")
Back to Journals » International Journal of Nanomedicine » Volume 14
Mitoxantrone-preloaded water-responsive phospholipid-amorphous calcium carbonate hybrid nanoparticles for targeted and effective cancer therapy
Authors Wang C, Han M, Liu X, Chen S, Hu F, Sun J, Yuan H
Received 9 November 2018
Accepted for publication 12 January 2019
Published 25 February 2019 Volume 2019:14 Pages 1503—1517
DOI https://doi.org/10.2147/IJN.S193976
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Linlin Sun
Cheng Wang,1,* Min Han,1,2,* Xuerong Liu,1 Shaoqing Chen,1 Fuqiang Hu,1 Jihong Sun,3 Hong Yuan1
1Department of Pharmaceutics, College of Pharmaceutical Sciences, Zhejiang University, Hangzhou 310058, China; 2Hangzhou Zhongmei Huadong Pharmaceutical Co, Ltd, Hangzhou 310011, China; 3Department of Radiology, Sir Run Run Shaw Hospital, School of Medicine, Zhejiang University, Hangzhou 310016, China
*These authors contributed equally to this work
Background: The application of mitoxantrone (MIT) in cancer therapy has been severely limited by its inherent drawbacks. In addition, effective cancer therapy calls for drug release systems capable of enforcing drug release within cancer cells in response to infinite stimulant with enhanced drug penetration capability.
Methods: MIT-preloaded phospholipid-amorphous calcium carbonate hybrid nanoparticles (PL/ACC-MIT) that surface modified with PL shell (containing shielding polymer polyethylene glycol and targeting moiety folic acid) were prepared by a facile solvent-diffusion method.
Results: It has been proven that the resulting PL/ACC-MIT nanoparticles demonstrated satisfactory stability against various aqueous environments with minimal drug leakage and exerted strong targeting capability but selective preference to the folate receptor-overexpressing cell line. In contrast, once exposed to the enzyme-abundant and acidic environments of cancer cells, the PL/ACC-MIT nanoparticles can readily decompose to facilitate quick drug release and enhanced drug penetration to yield preferable antitumor effect both in vitro and in vivo.
Conclusion: In this study, MIT-preloaded water-responsive hybrid nanoparticles with increased stability, targetability, controlled drug release, and enhanced drug penetration were successfully developed, which might be a candidate for targeted and effective cancer therapy.
Keywords: mitoxantrone, water responsive, hybrid nanoparticles, amorphous calcium carbonate, cancer therapy
Introduction
Currently, cancer remains one of the most lethal diseases around the world requiring unremitting efforts.1 Among the most widely adopted antitumor drugs, mitoxantrone (MIT) is a chemotherapeutic agent that can intercalate into DNA or RNA through hydrogen bonding to induce cross-links and strand breaks. Furthermore, MIT can also facilitate topoisomerase II inhibition as well as mitochondrial pathway inhibition.2 However, nonspecific cell death or apoptosis induced by MIT can injure both healthy and cancer cells. As a result, despite the utilization of MIT for the treatment of a wide range of cancers,3 inadvertent side effects, especially cardiotoxicity, limit its further clinical application in cancer therapy.4
The generally recognized approach to minimize the side effects of chemotherapy is to introduce drug delivery systems (DDSs).5 Although selective accumulation of DDSs in the neoplastic area can be easily satisfied by tumor homing or in situ injection technologies,6,7 insufficient drug release within targeted sites remains a pending issue that requires additional efforts.8,9 Many currently available DDSs usually suffer from ineffective drug release whereby high local drug levels cannot translate into sufficiently available drug levels.10 For some responsive ones, drug release might be impaired due to the consumption of corresponding stimulants (such as H+ and glutathione)11,12 during the release process. This calls for a new and effective approach that enforces drug release in the cancer cells in response to some infinite stimulants. Moreover, it has been found that normal DDSs are predominantly located near the blood vessels and only delivered to the cells on the tumor periphery, which is primarily attributed to the physiological barriers of the solid tumor that impedes the uniform distribution of anticancer drugs throughout the tumor in a therapeutic concentration.13
Calcium carbonate as a naturally nontoxic inorganic biomaterial has multiple intrinsic advantages that are suitable for broad biomedical applications.14 It can be divided into crystallized and amorphous categories based on its polymorph. Unlike crystallized calcium carbonate which usually is in the micrometer range, nanosized amorphous calcium carbonate (ACC) can be more readily obtained by various simple methods.15,16 The phenomenon that ACC will dissolve and/or transfer polymorph in water has generally been recognized as the major obstacle for its extended application in nanomedicine.16 However, in our view, if this characteristic can be preserved and yet be realized specifically within targeted sites, it would be beneficial to enforce drug release within cells in a water-responsive manner.
The use of organic-inorganic hybrid nanocarriers for controlled release of antitumor drugs has gained great interest, in particular to improve the selectivity and efficacy of drugs.17–19 Compared with single carriers, hybrid nanocarriers built upon the success of traditional platforms with further optimizations in material, size, and structural properties could greatly improve the therapeutic potential.20,21 The development of phospholipid (PL)-calcium carbonate hybrid nanoparticles would be one such example, as it combines the merits of nanostructured lipid carriers and calcium carbonate, both of which have been extensively evaluated in many previous studies.14,22 Hybrid nanoparticles comprise a drug-preloaded ACC core surrounded by a lipid layer that might be a promising platform to satisfy the requirements for employing ACC as a DDS. The lipid layer provides a biocompatible and water-resistant coating that enhances residence time in the bloodstream and preserves the water-responsive nature of ACC, while also acting as a diffusional barrier against potential drug leakage. Moreover, the lipid layer could be further modified with various shielding or targeting moieties by self-assembly, which has unique advantages including flexibility, convenience, and mild preparation compared with chemical reactions.23,24
In our study, MIT was preloaded into ACC during the synthesis process (ACC-MIT). The obtained ACC-MIT nanoparticles were then coated with PL, during which shielding polymer polyethylene glycol (PEG) and targeting moiety folic acid (FA) were inserted into the PL shell of the as-prepared hybrid nanoparticles in one-pot approach to finally construct a hybrid platform (PL/ACC-MIT) for targeted and effective cancer therapy. Compared with the currently available targeted platforms,1,25 the PL/ACC-MIT nanoparticles were expected to possess similar tumor homing capability with controllable water-responsive nature that can degrade to burst release the loaded drug molecules specifically in tumor cells and achieve deep tumor penetration. The ability to synthesize a hybrid water-responsive version of the established lipid and calcium carbonate platforms can ultimately enhance their applicability across a wide range of scientific fields.
Materials and methods
Materials
Ammonium carbonate ((NH4)2CO3) and anhydrous calcium chloride (CaCl2) were purchased from Sinopharm Chemical Reagent Co, Ltd (Shanghai, China). MIT was a gift from Dalian Meilun Biotech Co, Ltd (Dalian, China). PL (S100) and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-(methoxy[polyethylene glycol]-2000) (DSPE-PEG2000) were obtained from Lipoid GmbH (Ludwigshafen, German). DSPE-PEG-FA (35,000 Da) was obtained from Ponsure Biotechnology (Shanghai, China). LysoTracker Blue, Fluo-4 AM, fluorescein diacetate (FDA), propidium iodide (PI), and 1,1′-dioctadecyl-3,3,3′,3′-tetramethyl indotricarbocyanine iodide (DiR) were obtained from Thermo Fisher Scientific (Waltham, MA, USA). All other chemicals and reagents otherwise stated were from Sinopharm Chemical Reagent Co, Ltd and of analytical grade.
Cell culture
HeLa (human cervical carcinoma) was a gift from Dr Pengfei Cui (China Pharmaceutical University) and use of the gifted cell was approved by the institutional review board of the College of Pharmaceutical Sciences (Zhejiang University, China). A549 (human lung carcinoma) and NIH3T3 (mouse embryonic fibroblast) cell lines were purchased from the Institute of Biochemistry and Cell Biology (Shanghai, China). All cells were cultured in folate-free DMEM (Sigma-Aldrich Co, St Louis, MO, USA) supplemented with 10% (v/v) FBS (Hyclone Laboratories, Logan, UT, USA), 100 U/mL penicillin, and 100 μg/mL streptomycin (Beyotime Institute of Biotechnology, Jiangsu, China) in a humidified atmosphere of 95% air/5% CO2 in an incubator (Thermo Forma 311; Thermo Fisher Scientific) at 37°C. All experiments were performed on cells in the logarithmic phase of growth.
Multicellular tumor spheroid (MCTS) model
A 96-well plate (Corning Incorporated, Corning, NY, USA) was first covered with autoclaved agarose solution (1.5%, w/v) at 50 μL/well and then cooled to room temperature. Mixed HeLa and NIH3T3 cells (1:1) were seeded at a density of 2×103 cells/well and incubated for 4 days to grow into MCTS. The formation of MCTS was monitored using an optical microscope (TE2000-S; Nikon Corporation, Tokyo, Japan).
Animal model
Female New Zealand rabbits (5–7 months, 2.5 kg) and BALB/c nude mice (4–5 weeks, 16 g) purchased from Shanghai Laboratory Animal Center (China) were maintained at 22°C±2°C with access to food and water ad libitum. All animal experiments were approved by the Animal Care and Use Committee of Zhejiang University in accordance with the guidelines for the care and use of laboratory animals. Single HeLa or HeLa/A549 co-bearing tumor xenograft model was established according to a previous report with minor modifications.26 Briefly, suspensions of HeLa (1×107) or A549 (2×106) cells in 100 μL of PBS (0.01 M, pH 7.4) were inoculated subcutaneously in the flank of nude mice. Tumor sizes were measured using a vernier caliper, and tumor volumes were calculated as V=a2×b/2 mm3 (a: minor axis; b: major axis).
Synthesis of ACC-MIT nanoparticles
ACC-MIT nanoparticles were synthesized in accordance with a previous report with some modifications.16 Briefly, CaCl2 (200 mg) and MIT (4 mg/mL) were dissolved in absolute ethanol (100 mL). The mixture was transferred into a glass bottle and covered by parafilm with several pores. The bottle was then left in a desiccator along with two glass bottles of (NH4)2CO3 and one glass bottle of concentrated sulfuric acid at 20°C. After vapor diffusion reaction overnight, the products were centrifuged (Allegra 64R; Beckman Coulter, Brea, CA, USA; 8,000 rpm, 10 minutes), rinsed several times, and then re-dispersed in a proper amount of absolute ethanol. Drug-free ACC nanoparticles were prepared in the same way without MIT.
Preparation of PL/ACC-MIT
The ACC-MIT nanoparticles were then dispersed into mixed solution (ethanol:chloroform =1, v/v) containing PL (PL:ACC-MIT =1, w/w) and stirred at 37°C for 24 hours. After that, the mixture was centrifuged at 3,000 rpm for 10 minutes to remove unbounded free PL. The mixture was then subjected to rotary evaporation under vacuum at 50°C for 30 minutes, followed by vacuum drying in a desiccator for 6 hours to form a dried film on the bottle wall and to thoroughly remove the organic solvent. The film was dissolved in absolute ethanol, to which 20% (w/w to PL) of DSPE-PEG and DSPE-PEG-FA (1:1, w/w) were added. The mixed solution was then injected (via an injection syringe) into water under moderate stirring at a volume ratio of 1:9 (ethanol:water). The obtained PL/ACC-MIT nanoparticles underwent further dialysis against distilled water (EMD Millipore, Billerica, MA, USA) for 4 hours (molecular weight cutoff [MWCO]: 7 kDa, 2 L×6) with gentle agitation to remove unloaded drugs, followed by storage at 4°C until further use. Drug-free PL/ACC nanoparticles were prepared in the same way using ACC nanoparticles. The nanoparticles adopted in the following experiments all incorporated FA modification unless otherwise stated.
Morphology, particle size, zeta potential, and drug loading
The morphology and particle size of different nanoparticles were observed by transmission electron microscopy (TEM). Briefly, a drop of nanoparticle solution was deposited on a lacy carbon-coated cupper grid. The excess solution was removed by a piece of filter paper. The sample was air dried and observed under a TEM system (JEM-1200; JEOL, Tokyo, Japan) at an accelerating voltage of 80 kV. Moreover, the comparative particle size and zeta potential were further determined by a particle sizer (Nano S90; Malvern Instruments Ltd, Malvern, UK) and zeta analyzer (Litesizer 500; Anton Paar, Austria), respectively. The obtained nanoparticles were dissolved in a mixed solution of ethanol and HCl (ethanol:0.6 M HCl, 1:1, v/v). The drug loading content was determined by HPLC (Agilent 1100 series; Agilent Technologies, Santa Clara, CA, USA) using the following conditions. The mobile phase was a mixed solution of 0.3% aqueous sodium 1-heptanesulfonate (adjusted to pH =2 by phosphoric acid) and acetonitrile (72:28, v/v). The flow rate was 1.0 mL/min, the detection wavelength was set at 240 nm, and the column was Agilent ZORBAX 300 SB-C18 column (4.6×250 mm, 5 μm) with a column temperature of 40°C.27 The drug loading content and encapsulation efficiency of PL/ACC-MIT were determined using the following equations:
|
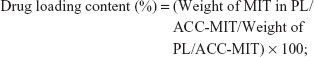
|
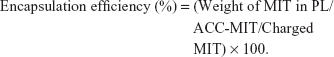
Stability and hemolysis studies
ACC-MIT and PL/ACC-MIT nanoparticles were incubated with mouse plasma at 37°C for 2 hours. Moreover, PL/ACC-MIT nanoparticles were incubated with PBS (0.01 M, pH 7.4) or mouse plasma at 37°C for 48 hours. Samples were measured at different time intervals and changes in particle size were recorded. For the hemolysis assay, red blood cells (RBCs) were first obtained from a New Zealand rabbit and diluted to 2% suspension with saline solution. The PL/ACC-MIT nanoparticles were added into 2% RBCs suspension with the same volume to achieve the designated concentrations (0.1, 0.25, 0.5, 0.75, 1 mg/mL) and incubated at 37°C for 1 hour. RBCs suspension was also incubated with saline and distilled water under the same condition as negative (0% hemolysis) and positive controls (100% hemolysis), respectively. Thereafter, all the samples were centrifuged at 3,000 rpm for 10 minutes and the absorption values (represented as the counts of released hemoglobin) of the same volume of supernatants were measured at 545 nm by an ultraviolet spectrophotometer (TU-1810; Purkinje General Instrument Co, Beijing, China).28
Drug release
The drug release profile of ACC-MIT and PL/ACC-MIT was investigated by dialysis method.29 Briefly, samples (containing 50 μg MIT) were placed into individual dialysis bags (MWCO: 7 KDa) and immersed in plastic tubes containing 50 mL of phosphate buffer with pH of 7.4 and 5.5, respectively. The plastic tubes were fixed in a thermostatic shaker (HZQ-C; Haerbin Dongming Medical Instrument Factory, Haerbin, China) at 37°C with a stirring speed of 100 rpm. At predetermined time intervals, 200 μL of the buffer solution within the tubes was withdrawn for analysis and replaced with an equal volume of fresh medium. The drug concentration was determined by HPLC as mentioned earlier.
In vitro targeting assay
To evaluate the cellular uptake of folate-modified PL/ACC-MIT nanoparticles in HeLa and A549 cell lines, 0.1% (w/w of PL) of the fluorescent probe coumarin-6 (C6) was loaded into the lipid matrix of PL/ACC-MIT nanoparticles during the preparation procedure. HeLa and A549 cells were seeded onto 35-mm glass-bottom culture dishes (Corning Incorporated) at a density of 1×105 cells/well and cultured overnight for 50%–60% confluence. The primary media were replaced by fresh serum-free medium containing PL/ACC-MIT nanoparticles. After 2, 4, and 8 hours of incubation, the cells were fixed with 4% paraformaldehyde (15 minutes), followed by staining with DAPI (1 μg/mL) for 15 minutes. Samples were subjected to confocal laser scanning microscopy (CLSM, IX83-FV3000-OSR; Olympus Corporation, Tokyo, Japan) observations.
Drug localization
HeLa cells were seeded onto 35-mm glass-bottom culture dishes and cultured as described earlier. Thereafter, the primary culture media were removed and replaced with an equal volume of serum-free medium containing PL/ACC-MIT nanoparticles (final MIT concentration: 1 μg/mL). At a predetermined co-incubation time, the cells were observed and imaged under CLSM.
Intracellular trafficking
Cells cultured under the same condition as described earlier were adopted. To observe the intracellular trafficking of PL/ACC-MIT nanoparticles, the cells were first incubated with Hank’s balanced salt solution (HBSS) containing PL/ACC-MIT nanoparticles at 37°C for 4 hours. At the end of incubation, Fluo-4 AM (5 μM) and LysoTracker Blue (50 nM) were added for incubation with cells for 30 and 15 minutes, respectively. Afterwards, the cells were rinsed with fresh HBSS and then subjected to CLSM observation.
Drug penetration study
Tumor spheroids were incubated with free MIT or PL/ACC-MIT nanoparticles at the MIT concentration of 2 μg/mL for 24 hours, respectively. Then, the tumor spheroids were washed thrice with ice-cold PBS, fixed with paraformaldehyde for 30 minutes, and placed in cavity microscope slides. The images of the tumor spheroids were acquired by tomoscan using Z-stack imaging with 5 μm intervals from the middle of the spheroid to the bottom by CLSM.
When the tumor volume reached 200 mm3, the HeLa tumor-bearing nude mice were intratumorally administered with free MIT or PL/ACC-MIT nanoparticles at the same dose of MIT at a fixed depth of needling insertion (2 mm under the tumor surface), respectively. At 24 hours postinjection, the tumors were collected, washed by PBS, and sliced from the top of the tumor to the end by cryotomy. The slices were stained with DAPI and then observed by CLSM.
In vitro cytotoxicity
For the cell viability measurements, 200 μL of DMEM containing HeLa cells (1×104 cells/mL) was seeded in 96-well plates (Corning Incorporated) and allowed to culture overnight. The media were discarded and all wells were washed twice with fresh PBS. Subsequently, 200 μL of serum-free medium was added to each well, in which MIT or nanoparticles were diluted to achieve the designated concentrations. After proper incubation, the cell viability was assessed with the standard MTT assay.30 Briefly, the medium was replaced with an equal volume of fresh medium containing 5 mg/mL MTT and incubated for 4 hours at 37°C. Then, MTT was removed, and cells were lysed with dimethyl sulfoxide with stirring for 15 minutes on a microtiter plate shaker. The cell viability was estimated according to the absorption values determined by a microplate reader (model 680; Bio-Rad Laboratories Inc, Hercules, CA, USA) at the wavelength of 570 nm. To visualize the cell viability, paralleled cells were incubated with FDA and PI (final concentration 50 μg/mL) for 10 minutes. Afterwards, the cells were rinsed with PBS carefully and then subjected to optical or fluorescence observation (TE2000-S; Nikon Corporation).
MCTSs with diameters of 300–400 μm were divided into four groups (n=3). The selected spheroids were treated with fresh medium containing free MIT or PL/ACC-MIT nanoparticles (MIT concentration 2 μg/mL). Untreated MCTS was employed as control. The spheroids were allowed to continue to incubate at 37°C for 6 days. The diameter of the spheroids was recorded at predetermined intervals using an optical microscope.
In vivo tumor targeting assay
For the in vivo imaging analysis, near-infrared (NIR) probe DiR (100 μg for 5 mg carriers) was dissolved in the organic solvent and loaded into the lipid matrix of PL/ACC-MIT nanoparticles during the preparation procedure. Subsequently, DiR-loaded PL/ACC-MIT nanoparticles were intravenously injected into the tumor-bearing nude mice (tumor volume 200 mm3) at a dose of 50 μg DiR/kg to investigate their biodistribution and tumor targeting efficacy (n=3). At 6, 12, and 36 hours postinjection, the NIR fluorescent images were captured at an excitation wavelength of 740 nm and an emission wavelength from 740 to 950 nm using an in vivo imaging system (Maestro In-vivo Imaging System, Cambridge Research & Instrumentation, Cambridge, MA, USA). After living imaging, mice were sacrificed, and the main organs and tumor tissues were excised for ex vivo imaging using the same imaging system.
In vivo antitumor assay
The in vivo antitumor efficacy of free PL/ACC-MIT was evaluated on HeLa tumor xenograft models. Mice were randomly divided into three groups (n=3), and intravenously injected with saline, free MIT, or PL/ACC-MIT nanoparticles (MIT concentration 2.5 mg/kg). The initial day of administration was defined as day 0, and administration was then repeated once every 2 days over a 14-day therapeutic period. The tumor volume of mice was measured before administration. On day 14, mice in each group were randomly selected and euthanized. The tumors were collected, weighed, washed with saline thrice, and fixed in 10% formalin. Formalin-fixed main organs and tumors were embedded in paraffin blocks to prepare sections and then subjected to TUNEL staining. Tissue images were captured with an optical microscope.
Results and discussion
Preparation of nanoparticles
ACC can readily be prepared using ethanol as the reaction medium and ammonium carbonate as the source of CO2. (NH4)2CO3 continuously decomposes into NH3 and CO2. The CO2 can again dissolve in ethanol in which it will react with Ca2+ to form ACC. MIT was preloaded into ACC to form ACC-MIT nanoparticles during the synthesis process. However, NH3 can also dissolve in ethanol to alkalize the solution. Our experience has shown that MIT can easily precipitate in alkaline ethanol. As a result, a glass bottle of sulfuric acid was added to adsorb the generated NH3 to maintain the stability of MIT and facilitate the reaction. Finally, the obtained ACC-MIT nanoparticles were modified with PL (including the proper amount of DSPE-PEG and/or DSPE-PEG-FA) in a one-pot approach to finally construct the hybrid nanoparticles (PL/ACC-MIT). The lipid layer provides a biocompatible and water-resistant coating that enhances the residence time in the bloodstream and preserves the water-responsive nature of ACC. The formation of PL/ACC-MIT nanoparticles from ACC nanoparticles was verified by Fourier-transform infrared spectroscopy (FTIR; Figure 1A). The FTIR spectrum of ACC, MIT, and ACC-MIT nanoparticles was first recorded. According to previous reports, bands at 864 and 1,075 cm−1 could be attributed to out-of-plane bending and symmetric stretch in the non-centrosymmetric structures of ACC.31 The splitting peaks at 1,417 and 1,474 cm−1 can be assigned to the asymmetric stretch of the carbonate ions. Those bands are considered to be typical characteristics of ACC vibrations.32 On the other hand, the band at around 1,635 cm−1 and the broad band at around above 3,000 cm−1 were attributed to vibrations of structural water molecules of ACC.33 Besides, the ACC nanoparticles prepared in our study have only one symmetric stretch at around 1,075 cm−1; as a result, it can be identified as proto-calcite ACC that is prone to transform to calcite upon water irritation.34 ACC-MIT showed both the characteristic peaks of ACC and MIT (the cycle in Figure 1A indicated the characteristic peak of MIT in ACC-MIT), indicating the successful loading of MIT into ACC. Pure PL displayed characteristics of asymmetric and symmetric methylene vibration bands at 2,926 and 2,850 cm−1.35 As expected, the same bands also appeared clearly at around 2,926 and 2,850 cm−1 in PL-modified ACC nanoparticles, which indicated the existence of long-chain aliphatic groups, suggesting the successful preparation of PL/ACC-MIT nanoparticles. The drug loading content calculated by HPLC was 4.58% for PL/ACC-MIT (encapsulation efficiency: 91.36%).
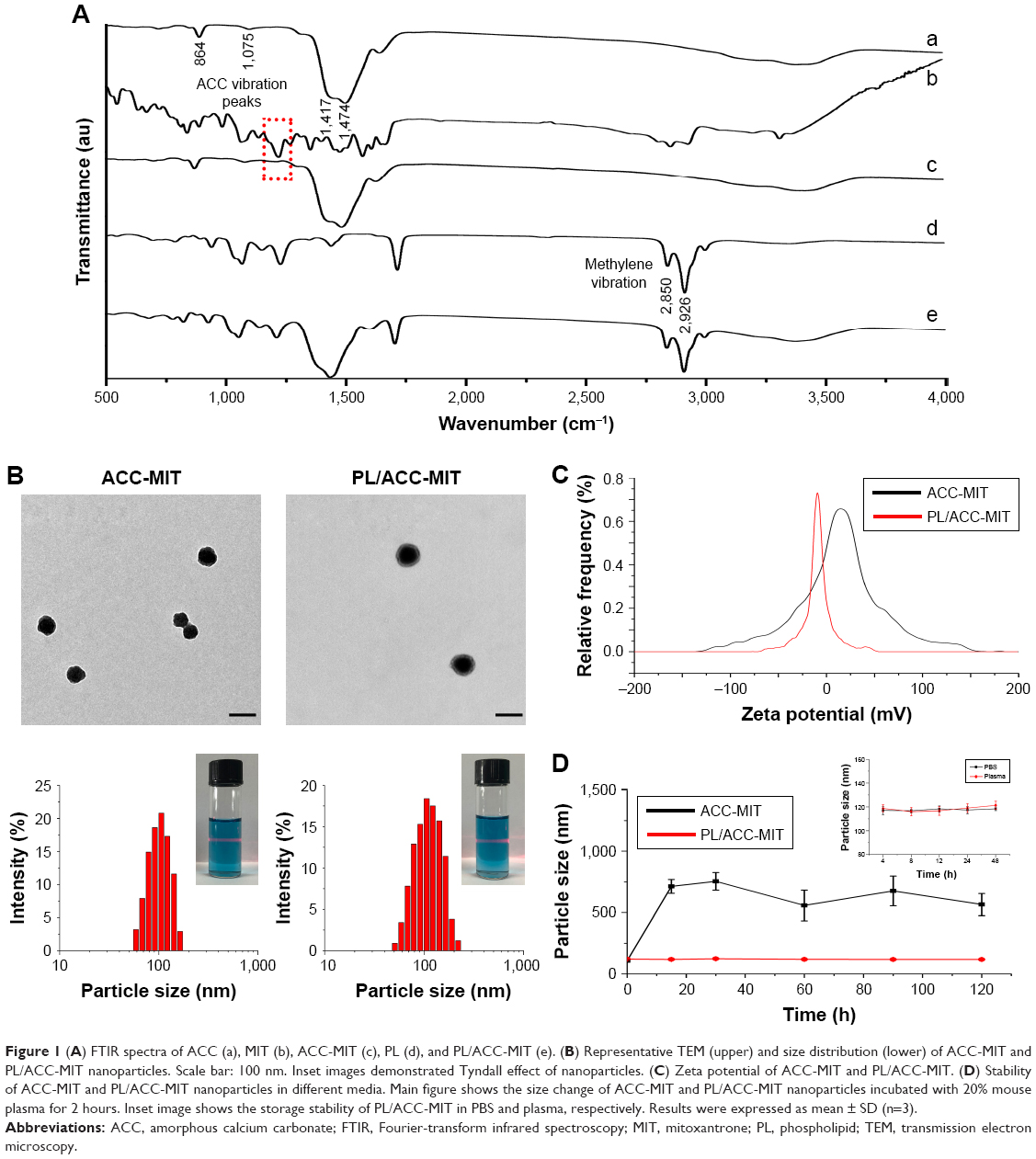 | Figure 1 (A) FTIR spectra of ACC (a), MIT (b), ACC-MIT (c), PL (d), and PL/ACC-MIT (e). (B) Representative TEM (upper) and size distribution (lower) of ACC-MIT and PL/ACC-MIT nanoparticles. Scale bar: 100 nm. Inset images demonstrated Tyndall effect of nanoparticles. (C) Zeta potential of ACC-MIT and PL/ACC-MIT. (D) Stability of ACC-MIT and PL/ACC-MIT nanoparticles in different media. Main figure shows the size change of ACC-MIT and PL/ACC-MIT nanoparticles incubated with 20% mouse plasma for 2 hours. Inset image shows the storage stability of PL/ACC-MIT in PBS and plasma, respectively. Results were expressed as mean ± SD (n=3). |
In our study, TEM observation (Figure 1B) revealed that the as-prepared PL/ACC-MIT particles were spherical and nanosized with distinct boundaries and a diameter of ~80 nm. The size distribution of nanoparticles dispersed in ethanol was measured by dynamic light scattering (DLS), which demonstrated that the obtained ACC nanoparticles were narrowly distributed at ~80 nm with a polydispersity index of 0.056. According to both TEM and DLS, after being modified with PL and DSPE-PEG, the resulting PL/ACC-MIT nanoparticles showed an increase in size compared to that of bare ACC-MIT nanoparticles. Both nanoparticles formed transparent solutions with an easily observed Tyndall effect (an incident light beam through the nanoparticle solution, inserted images), indicating that they were stable colloidal systems.
It has been demonstrated that the zeta potential of calcium carbonate particles is strongly related to the concentration of Ca2+ ions in the synthesis process.36 The as-prepared ACC-MIT nanoparticles were generated in the presence of excess Ca2+ ions. As a result, additional Ca2+ was absorbed on the surface of the ACC-MIT nanoparticles, resulting in a positive zeta potential.37 According to the results obtained by a Zetasizer (Figure 1C), the ACC-MIT nanoparticles were positively charged, with a mean zeta potential of +16.32±0.74 mV (n=3), which was consistent with a previous report.38 After locking and modification, the PL/ACC-MIT nanoparticles exhibited a negative zeta potential of −10.19±0.74 mV (n=3). This could be attributed to the presence of hydroxyl and carboxyl groups in PL and DSPE-PEG. The reversal in surface zeta potential from positive to negative was decisive evidence that proved that the ACC-MIT nanoparticles were successfully modified with PL and DSPE-PEG.
The current understanding of the fate of a drug delivery vector as a foreign material introduced into the circulatory system underlines the formation of surface protein corona with plasma proteins.39 The subsequent destiny of the drug delivery vectors is critically dependent on the amount and identity of the adsorbed proteins. In general, protein adsorption will result in increased particle size, which may in part alter their in vivo accumulation behavior. Strong adsorption may further lead to aggregation or even blockage. Moreover, opsonin-decorated nanoparticles usually suffer from premature elimination and/or excretion due to the triggering of phagocytosis.40,41 As a result, plasma protein adsorption should be avoided if successful drug delivery is to be realized. PL with amphipathicity can greatly improve the colloidal stability of the modified nanoparticles to prevent neighboring particles from aggregating, while PEG modification is among the most widely adopted approaches to overcome the adsorption of plasma proteins.42 The combination of both methods is expected to repel protein absorption via steric repulsion forces.41,43 In order to verify our assumption, both ACC-MIT and PL/ACC-MIT nanoparticles were incubated in mouse plasma at 37°C for 2 hours and the change in nanoparticle size was recorded. As displayed in Figure 1D, the size of the ACC-MIT nanoparticles immediately increased from an average size of ~100 nm to over 500 nm and unstably fluctuated with relatively large SD during the whole incubation time. Conversely, no aggregation was detected for PL/ACC-MIT nanoparticles and the average size of the particles remained unchanged at ~100 nm with small SD value. Further investigation of the long-term stability (inset image in Figure 1D) also confirmed this observation. It was noted that both the dissolution and crystal transition behaviors of bare ACC-MIT nanoparticles in aqueous solution, as well as their strong protein absorption ability (as demonstrated in the above assays), would compromise their capacity to deliver drugs. Furthermore, drastic variation in the particle size to the microscale also attests to their proposed drug delivery function as nanoparticles because the in vivo fate of microscale nanoparticles is quite different from that of nanoscale ones. As a result, the increased aqueous stability of PL/ACC-MIT nanoparticles was believed to be beneficial for their overall performance as a DDS in vivo.
Hemolysis and drug release
The first and foremost requirement of DDSs intended to be administered in vivo, by any mode, and for any biomedical application, is that they should be safe. Moreover, the DDSs should not induce any adverse reaction, such as hemolysis, during the whole process. As a result, hemolysis of PL/ACC-MIT nanoparticles was investigated as a safety guide for its potential medicinal application. As shown in Figure 2A, PL/ACC-MIT nanoparticles displayed neglectable hemolysis (merely 1.46%) at the highest concentration of 1 mg/mL. Considering that when applied in vivo, the nanoparticle concentration would be diluted to a much lower level compared with ones being tested in our study, we concluded that the as-prepared PL/ACC-MIT nanoparticles were relatively safe without inducing noticeable hemolysis, which holds great promise to benefit therapeutic effects in vivo.25
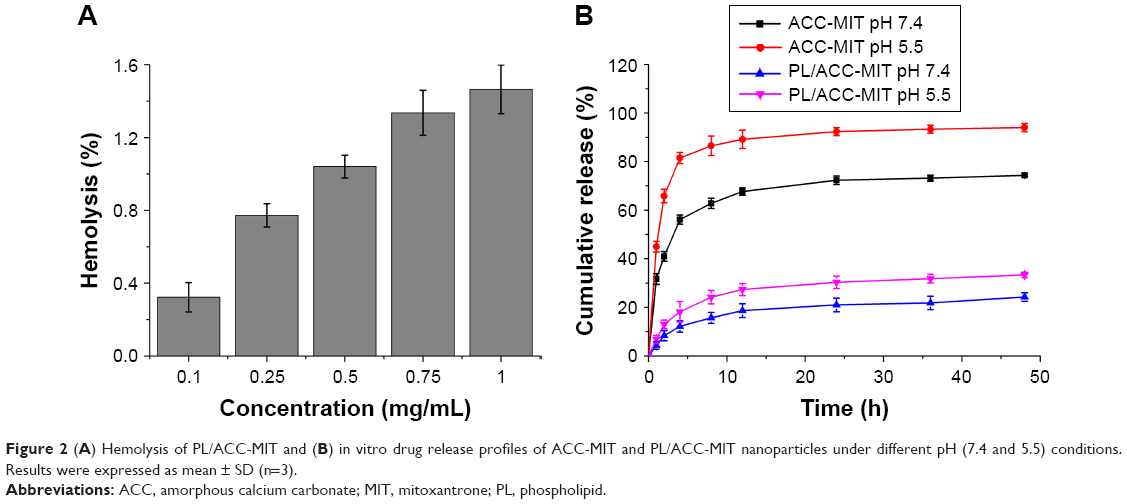 | Figure 2 (A) Hemolysis of PL/ACC-MIT and (B) in vitro drug release profiles of ACC-MIT and PL/ACC-MIT nanoparticles under different pH (7.4 and 5.5) conditions. Results were expressed as mean ± SD (n=3). |
Afterwards, a release experiment using phosphate buffers of different pH values was conducted to fully study the drug release profile of PL/ACC-MIT nanoparticles under different intracorporeal conditions. The classic dialysis method with time intervals up to 24 hours was employed. As shown in Figure 2B, in both aqueous solutions, burst MIT release from ACC-MIT nanoparticles within the initiation phase was observed, which might be ascribed to water-induced dissolution and/or phase transition, as reported previously.16 Furthermore, the cumulative release percentage increased with a decrease in pH value, as acidic solutions (pH 5.5) resulted in more drastic drug release than the neutral one (pH 7.4), which indicated that ACC nanoparticles still possess the acid-sensitive nature of calcium carbonate. It was noted that compared with ACC-MIT nanoparticles under the same conditions, PL/ACC-MIT nanoparticles showed a much more retarded drug release profile. For example, in phosphate buffer of pH 7.4, merely 21% of the drug was released over 24 hours, suggesting that the proposed drug locking and ACC preserving effect was well realized. In addition, unlike ACC-MIT nanoparticles, decreasing the pH value to 5.5 increased the drug release in PL/ACC-MIT nanoparticles to merely 30.3%, revealing that the acid-sensitive nature of ACC might also be suppressed due to its isolation from the exterior medium. This can be an advantage for PL/ACC-MIT nanoparticles to serve as a versatile DDS in extended applications (for example, withstanding the harsh environments of oral administration).
In vitro targeting
DDSs that are intended for cancer therapy require versatile capabilities to bypass the multiple extracellular barriers that lie between administration and the target sites. Despite satisfactory stability, the facile modification of lipid carriers has elicited research toward integrating active-targeting moieties into the system for improved tumor homing ability both in vitro and in vivo. The membrane-bound folate receptor (FR) is overexpressed in a wide range of human cancers but is generally absent in most normal tissues.26 FA is a small, high-affinity ligand of FR. As one of the most widely studied targeting moieties, FA is considered a reliable choice over other counterparts due to its ease of modification, favorable tumor homing properties, and reduced probability of immunogenicity.44 To investigate the in vitro targeting potential of DSPE-PEG-FA-modified hybrid nanoparticles, HeLa as an FR-overexpressing cell line and A549 as an FR-deficient cell line45 were employed in our study. C6 as a model fluorescent molecule was loaded to the nanoparticles, and the endocytosis kinetics were monitored by CLSM. As shown in Figure 3A, CLSM revealed bright green fluorescence signals in the cytoplasm of both HeLa and A549 cells, which was attributed to C6, indicating that the C6-loaded nanoparticles successfully entered the cells. The fluorescence signal gradually became stronger as a function of time, indicating that the intracellular uptake of the nanoparticles was time dependent and positively related to the incubation duration. It was noted that at all tested time intervals, the HeLa cells exerted stronger fluorescence intensity than the A549 cells. Considering the different FR levels in these two cell lines, the positively correlated fluorescence results indicated that FA modification facilitated the internalization of PL/ACC-MIT nanoparticles via FR-mediated endocytosis, which was beneficial for accurate tumor targeting in the case of in vivo administration.
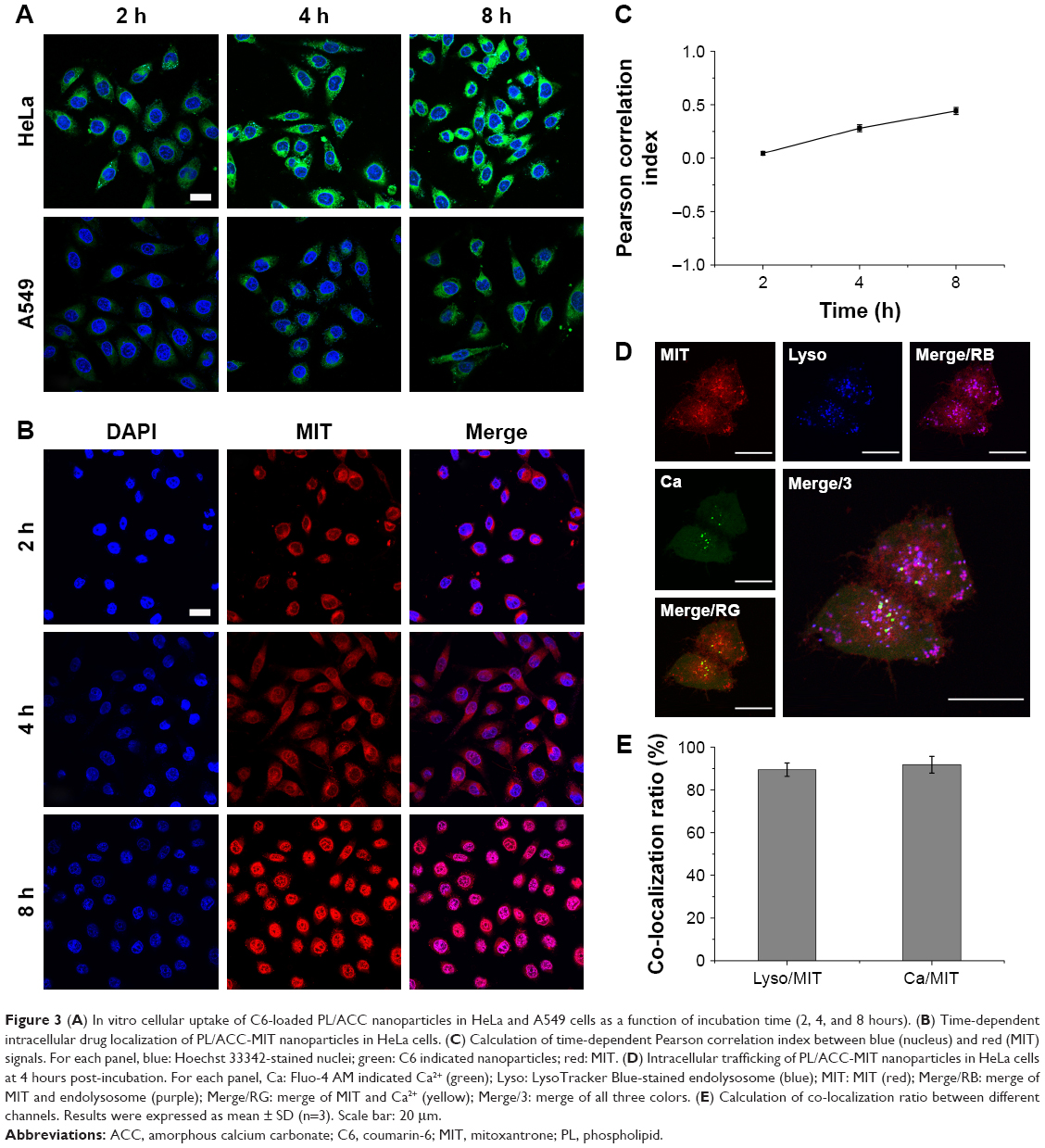 | Figure 3 (A) In vitro cellular uptake of C6-loaded PL/ACC nanoparticles in HeLa and A549 cells as a function of incubation time (2, 4, and 8 hours). (B) Time-dependent intracellular drug localization of PL/ACC-MIT nanoparticles in HeLa cells. (C) Calculation of time-dependent Pearson correlation index between blue (nucleus) and red (MIT) signals. For each panel, blue: Hoechst 33342-stained nuclei; green: C6 indicated nanoparticles; red: MIT. (D) Intracellular trafficking of PL/ACC-MIT nanoparticles in HeLa cells at 4 hours post-incubation. For each panel, Ca: Fluo-4 AM indicated Ca2+ (green); Lyso: LysoTracker Blue-stained endolysosome (blue); MIT: MIT (red); Merge/RB: merge of MIT and endolysosome (purple); Merge/RG: merge of MIT and Ca2+ (yellow); Merge/3: merge of all three colors. (E) Calculation of co-localization ratio between different channels. Results were expressed as mean ± SD (n=3). Scale bar: 20 μm. |
In vitro drug localization
After entry into the cells, drug delivery vectors without responsive properties are usually retarded within the cytoplasm, and their drug release follows slow gradient-diffusion behavior.46 As an antitumor effect can only be realized above the minimal therapeutic concentration, drug and vector overdose are usually required for these drug delivery vectors in order to reach the minimal therapeutic concentration, which further causes metabolic burden and even potential toxicity to the body. To determine the intracellular drug release profile of PL/ACC-MIT nanoparticles, the time-dependent drug localization in HeLa cells was investigated. As illustrated in Figure 3B, the accumulation of MIT in the cell nucleus occurred in a time-dependent manner, while significantly enhanced signal in the nucleus was obtained at 4 hours post-incubation (Figure 4B), which was further confirmed by the calculation of Pearson correlation index. As shown in Figure 3C, the Pearson correlation index increased as a function of incubation time, which suggested that MIT in the nucleus was positively related to time. Only free MIT molecules can enter the nuclei and are prone to remaining in the nuclei instead of the plasma,47 suggesting that quite a number of MIT molecules loaded to PL/ACC-MIT nanoparticles were released in the free form after internalization. The clearly observed MIT signals in the nuclei, in contrast to the negligible extracellular drug release within 4 hours in the drug release assay, also suggested that intracellular drug release of PL/ACC-MIT nanoparticles might follow a different and faster mechanism.
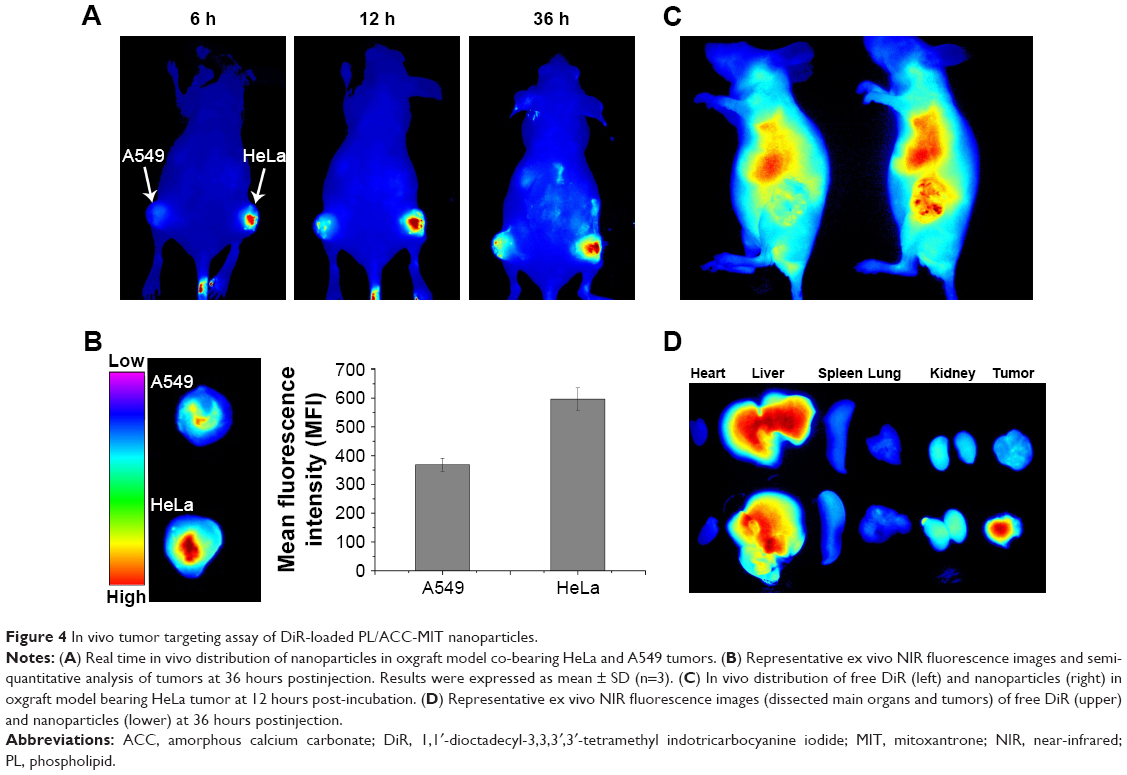 | Figure 4 In vivo tumor targeting assay of DiR-loaded PL/ACC-MIT nanoparticles. |
To seek the reasons responsible for the change in release behavior of the PL/ACC-MIT nanoparticles, intracellular trafficking of PL/ACC-MIT nanoparticles in HeLa (Figure 3D) cells at 4 hours post-incubation was investigated. The endolysosomes stained with LysoTracker Blue were perfectly merged with red dots indicating the location of PL/ACC-MIT nanoparticles, suggesting that endocytosis of the PL/ACC-MIT nanoparticles was regulated by the endolysosomes (Figure 3E). We also noted that the green dots indicating Fluo-4 AM, which implied the decomposition of calcium carbonate, were observed in some endolysosomes, revealing the preliminary conclusion that the decomposition of PL/ACC-MIT nanoparticles occurred as early as in the endolysosomes. According to an earlier report, the endolysosome is responsible for the transport and degradation of exogenous components. The maturation of endolysosomes is always accompanied by successive decreases in pH and the activation of various enzymes.26 It was inferred that the acid-activated esterase within endolysosomes might have deshielded the lipid components (PL and DSPE-PEG) from the PL/ACC-MIT nanoparticles and exposed the ACC-MIT core. Afterwards, the ACC-MIT core readily decomposed and dramatically released the entrapped MIT in a water- and acid-responsive manner as illustrated in the drug release assay.
In vivo targeting
To monitor the in vivo distribution of PL/ACC-MIT nanoparticles, the real-time performance of DiR-loaded PL/ACC-MIT nanoparticles was investigated on a xenograft model co-bearing HeLa and A549 tumors. As displayed in Figure 4A, the fluorescence signal at the tumor tissue was clearly observed at merely 6 hours after injection. The accumulation of nanoparticles in the tumor tissues was further enhanced as a function of time. It was inferred that the PEG-modified nanoparticles (PL/ACC-MIT) had a long-circulating property, preventing premature excretion and allowing them to take advantage of the abnormal tumor vasculature, which contributed to consistently targeting the tumor tissue via the enhanced permeability and retention (EPR) effect. Moreover, PL/ACC-MIT nanoparticles modified with FA showed preferable accumulation in the HeLa tumor over the A549 tumor, which was consistent with previous reports that HeLa cells overexpressing FR can have higher affinity for FA-decorated nanoparticles.26,45 An ex vivo study of nanoparticle distribution (Figure 4B) in the main organs and tumor tissues also confirmed the conclusion that PL/ACC-MIT nanoparticles showed a strong tumor targeting property with preferable accumulation in FR expressing tumor tissues. The comparative in vivo distribution of free DiR and DiR-loaded PL/ACC-MIT nanoparticles in the HeLa tumor-bearing model further provided additional proof for the preferable tumor targeting property of PL/ACC-MIT (Figure 4C and D).
Tumor penetration
Solid tumors are characterized by an abnormal tumor vasculature and a dense tumor extracellular matrix.48,49 It has been found that nanoparticles accumulated via the EPR effect are predominantly located near the blood vessels.50 As a result, the loaded drugs are usually delivered to the cells in the tumor periphery, which impedes uniform distribution of the antitumor drugs throughout the tumor in a therapeutic concentration. As a result, the tumor penetration capability of DDSs is vital for the final antitumor outcome. The MCTS mimicking a solid tumor in vivo was employed as a model. The MIT fluorescence at different depths of MCTS (z-axis) was scanned and recorded by CLSM (Figure 5A). The drug penetration of free MIT vs PL/ACC-MIT nanoparticles was investigated. As shown in Figure 5A, the drug in the PL/ACC-MIT group penetrates more deeply into the MCTS, as the MIT signal could still be clearly observed even at 60 μm below the maximum cross-section. In contrast, the free MIT group only accumulated at the periphery of the MCTS with greatly declined intensity when the depth exceeded 15 μm. This conclusion was further verified by studying the distribution of nanoparticles from superficial to in-depth of the tumor tissue (Figure 5B). PL/ACC-MIT nanoparticles were observed to potentiate MIT distribution into the deep tumor tissue, as the MIT signal could still be observed even at 600 μm below the superficial layer. Conversely, free MIT fluorescence decreased significantly after applying free MIT from the surface layer to 400 μm, and barely no MIT signal could be observed below a depth of 600 μm. Overall, PL/ACC-MIT nanoparticles were capable of delivering their drug cargo from the exterior to the interior of the solid tumor. It was inferred that when the PL/ACC-MIT nanoparticles entered the tumor cells, the enzymes within could disintegrate the PL corona and expose their ACC-MIT core, which consequently exerted its water-responsive function to realize effective MIT release in cancer cells. A high dose of MIT was expected to induce apoptosis and cytotoxicity to kill cancer cells. Afterwards, the exposed PL/ACC-MIT nanoparticles that were liberated from the dead or dying cells continued to respond to the ubiquitous aqueous environment in the MCTS and released MIT, inducing severe cytotoxicity in the neighboring cells, which might contribute to the enhanced drug penetration into the deep MCTS.
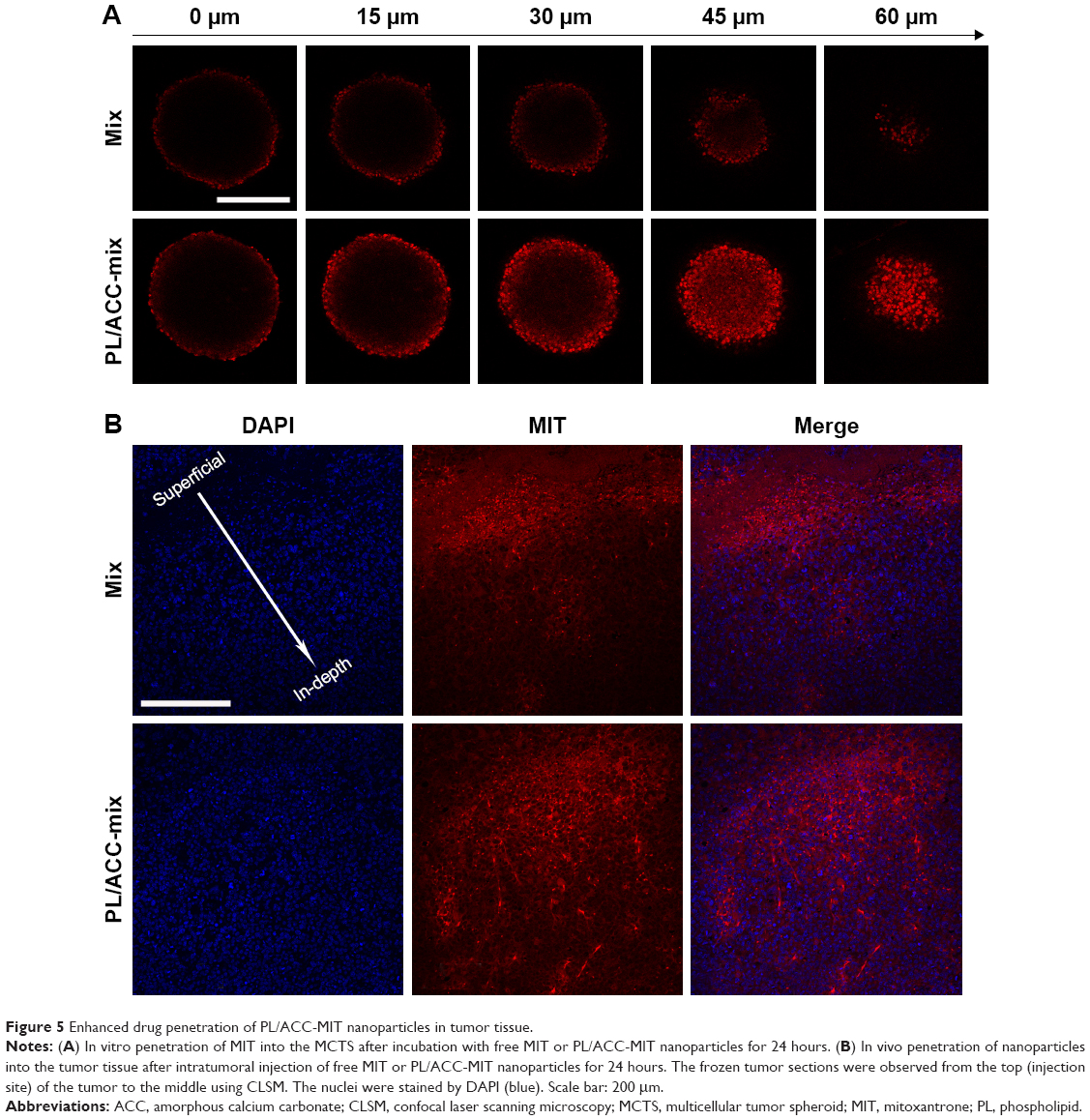 | Figure 5 Enhanced drug penetration of PL/ACC-MIT nanoparticles in tumor tissue. |
In vitro antitumor activity
In an attempt to investigate the antitumor effect of the PL/ACC-MIT nanoparticles, the cell viability of HeLa cells treated with different formulations at various MIT concentrations was recorded using the MTT assay for up to 48 hours. Prior to this, the cell viability of drug-free nanoparticles was assessed to exclude the influence of the carriers. In line with our expectation, PL/ACC (Figure 6A) nanoparticles showed negligible cytotoxicity (cell viability over 80%) even after being incubated with HeLa cells for 48 hours at a concentration of 200 μg/mL, which suggested that the cytotoxic effect in the following assay was caused by the released MIT. As shown in Figure 6B, in the case of free MIT, due to its fast intercalation with nuclear DNA base pairs, a strong cytotoxic effect was achieved in a short time at a low drug concentration, and this effect was further enhanced along with decreased cell viability, which positively correlated with incubation time and concentration.51 The PL/ACC-MIT nanoparticles exerted a comparable cytotoxic effect to that of free MIT under the same conditions, which might be due to their effective drug release ability (water responsive + pH responsive) within cells. This conclusion was further supported by optical and fluorescence observations (Figure 6C).
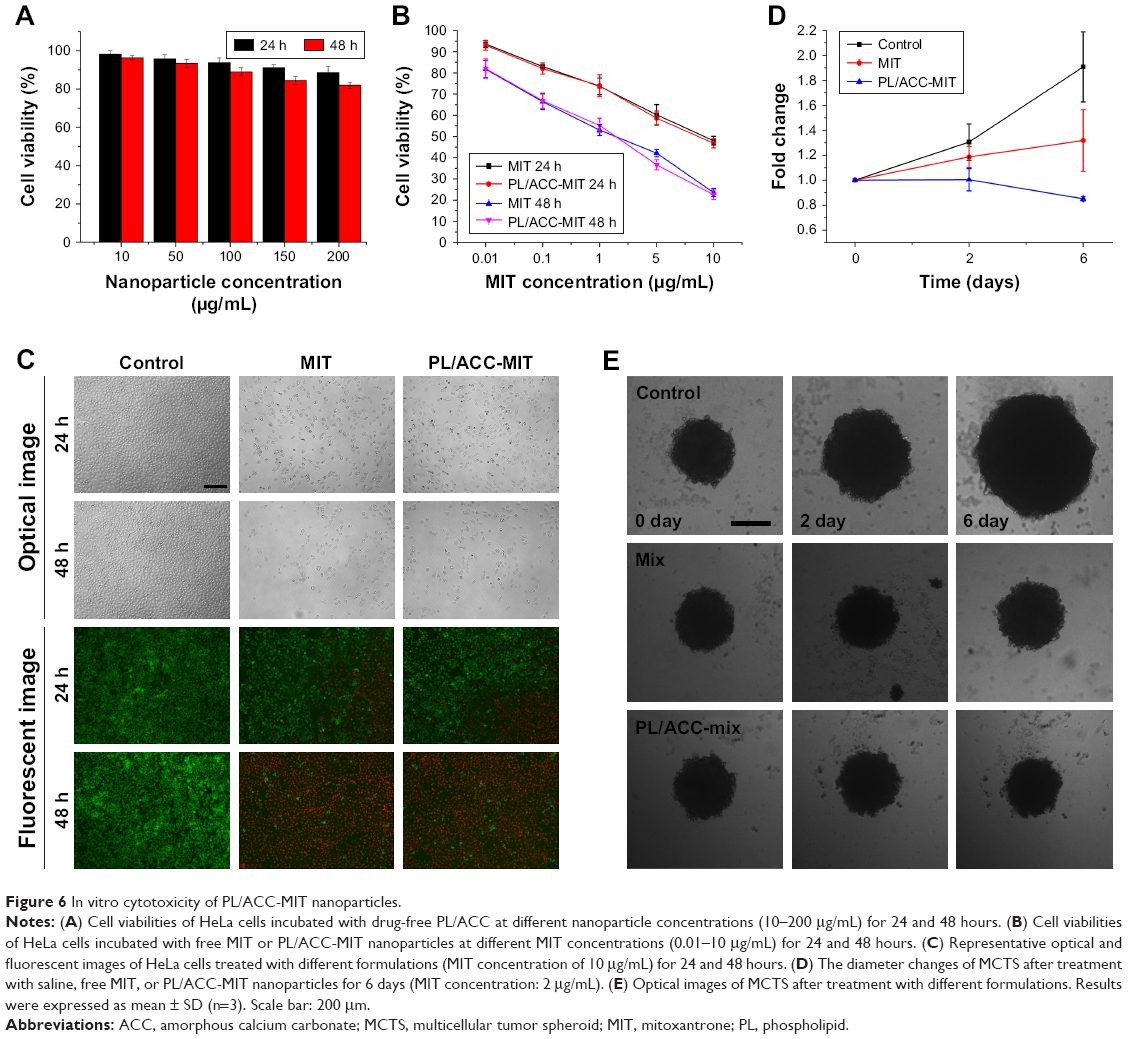 | Figure 6 In vitro cytotoxicity of PL/ACC-MIT nanoparticles. |
The cytotoxicity to the MCTS model was further assessed to demonstrate the desirable in vitro cytotoxicity of PL/ACC-MIT nanoparticles. Unlike the saline group, which showed continuous growth in the MCTS volume, the group treated with PL/ACC-MIT nanoparticles showed the best cytotoxicity effect with decreased MCTS volume at the end of the test, which was even better than that in the free MIT group (Figure 6D and E). It was inferred that the strong cytotoxicity of the PL/ACC-MIT nanoparticles could induce immediate apoptosis to the superficial cells and then infiltrate into the next layer to repeat this process, similar to “peeling an onion” layer by layer, which offered an efficient approach to penetrate into deep tissue to achieve uniform distribution of MIT and was beneficial for enhanced antitumor outcomes. The above-mentioned in vitro cytotoxicity assays added additional evidence to prove that the rapid drug release of PL/ACC-MIT nanoparticles within cancer cells was successfully realized and was advantageous in cancer therapy since it enables the drug to fully exert its cytotoxicity function.
In vivo antitumor assay
The in vivo antitumor assay of PL/ACC-MIT nanoparticles against different formulations was conducted on HeLa oxgraft with saline as a control. As expected, the tumor volume in the saline group showed unmanageable growth that finally crossed 1,000 mm3 at the end of the test (Figure 7A). In contrast, the other three MIT formulations showed certain antitumor effect as the growth curve of the tumors was suppressed. In detail, PL/ACC-MIT nanoparticles exerted the most preferable outcome with a final tumor volume of 302.09±16.13 mm3, which was superior to that of free MIT. As illustrated in Figure 7B, the histologic sections assessed by the TUNEL assay displayed the highest level of cell apoptosis compared to those of other groups. It was interesting to note that unlike their powerful in vitro effect, free MIT showed only moderately evaluated antitumor efficacy. It was suggested that free MIT had a short half-value period wherein most of the drug would be metabolized and/or eliminated and only a small portion of injected dose could come into effect. In addition, MIT also suffered from untargeted distribution upon being introduced into the circulation system, which raises the concern that free MIT might exert undesirable side effects on the subjects.
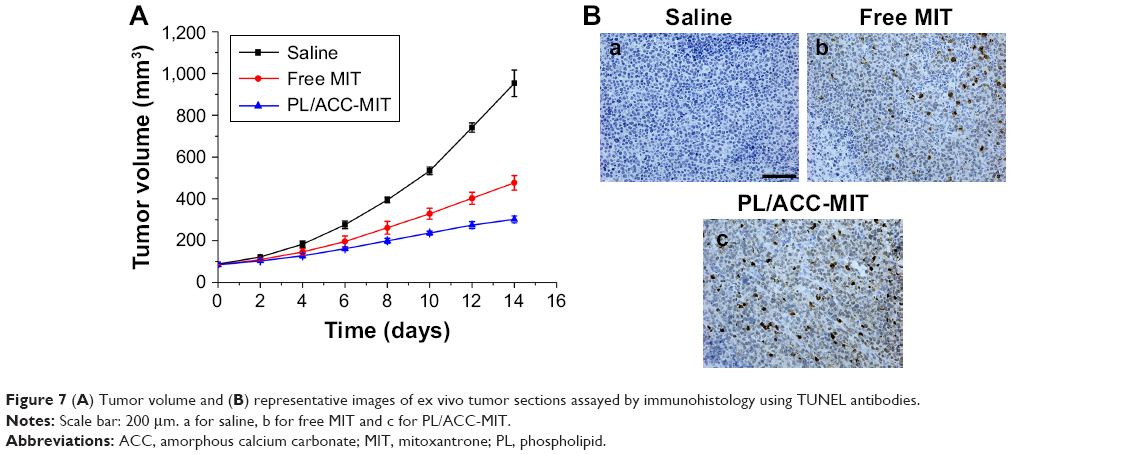 | Figure 7 (A) Tumor volume and (B) representative images of ex vivo tumor sections assayed by immunohistology using TUNEL antibodies. |
Conclusion
In summary, we successfully prepared MIT-preloaded water-responsive PL-calcium carbonate hybrid nanoparticles (PL/ACC-MIT). The well-formed PL/ACC-MIT nanoparticles exhibited enhanced stability in various aqueous conditions and were capable of suppressing unwanted drug leakage at the extracellular level. After targeting FR-overexpressed HeLa cells, by selectively taking advantage of the high aqueous instability of ACC upon exposure to water, the PL/ACC-MIT nanoparticles could respond to the specific environment within the cancer cells to release their payload in an accelerated way, which achieved desirable tumor penetration and antitumor effects compared to those of the free drug, both in vitro and in vivo.
Acknowledgments
We acknowledge the financial support from the National Key Research and Development Program of China (2016YFA0100900) and National Natural Science Foundation of China (81773644, 81773648, 81573366, 81473144, 81871403, and 81571738).
Disclosure
The authors report no conflicts of interest in this work.
References
Xiong H, Wu Y, Jiang Z, Zhou J, Yang M, Yao J. pH-activatable polymeric nanodrugs enhanced tumor chemo/antiangiogenic combination therapy through improving targeting drug release. J Colloid Interface Sci. 2019;536:135–148. | ||
Heidari Majd M, Asgari D, Barar J, et al. Specific targeting of cancer cells by multifunctional mitoxantrone-conjugated magnetic nanoparticles. J Drug Target. 2013;21(4):328–340. | ||
Sargazi A, Shiri F, Keikha S, Majd MH. Hyaluronan magnetic nanoparticle for mitoxantrone delivery toward CD44-positive cancer cells. Colloids Surf B Biointerfaces. 2018;171:150–158. | ||
Rossato LG, Costa VM, de Pinho PG, et al. The metabolic profile of mitoxantrone and its relation with Mitoxantrone-induced cardiotoxicity. Arch Toxicol. 2013;87(10):1809–1820. | ||
Xiong H, Ni J, Jiang Z, Tian F, Zhou J, Yao J. Intracellular self-disassemble polysaccharide nanoassembly for multi-factors tumor drug resistance modulation of doxorubicin. Biomater Sci. 2018;6(9):2527–2540. | ||
Wang C, Li M, Yang T, et al. A self-assembled system for tumor-targeted co-delivery of drug and gene. Mater Sci Eng C Mater Biol Appl. 2015;56:280–285. | ||
Cheng C, Zhang X, Meng Y, Chen L, Zhang Q. Development of a dual drug-loaded hydrogel delivery system for enhanced cancer therapy: in situ formation, degradation and synergistic antitumor efficiency. J Mater Chem B. 2017;5(43):8487–8497. | ||
Yang G, Sun X, Liu J, Feng L, Liu Z. Light-responsive, singlet-oxygen-triggered on-demand drug release from photosensitizer-doped mesoporous silica nanorods for cancer combination therapy. Adv Funct Mater. 2016;26(26):4722–4732. | ||
Yu H, Cui Z, Yu P, et al. pH- and NIR light-responsive micelles with Hyperthermia-Triggered tumor penetration and cytoplasm drug release to reverse doxorubicin resistance in breast cancer. Adv Funct Mater. 2015;25(17):2489–2500. | ||
Oude Blenke E, Mastrobattista E, Schiffelers RM. Strategies for triggered drug release from tumor targeted liposomes. Expert Opin Drug Deliv. 2013;10(10):1399–1410. | ||
Chiang YT, Yen YW, Lo CL. Reactive oxygen species and glutathione dual redox-responsive micelles for selective cytotoxicity of cancer. Biomaterials. 2015;61:150–161. | ||
Yamamoto S-Ichi, Pietrasik J, Matyjaszewski K. Temperature- and pH-responsive dense copolymer brushes prepared by ATRP. Macromolecules. 2008;41(19):7013–7020. | ||
Wang C, Chen S, Wang Y, et al. Lipase-triggered water-responsive “Pandora’s Box” for cancer therapy: toward induced neighboring effect and enhanced drug penetration. Adv Mater. 2018;30(14):1706407. | ||
Maleki Dizaj S, Barzegar-Jalali M, Zarrintan MH, Adibkia K, Lotfipour F. Calcium carbonate nanoparticles as cancer drug delivery system. Expert Opin Drug Deliv. 2015;12(10):1649–1660. | ||
Li M, Mann S. Emergent nanostructures: water-induced mesoscale transformation of Surfactant-Stabilized amorphous calcium carbonate nanoparticles in reverse microemulsions. Adv Funct Mater. 2002;12(1112):773–779. | ||
Zhao Y, Luo Z, Li M, et al. A preloaded amorphous calcium carbonate/doxorubicin@silica nanoreactor for pH-responsive delivery of an anticancer drug. Angew Chem Int Ed. 2015;54(3):919–922. | ||
Dehaini D, Fang RH, Luk BT, et al. Ultra-small lipid-polymer hybrid nanoparticles for tumor-penetrating drug delivery. Nanoscale. 2016;8(30):14411–14419. | ||
Huang P, Chen Y, Lin H, et al. Molecularly organic/inorganic hybrid hollow mesoporous organosilica nanocapsules with tumor-specific biodegradability and enhanced chemotherapeutic functionality. Biomaterials. 2017;125:23–37. | ||
Timin AS, Muslimov AR, Lepik KV, et al. Triple-responsive inorganic–organic hybrid microcapsules as a biocompatible smart platform for the delivery of small molecules. J Mater Chem B. 2016;4(45):7270–7282. | ||
Li M, Luo Z, Zhao Y. Self-assembled hybrid nanostructures: versatile multifunctional nanoplatforms for cancer diagnosis and therapy. Chem Mater. 2017;30(1):25–53. | ||
Sun L, Liu T, Li H, et al. Fluorescent and cross-linked organic-inorganic hybrid nanoshells for monitoring drug delivery. ACS Appl Mater Interfaces. 2015;7(8):4990–4997. | ||
Puri A, Loomis K, Smith B, et al. Lipid-based nanoparticles as pharmaceutical drug carriers: from concepts to clinic. Crit Rev Ther Drug Carrier Syst. 2009;26(6):523–580. | ||
Chen WH, Lecaros RL, Tseng YC, Huang L, Hsu YC. Nanoparticle delivery of HIF1α siRNA combined with photodynamic therapy as a potential treatment strategy for head-and-neck cancer. Cancer Lett. 2015;359(1):65–74. | ||
Yuan H, Miao J, Du YZ, You J, Hu FQ, Zeng S. Cellular uptake of solid lipid nanoparticles and cytotoxicity of encapsulated paclitaxel in A549 cancer cells. Int J Pharm. 2008;348(1–2):137–145. | ||
Tang D, Zhao X, Yang T, Wang C. Paclitaxel prodrug based mixed micelles for tumor-targeted chemotherapy. RSC Adv. 2018;8(1):380–389. | ||
Wang C, Bao X, Ding X, et al. A multifunctional self-dissociative polyethyleneimine derivative coating polymer for enhancing the gene transfection efficiency of DNA/polyethyleneimine polyplexes in vitro and in vivo. Polym Chem. 2015;6(5):780–796. | ||
Xin Y, Qi Q, Mao Z, Zhan X. PLGA nanoparticles introduction into mitoxantrone-loaded ultrasound-responsive liposomes: in vitro and in vivo investigations. Int J Pharm. 2017;528(1–2):47–54. | ||
Zhao X, Tang D, Yang T, Wang C. Facile preparation of biocompatible nanostructured lipid carrier with ultra-small size as a tumor-penetration delivery system. Colloids Surf B Biointerfaces. 2018;170:355–363. | ||
Wang C, Chen S, Yu Q, Hu F, Yuan H. Taking advantage of the disadvantage: employing the high aqueous instability of amorphous calcium carbonate to realize burst drug release within cancer cells. J Mater Chem B. 2017;5(11):2068–2073. | ||
Han D, Fang W, Zhang R, et al. Clematichinenoside protects blood brain barrier against ischemic stroke superimposed on systemic inflammatory challenges through up-regulating A20. Brain Behav Immun. 2016;51:56–69. | ||
Farmer VC. Infrared Spectra of Minerals. London: Mineralogical Society; 1974. | ||
Addadi L, Raz S, Weiner S. Taking advantage of disorder: amorphous calcium carbonate and its roles in biomineralization. Adv Mater. 2003;15(12):959–970. | ||
Kellermeier M, Melero-García E, Glaab F, et al. Stabilization of amorphous calcium carbonate in inorganic silica-rich environments. J Am Chem Soc. 2010;132(50):17859–17866. | ||
Chen SF, Cölfen H, Antonietti M, Yu SH. Ethanol assisted synthesis of pure and stable amorphous calcium carbonate nanoparticles. Chem Commun. 2013;49(83):9564–9566. | ||
Wan C-Q, Li AM, Al-Thabaiti SA, El-Mosslamy ESH, Mak TCW. Efficient solvent-controlled crystallization of pure polymorphs of 1-nitro-4-(4-nitrophenylmethylthio)benzene. CrystEngComm. 2014;16(38):8960–8968. | ||
Moulin P, Roques H. Zeta potential measurement of calcium carbonate. J Colloid Interface Sci. 2003;261(1):115–126. | ||
Chibowski E, Hotysz L, Szcześ A. Time dependent changes in zeta potential of freshly precipitated calcium carbonate. Colloids Surf A Physicochem Eng Asp. 2003;222(1–3):41–54. | ||
Xu X, Han JT, Cho K. Deposition of amorphous calcium carbonate hemispheres on substrates. Langmuir. 2005;21(11):4801–4804. | ||
Sarparanta M, Bimbo LM, Rytkönen J, et al. Intravenous delivery of hydrophobin-functionalized porous silicon nanoparticles: stability, plasma protein adsorption and biodistribution. Mol Pharm. 2012;9(3):654–663. | ||
Mirshafiee V, Kim R, Park S, Mahmoudi M, Kraft ML. Impact of protein pre-coating on the protein corona composition and nanoparticle cellular uptake. Biomaterials. 2016;75:295–304. | ||
Owens DE, Peppas NA, Owensiii D, Opsonization PNA. Opsonization, biodistribution, and pharmacokinetics of polymeric nanoparticles. Int J Pharm. 2006;307(1):93–102. | ||
Gref R, Lück M, Quellec P, et al. ‘Stealth’ corona-core nanoparticles surface modified by polyethylene glycol (PEG): influences of the corona (PEG chain length and surface density) and of the core composition on phagocytic uptake and plasma protein adsorption. Colloids Surf B Biointerfaces. 2000;18(3–4):301–313. | ||
Needham D, Mcintosh TJ, Lasic DD. Repulsive interactions and mechanical stability of polymer-grafted lipid membranes. Biochim Biophys Acta Biomembr. 1992;1108(1):40–48. | ||
Sudimack J, Lee RJ. Targeted drug delivery via the folate receptor. Adv Drug Deliv Rev. 2000;41:147–162. | ||
Lv Y, Cao Y, Li P, et al. Ultrasound-triggered destruction of Folate-Functionalized mesoporous silica Nanoparticle-Loaded microbubble for targeted tumor therapy. Adv Healthc Mater. 2017;6(18):1700354. | ||
Xiong H, Du S, Ni J, Zhou J, Yao J. Mitochondria and nuclei dual-targeted heterogeneous hydroxyapatite nanoparticles for enhancing therapeutic efficacy of doxorubicin. Biomaterials. 2016;94:70–83. | ||
Alev M, Egenberger L, Mühleisen L, et al. Targeting of drug-loaded nanoparticles to tumor sites increases cell death and release of danger signals. J Control Release. 2018;285:67–80. | ||
Carmeliet P, Jain RK. Angiogenesis in cancer and other disease. Nature. 2000;407:249–257. | ||
Netti PA, Berk DA, Swartz MA, Grodzinsky AJ, Jain RK. Role of extracellular matrix assembly in interstitial transport in solid tumors. Cancer Res. 2000;60(9):2497–2503. | ||
Ju C, Mo R, Xue J, et al. Sequential Intra-Intercellular nanoparticle delivery system for deep tumor penetration. Angew Chem Int Ed. 2014;53(24):6253–6258. | ||
Fisichella M, Dabboue H, Bhattacharyya S, et al. Mesoporous silica nanoparticles enhance MTT formazan exocytosis in HeLa cells and astrocytes. Toxicol In Vitro. 2009;23(4):697–703. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.