical and clinical strategies via in vitro high-throughput screening for prediction of new drug analogs against Brucella.
Conclusion
Overall, this work suggests that the complete restrained 3D structure of the TtFRho protein is reliable for the binding of inhibitors. Herein, we identified the two best inhibitors – ZINC24934545 and ZINC72319544 – which are compounds with similar binding affinity and an enzyme-inhibition property, by structure-based virtual screening and molecular-docking approaches and identification of their inhibitor specificity with functional cleft by further MD simulations.
The binding-pathway analysis revealed that ZINC72319544 has higher binding affinity and fewer error fluctuations than ZINC24934545. The unbinding routes of both ligands were found to be similar, extending from the active site and down toward the cytoplasm. These inhibitors bind to the active site by one possible direction, owing to the conformational change to the binding pocket in the loop of the active site. The inhibitors then leave the active site through the base of the binding site by one pathway that remains unobstructed after the movement to the cytoplasm. This places the ligand at some detachment from the binding site, and the protein goes through a conformational alteration back into the closed, inactive state. The key amino acids – Arg180, Arg184, Thr185, Gly186, Lys187, Thr188, Asp213, Arg215, Phe235, and Arg272 – may play a vital role in binding and unbinding of inhibitors.
Thus, both identified compounds may serve as a lead for developing potential candidates against B. melitensis 16M. Additionally, the rotatable axis of the lead molecules with functional amino acids provides clues for the discovery of inhibitors for other Brucella spp. targets in the drug-discovery process.
Acknowledgment
We would like to thank Dr SB Sainath, Vikrama Simhapuri University, India, for his valuable suggestions.
Disclosure
The authors declare no conflicts of interest in this work.
References
Espinosa EJ, Calero M, Sridevi K, Pfeffer SR. RhoBTB3: a Rho GTPase-family ATPase required for endosome to Golgi transport. Cell. 2009;137(5):938–948. | ||
Banerjee S, Chalissery J, Bandey I, Sen R. Rho-dependent transcription termination: more questions than answers. J Microbiol. 2006;44(1):11–22. | ||
Caldelari I, Chao Y, Romby P, Vogel J. RNA-mediated regulation in pathogenic bacteria. Cold Spring Harb Perspect Med. 2013;3(9):a010298. | ||
Schmidt MC, Chamberlin MJ. Binding of Rho factor to Escherichia coli RNA polymerase mediated by nusA protein. J Biol Chem. 1984;259(24):15000–15002. | ||
Ingham CJ, Dennis J, Furneaux PA. Autogenous regulation of transcription termination factor Rho and the requirement for Nus factors in Bacillus subtilis. Mol Microbiol. 1999;31(2):651–663. | ||
Fiorentini C, Falzano L, Travaglione S, Fabbri A. Hijacking Rho GTPases by protein toxins and apoptosis: molecular strategies of pathogenic bacteria. Cell Death Differ. 2003;10(2):147–152. | ||
Veeranagouda Y, Husain F, Tenorio EL, Wexler HM. Identification of genes required for the survival of B. fragilis using massive parallel sequencing of a saturated transposon mutant library. BMC Genomics. 2014;15:429. | ||
Skordalakes E, Berger JM. Structural insights into RNA-dependent ring closure and ATPase activation by the Rho termination factor. Cell. 2006;127(3):553–564. | ||
Paulsen IT, Seshadri R, Nelson KE, et al. The Brucella suis genome reveals fundamental similarities between animal and plant pathogens and symbionts. Proc Natl Acad Sci U S A. 2002;99(20):13148–13153. | ||
Slabinski L, Jaroszewski L, Rychlewski L, Wilson IA, Lesley SA, Godzik A. XtalPred: a web server for prediction of protein crystallizability. Bioinformatics. 2007;23(24):3403–3405. | ||
Slabinski L, Jaroszewski L, Rodrigues AP, et al. The challenge of protein structure determination-lessons from structural genomics. Protein Sci. 2007;16(11):2472–2482. | ||
Lupas A, Van Dyke M, Stock J. Predicting coiled coils from protein sequences. Science. 1991;252(5009):1162–1164. | ||
Ward JJ, McGuffin LJ, Bryson K, Buxton BF, Jones DT. The DISOPRED server for the prediction of protein disorder. Bioinformatics. 2004;20(13):2138–2139. | ||
Krogh A, Larsson B, von Heijne G, Sonnhammer EL. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol. 2001;305(3):567–580. | ||
Plewczynski D, Slabinski L, Ginalski K, Rychlewski L. Prediction of signal peptides in protein sequences by neural networks. Acta Biochim Pol. 2008;55(2):261–267. | ||
Wootton JC. Non-globular domains in protein sequences: automated segmentation using complexity measures. Comput Chem. 1994;18(3):269–285. | ||
Jones DT. Protein secondary structure prediction based on position-specific scoring matrices. J Mol Biol. 1999;292(2):195–202. | ||
Chou KC, Shen HB. Cell-PLoc: a package of Web servers for predicting subcellular localization of proteins in various organisms. Nat Protoc. 2008;3(2):153–162. | ||
Yu NY, Wagner JR, Laird MR, et al. PSORTb 3.0: improved protein subcellular localization prediction with refined localization subcategories and predictive capabilities for all prokaryotes. Bioinformatics. 2010;26(13):1608–1615. | ||
Petersen TN, Brunak S, von Heijne G, Nielsen H. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods. 2011;8(10):785–786. | ||
Altschul SF, Madden TL, Schaffer AA, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25(17):3389–3402. | ||
Berman HM, Battistuz T, Bhat TN, et al. The Protein Data Bank. Acta Crystallogr D Biol Crystallogr. 2002;58(Pt 6 No 1):899–907. | ||
Skordalakes E, Berger JM. Structure of the Rho transcription terminator: mechanism of mRNA recognition and helicase loading. Cell. 2003;114(1):135–146. | ||
Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol. 1993;234(3):779–815. | ||
Fiser A, Do RK, Sali A. Modeling of loops in protein structures. Protein Sci. 2000;9(9):1753–1773. | ||
Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr. 1993;26(Pt 2):283–291. | ||
Colovos C, Yeates T. Verification of protein structures: patterns of nonbonded atomic interactions. Protein Sci. 1993;2(9):1511–1519. | ||
Lüthy R, Bowie JU, Eisenberg D. Assessment of protein models with three-dimensional profiles. Nature. 1992;356(6364):83–85. | ||
Wiederstein M, Sippl MJ. ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007;35(Web Server issue):W407–W410. | ||
Maiti R, Van Domselaar GH, Zhang H, Wishart DS. SuperPose: a simple server for sophisticated structural superposition. Nucleic Acids Res. 2004;32(Web Server issue):W590–W594. | ||
Castrignanò T, De Meo PD, Cozzetto D, Talamo IG, Tramontano A. The PMDB Protein Model Database. Nucleic Acids Res. 2006;34 (Database issue):D306–D309. | ||
Sarma K, Sen P, Barooah M, Choudhury MD, Roychoudhury S, Modi MK. Structural comparison, substrate specificity, and inhibitor binding of AGPase small subunit from monocot and dicot: present insight and future potential. Biomed Res Int. 2014;2014:583606. | ||
Dundas J, Ouyang Z, Tseng J, Binkowski A, Turpaz Y, Liang J. CASTp: computed atlas of surface topography of proteins with structural and topographical mapping of functionally annotated residues. Nucleic Acids Res. 2006;34(Web Server issue):W116–W118. | ||
Irwin JJ, Shoichet BK. ZINC – a free database of commercially available compounds for virtual screening. J Chem Inf Model. 2005;45(1):177–182. | ||
O’Boyle NM, Banck M, James CA, Morley C, Vandermeersch T, Hutchison GR. Open Babel: An open chemical toolbox. J Cheminform. 2011;3:33. | ||
Morris GM, Huey R, Lindstrom W, et al. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J Comput Chem. 2009;30(16):2785–2791. | ||
Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31(2):455–461. | ||
Laskowski RA, Swindells MB. LigPlot+: multiple ligand-protein interaction diagrams for drug discovery. J Chem Inf Model. 2011;51(10):2778–2786. | ||
Kay HY, Wu H, Lee SI, Kim SG. Applications of genetically modified tools to safety assessment in drug development. Toxicol Res. 2010;26(1):1–8. | ||
Sander T, Freyss J, von Korff M, Reich JR, Rufener C. OSIRIS, an entirely in-house developed drug discovery informatics system. J Chem Inf Model. 2009;49(2):232-246. | ||
Keiser M, Roth B, Armbruster B, Ernsberger P, Irwin J, Shoichet B. Relating protein pharmacology by ligand chemistry. Nat Biotechnol. 2007;25(2):197–206. | ||
Nelson MT, Humphrey W, Gursoy A, et al. NAMD: a parallel, object-oriented molecular dynamics program. Int J Supercomput Appl. 1996;10(4):251–268. | ||
Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph. 1996;14(1):33–38, 27–28. | ||
Lopes PE, Huang J, Shim J, et al. Polarizable force field for peptides and proteins based on the classical Drude oscillator. J Chem Theory Comput. 2013;9(12):5430–5449. | ||
Essmann U, Perera L, Berkowitz ML, Darden T, Hsing L, Pedersen LG. A smooth particle mesh Ewald method. J Chem Phys. 1995;103(19):8577–8593. | ||
Feller S, Zhang Y, Pastor R, Brooks B. Constant pressure molecular dynamics simulation: the Langevin piston method. J Chem Phys. 1995;103:4613–4621. | ||
Pettersen EF, Goddard TD, Huang CC, et al. UCSF Chimera – a visualization system for exploratory research and analysis. J Comput Chem. 2004;25(13):1605–1612. | ||
Zoete V, Cuendet MA, Grosdidier A, Michielin O. SwissParam, a Fast Force Field Generation Tool For Small Organic Molecules. J Comput Chem. 2011;32(11):2359–2368. | ||
Bishop TC, Kosztin D, Schulten K. How hormone receptor-DNA binding affects nucleosomal DNA: the role of symmetry. Biophys J. 1997;72(5):2056–2067. | ||
Kosztin D, Bishop TC, Schulten K. Binding of the estrogen receptor to DNA: the role of waters. Biophys J. 1997;73(2):557–570. | ||
Jarzynski C. Nonequilibrium equality for free energy differences. Phys Rev Lett. 1997;78(14):2690–2693. | ||
Phillips JC, Braun R, Wang W, et al. Scalable molecular dynamics with NAMD. J Comp Chem. 2005;26(16):1781–1802. | ||
Izrailev S, Stepaniants S, Balsera M, Oono Y, Schulten K. Molecular dynamics study of unbinding of the avidin-biotin complex. Biophys J. 1997;72(4):1568–1581. | ||
Balsera M, Stepaniants S, Izrailev S, Oono Y, Schulten K. Reconstructing potential energy functions from simulated force-induced unbinding processes. Biophys J. 1997;73(3):1281–1287. | ||
Evans E, Ritchie K. Dynamic strength of molecular adhesion bonds. Biophys J. 1997;72(4):1541–1555. | ||
Kabsch W. A solution for the best rotation to relate two sets of vectors. Acta Crystallogr A Found Adv. 1976;A32:922–923. | ||
Lodish H, Berk A, Zipursky SL, Matsudaira P, Baltimore D, Darnell J. Functional design of proteins. Molecular Cell Biology. 4th ed. New York, NY: WH Freeman; 2000: Section 3.3. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.