")
Back to Journals » Journal of Blood Medicine » Volume 7
Midostaurin: an emerging treatment for acute myeloid leukemia patients
Received 10 November 2015
Accepted for publication 22 December 2015
Published 19 April 2016 Volume 2016:7 Pages 73—83
DOI https://doi.org/10.2147/JBM.S100283
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Martin Bluth
Molly Megan Gallogly, Hillard M Lazarus
Department of Medicine, University Hospitals Case Medical Center, Case Comprehensive Cancer Center, Case Western Reserve University, Cleveland, OH, USA
Abstract: Acute myeloid leukemia (AML) is a hematologic malignancy that carries a poor prognosis and has garnered few treatment advances in the last few decades. Mutation of the internal tandem duplication (ITD) region of fms-like tyrosine kinase (FLT3) is considered high risk for decreased response and overall survival. Midostaurin is a Type III receptor tyrosine kinase inhibitor found to inhibit FLT3 and other receptor tyrosine kinases, including platelet-derived growth factor receptors, cyclin-dependent kinase 1, src, c-kit, and vascular endothelial growth factor receptor. In preclinical studies, midostaurin exhibited broad-spectrum antitumor activity toward a wide range of tumor xenografts, as well as an FLT3-ITD-driven mouse model of myelodysplastic syndrome (MDS). Midostaurin is orally administered and generally well tolerated as a single agent; hematologic toxicity increases substantially when administered in combination with standard induction chemotherapy. Clinical trials primarily have focused on relapsed/refractory AML and MDS and included single- and combination-agent studies. Administration of midostaurin to relapsed/refractory MDS and AML patients confers a robust anti-blast response sufficient to bridge a minority of patients to transplant. In combination with histone deacetylase inhibitors, responses appear comparable to historic controls, while the addition of midostaurin to standard induction chemotherapy may prolong survival in FLT3-ITD mutant patients. The response of some wild-type (WT)-FLT3 patients to midostaurin therapy is consistent with midostaurin's ability to inhibit WT-FLT3 in vitro, and also may reflect overexpression of WT-FLT3 in those patients and/or off-target effects such as inhibition of kinases other than FLT3. Midostaurin represents a well-tolerated, easily administered oral agent with the potential to bridge mutant and WT-FLT3 AML patients to transplant and possibly deepen response to induction chemotherapy. Ongoing studies are investigating midostaurin's role in pretransplant induction and posttransplant consolidation therapy.
Keywords: midostaurin, acute leukemia, FLT3, myelodysplastic syndrome, clinical trial
Introduction
Acute myeloid leukemia (AML) is the most common acute leukemia in adults, with an annual US incidence over 20,000 and death rate of over 10,000 per year.1 Despite good initial response to conventional induction therapy, relapse is common, contributing to 5-year remission rates of only 40% in patients under 60 years of age and 10%–20% in older patients.2 Although understanding of the pathogenesis and evolution of AML is increasing, standard induction therapy has remained essentially unchanged over the past 40 years, with increases in survival influenced significantly by improvements in supportive care.
AML is a heterogeneous disease, with multiple factors influencing treatment response, including patient age, performance status, antecedent hematologic conditions and cytotoxic treatments, and cytogenetic and molecular features. The importance of cytogenetic and molecular features in predicting prognosis and guiding treatment decisions is reflected in risk stratification at the time of diagnosis into high-, intermediate-, and low-risk disease. FLT3 (fms-like tyrosine kinase 3) is a Type III receptor tyrosine kinase expressed primarily in hematopoietic cell precursors, where it regulates cell development, differentiation, survival, and expansion.2 Overexpression of FLT3 in cell lines results in increased proliferation and decreased apoptosis. FLT3 mutations occur in approximately 30% of AMLs, with the majority of mutations occurring via internal tandem duplication (ITD, 23%) in the juxtamembrane domain and others via point mutation (usually the Asp835 residue within the activation loop, 7%). Both mutations result in constitutive activation of the kinase.3 FLT3-ITD mutation is associated with a lower complete remission (CR) rate, higher induction death rate, increased risk of relapse, and adverse disease-free survival (DFS), event-free survival, and overall survival (OS).4–9 Because of its association with poor outcomes, detection of the FLT3-ITD mutation classifies any newly diagnosed AML as poor risk.
FLT3’s role as a regulator of hematopoietic cell survival and the deleterious effects of its mutation on AML prognosis make it an attractive therapeutic target in the treatment of AML. Here, we will review the pharmacologic activity, safety, and clinical efficacy of the FLT3 inhibitor midostaurin, compare it to other emerging FLT3 inhibitors, and evaluate its role in the current therapeutic landscape of AML.
Methods
We reviewed data published between 1997 and 2015. We searched the PubMed database using the terms “midostaurin” and “acute myeloid leukemia” as well as topics relevant to each particular discussion section. Only finalized, peer-reviewed publications and abstracts were included for review. Ongoing clinical trials using midostaurin were identified by searching ClinicalTrials.gov for “midostaurin”, and by reviewing our institution’s active clinical studies.
Preclinical studies
Midostaurin, the N-benzoyl derivative of staurosporine (an alkaloid derived from the bacterium Streptomyces staurosporeus), initially was characterized as an inhibitor of protein kinase C (PKC). Subsequently, it was found to inhibit multiple other kinases, including platelet-derived growth factor receptors (PDGFRs) alpha and beta, cyclin-dependent kinase 1 (cdk1), src, Fgr, Syk (spleen tyrosine kinase), c-kit, and the major vascular endothelial growth factor (VEGF) receptor, KDR.10 Midostaurin exhibits antiproliferative activity toward multiple cancer cell lines and xenografts, and its administration has been associated with decreased activities of PKC, PDGFR, c-kit, and VEGF. On the basis of its antikinase activity, midostaurin initially was characterized as a potential broad-spectrum antineoplastic agent, with activity toward diverse solid and hematopoietic tumors.11
Midostaurin’s inhibitory activity toward FLT3 was discovered in a drug screen designed to identify molecules that induce apoptosis in Ba/F3 (interleukin [IL]-3-dependent murine pro-B) cells transduced with FLT3-ITD. In subsequent in vitro kinase assays, this agent was found to inhibit autophosphorylation of FLT3’s cytoplasmic tail with an half-maximal inhibitory concentration (IC50) in the submicromolar range. Midostaurin treatment inhibited proliferation of Ba/F3 cells expressing either FLT3-ITD or FLT3-D835Y with IC50s of less than 10 nM, which correlated with decreased FLT3 autophosphorylation in both mutant cell lines.3
Midostaurin was also found to inhibit FLT3 activity in cells not specifically engineered to overexpress mutant FLT3. In untransfected Ba/F3 cells, midostaurin inhibited ligand-dependent autophosphorylation of endogenous, wild-type (WT)-FLT3, though an IC50 was not reported.3 Pratz et al12 directly compared midostaurin-induced inhibition of FLT3 phosphorylation in cells expressing WT-FLT3 (SEMK2) to those expressing FLT3-ITD (MOLM-14) and reported a tenfold lower IC50 in the mutant cell line (3 versus 30 nM). Midostaurin also depleted phospho-FLT3 and downstream STAT5 in MOLM-13 (AML M5a cell line), MV4-11 (B-myelomonocytic leukemia cell line), and primary FLT3-ITD AML blast progenitor cells.13
The efficacy of midostaurin in FLT3-driven myeloid disease was tested in a mouse model of FLT3-ITD-induced myeloproliferative disorder (MPN). Administration of midostaurin by oral gavage prolonged survival and decreased white blood cell count, myeloid hyperplasia, and spleen weight in mice transplanted with hematopoietic stem cells (HSCs) expressing FLT3-ITD.3
Determining the relative contribution of FLT3 inhibition to midostaurin’s antiproliferative effects on myeloid cells can be challenging given its diverse kinase inhibition profile. Ideally, experiments in myeloid cell lines in which FLT3 and other, midostaurin-sensitive kinase pathways are manipulated separately could further elucidate the relative importance of FLT3 inhibition in midostaurin-mediated growth inhibition. Weisberg et al3 described several indirect measures of specificity of midostaurin for FLT3, including: 1) decreased sensitivity to midostaurin in cells overexpressing bcr-abl in comparison to those overexpressing FLT3-ITD; 2) rescue of midostaurin-treated Ba/F3 FLT3-ITD cells with IL-3 (suggesting IL-3-dependent pathways are not affected); and 3) upregulation of FLT3 in resistant cells cultured in midostaurin to generate resistance. Taken together, these observations support a substantial contribution of FLT3 inhibition toward midostaurin’s growth-inhibitory effects on myeloid cells.3
Combination studies of midostaurin with chemotherapeutic agents were reported in colon cancer cell lines and large-cell lung tumor xenografts.10,11 In the xenograft studies, combination of midostaurin with cisplatin, carboplatin, gemcitabine, doxorubicin, or paclitaxel did not result in antagonism or exacerbate chemotherapy toxicity. Combination effects were observed when midostaurin was combined with doxorubicin or paclitaxel in lung cancer xenografts, and with 5-FU in colon cancer xenografts. Although these additive effects may be specific to xenograft cell type, they established midostaurin as a candidate for combination therapy, particularly with anthracyclines, in treatment of AML.
Midostaurin was studied in combination with the demethylating agent decitabine in cultured AML blast progenitor cells expressing FLT3-ITD.13 Treatment with both agents resulted in significantly greater apoptosis than either agent alone (twofold greater versus decitabine, 0.5-fold versus midostaurin); further, inhibition of phosphorylation of FLT3 and its downstream targets Akt and STAT5 were increased implicating an FLT3-dependent mechanism of inhibition. A midostaurin–decitabine regimen has been tested clinically, but was not designed to demonstrate synergy as there was no control arm (see “Efficacy” section).13
In summary, midostaurin is a potent inhibitor of FLT3 (and other kinases) in vitro, exhibits antiproliferative effects toward cells expressing mutant and WT-FLT3, and demonstrates preclinical efficacy in a mouse model of FLT3-ITD-driven MPN. Given its diverse kinase inhibition profile, demonstrating specificity for FLT3 is challenging; however, several indirect lines of evidence support a role for FLT3 inhibition in midostaurin’s growth-inhibitory effects on myeloid cells. Taken together, these observations identified midostaurin as a promising agent for clinical evaluation in the treatment of FLT3-mutant (and WT) AML.
Pharmacokinetics
Exploratory clinical trials were initiated to better define the optimal route, dosing, and administration of this potentially effective agent. Oral doses ranging from 12.5 mg daily to 100 mg TID have been evaluated in patients with advanced solid tumors, and doses from 25 mg BID to 75 mg TID were studied in diabetic patients.14,15 After oral administration, midostaurin rapidly is absorbed, with Cmax reached within 1–1.5 hours.16 In serum, it binds primarily to α-1 acidic glycoprotein (AAG), which significantly increases its IC50 (in vitro IC50 for PKC increased tenfold in the presence of 10% human serum and 100-fold in physiologic concentrations of AAG).11 When dosed daily, midostaurin serum concentration reaches a maximum after approximately 7 days, after which it slowly decreases despite continued regular dosing.16–18 Modeling studies suggest this time-dependent drop in concentration (and increase in metabolite concentration) can be explained by enzyme autoinduction.19 Median elimination half-life was determined to be 1.6 (range: 0.9–4) days in subjects with refractory solid tumors, approximately twice the t1/2 of 0.7 (range: 0.2–1.3) days determined for healthy volunteers.16 Midostaurin does not accumulate, and its plasma concentration drops to near zero in most patients during treatment-free periods.16,20
Midostaurin is metabolized primarily by hepatic CYP3A4 into two major metabolites: CGP 52421, a 7-hydroxyl derivative that exists as a mixture of two epimers; and CGP 62221, the O-demethylation product.11 In vitro, CGP 62221 exhibits comparable potency to midostaurin in the inhibition of PKCα, and equal potency in the inhibition of cancer cell proliferation. CGP 52421, on the other hand, is 2–4 times less potent than midostaurin in the inhibition of PKC and cancer cell growth, but equivalent in activity to midostaurin in the inhibition of FLT3 kinase in vitro.3 Its selectivity for FLT3 inhibition (versus off-target kinases) is also comparable to the parent compound.21
In the majority of pharmacokinetic analyses, CGP 62221 exhibits a similar concentration–time dependence to midostaurin, initially increasing before reaching a peak concentration and decreasing again before drug cessation.13,17,18,20 In contrast, CGP 52421 concentrations increase steadily throughout the treatment cycle (regardless of cycle duration or concurrently administered agents) without decrements during weeks off treatment; its terminal elimination half-life is much longer than that of the parent compound (eg, median 36 days, range: 27–164 days).16 The greater affinity of CGP 52421 for AAG may account for its prolonged t1/2 in vivo, although CYP3A4 autoinduction also may contribute.11
In summary, midostaurin can be administered orally at a wide range of doses, allowing flexibility in dose adjustment. Its two primary metabolites (both active) demonstrate distinct pharmacokinetic profiles, with CGP 52421 remaining in circulation after cessation of the parent compound. The contributions to toxicity and efficacy of these metabolites are unknown. Midostaurin’s metabolism by CYP3A4 results in significant drug–drug interactions with CYP3A4 inhibitors and inducers in healthy volunteers.22 Thus, most clinical trials (Table 1) have not allowed concomitant use of CYP3A4-interacting medications. Dose adjustments for renal or hepatic impairment have not yet been determined, although an open trial is comparing midostaurin pharmacokinetics in patients with normal and impaired hepatic function (NCT 01429337, Table 2). To our knowledge, central nervous system penetration by midostaurin has not been reported. Midostaurin may synergize with other chemotherapeutic agents, providing opportunities for dose combinations with the caveat of increased toxicity (see “Efficacy” section).
 | Table 1 Published clinical trials of midostaurin: single-agent trials and combination-agent trials |
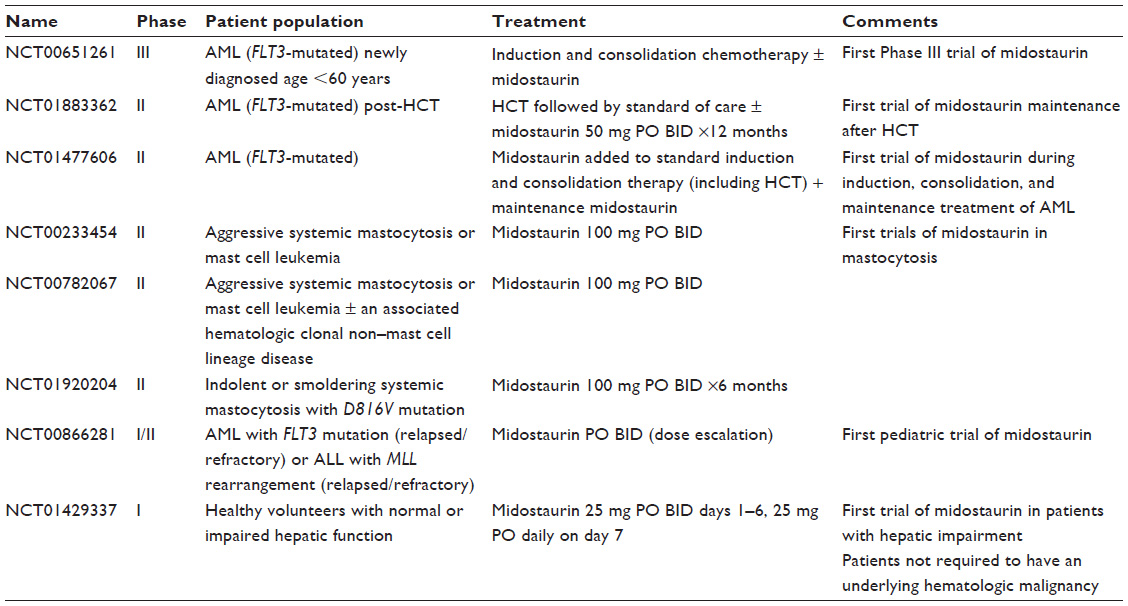 | Table 2 Selected ongoing clinical trials of midostaurin |
Safety and tolerability
Most frequent toxicities
Midostaurin has been characterized as generally well tolerated when administered as a single agent.16,17,23 The first Phase I midostaurin study explored oral doses ranging 12.5 mg daily to 100 mg TID in 32 patients who had refractory or unresponsive solid tumors.16 Although the maximum tolerated dose (MTD) was not reached, the highest doses (225 and 300 mg per day) were associated with sufficient symptoms to exclude them from further study. Subsequent studies utilized total daily oral doses of 100–225 mg when administered as a single agent and 100–200 mg daily when administered in combination with other agents (Table 1). Among all single-agent studies, nausea and vomiting were reported as the most common adverse events (AEs), and rarely was a cause for discontinuation, especially at lower doses.
Drug discontinuation rates increased substantially when midostaurin was combined with conventional cytarabine and daunorubicin, particularly with higher doses of midostaurin (79% discontinuation rate when administered 100 mg BID, 45% for 50 mg BID).20 Midostaurin’s AE profile also shifted when administered with other agents; gastrointestinal toxicity increased when administered with the cytarabine/daunorubicin combination or azacitidine, and hematologic toxicity increased with all combinations.20,24 Whether the increase in AEs with drug combinations is due to inherent toxicities of the additional agents or due to pharmacokinetic interactions between these drugs and midostaurin has been addressed in limited correlative pharmacokinetic studies. These investigations have suggested a possible interaction between daunorubicin and midostaurin, but no apparent interactions between midostaurin and decitabine or azacitidine.13,18,20 In general, sequential administration is better tolerated than concomitant administration when midostaurin is combined with standard induction therapy.20
Hematologic toxicity
When used as a single agent, hematologic toxicity is uncommon with midostaurin.13,20 In combination with cytarabine and an anthracycline, 73% of patients given midostaurin 100 mg BID developed Grade 3–4 neutropenia and thrombocytopenia; 43% had Grade 3–4 anemia. When combined with decitabine, 94% of patients had Grade 3–4 neutropenia, and 50% had Grade 3–4 thrombocytopenia. A significant subset of these toxicities are likely disease related, and/or reflective of the combination agent(s). Strati et al18 reported an increase in Grade 3–4 hematologic AEs from 81% before enrollment to 100% in patients receiving the combination of midostaurin and azacitidine trial.
Renal and hepatic toxicities
Few renal or hepatic toxicities have been reported in single- or combination-agent trials, possibly related to the fact that eligibility criteria required adequate baseline renal and hepatic function. A current study is focused on the pharmacokinetics and safety of midostaurin in subjects with impaired hepatic function (NCT01429337, Table 2).
Cardiac toxicity
In some trials, (QTc)>450 ms was an exclusion criterion for enrollment.13,24 In two trials, patients were observed using telemetry during the first 24 hours of administration; however, no cardiac conduction abnormalities were reported (total of 49 patients).16,24 One dose limiting toxicity (DLT) in the combination study of midostaurin with decitabine was reported as a cardiac event, but no further details were provided. Decreased ejection fraction was reported in 11% of patients receiving combination therapy with midostaurin and azacitidine, and QTc prolongation was reported in one patient (2%).18
Pulmonary toxicity
Three fatal pulmonary events were reported by Stone et al17 in their study of midostaurin 75 mg TID in relapsed/refractory AML or myelodysplastic syndrome (MDS) patients. One event was associated with progressive leukocytosis; the others were not definitively associated with infection and were hypothesized to relate to midostaurin’s antiangiogenic properties (see “Preclinical” section and Table 3). In a combination study with decitabine, two patients died from complications from viral pneumonia, and two others (in the cohort receiving concomitant treatment) died from “pulmonary failure”, though further details regarding their disease course were not reported.13 One patient receiving midostaurin with azacitidine died from complications of pneumonia and hepatotoxicity.24
 | Table 3 Midostaurin’s major pharmacokinetic and pharmacodynamic properties |
Efficacy
Overview of published trials
Midostaurin initially was evaluated in the setting of relapsed or refractory AML or MDS, with a focus on elderly patients in whom outpatient, primarily oral treatment regimens are preferred (Table 1). Later, it was added to standard induction regimens, including cytarabine/daunorubicin, decitabine, and azacitidine both concomitantly and sequentially. It is currently being evaluated in FLT3-mutant AML after hematopoietic cell transplant (HCT), pediatric leukemia, mastocytosis, mast cell leukemia, and AML or MDS in combination with bortezomib and chemotherapy, cladribine and all-trans retinoic acid (ATRA), and a variety of novel agents (Table 2). The only Phase III study of midostaurin published to date compares standard induction chemotherapy with and without midostaurin in newly diagnosed FLT3-ITD AML patients, and is discussed further below.
Single-agent trials
In a Phase IIB trial of single-agent midostaurin (75 mg by mouth (PO) TID) in FLT3-mutated, relapsed/refractory AML or MDS, or in newly diagnosed FLT3-mutated AML patients ineligible for standard induction chemotherapy, 70% achieved a blast response (BR, blast decrease ≥50%) in the peripheral blood and 30% achieved a BR in the bone marrow.17 Although only one patient (5%) had a partial response (PR), seven (35%) demonstrated significant clinical benefit, and two went on to undergo an HCT. A subsequent Phase IIB study enrolled a similar cohort of patients but included FLT3 WT patients (63%).23 BR was achieved in 71% of patients with FLT3 mutations (100% of those previously untreated versus 69% of pretreated patients) and 42% of patients with WT-FLT3. Interestingly, BR correlated to serum concentrations of midostaurin as well as the sum of the drug and its primary metabolites, CGP 62221 and CGP 52421. The best response was a PR in one patient (6%) with mutant FLT3 who received the higher dose of midostaurin (100 mg BID).
Midostaurin plus standard induction chemotherapy
Stone et al20 added midostaurin to standard induction daunorubicin plus cytarabine in newly diagnosed AML patients age 18–60 years whose malignancy did or did not express the FLT3 mutation. In the initial dosing protocol, midostaurin was administered 100 mg PO BID starting the same day as induction chemotherapy (concomitant) or the day after induction completion (sequential); owing to toxicity in both arms, the treatment plan was amended to two midostaurin dose levels (50–100 mg PO BID) administered for 2 of 4 weeks per treatment cycle, starting during (concomitant) or after (sequential) induction. The CR rate was 45% in the higher-dose group (35% WT, 83% FLT3 mutaion) compared to 80% (74% WT, 92% FLT3 mutation) in the lower-dose group. Nine of those receiving a lower dose proceeded to undergo HCT (31% of the FLT3 mutant patients and 19% of the FLT3 WT patients). The lower CR rate in the higher-dosed cohort may reflect the higher discontinuation rate due to toxicity (79% versus 45%).
The added benefit of midostaurin in combination with standard induction chemotherapy is evaluated best in a trial with a control arm (ie, without midostaurin). A randomized, double-blind, Phase III international study (NCT00651261, Table 2) compared the addition of midostaurin versus placebo to standard induction cytarabine and daunorubicin, high-dose cytarabine consolidation, and postconsolidation continuation therapy in 717 patients <60 years old with newly diagnosed, FLT3-mutant AML (ITD or point mutation, Table 2). Initial results demonstrated a survival benefit in the midostaurin arm (hazard ratio for midostaurin to placebo of 0.77 for OS), with no increase in Grade 3 or higher AEs with midostaurin.25
Midostaurin plus hypomethylating agents
On the basis of preclinical data demonstrating additive effects of midostaurin and decitabine on AML cell lines and patient-derived primary AML cells, midostaurin was evaluated in combination with decitabine in a Phase I dose escalation study in relapsed/refractory AML and newly diagnosed AML patients ineligible for standard induction chemotherapy regardless of FLT3 mutation status.13 Twenty-five percent of patients achieved CR or CRi (duration 28–331 days), and half of those went on to matched-unrelated donor HCT.
Midostaurin in conjunction with azacitidine in patients irrespective of FLT3 mutation status was analyzed in two recent trials. The first compared two doses of midostaurin (25–50 mg PO BID) with standard dose azacitidine, and enrolled untreated and previously treated patients with AML or high risk MDS (74% FLT3 mutant, 26% FLT3-WT).18 Despite a 96% BR in peripheral blood and 79% BR in bone marrow, CR rate was only 2%. FLT3 mutation status did not affect the overall response rate (ORR). Median response duration (RD) was 20 weeks, which was significantly longer in patients with FLT3 mutations who had not been previously exposed to a FLT3 inhibitor or patients who had not previously received a stem cell therapy.
The second midostaurin/azacitidine trial was unique in its enrollment of only FLT3-WT patients, despite being open to untreated, elderly patients with AML or relapsed AML at any age regardless of FLT3 mutant status.24 This study tested a midostaurin dose range of 25–75 mg PO BID in combination with 75 mg/m2 azacitidine IV daily for 7 days and reported an 18% CR rate. The higher response rate compared to the former trial is tempered by the increased Grade 3 gastrointestinal toxicities (see “Safety and tolerability” section).
Posttransplant maintenance
Midostaurin is being evaluated in the posttransplant setting following allogeneic HSCT in FLT3-ITD AML patients (NCT01883362). The drug is administered as 50 mg PO BID continuously for 12 months starting within 60 days of HCT; outcomes include relapse free survival (primary), DFS, OS, nonrelapse mortality, and plasma concentrations of midostaurin and its primary metabolites (secondary).
Discussion
Disease targets
Patients with relapsed/refractory AML or MDS, and newly diagnosed AML patients with significant comorbidities have limited treatment options for their diseases, and response rates and durations are limited. The use of midostaurin in such patients (as a single agent or combined with a histone deacetylase (HDAC) inhibitor) may achieve robust BRs, rare CRs, and can serve as a bridge to HCT in patients who previously had no potentially curative treatment options. Given the increased hematologic, gastrointestinal, and pulmonary toxicities observed with the addition of HDAC inhibitors to midostaurin monotherapy, the choice of single agent versus combination therapy should be individualized to each patient. In general, CR rates were higher with administration of an HDAC inhibitor plus midostaurin (Table 1), but this result may not be required to achieve candidacy for HCT.
For patients with newly diagnosed AML, the role of supplementing midostaurin is emerging. Toxicity increased substantially in initial combination studies of midostaurin with induction chemotherapy, but a subsequent Phase III trial showed comparable toxicity between arms with and without midostaurin.25 Moreover, the addition of midostaurin to standard induction chemotherapy conferred a survival benefit to a population including FLT3-ITD and point mutations.20,25
The role of midostaurin in post-HCT consolidation therapy for FLT3-ITD AML is under active investigation (Table 2). Midostaurin also is being evaluated as a single agent in aggressive, indolent, and smoldering mastocytosis, presumably due to its in vitro inhibitory activity toward c-kit. Finally, it is being studied in relapsed/refractory pediatric ALL and FLT3-mutant AML (Table 2).
Efficacy
As a single agent, midostaurin induces a BR in the majority of relapsed/refractory or vulnerable/frail AML/MDS patients, but CRs are rare and transient. Combination with HDAC inhibitors results in higher reported CR rates in general, but these treatment regimens have not been compared head to head in the same trial. In patients fit enough to tolerate induction chemotherapy, the potential benefit of the addition of midostaurin to cytarabine and daunorubicin is supported by the recent Phase III abstract demonstrating improved OS in the midostaurin arm, without significant increases in toxicity.25
What could explain the modest, transient responses to midostaurin in single-agent trials? One possibility is pharmacokinetic modification. For example, the midostaurin concentration may be subtherapeutic at the site of interest (ie, bone marrow, which is not readily accessible), or cyclic administration of the drug could allow recovery of FLT3 activity during treatment breaks. Another possibility is pharmacodynamic, that is, patients who demonstrate a modest (or absent) response to midostaurin could have driver mutations in addition to FLT3, as AML is a genetically heterogeneous disease with the potential to acquire multiple mutations during leukemogenesis.26,27 Such mutations could offset the inhibition conferred by midostaurin treatment, and disease progression could occur through clonal evolution.
Transient responses to midostaurin – particularly in patients previously treated with FLT3 inhibitors – could reflect development of resistance, which has been explored in FLT3-ITD cell lines exposed to chronic, low levels of midostaurin as well as analysis of primary blast samples from midostaurin-resistant patients.13,18,28,29 Multiple mechanisms of resistance have been identified in midostaurin resistance (Table 3), from point mutations to global alterations in gene expression. The point mutations identified to date confer differential resistance patterns to midostaurin (and to other FLT3 inhibitors), suggesting that mutation analysis by PCR could play a critical role in predicting drug response in the future.13
Role of FLT3 mutation status
How important is the FLT3 mutation in understanding response to midostaurin therapy? Studies to date do not consistently demonstrate better responses to midostaurin in FLT3-mutant compared to FLT3-WT patients. In a Phase I/II trial of azacitidine with or without midostaurin in untreated and previously treated AML or high-risk MDS, FLT3 mutation status was not associated with improved ORR; the lack of prior exposure to an FLT3 inhibitor was associated with better response among FLT3-mutated patients.18 In a similar population of patients treated with single-agent midostaurin, median OS was worse in the FLT3-mutated population, presumed to reflect the overall poor risk associated with the FLT3 mutation.23 In newly diagnosed AML patients receiving midostaurin in addition to standard induction chemotherapy, CR rates were higher in FLT3-mutated than FLT3-WT patients, but no error ranges were reported (Table 1), and Kaplan–Meier curves were comparable between the two populations.20
Response of FLT3-WT AML to midostaurin may reflect overexpression of WT-FLT3 in responsive patients and/or off-target, anti-AML effects of midostaurin (such as inhibition of other kinases). An open trial of midostaurin in FLT3-mutated or c-kit-mutated, t(8;21) AML (NCT01830361) may provide insight into the potential contribution of c-kit inhibition (an alternative kinase target) to midostaurin response in FLT3-WT disease.
Other FLT3 inhibitors
Several FLT3 inhibitors in addition to midostaurin have been characterized and many have been tested in clinical trials. The multikinase inhibitor sorafenib, currently approved for the treatment of renal cell carcinoma, hepatocellular carcinoma, and radioactive iodine-refractory thyroid cancer, inhibits FLT3 (and FLT3-driven cell proliferation) with approximately tenfold lower IC50 than midostaurin in vitro.21 In single-agent trials, sorafenib exhibited a high clinical response rate in FLT3-ITD-positive relapsed/refractory AML.30 Sorafenib has been studied in combination with clofarabine, azacitidine, and cytarabine (alone and with anthracyclines) and is not associated with as frequent hematologic AEs compared to midostaurin combination therapy. A small, randomized Phase II trial comparing placebo versus sorafenib with induction cytarabine and daunorubicin did not show a benefit in sorafenib-treated patients; however, the study may not have been powered to detect a true difference (just 15 of 102 patients had a FLT3-ITD mutation).31 An open Phase III trial is comparing placebo, sorafenib, and velcade added to induction chemotherapy in newly diagnosed AML patients with a high allelic ratio of FLT3-ITD (NCT01371981, ClinicalTrials.gov). A Phase IV study of sorafenib as maintenance therapy after HCT for FLT3-ITD AML is planned but is not yet recruiting (NCT02474290, ClinicalTrials.gov).
Several other FLT3 inhibitors are being investigated in FLT3-mutant AML, including lestaurtinib, tandutinib, sunitinib, ponatinib, crenolanib, and gilteritinib.32–35 Although a comprehensive analysis of these inhibitors is beyond the scope of this review, selected agents’ distinguishing characteristics and active trials are summarized in Table 4. Of the emerging FLT3 inhibitors, quizartinib appears to be the most potent and selective, although its side effect profile is notable for prolonged QTc.21,36 Gilteritinib is distinguished by a particularly favorable toxicity profile, and showed promise in a Phase I/II study in relapsed/refractory AML patients, with ORR of 57%–65% (depending on dose level) and CRs in a minority of patients.37 Crenolanib retains activity toward all identified resistance-conferring FLT3 kinase domain mutations, which could be of critical importance for patients who develop resistance to other FLT3 inhibitors. It is currently being studied as a single agent and in combination in FLT3-ITD AML.38
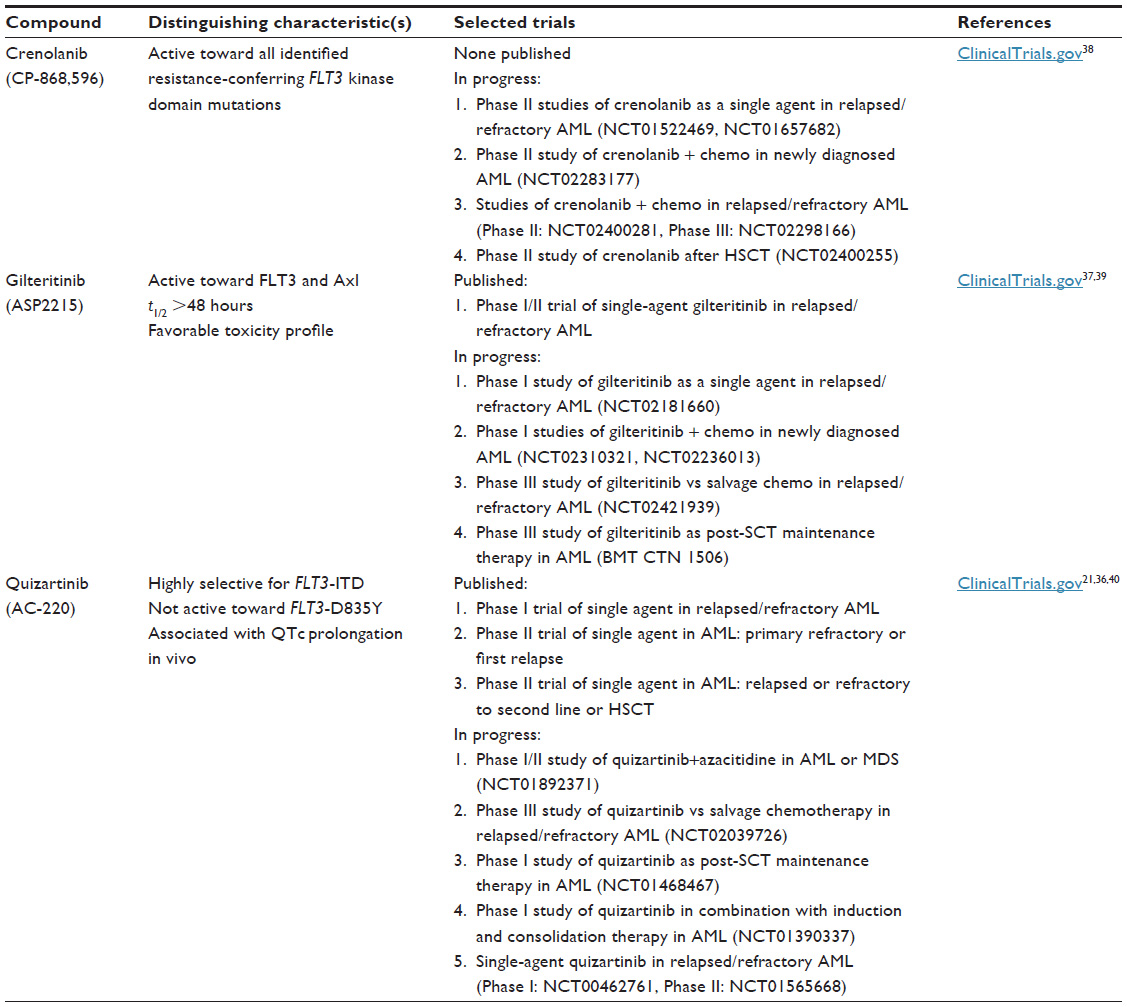 | Table 4 Selected emerging FLT3 inhibitors |
Conclusion
Midostaurin’s inhibition of WT and mutant FLT3, ease of administration, and general good tolerance make it an attractive agent in the limited armamentarium of treatments for FLT3-mutant (and WT) AML. The preclinical and clinical studies published to date provide a starting point for the determination of midostaurin’s potential clinical utility in AML treatment. As a single agent, midostaurin has the potential to induce responses in relapsed/refractory AML or MDS that carry a minority of patients to a potentially curative HCT. When administered in combination with induction chemotherapy, midostaurin has the potential to deepen and/or prolong response. Whether midostaurin improves relapse-free survival after transplant remains an open question, which is under active investigation.
On the basis of the observations discussed earlier, we recommend that midostaurin be regarded as a single agent in relapsed/refractory AML or MDS patients who are HCT candidates as a potential bridge to curative therapy. This recommendation is independent of FLT3 mutation status given the documented response of FLT3-WT patients (though sometimes inferior to FLT3-mutated patients) to treatment. We also recommend enrollment in clinical trials of midostaurin for induction or consolidation therapy. Hopefully, the answer to important clinical questions such as how deeply midostaurin improves response to induction chemotherapy, if its use can extend duration of response after HCT, and how it compares to other emerging FLT3 inhibitors in efficacy and safety will be forthcoming.
Disclosure
The authors report no conflicts of interest in this work.
References
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65(1):5–29. | |
Sanz M, Burnett A, Lo-Coco F, Lowenberg B. FLT3 inhibition as a targeted therapy for acute myeloid leukemia. Curr Opin Oncol. 2009;21(6):594–600. | |
Weisberg E, Boulton C, Kelly LM, et al. Inhibition of mutant FLT3 receptors in leukemia cells by the small molecule tyrosine kinase inhibitor PKC412. Cancer Cell. 2002;1(5):433–443. | |
Whitman SP, Archer KJ, Feng L, et al. Absence of the wild-type allele predicts poor prognosis in adult de novo acute myeloid leukemia with normal cytogenetics and the internal tandem duplication of FLT3: a cancer and leukemia group B study. Cancer Res. 2001;61(19):7233–7239. | |
Frohling S, Schlenk RF, Breitruck J, et al. Prognostic significance of activating FLT3 mutations in younger adults (16 to 60 years) with acute myeloid leukemia and normal cytogenetics: a study of the AML Study Group Ulm. Blood. 2002;100(13):4372–4380. | |
Schlenk RF, Dohner K, Krauter J, et al. Mutations and treatment outcome in cytogenetically normal acute myeloid leukemia. N Engl J Med. 2008;358(18):1909–1918. | |
Kayser S, Schlenk RF, Londono MC, et al. Insertion of FLT3 internal tandem duplication in the tyrosine kinase domain-1 is associated with resistance to chemotherapy and inferior outcome. Blood. 2009;114(12):2386–2392. | |
Rockova V, Abbas S, Wouters BJ, et al. Risk stratification of intermediate-risk acute myeloid leukemia: integrative analysis of a multitude of gene mutation and gene expression markers. Blood. 2011;118(4):1069–1076. | |
Patel JP, Gonen M, Figueroa ME, et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med. 2012;366(12):1079–1089. | |
Fabbro D, Buchdunger E, Wood J, et al. Inhibitors of protein kinases: CGP 41251, a protein kinase inhibitor with potential as an anticancer agent. Pharmacol Ther. 1999;82(2–3):293–301. | |
Fabbro D, Ruetz S, Bodis S, et al. PKC412 – a protein kinase inhibitor with a broad therapeutic potential. Anticancer Drug Des. 2000;15(1):17–28. | |
Pratz KW, Sato T, Murphy KM, Stine A, Rajkhowa T, Levis M. FLT3-mutant allelic burden and clinical status are predictive of response to FLT3 inhibitors in AML. Blood. 2010;115(7):1425–1432. | |
Williams CB, Kambhampati S, Fiskus W, et al. Preclinical and phase I results of decitabine in combination with midostaurin (PKC412) for newly diagnosed elderly or relapsed/refractory adult patients with acute myeloid leukemia. Pharmacotherapy. 2013;33(12):1341–1352. | |
McDonald AC, Popper D, King D, Champain K, Graf P, Man A. Phase I and pharmacokinetic study of CGP 41521, an inhibitor of protein kinase (abstract). Proc Am Soc Clin Oncol. 1997;15(212a). | |
Wang Y, Yin OQ, Graf P, Kisicki JC, Schran H. Dose- and time-dependent pharmacokinetics of midostaurin in patients with diabetes mellitus. J Clin Pharmacol. 2008;48(6):763–775. | |
Propper DJ, McDonald AC, Man A, et al. Phase I and pharmacokinetic study of PKC412, an inhibitor of protein kinase C. J Clin Oncol. 2001;19(5):1485–1492. | |
Stone RM, DeAngelo DJ, Klimek V, et al. Patients with acute myeloid leukemia and an activating mutation in FLT3 respond to a small-molecule FLT3 tyrosine kinase inhibitor, PKC412. Blood. 2005;105(1):54–60. | |
Strati P, Kantarjian H, Ravandi F, et al. Phase I/II trial of the combination of midostaurin (PKC412) and 5-azacytidine for patients with acute myeloid leukemia and myelodysplastic syndrome. Am J Hematol. 2015;90(4):276–281. | |
Yin OQ, Wang Y, Schran H. A mechanism-based population pharmacokinetic model for characterizing time-dependent pharmacokinetics of midostaurin and its metabolites in human subjects. Clin Pharmacokinet. 2008;47(12):807–816. | |
Stone RM, Fischer T, Paquette R, et al. Phase IB study of the FLT3 kinase inhibitor midostaurin with chemotherapy in younger newly diagnosed adult patients with acute myeloid leukemia. Leukemia. 2012;26(9):2061–2068. | |
Zarrinkar PP, Gunawardane RN, Cramer MD, et al. AC220 is a uniquely potent and selective inhibitor of FLT3 for the treatment of acute myeloid leukemia (AML). Blood. 2009;114(14):2984–2992. | |
Dutreix C, Munarini F, Lorenzo S, Roesel J, Wang Y. Investigation into CYP3A4-mediated drug-drug interactions on midostaurin in healthy volunteers. Cancer Chemother Pharmacol. 2013;72(6):1223–1234. | |
Fischer T, Stone RM, Deangelo DJ, et al. Phase IIB trial of oral Midostaurin (PKC412), the FMS-like tyrosine kinase 3 receptor (FLT3) and multi-targeted kinase inhibitor, in patients with acute myeloid leukemia and high-risk myelodysplastic syndrome with either wild-type or mutated FLT3. J Clin Oncol. 2010;28(28):4339–4345. | |
Cooper BW, Kindwall-Keller TL, Craig MD, et al. A phase I study of midostaurin and azacitidine in relapsed and elderly AML patients. Clin Lymphoma Myeloma Leuk. 2015;15(7):428–432, e422. | |
Stone RM MS, Sanford BL, Geyer S, et al. The multi-kinase inhibitor midostaurin (M) prolongs survival compared with placebo (P) in combination with daunorubicin (D)/cytarabine (C) induction (ind), high-dose C consolidation (consol), and as maintenance (maint) therapy in newly diagnosed acute myeloid leukemia (AML) patients (pts) age 18–60 with FLT3 mutations (muts): An international prospective randomized (rand) P-controlled double-blind trial (CALGB 10603/RATIFY [Alliance]) (abstract). Paper presented at: American Society of Hematology (ASH) 57th Annual Meeting, December 3–6, 2015, Orlando, FL, USA. | |
Corces-Zimmerman MR, Majeti R. Pre-leukemic evolution of hematopoietic stem cells: the importance of early mutations in leukemogenesis. Leukemia. 2014;28(12):2276–2282. | |
Dohner H, Weisdorf DJ, Bloomfield CD. Acute myeloid leukemia. N Engl J Med. 2015;373(12):1136–1152. | |
Stolzel F, Steudel C, Oelschlagel U, et al. Mechanisms of resistance against PKC412 in resistant FLT3-ITD positive human acute myeloid leukemia cells. Ann Hematol. 2010;89(7):653–662. | |
Heidel F, Solem FK, Breitenbuecher F, et al. Clinical resistance to the kinase inhibitor PKC412 in acute myeloid leukemia by mutation of Asn-676 in the FLT3 tyrosine kinase domain. Blood. 2006;107(1):293–300. | |
Giri S, Hamdeh S, Bhatt VR, Schwarz JK. Sorafenib in relapsed AML with FMS-like receptor tyrosine kinase-3 internal tandem duplication mutation. J Natl Compr Canc Netw. 2015;13(5):508–514. | |
Serve H, Krug U, Wagner R, et al. Sorafenib in combination with intensive chemotherapy in elderly patients with acute myeloid leukemia: results from a randomized, placebo-controlled trial. J Clin Oncol. 2013;31(25):3110–3118. | |
Leung AY, Man CH, Kwong YL. FLT3 inhibition: a moving and evolving target in acute myeloid leukaemia. Leukemia. 2013;27(2):260–268. | |
Sudhindra A, Smith CC. FLT3 inhibitors in AML: are we there yet? Curr Hematol Malig Rep. 2014;9(2):174–185. | |
Grunwald MR, Levis MJ. FLT3 tyrosine kinase inhibition as a paradigm for targeted drug development in acute myeloid leukemia. Semin Hematol. 2015;52(3):193–199. | |
Kiyoi H. Flt3 inhibitors: recent advances and problems for clinical application. Nagoya J Med Sci. 2015;77(1–2):7–17. | |
Cortes JE, Kantarjian H, Foran JM, et al. Phase I study of quizartinib administered daily to patients with relapsed or refractory acute myeloid leukemia irrespective of FMS-like tyrosine kinase 3-internal tandem duplication status. J Clin Oncol. 2013;31(29):3681–3687. | |
Levis MJ PA, Altman JK, Cortes JE, et al. Results of a first-in-human, phase I/II trial of ASP2215, a selective, potent inhibitor of FLT3/Axl in patients with relapsed or refractory (R/R) acute myeloid leukemia (AML) (Abstr 7003). J Clin Oncol. 2015;33:abstr 7003. | |
Zimmerman EI, Turner DC, Buaboonnam J, et al. Crenolanib is active against models of drug-resistant FLT3-ITD-positive acute myeloid leukemia. Blood. 2013;122(22):3607–3615. | |
Thom C. Preliminary data on ASP2215: tolerability and efficacy in acute myeloid leukemia patients. Future Oncol. 2015;11(18):2499–2501. | |
Levis MJ PA, Dombret H, Dohner H, et al. Final results of a phase 2 open-label, monotherapy efficacy and safety study of quizartinib (AC220) in patients with FLT3-ITD positive or negative relapsed/refractory myeloid leukemia after second-line chemotherapy or hematopoietic stem cell transplantation. Blood. 2012;120:673. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.