")
Back to Journals » International Journal of Nanomedicine » Volume 14
Micelles Loaded With Puerarin And Modified With Triphenylphosphonium Cation Possess Mitochondrial Targeting And Demonstrate Enhanced Protective Effect Against Isoprenaline-Induced H9c2 Cells Apoptosis
Authors Li W, Wu J, Xiang D, Luo S, Hu X, Tang T, Sun T, Liu X
Received 16 June 2019
Accepted for publication 16 September 2019
Published 21 October 2019 Volume 2019:14 Pages 8345—8360
DOI https://doi.org/10.2147/IJN.S219670
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Lei Yang
Wen-qun Li,1–3,* Jun-yong Wu,1–3,* Da-xiong Xiang,1–3 Shi-lin Luo,1–3 Xiong-bin Hu,1–3 Tian-tian Tang,1–3 Tao-li Sun,4 Xin-yi Liu1–3
1Department of Pharmacy, The Second Xiangya Hospital, Central South University, Changsha 410011, People’s Republic of China; 2Institution of Clinical Pharmacy, Central South University, Changsha 410011, People’s Republic of China; 3Hunan Provincial Engineering Research Center of Translational Medicine and Innovative Drugs, Changsha 410011, People’s Republic of China; 4Key Laboratory Breeding Base of Hu’nan Oriented Fundamental and Applied Research of Innovative Pharmaceutics, College of Pharmacy, Changsha Medical University, Changsha 410219, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Xin-yi Liu
Department of Pharmacy, The Second Xiangya Hospital, Central South University, Changsha, People’s Republic of China
Tel +86-731-8529-2093
Email [email protected]
Background: The protective role of puerarin (PUE) against myocardial infarction is closely related to its regulation on mitochondria. However, free PUE can hardly reach the mitochondria of ischemic cardiomyocytes due to the lack of mitochondrial targeting of PUE. Here PUE was loaded into mitochondria-targeted micelles (PUE@TPP/PEG-PE) for precisely delivering PUE into mitochondria with the aim of enhancing the anti-apoptosis effect.
Methods: The mitochondriotropic polymer TPP-PEG-PE was synthesized for the preparation of PUE@TPP/PEG-PE micelles modified with triphenylphosphonium (TPP) cation. The physicochemical properties and anti-apoptosis effect of PUE@TPP/PEG-PE micelles were investigated. The coumarin 6 (C6)-labeled TPP/PEG-PE (C6@TPP/PEG-PE) micelles were used to observe the enhanced cellular uptake, mitochondrial targeting and lysosomes escape. Moreover, in vivo and ex vivo biodistribution of lipophilic near-infrared dye 1,1ʹ-dioctadecyl-3,3,3′,3ʹ-tetramethylindotricarbocyanine iodide (DiR)-labeled PUE@TPP/PEG-PE (DiR@TPP/PEG-PE) micelles were detected through fluorescence imaging.
Results: The successful synthesis of TPP-PEG-PE conjugate was confirmed. PUE@TPP/PEG-PE micelles had a particle size of 17.1 nm, a zeta potential of −6.2 mV, and a sustained-release behavior. The in vitro results showed that the intracellular uptake of C6@TPP/PEG-PE micelles was significantly enhanced in H9c2 cells. C6@TPP/PEG-PE micelles could deliver C6 to mitochondria and reduce the capture of lysosomes. In addition, compared with the PUE@PEG-PE micelles and free PUE, the PUE@TPP/PEG-PE micelles exerted an enhanced protective effect against isoprenaline-induced H9c2 cell apoptosis, as evident by the decreased percentage of apoptotic cells, Caspase-3 activity, ROS level, Bax expression, and increased Bcl-2 expression. The in vivo detecting results of the targeting effect using DiR probe also indicated that TPP/PEG-PE micelles could accumulate and retain in the ischemic myocardium.
Conclusion: The results of this study demonstrate the promising potential of applying PUE@TPP/PEG-PE micelles in mitochondria-targeted drug delivery to achieve maximum therapeutic effects of PUE.
Keywords: mitochondrial targeting, anti-apoptosis, ischemic myocardium, TPP
Introduction
Acute myocardial infarction (AMI) is a major cause of hospital admissions and mortality in the world.1 In clinical practice, revascularization strategies followed by palliative care are the standard care for AMI.2 These clinical treatments can only relieve the clinical symptoms of AMI in a short period of time, but fail to effectively treat the pathogenesis.3 In addition, these clinical treatments are highly invasive and require the operation of trained surgical personnel, with high associated cost of treatment and the risk of surgical complications.4 Therefore, there is an urgent need for improving the treatment of AMI.
Mitochondrial dysfunction plays a vital role in the pathogenesis of AMI.5 Under the normal physiological conditions, the mitochondrial permeability transition pore (mPTP) within the inner mitochondrial membrane is closed.6 In the early stages of AMI, the principal cardiomyocyte death pathways are programmed necrosis and apoptosis through the intrinsic pathway, initiated by the opening of the mPTP and external mitochondrial membrane permeabilization, respectively.7 Furthermore, the mitochondrial membrane potential is decreased, and pro-apoptotic factors such as cytochrome c are released from the mitochondria into the cytosol, resulting in initial apoptosis of myocardial cell.8 mPTP acts as a key regulatory nodal point in mediating cardiac dysfunction and cell death. Therefore, the inhibition of the opening of the mPTP at the onset of myocardial infarction can be considered as a very precise treatment of AMI. The urgent problem at present is how to specifically deliver drugs into mitochondria of ischemic cardiomyocyte.
Puerarin (PUE), a major bioactive ingredient isolated from the Chinese medicine Ge-gen (Radix Puerariae), has been used for the treatment of myocardial ischemia.9 PUE could amelioratemitochondrial dysfunction and exhibit cardio-protective effects.10 Recent studies have indicated that PUE can protect the myocardium against ischemia and reperfusion injury by the opening of mitochondrial ATP-sensitive potassium channel and the inhibition of mPTP opening.11 In addition, experimental studies have shown that PUE can inhibit cardiomyocyte apoptosis as evidenced by the increased expression of anti-apoptotic protein Bcl-2 and decreased expression of pro-apoptotic protein Bax.12 However, owing to the poor solubility, low bioavailability, especially lack of mitochondrial targeting of PUE, only little amount of drug could be delivered to the mitochondria of ischemic cardiomyocyte. Therefore, exploring new sub-cellular targeting strategies for targeted delivery of PUE to mitochondria, taking full advantage of the precise regulation of mitochondrial function will likely greatly improve the efficacy of the drug.
Nowadays, various mitochondrial drug delivery systems have been developed to introduce mitochondriotropic molecules into carriers to target mitochondria.13 Triphenylphosphonium (TPP), a polar cationic molecule, was widely used as an efficient mitochondrial targeting moiety.14 The mitochondria-targeted nanoparticles carrying the TPP moiety can penetrate through the mitochondrial membrane due to the cationic property and lipophilicity of TPP, driving the molecules with positive charges and high lipophilicity to accumulate within the negative charge of mitochondria.15 Hence, TPP has been conjugated to various vectors such as dendrimers, liposomes, and nanoparticles for mitochondrial-targeted drug delivery. These mitochondria-targeted drug delivery systems have achieved promising anticancer effects in cancer chemotherapy.16 In addition (18F-Fluoropentyl) Triphenylphosphonium cation (18F-FPTP) carrying the TPP moiety has been used as a mitochondrial voltage sensor for myocardial imaging by positron emission tomography (PET).17 In vivo biodistribution and imaging studies showed that 18F-FPTP could accumulate in the myocardium with rapid clearance from the liver and lung.18 However, these nanostructures with mitochondria-targeting properties have rarely been studied on cardiomyocyte apoptosis in myocardial ischemia.
Recently, a growing number of studies have reported that the 1,2-Distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000] (PEG-PE) micelles are promising drug delivery carriers for its enhanced permeation and retention (EPR) effect at ischemic myocardium,19 enhanced transmembrane transport of drugs, and altered drug internalization route and subcellular localization properties.20 Moreover, PEG forming the hydrophilic corona in the PEG-PE micelles could also reduce uptake of the reticuloendothelial system (RES) in the liver and spleen and increase the retention time in the blood, which could deliver cargoes to the targets.21 Our previous research found that PEG-PE micelles exhibited favorable cellular uptake efficiency on H9c2 cells.22 On this basis, we synthesized a PEG-PE copolymer with a single terminal lipophilic TPP cation, which was then incorporated into PEG-PE to form mixed micelles with mitochondrial targeting ligand TPP. The cationic TPP could facilitate nanoparticle penetration through the mitochondrial membrane. Thus, it is reasonable to speculate that PUE-loaded TPP/PEG-PE (PUE@TPP/PEG-PE) micelles bearing several TPP residues could effectively deliver PUE into mitochondria and enhance the anti-apoptosis effect of PUE by precisely regulating mitochondrial function.
Materials
1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethyleneglycol)-2000] (PEG2000-PE) was acquired from Lipoid GmbH (Ludwigshafen, Germany). 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[amino (polyethylene glycol)-2000] (ammonium salt) (DSPE-PEG2000-NH2) was acquired from Avanti Polar Lipids Inc. (Alabaster, AL) and used without further purification. PUE was purchased from Shandong Fangming Pharmaceutical Group Co., Ltd. (Shandong, China). Triphenylphosphonium (TPP), N-Hydroxysuccinimide (NHS), triethylamine (TEA), and 1-(3-Dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDCI) were obtained from Sigma Chemical Co. (St Louis, MO). Coumarin-6 (C6) and isoprenaline (ISO) were purchased from J&K Chemical Ltd. (Shanghai, China). 4ʹ6-diamidino-2-phenylindole (DAPI) was provided by Beyotime Biotech (Jiangsu, China). Mitotracker Red, Lysotracker Red, Hoechst 33342, and 1,1ʹ-dioctadecyl-3,3,3′,3ʹ-tetramethylindotricarbocyanine iodide (DiR) were obtained from Invitrogen (Carlsbad, USA). H9c2 cells derived from rat myocardium were purchased from the American Type Culture Collection (Manassas, VA, USA). Cell cultures were maintained in a humidified atmosphere of 5% CO2 at 37°C. Dulbecco’s modified Eagle’s medium (DMEM) and fetal bovine serum (FBS) were purchased from Gibico (Gibco BRL Co. Ltd., USA). The antibodies (Bcl-2, Bax, and GAPDH) were obtained from Abcam (Abcam, Hong Kong, China). All other chemicals and reagents were analytical grade or chromatography grade.
Synthesis And Identification Of TPP-PEG-PE Copolymer
The synthesis of TPP-PEG-PE conjugate was performed following the previously published procedure.23 Briefly, 18.4 mg of TPP was dissolved in 2 mL of chloroform, and then 40 µL of TEA, 24.8 mg of EDCI, and 13 mg of NHS were added to react for 4 hrs to activate the carboxyl group of TPP. Then, 100 mg of NH2-PEG-PE was subsequently added to the reaction system. The mixture was stirred overnight at room temperature under argon, and chloroform was evaporated. The reaction product was diluted with distilled water and transferred into a dialysis bag (2kDa) and dialyzed in 1 L of pure water for 48 hrs to remove the unreacted materials and byproducts. Finally, the dialysate was freeze-dried to obtain pure TPP-PEG-PE polymer. The structures of the synthesized TPP-PEG-PE polymer were confirmed by 1H NMR and FT-IR spectroscopy. 1H NMR spectroscopy analysis was carried out using chloroform-d as the solvent at 400 MHz. FT-IR spectroscopy was implemented to scan TPP-PEG-PE and TPP with KBr at a wave number range of 400~4000 cm−1.
Preparation Of Drug-Loaded Micelles
PUE was entrapped into PEG-PE micelles and mitochondria-targeted micelles by the thin-film hydration method. Briefly, 4 mg of PUE was dissolved in 10 mL chromatographic methanol along with 40 mg of PEG-PE or a mixture of 40 mg of PEG-PE and TPP-PEG-PE (9:1 molar ratio). The organic solvent was removed by the rotary evaporation to form a thin film at the bottom of the vials. The film was further dried under high vacuum overnight to remove any remaining organic solvents. Then, the dried film was hydrated with 4 mL of distilled water and incubated in water bath at 37°C for 30 mins. The mixture was vortexed for 2 mins and ultrasonicated for 5 mins to ensure proper resuspension of the film. Finally, the obtained clear micellar solution was filtrated through a 0.22-μm syringe filter to afford a clear solution and remove any non-incorporated drug and copolymer. Spontaneous formation of micelles was initially confirmed by the observation of a transparent reddish solution and Tyndall phenomenon. To study the cellular uptake of drug-loaded micelles by H9c2 cells, the coumarin-6 (fluorescent material) loaded unmodified or TPP-modified micelles (C6@TPP/PEG-PE) were prepared by the same method as that for PUE-loaded micelles.
Characterization
The morphology and structure of micelles were observed by a transmission electron microscope (TEM, JEM 2100, JEOL Ltd., Japan) with an accelerated voltage of 200 kV. The particle size (hydrodynamic diameter) and zeta potentials of the samples were determined by dynamic light scattering (DLS) using Malvern Zetasizer Nano ZS (Malvern Instruments Ltd, UK) at 25°C. Each sample was diluted with distilled water appropriately before analysis and all samples were measured in triplicate.
Entrapment Efficiency (EE) And Drug Loading (DL)
HPLC was used to analyze the drug loading efficiency (DL%) and entrapment efficiency (EE%) for PUE in micelles. The drug DL% and EE% of micelles were calculated by the following equations. The content assay of the drugs was determined by LC-20AT HPLC system equipped with a UV detector set at a wavelength of 250 nm.
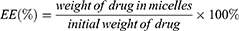
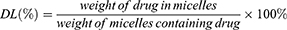
Micelles Stability Studies
The PUE@TPP/PEG-PE micelles were stored as a micelles suspension in the dark at 4°C and as a lyophilized powder in a −20°C freezer for 2 months. The stability of the PUE@TPP/PEG-PE micelles was monitored by the signs of precipitation or crystal growth, particle size analysis, and drug content in the samples during the storage period. Moreover, the serum stability of PUE@PEG-PE micelles was incubated with 10% fetal bovine serum (FBS). The changes in particle size at 0, 4, 8, 12, and 24 hrs were monitored by DLS.
In Vitro Release Kinetics
The in vitro release study of PUE from drug-loaded micelles was performed via dialysis technology. Briefly, free PUE, PUE@PEG-PE, or PUE@TPP/PEG-PE micelles at the same amount of 100 μg PUE were placed in a dialysis bag (Spectrum Labs Inc., USA) having a molecular cut-off 2kDa. The bag was immersed in 50 mL of PBS (pH 7.4) while being shaken at 100 rpm. The temperature was maintained at 37°C during the experiment. At set time intervals, the solution outside the dialysis bag (0.5 mL) was withdrawn from the release medium. The removed solution was immediately replaced with the same volume of fresh PBS. The content of PUE in each sample was then analyzed by HPLC.
Cellular Uptake Assay
Considering the no fluorescent property of PUE, C6 was used as the classic fluorescence model incorporated into the micelles for observation of cellular uptake. A fluorescent inverted microscope was used to qualitatively probe into cellular uptake efficiency of free C6, C6@PEG-PE, and C6@TPP/PEG-PE micelles. H9c2 cells were seeded into 12-well culture plates at 1.0×105 cells per well and cultured for 24 hrs. Then, the cells were incubated with free C6, C6@PEG-PE, or C6@TPP/PEG-PE micelles at an equivalent C6 concentration (100 ng/mL) in a medium supplement with 10% FBS. After 1 and 4 hrs incubation, cells were washed with PBS (pH 7.4) three times to eliminate residual C6 outside the cells. After rinsing with PBS, the cell nuclei were stained with DAPI. Fluorescent images of the cells were analyzed using a TCS SP2 confocal microscope (Leica, Germany) in order to investigate quantitatively cellular uptake of free C6, C6@PEG-PE, and C6@TPP/PEG-PE micelles. The cell processing process was the same as the above-mentioned intracellular fluorescence qualitative test. The intracellular C6 fluorescent intensity in each H9c2 cells was quantitatively analyzed by flow cytometry using a BD LSRFortessaTM cell analyzer (BD Biosciences).
Confocal Laser Scanning Microscopy (CLSM)
H9c2 cells were grown to 75–85% confluence on 22 mm coverslips in 6-well cell culture plates. After incubation overnight, the cells were treated with 100 ng/mL of C6@PEG-PE and C6@TPP/PEG-PE micelles followed by incubation at 37°C in a 5% CO2 atmosphere for time periods of 1 and 4 hrs. Then, the cells were washed several times with PBS, fixed with 4% paraformaldehyde for 20 mins and stained with Mitotracker Red (150 nM) or Lysotracker Red (250 nM). Nuclei were stained blue with Hoechst 33342 for 10 mins. The images were captured in individual channels and overlapped as composite pictures. The Person’s correlation coefficient (R) of mitochondria or lysosomes with micelles were obtained through Image J software.
Cell Culture And Treatment
H9c2 cells were grown at 37°C under 5% CO2 in DMEM containing 10% FBS. H9c2 cells were divided into 5 groups as follows: 1) control, cells were incubated with phosphate-buffered saline (PBS). 2) ISO, ISO (10 μM, dissolved in PBS) for 24 hrs. 3) ISO + PUE, pre-incubated with free PUE (20 μM) for 0.5 hr before treated with ISO. 4) ISO + PUE@PEG-PE, pre-incubated with PUE@PEG-PE micelles (20 μM, PUE equivalent) for 0.5 hr before treated with ISO. 5) ISO + PUE@TPP/PEG-PE, pre-incubated with PUE@TPP/PEG-PE micelles (20 μM, PUE equivalent) for 0.5 hr before treated with ISO. All assays were conducted in triplicate and repeated for three times.
Cell Apoptosis Assay
Following treatment, the apoptosis rate of H9c2 cells was determined by flow cytometry with the Annexin V-FITC/PI apoptosis detection kit according to the manufacturer’s protocol (BD Biosciences Inc.; San Jose, CA, USA). Briefly, cells were washed with ice-cold PBS, and then resuspended in binding buffer at a concentration of 1×106 cells/mL. Five microliter FITC Annexin V and 5 µL propidium iodide (PI) were added into each well for 15 mins at room temperature (25°C) in the dark, followed by the addition of 400 µL of 1X binding buffer to each tube. Analysis by flow cytometry was performed within 1 hr. Apoptosis in H9c2 cells was determined using flow cytometry (FACSCalibur; BD Biosciences) and analyzed using the FCS Express v2.0 software (De Novo Software, Los Angeles, CA, USA). The cells stained with FITC but not with PI (FITC+, PI−) were considered as early apoptosis, and the cells double-stained with both dyes (FITC+, PI+) were regarded as late apoptosis.
ROS Level Assay
The fluorescent probe dihydroethidium (DHE; Beyotime, Shanghai, China) was used to monitor intracellular ROS levels. Intracellular DHE is oxidized to ethidium, which binds to DNA and stains the nuclei bright fluorescent red. The detection of ROS level in H9c2 cells was conducted according to the manufacturer’s instructions and was similar to our previous study.24
Caspase-3 Activity Assay
The activity of Caspase-3 was determined by using a Caspase-3 activity kit (Beyotime Institute of Biotechnology, China). The total protein of cells was obtained through lysis buffer. A total of 40 µg of protein was diluted to 50 µL of final volume that was then mixed with 75 µL of caspase-3 substrate for 3 hrs; the hydrolysis of Ac-DEVD-pNA by caspase-3 released free pNA (yellow formazan product) that was detected at 405 nm. Caspase-3 activity was expressed as the fold of enzyme activity compared to that of synchronized cells. The detailed protocol was conducted according to the manufacturer’s instructions and was similar to our previous study.22
Western Blot Analysis
Following treatment, the total protein was extracted from each group. Proteins were resolved on 10% SDS-PAGE and electroblotted onto nitrocellulose membranes. Membranes were blocked with 2.5% milk solution, followed by incubating overnight at 4°C with primary antibodies. The blots were then incubated with horseradish peroxidase (HRP)-coupled secondary antibody. The primary antibodies as following: Bcl2 (ab692, 1:1000, Abcam), Bax (ab32503, 1:1000, Abcam), and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (ab9484, 1:1000, Abcam).
In Vivo And Ex Vivo Biodistribution Determined By Fluorescence Imaging
The animal experiment was approved by the Animal Ethics Committee at the Second Xiangya Hospital of Central South University and followed the guidelines of the National Act on the Use of Experimental Animals (China). Female Bab/c mice were used and supplied by Hunan Slack Scene of Laboratory Animal Co., Ltd. According to the existing literature,25 the induction of myocardial ischemia in mice was performed by subcutaneous injection of ISO (100 mg/kg) at an interval of 24 hrs for 2 days. The model of the ISO-induced myocardial ischemia in mice was confirmed by echocardiography and hematoxylin and eosin (H&E) staining. Echocardiography was performed using a Vevo 770 High-Resolution Imaging System. Animals were anesthetized with isoflurane and placed in a supine position. The chests were shaved, and the parasternal short- and long-axis views were performed to obtain two-dimensional and M-mode images. The ejection fraction (EF) and fractional shortening (FS) were calculated and expressed as a percentage (%). At least 10 independent cardiac cycles per each experiment were obtained. DiR, a near-infrared fluorescent probe, was encapsulated into the PEG-PE and TPP/PEG-PE micelles according to the similar method described earlier. Free DiR solution and DiR-loaded micelles at DiR-eq dose were injected into the ISO-induced myocardial ischemia mice tail veins. Then, the mice were scanned at 6- and 12-hr post injection using an FXPRO imaging device (Bruker, USA) at appropriate wavelengths (DiR, EX: 720nm, EM: 790 nm) and then were sacrificed. Their major organs (heart, liver, spleen, lung, and kidney) were excised, followed by washing the surface with 0.9% normal saline for the ex vivo imaging. Images were analyzed using the Bruker MI software.
Statistical Analysis
The results were reported as the mean ± SEM from triplicate determinations. The mean values of the different treatment groups were then statistically compared to that of the control groups using the Student’s t-test and one-way analysis of variance (ANOVA). The criterion for statistical significance was at *p < 0.05 and **p < 0.01.
Results And Discussion
Synthesis And Identification Of TPP-PEG-PE Copolymer
Cell Apoptosis AssayThe characteristic signals of aromatic protons of the TPP group at 7.5–7.9 ppm appeared in the 1H NMR of TPP-PEG-PE (Figure 1A), suggesting that the TPP had been conjugated to NH2-PEG-PE to form TPP-PEG-PE. Compared with the FT-IR spectrum of PEG-PE, a new peak occurred at 1652.82 cm−1 representing amide-bond formation appeared in the TPP-PEG-PE spectrum (shown as arrows), suggesting the carbonyl group should have converted into amide group. Moreover, the characteristic peaks of TPP at 755.10 cm−1 and 690.03 cm−1 can also be shown in TPP-PEG-PE (Figure 1B). Taken together, these results indicated that the successful synthesis of TPP-PEG-PE conjugate.
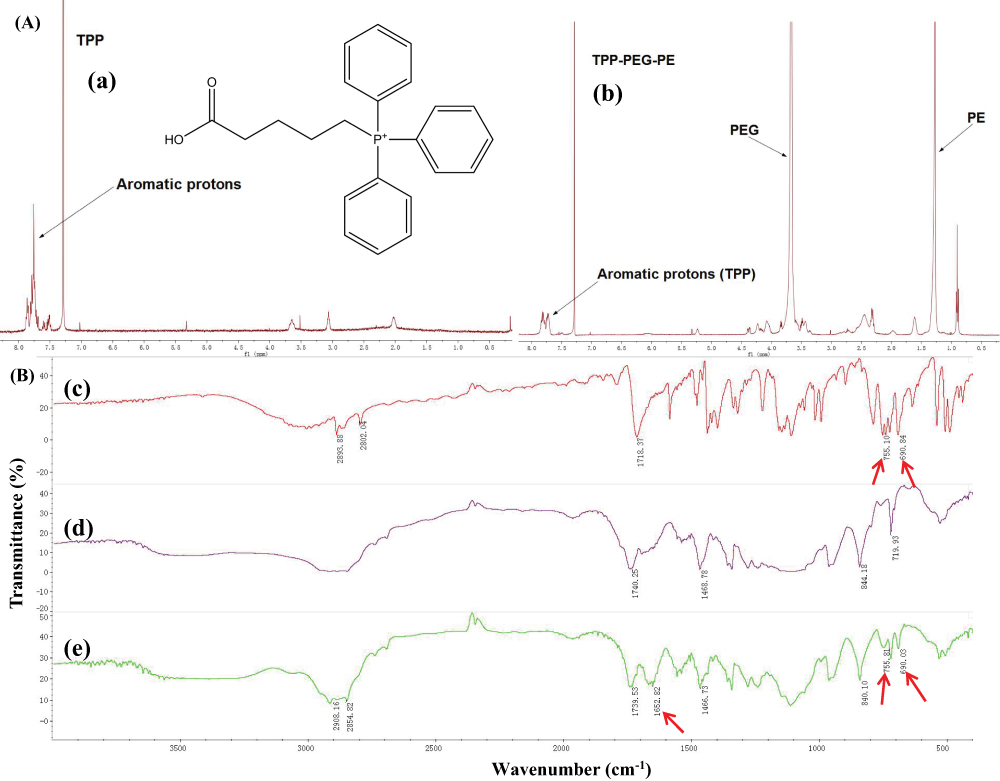 |
Figure 1 Synthesis of characterization of TPP-PEG-PE copolymer. (A) The 1H NMR spectrum of (a) TPP and (b) TPP-PEG-PE copolymer. (B) FT-IR analysis of (c) TPP, (d) NH2-PEG-PE, and (e) TPP-PEG-PE. The red arrows point out the typical functional groups of above-mentioned three compounds, TPP-PEG-PE is synthesized from the combination of TPP and NH2-PEG-PE with the amide-bond. The peaks of 755.10-1 cm, 690.84 cm-1, 755.81 cm-1 and 690.03-1indictaed by the red arrowsrepresent the aromatic ring characteristic peaks of TPP and the TPP-PEG-PE, respectiveley. The peaks of 1652.82 cm-1indicated by the red arrows represents the amide-bond at the spectra of TPP-PEG-PE. |
Micelle Formation, Nanoparticle Size, Zeta Potential, And Morphology
Amphiphatic polymer could self-assemble to form micelles in an aqueous solution by the filming-rehydration method (Figure S1A). A transparent reddish solution and Tyndall phenomenon shown in Figure S1B initially indicated the formation of micelles. The micelle-forming nanoparticles can produce good scattering of the irradiated light and behave as a beam of light; thus, the Tyndall effect is a common physical method for initially identifying the formation of micelles. Under TEM, the morphology of PUE@TPP/PEG-PE micelles was illustrated in Figure 2A, showing approximately core-shell structure. The periphery of the nanoparticles was covered by a thin coating layer. DLS displayed that the polydispersity index (PDI) values of PUE@TPP/PEG-PE micelles were under 0.2, meaning homogenous size distribution in the formulation. Moreover, the hydrodynamic diameter of PUE@TPP/PEG-PE micelles was 17.1 nm (Figure 2B), which was slightly larger than the particle diameter of PUE@PEG-PE micelles. It indicated that the embedding of the TPP cations did not substantially destroy the morphological structure of the micelles. The small size of PUE@TPP/PEG-PE micelles is beneficial to be easily taken up by cells mainly through the caveolae-mediated endocytosis.26 Zeta potential of micelles increased significantly upon the introduction of the TPP cations. PUE@TPP/PEG-PE micelles containing highly positively charged TPP exhibited a zeta potential of −6.24 mV, whereas the zeta potential of PUE@PEG-PE micelles was −23.8 mV (Figure 2C). This result also confirmed that TPP was successfully modified on the outer layer of the micelles. Although TPP was a highly cationic molecule, the PUE@TPP/PEG-PE micelles had a slightly negative zeta potential, which was attributed to the low surface density of TPP-PEG-PE copolymer. At the physiological environment, the red blood cells and plasma protein have a negative charge, which could reduce the nonspecific interaction with the negatively charged PUE@TPP/PEG-PE micelles. The result is benefited by reducing their rapid elimination from the blood circulation and promoting the passive accumulation at the infarcted myocardium.27
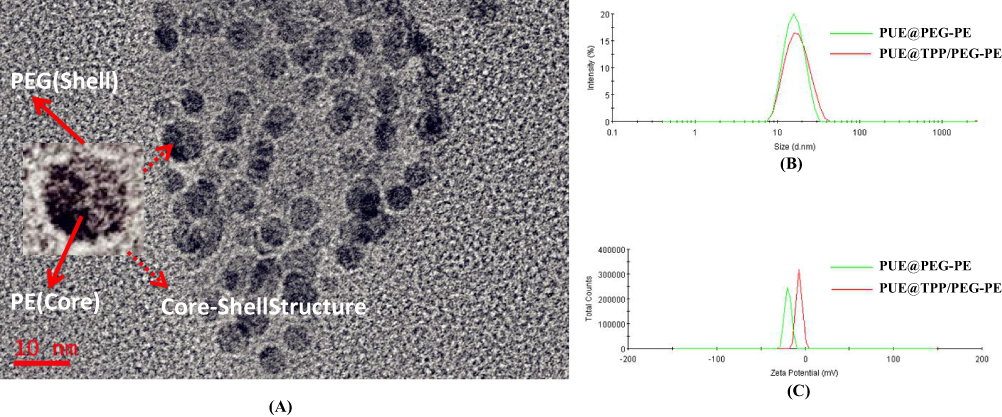 |
Figure 2 Characterization of PUE@TPP/PEG-PE micelles. (A) TEM photographs. (B) Size and (C) zeta potential distribution of PUE@PEG-PE and PUE@TPP-PEG-PE micelles. |
Entrapment Efficiency (EE%), Drug Loading (DL%) And In Vitro Stability
Owing to the hydrophobic PE core and the hydrophilic PEG shell in the PUE@TPP/PEG-PE micelles, PUE can be loaded in the TPP/PEG-PE micelles with the high EE% and DL% of 85.5% and 5.3%, respectively. The HPLC detection and DLS results indicated that PUE@TPP/PEG-PE micelles remained neither decrease of drug content nor micelles size distribution changes for at least 2 months at 4°C and as a freeze-dried powder for 2 months at −20°C. This high EE% and favorable stability of PUE@TPP/PEG-PE micelles were attributed to the excellent fusion of PUE with the lipophilic core of micelles. Thus, PUE could not easily escape from the tightly packed core of micelles and enter into the aqueous solution during the self-assembling process.28 To enable the micelles to be applicable in intracellular delivery, stability of the micelles in cell media is a major concern. As reported in previous literature,29 micelle incubation in the presence of 10% FBS at 37°C for 24 hrs also exhibited excellent stability, and there was no significant difference in size or size distribution. Micelles maintained the same size and showed minimal PUE leakage in a serum-containing medium, which indicated high suitability of these micelles for intracellular uptake, allowing for desired concentrations of drug accumulation in the ischemic zone by the enhanced permeation and retention effect.30
In Vitro Release Of PUE From Micelles
As shown in Figure 3, free PUE was released rapidly, more than 90% of the drug has been released within the first 12 hrs. However, the release of two PUE-loaded micellar formulations, both showed an initial rapid release within the first 18 hrs, followed by a slow and sustained release of PUE over the course of almost 72 hrs. The effect of incorporation of the cationic TPP into PEG-PE on slow release of PUE was more obvious than that of PEG-PE micelles during the whole release test. PEG-PE micelles released 72.8% of PUE at 72 hrs, but there were 64.5% of PUE eventually released from TPP/PEG-PE micelles. This sustained release profile of PUE@TPP/PEG-PE micelles was desired to maintain the desired drug concentrations at the site of action.
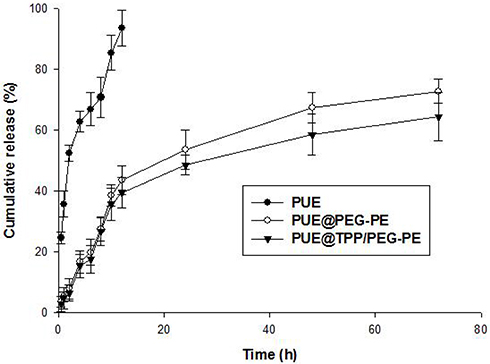 |
Figure 3 In vitro release curve of free PUE, PUE@PEG-PE, and PUE@TPP/PEG-PE micelles in PBS (pH = 7.4) at 37°C. |
Cellular Uptake Assay
As shown in Figure 4A, after 1 hr of incubation, compared with the C6@PEG-PE micelles, the fluorescence of C6 in TPP/PEG-PE micelles treated cells was significantly brighter and clustered around the nucleus, suggesting that the C6@TPP/PEG-PE micelles were rapidly internalized into the cells within 1 hr of incubation. As the incubation time was prolonged from 1 to 4 hrs, the fluorescent intensity of C6 entering into the cells showed an obvious increasing trend. Compared with the C6@PEG-PE micelles, the green fluorescent intensity was slightly stronger following 4 hrs incubation of C6@TPP/PEG-PE micelles. Consistent with the qualitative analysis of the cellular uptake under fluorescence microscope, quantitative analysis via flow cytometry indicated the same time-dependent cellular internalization manner. The fluorescence intensity reflecting cellular uptakes of C6@TPP/PEG-PE micelles at 1 hr post-treatment was significantly higher than that of the corresponding C6@PEG-PE micelles as seen from Figure 4B(a). After 4 hrs of incubation, the greatest fluorescence intensity was found in the cells exposed to C6@TPP/PEG-PE micelles, which was almost 1.62-fold and 1.15-fold higher than cells exposed to free C6 and C6@PEG-PE micelles, respectively (Figure 4B(b)). Although no obvious difference was observed between C6@PEG-PE and C6@TPP/PEG-PE micelles at 4 hr post-treatment. Overall, the cellular uptake of C6 in TPP/PEG-PE micelles exhibited a higher level of internalization compared with that of C6@PEG-PE micelles and free C6. The results indicated that the TPP cation could be used to enhance the cellular uptake in the H9c2 cells, which might result from the fact that cationic TPP could enable the micelles to interact with the negatively charged cell membrane, leading to absorptive endocytosis.31
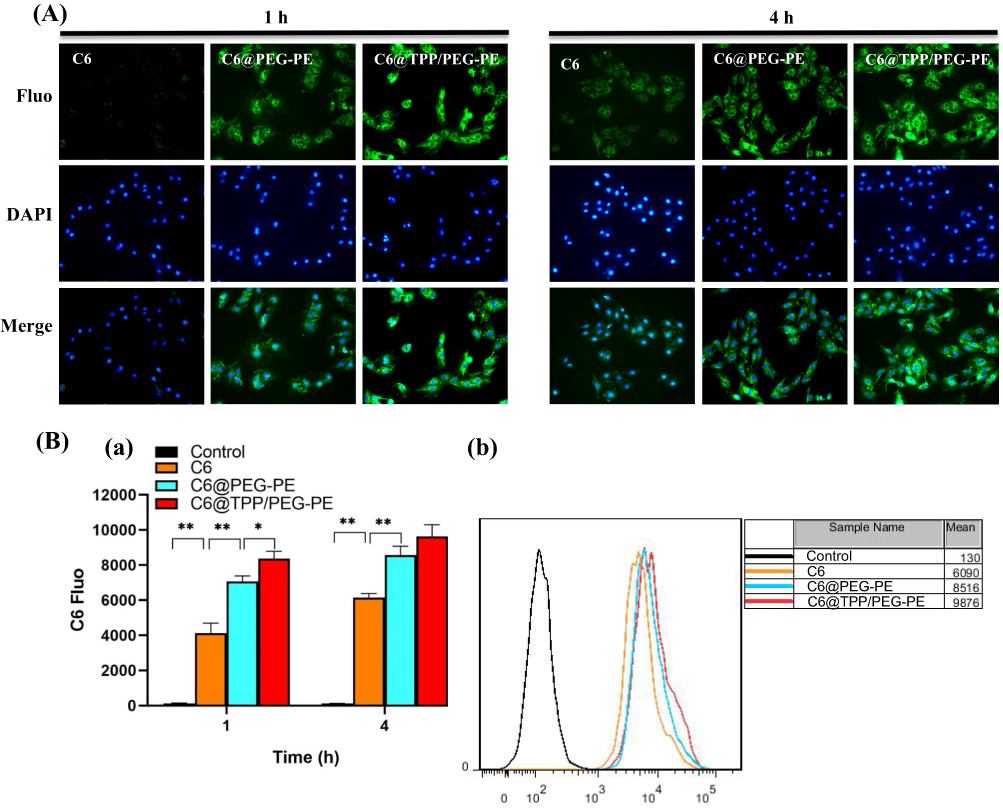 |
Figure 4 Fluorescent images for qualitative cellular uptake of free C6, C6@PEG-PE, and C6@TPP/PEG-PE micelles incubation in H9c2 cells at 1 and 4 hrs (A). H9c2 cells were incubated with free C6, C6@PEG-PE, and C6@TPP/PEG-PE micelles and the intracellular fluorescence intensity was measured by flow cytometry (B). (a) Quantification of cellular uptake after incubation with free C6, C6@PEG-PE, and C6@TPP/PEG-PE micelles for 1 and 4 hrs. (b) Intracellular fluorescence intensity after treated with free C6, C6@PEG-PE, and C6@TPP/PEG-PE micelles for 4 hrs. Data were expressed as the mean ± SEM from triplicate determinations. *p <0.05, **p <0.01 |
Confocal Laser Scanning Microscopy (CLSM)
C6@TPP/PEG-PE micelles were functionalized with mitochondrial targeting ligand TPP, which facilitates micelles penetration through the mitochondrial membrane. We speculated that C6@TPP/PEG-PE micelles would effectively deliver C6 into mitochondria. The mitochondrial targeting effect of C6@TPP/PEG-PE micelles by H9c2 Cells was evaluated using confocal laser scanning microscope (CLSM) with Mitotracker red staining methods. C6 and Mitotracker red emit green and red fluorescence, respectively. The CLSM photograph of H9c2 cells showed that the co-localization of the green fluorescence of the micelles and the red fluorescence of the stained mitochondria appeared yellow. A comparison of the yellow fluorescence intensities indicated a significantly greater overall uptake of the C6@TPP/PEG-PE micelles than that of C6@PEG-PE micelles in the mitochondria of H9c2 cells (Figure 5). A strong yellow fluorescence (the overlap of C6@TPP/PEG-PE with mitochondria) was found after incubation for 4 hrs, indicating maximum accumulation. Quantitative analysis using the Image J software showed that the significant co-localization of C6@TPP/PEG-PE micelles and mitochondria with the Pearson’s correlation coefficients (R) of 0.52 (1 hr) and 0.69 (4 hrs) can be observed. On the contrary, the C6@PEG-PE micelles showed a much lower R value of 0.44 (1 hr) and 0.50 (4 hrs). The above results indicated that the TPP/PEG-PE micelles effectively delivered drugs to the mitochondria and may greatly improve the efficacy of the drugs. This may be due to the highly lipophilic ligand of TPP with three phenyl groups and a positive charge on phosphorous, which enhanced its cell association and mitochondrial targeting.32
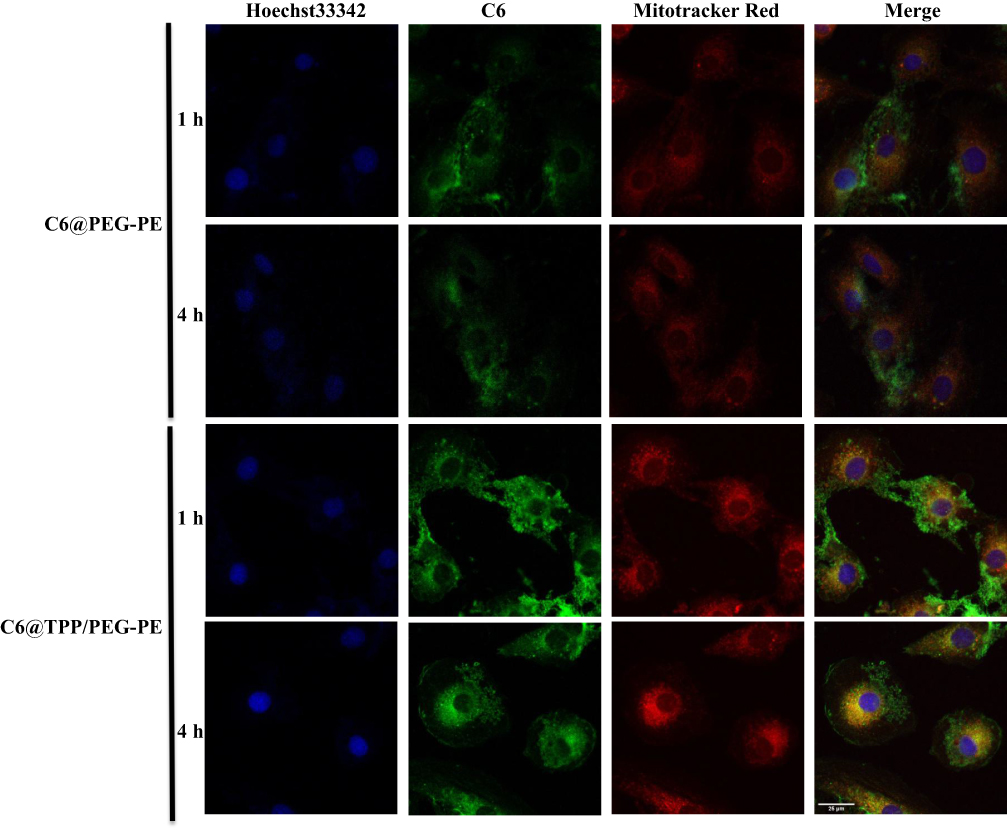 |
Figure 5 CLSM images of H9c2 cells incubated with C6@PEG-PE and C6@TPP/PEG-PE micelles for 1 and 4 hrs (Green: C6, Red: Mitotracker red stained mitochondria, Blue: Hoechst 33342 stained nuclei). |
The use of nanoparticles for mitochondrial targeting is often limited by the fact that nanoparticles are often absorbed by lysosomes, and lysosomes act as a barrier to mitochondrial transport. Thus, degradation of the encapsulated drug in lysosomes should be reduced as much as possible for efficient mitochondria-targeting delivery. As shown in Figure 6, in the merged images, the yellow fluorescence is overlaid by the red fluorescence of Lysotracker Red and green fluorescence of the micelles. We studied a time-dependent colocalization of the micelles with lysosomes. C6@TPP/PEG-PE micelles exhibited a much lower level of colocalization with lysosomes as compared to C6@PEG-PE micelles. Analysis of colocalization (based on Image J software) clearly showed that the R of C6@TPP/PEG-PE micelles decreased significantly over time, especially at 4 hrs, the calculated Pearson’s correlation coefficients (R) for C6@PEG-PE and C6@TPP/PEG-PE micelles in lysosomes were 0.14 and 0.03, respectively. It implied the highly efficient lysosomal escape ability of the C6@TPP/PEG-PE micelles due to the presence of cationic TPP on the surface of nanoparticles. Due to the positive charge on phosphorous, the high cationic property of TPP was easily internalized in the acidic lumen of the lysosomal vesicle. These nanoparticles containing highly positively charged TPP become protonated and cause the entrance of protons in the vesicle in order to maintain the lysosomal physiologic condition.33 Cationic TPP can act as “proton sponges”.34 It causes an increase in the pH of the lysosomes, thereby promoting the influx of extracellular counter ions and water molecules, resulting in osmotic swelling, which in turn leads to lysosome membrane rupture, and eventual leakage of the nanoparticles into the cytosol, making them accessible for mitochondrial uptake.35 It can also be seen from the photograph that the red fluorescence of Lysotracker Red became very weak after 4 hrs of C6@TPP/PEG-PE micelles incubation with the H9c2 cells. It was speculated that the “proton sponges” effect of cationic TPP may lead to swelling and rupture of lysosomes.
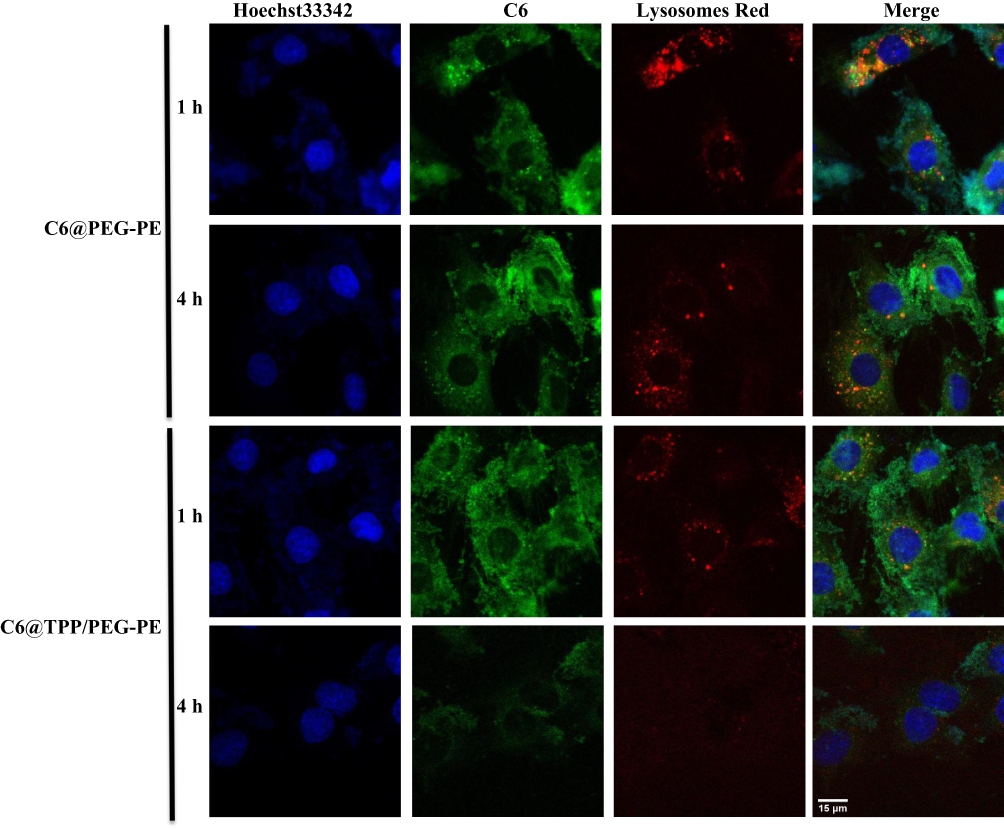 |
Figure 6 CLSM images of H9c2 cells incubated with C6@PEG-PE and C6@TPP/PEG-PE micelles for 1 and 4 hrs (Green: C6, Red: Lysotracker red stained with lysosomes, Blue: Hoechst 33342 stained nuclei). |
Cell Apoptosis Assay
As mentioned in the introduction, programmed necrosis and apoptosis act as the principal cardiomyocyte death pathways and play key roles in the process of myocardial infarction.36 Both of them are closely related to mitochondrial function, especially the apoptosis. The TPP/PEG-PE micelles in the present study were used to increase the mitochondrial targeting of PUE, and improve the protective effect of PUE against ISO-induced H9c2 cell apoptosis. Considering a clearer molecular biological approach for determining apoptosis than necrosis,37 the pharmacological effect of PUE@TPP/PEG-PE micelles to regulate mitochondrial function was investigated in ISO-induced H9c2 cell apoptosis.
Exposure of H9c2 cells to 10 μM ISO for 24-hr-induced apoptosis model, as evidenced by the increase in the number of Annexin V-FITC positive fluorescent cells from 3% (control cells) to 38% (ISO-treated cells). As shown in Figure 7A, compared with the model group, all drug pretreatment groups could reduce the ISO-induced cell apoptosis rate. Among them, the lowest percentage of cell apoptosis was observed after pretreatment with PUE@TPP/PEG-PE micelles (Figure 7B). Both experiments of ROS level and caspase-3 activity further confirmed the results of flow cytometry, PUE@TPP/PEG-PE micelles reduced the ROS level and Caspase-3 activity more significantly when compared to the PUE@PEG-PE micelles and free PUE (Figure 7C and D). Moreover, we further investigated the effects of PUE@TPP/PEG-PE micelles on the expression of apoptosis-related proteins. As shown in Figure 7E, ISO induced the increase of Bax expression and decrease of Bcl-2 expression. Pretreatment with drugs could markedly reduce the expression of Bax but increase the expression of Bcl-2. Both PUE@ PEG-PE and PUE@TPP/PEG-PE micelles inhibited the effect of ISO, and PUE@TPP/PEG-PE micelles exhibited stronger effect than PUE@PEG-PE micelles. These results collectively suggested that PUE@TPP/PEG-PE micelles can enhance the protective effects of PUE against ISO-induced H9c2 cell apoptosis.
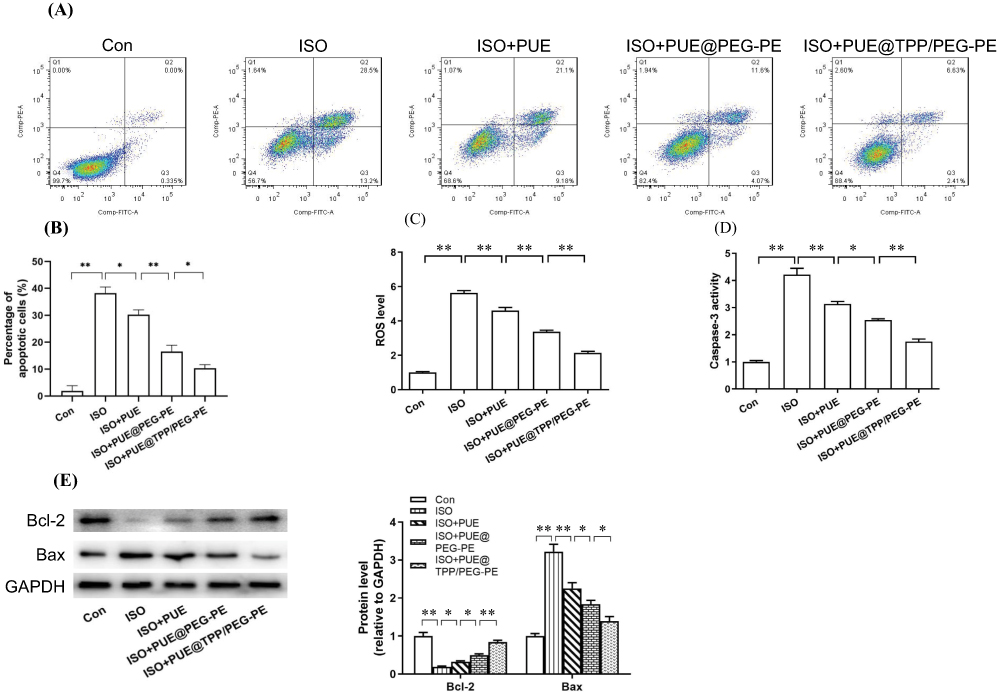 |
Figure 7 Protective effect of free PUE, PUE@PEG-PE, and PUE@TPP/PEG-PE micelles against ISO-induced H9c2 cell apoptosis. (A) Representative flow cytometric graphs of H9c2 cells after stained with Annexin V-FITC/PI-labeled. (B) ROS level. (C) Caspase-3 activity. (D) The percentage of apoptotic cells. (E) Apoptosis-related protein (Bcl-2 and Bax) expression. Data are mean ± SEM (n = 3) (*p <0.05, **p <0.01). |
In Vivo And Ex Vivo Biodistribution Determined By Fluorescence Imaging
According to the methods reported in the literature, we adopted female mice as a model for myocardial ischemia.38 In addition, female mice are relatively temperate and are not easy to fight during the same cage feeding process. In the model of myocardial ischemia induced by subcutaneous injection of ISO, the skin of the mouse has been damaged. Considering that males are relatively aggressive, they may bite each other during feeding, causing damage to the skin again. Therefore, we finally chose female mice as a model for myocardial ischemia. Although left coronary artery (LCA) ligation is most commonly used to induce myocardial ischemia in mice, this surgical procedure has disadvantages of a high mortality rate and a large variation in infarct size.39 In contrast, ISO-induced myocardial ischemia is a simple, easy-to-operate modeling method that can reduce inconsistency in infarct size due to anatomic diversity of the LCA in mice.40 Recently, it has been reported that ISO has deleterious cardiac effects in rat, including apoptosis, mitochondrial alterations, fibrosis, oxidative damage, inflammatory cell infiltration, and morphological changes of cardiomyocytes, which is similar to that in the infarcted human heart.41,42 Based on this, we adopted experimental model of myocardial infarction induced by isoproterenol in mice. Moreover, the successful modeling method was verified by echocardiography and H&E staining. As indicated by Figure 8A, the results of echocardiography showed significant decrease in ejection fraction (EF%) and fractional shortening (FS%) in the model group when compared with the control group. H&E staining results further confirmed the difference between the two groups; the cardiac muscle fibers of the control group (without any operation) were relatively uniform, while myocardial fibers of the myocardial ischemia model group were in disordered and wavy arrangement (Figure 8B). Collectively, the results of echocardiography and H&E staining implied that ISO-induced myocardial infarction in mice was successful.
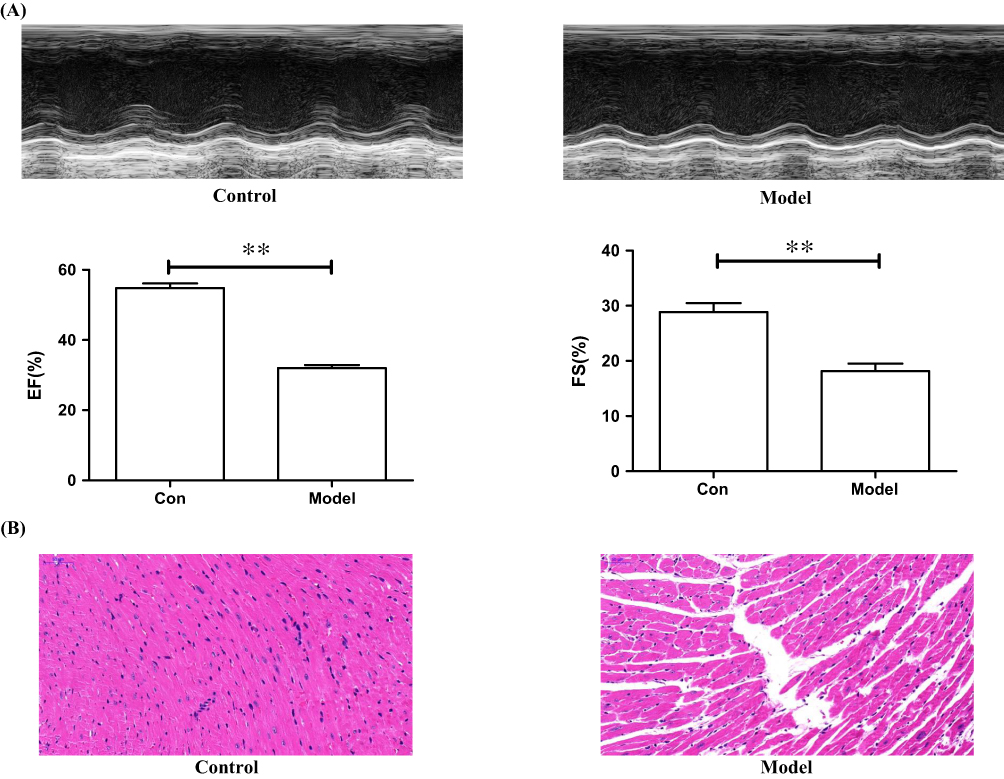 |
Figure 8 Representative echocardiography (A) and microscopic images of H&E staining of myocardial tissue sections (B) from the control group and the model of the ISO-induced myocardial ischemia group. **p<0.01. |
As clearly shown in Figure 9 (A and B), DiR-incorporated PEG-PE and TPP/PEG-PE micelles showed decreased fluorescent intensity in the liver compared with the free DiR due to the long circulation time of micelles and the reduction of reticuloendothelial system (RES) uptake in vivo.43 After 6 hrs, the fluorescent intensity from the thoracic region of the mice in the DiR@TPP/PEG-PE micelles was significantly greater than that in the DiR@PEG-PE micelles. In addition, the significant fluorescence intensity in the chest area of the mice treated with DiR@TPP/PEG-PE micelles can remain even after 12 hrs of injection, while the fluorescence signal of DiR@PEG-PE micelles in this heart region was very weak at this time. These results were further confirmed with the ex vivo studies of fluorescence intensity of heart carried out after 6 and 12 hrs (Figure 9C). These findings suggested that TPP/PEG-PE micelles permitted preferential accumulation and retention in the ischemic myocardium. This is due to the synergistic targeting effect of TPP/PEG-PE micelles. On the one hand, PEG-PE micelles can accumulate in the ischemic myocardium through the EPR effect. On the other hand, TPP cations can also accumulate several hundred-fold within mitochondria due to the high negative mitochondrial membrane.44 Therefore, 18F-FPTP, as mitochondrial voltage sensors in PET myocardial imaging, demonstrated the higher accumulation and retention in cardiac cells owing to the higher mitochondrial membrane potential and a huge bulk of mitochondria in cardiomyocytes,45 which could enhance their electrostatic attraction to the TPP moiety of 18F-FPTP. Thus, it could be further inferred that TPP/PEG-PE micelles could deliver more amount of drugs into ischemic myocardium.
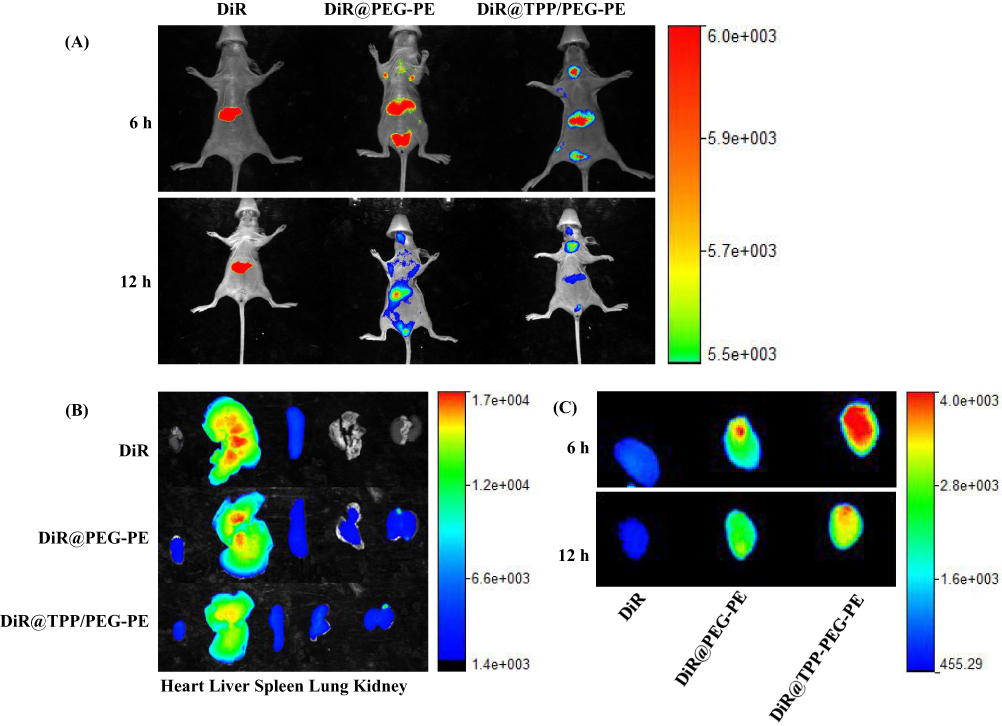 |
Figure 9 In vivo real-time imaging of ISO-induced myocardial infarction model mice administrated with free DiR, DiR@PEG-PE, and DiR@TPP/PEG-PE micelles (A); Ex vivo optical images of the major organs (heart, liver, spleen, lung, and kidney), which were taken after the mice were sacrificed at 12-hr post injection (B). Ex vivo optical images of the heart which were taken after the mice were sacrificed at 6 and 12 hrs after injection (C). |
Conclusion
In summary, the TPP/PEG-PE micelles decorated with the classic mitochondriotropic ligand TPP were successfully developed and achieved mitochondria-targeted delivery of PUE. The TPP/PEG-PE micelles showed improved cellular uptake and increased drug concentration in the mitochondria and reduced the capture of lysosomes. Compared with free PUE and PUE@PEG-PE micelles, the PUE@TPP/PEG-PE micelles exhibited better protective effect against ISO-induced H9c2 cell apoptosis, as evident by the decreased percentage of apoptotic cells, Caspase-3 activity, ROS level, and Bax expression, as well as increased Bcl-2 expression. Moreover, TPP/PEG-PE micelles had a powerful ability for delivering drugs into ischemic myocardium. Taken together, these results demonstrated that PUE@TPP/PEG-PE micelles had great potential for the treatment of AMI.
Acknowledgments
Financial support for this work is provided by the National Natural Science Foundation of China (81673614, 81703518), Hunan Provincial Science and Technology Plan (2016TP2002), Scientific Research Project of Hunan Provincial Health and Family Planning Commission (No. B20180253, B20180328, C20180059), Hunan Provincial Natural Scientific Foundation (No. 2019JJ50849, 2018JJ3571), Hunan Traditional Chinese Medicine Science and Technology Project (201579), and Open-End Fund for the Valuable and Precision Instruments of Central South University (No. CSUZC201937, CSUZC201942).
Author Contributions
All authors contributed to data analysis, drafting or revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Hong G, Rui G, Zhang D, et al. A smartphone-assisted pressure-measuring-based diagnosis system for acute myocardial infarction diagnosis. Int J Nanomedicine. 2019;14:2451–2464. doi:10.2147/IJN.S197541
2. Kavalieratos D, Gelfman LP, Tycon LE, et al. Palliative care in heart failure: rationale, evidence, and future priorities. J Am Coll Cardiol. 2017;70(15):1919–1930. doi:10.1016/j.jacc.2017.08.036
3. Reddy K, Khaliq A, Henning RJ. Recent advances in the diagnosis and treatment of acute myocardial infarction. World J Cardiol. 2015;7(5):243–276.
4. Miyagi Y, Zeng F, Huang XP, et al. Surgical ventricular restoration with a cell- and cytokine-seeded biodegradable scaffold. Biomaterials. 2010;31(30):7684–7694. doi:10.1016/j.biomaterials.2010.06.048
5. Jiang HK, Wang YH, Sun L, et al. Aerobic interval training attenuates mitochondrial dysfunction in rats post-myocardial infarction: roles of mitochondrial network dynamics. Int J Mol Sci. 2014;15(4):5304–5322. doi:10.3390/ijms15045304
6. Morciano G, Bonora M, Campo G, et al. Mechanistic role of mPTP in ischemia-reperfusion injury. Adv Exp Med Biol. 2017;982:169–189. doi:10.1007/978-3-319-55330-6_9
7. Webster KA. Mitochondrial membrane permeabilization and cell death during myocardial infarction: roles of calcium and reactive oxygen species. Future Cardiol. 2012;8(6):863–884. doi:10.2217/fca.12.58
8. Narula J, Pandey P, Arbustini E, et al. Apoptosis in heart failure: release of cytochrome c from mitochondria and activation of caspase-3 in human cardiomyopathy. Proc Natl Acad Sci U S A. 1999;96(14):8144–8149. doi:10.1073/pnas.96.14.8144
9. Wenjun H, Jing W, Tao L, et al. The protective effect of puerarin on Myocardial Infarction Reperfusion Injury (MIRI): a meta-analysis of randomized studies in rat models. Med Sci Monit. 2015;21:1700–1706. doi:10.12659/MSM.894312
10. Yuan Y, Zong J, Zhou H, et al. Puerarin attenuates pressure overload-induced cardiac hypertrophy. J Cardiol. 2014;63(1):73–81. doi:10.1016/j.jjcc.2013.06.008
11. Yang B, Gao Q, Yao H, Xia Q. Mitochondrial mechanism of cardioprotective effect of puerarin against H202-stress in rats. Zhongguo Ying Yong Sheng Li Xue Za Zhi. 2008;24(4):399–404.
12. Ma Y, Gai Y, Yan J, Li J, Zhang Y. Puerarin attenuates anoxia/reoxygenation injury through enhancing Bcl-2 associated athanogene 3 expression, a modulator of apoptosis and autophagy. Med Sci Monit. 2016;22:977–983. doi:10.12659/msm.897379
13. Battogtokh G, Choi YS, Kang DS, et al. Mitochondria-targeting drug conjugates for cytotoxic, anti-oxidizing and sensing purposes: current strategies and future perspectives. Acta Pharm Sin B. 2018;8(6):862–880. doi:10.1016/j.apsb.2018.05.006
14. Zielonka J, Joseph J, Sikora A, et al. Mitochondria-targeted triphenylphosphonium-based compounds: syntheses, mechanisms of action, and therapeutic and diagnostic applications. Chem Rev. 2017;117(15):10043–10120. doi:10.1021/acs.chemrev.7b00042
15. Porteous CM, Logan A, Evans C, et al. Rapid uptake of lipophilic triphenylphosphonium cations by mitochondria in vivo following intravenous injection: implications for mitochondria-specific therapies and probes. Biochim Biophys Acta. 2010;1800(9):1009–1017. doi:10.1016/j.bbagen.2010.06.001
16. Modica-Napolitano JS, Weissig V. Treatment strategies that enhance the efficacy and selectivity of mitochondria-targeted anticancer agents. Int J Mol Sci. 2015;16(8):17394–17421. doi:10.3390/ijms160817394
17. Park H, Kim HS, Hong YJ, et al. Therapeutic effect of fimasartan in a rat model of myocardial infarction evaluated by cardiac positron emission tomography with [(18)F]FPTP. Chonnam Med J. 2019;55(2):109–115. doi:10.4068/cmj.2019.55.2.109
18. Kim DY, Kim HS, Min JJ. Radiosynthesis and evaluation of 18F-labeled aliphatic phosphonium cations as a myocardial imaging agent for positron emission tomography. Nucl Med Commun. 2015;36(7):747–754. doi:10.1097/MNM.0000000000000315
19. Lukyanov AN, Hartner WC, Torchilin VP. Increased accumulation of PEG-PE micelles in the area of experimental myocardial infarction in rabbits. J Control Release. 2004;94(1):187–193. doi:10.1016/j.jconrel.2003.10.008
20. Kohay H, Sarisozen C, Sawant R, Jhaveri A, Torchilin VP, Mishael YG. PEG-PE/clay composite carriers for doxorubicin: effect of composite structure on release, cell interaction and cytotoxicity. Acta Biomater. 2017;55:443–454. doi:10.1016/j.actbio.2017.04.008
21. Gao H, Liu J, Yang C, et al. The impact of PEGylation patterns on the in vivo biodistribution of mixed shell micelles. Int J Nanomedicine. 2013;8:4229–4246. doi:10.2147/IJN.S51566
22. Li W, Wu J, Zhang J, et al. Puerarin-loaded PEG-PE micelles with enhanced anti-apoptotic effect and better pharmacokinetic profile. Drug Deliv. 2018;25(1):827–837. doi:10.1080/10717544.2018.1455763
23. Biswas S, Dodwadkar NS, Deshpande PP, Torchilin VP. Liposomes loaded with paclitaxel and modified with novel triphenylphosphonium-PEG-PE conjugate possess low toxicity, target mitochondria and demonstrate enhanced antitumor effects in vitro and in vivo. J Control Release. 2012;159(3):393–402. doi:10.1016/j.jconrel.2012.01.009
24. Wei S, Sun T, Du J, Zhang B, Xiang D, Li W. Xanthohumol, a prenylated flavonoid from Hops, exerts anticancer effects against gastric cancer in vitro. Oncol Rep. 2018;40(6):3213–3222. doi:10.3892/or.2018.6723
25. Cheng Y, Zhao J, Tse HF, Le XC, Rong J. Plant natural products calycosin and gallic acid synergistically attenuate neutrophil infiltration and subsequent injury in isoproterenol-induced myocardial infarction: a possible role for leukotriene B4 12-hydroxydehydrogenase? Oxid Med Cell Longev. 2015;2015:434052. doi:10.1155/2015/659750
26. Bi Y, Liu L, Lu Y, et al. T7 peptide-functionalized PEG-PLGA micelles loaded with carmustine for targeting therapy of glioma. ACS Appl Mater Interfaces. 2016;8(41):27465–27473. doi:10.1021/acsami.6b05572
27. Zuo T, Guan Y, Chang M, et al. RGD(Arg-Gly-Asp) internalized docetaxel-loaded pH sensitive liposomes: preparation, characterization and antitumor efficacy in vivo and in vitro. Colloids Surf B Biointerfaces. 2016;147:90–99. doi:10.1016/j.colsurfb.2016.07.056
28. Abouzeid AH, Patel NR, Torchilin VP. Polyethylene glycol-phosphatidylethanolamine (PEG-PE)/vitamin E micelles for co-delivery of paclitaxel and curcumin to overcome multi-drug resistance in ovarian cancer. Int J Pharm. 2014;464(1–2):178–184. doi:10.1016/j.ijpharm.2014.01.009
29. Roby A, Erdogan S, Torchilin VP. Solubilization of poorly soluble PDT agent, meso-tetraphenylporphin, in plain or immunotargeted PEG-PE micelles results in dramatically improved cancer cell killing in vitro. Eur J Pharm Biopharm. 2006;62(3):235–240. doi:10.1016/j.ejpb.2005.09.010
30. Dong Z, Guo J, Xing X, Zhang X, Du Y, Lu Q. RGD modified and PEGylated lipid nanoparticles loaded with puerarin: formulation, characterization and protective effects on acute myocardial ischemia model. Biomed Pharmacother. 2017;89:297–304. doi:10.1016/j.biopha.2017.02.029
31. Choi YS, Kwon K, Yoon K, Huh KM, Kang HC. Photosensitizer-mediated mitochondria-targeting nanosized drug carriers: subcellular targeting, therapeutic, and imaging potentials. Int J Pharm. 2017;520(1–2):195–206. doi:10.1016/j.ijpharm.2017.02.013
32. Zhang E, Zhang C, Su Y, Cheng T, Shi C. Newly developed strategies for multifunctional mitochondria-targeted agents in cancer therapy. Drug Discov Today. 2011;16(3–4):140–146.
33. Galliani M, Tremolanti C, Signore G. Nanocarriers for protein delivery to the cytosol: assessing the endosomal escape of Poly(Lactide-co-Glycolide)-Poly(Ethylene Imine) nanoparticles. Nanomaterials. 2019;9(4):Apr. doi:10.3390/nano9071000
34. Sonawane ND, Szoka FC
35. Marrache S, Dhar S. Engineering of blended nanoparticle platform for delivery of mitochondria-acting therapeutics. Proc Natl Acad Sci U S A. 2012;109(40):16288–16293. doi:10.1073/pnas.1210096109
36. Moe GW, Marin-Garcia J. Role of cell death in the progression of heart failure. Heart Fail Rev. 2016;21(2):157–167. doi:10.1007/s10741-016-9532-0
37. Jose Corbalan J, Vatner DE, Vatner SF. Myocardial apoptosis in heart disease: does the emperor have clothes? Basic Res Cardiol. 2016;111(3):31. doi:10.1007/s00395-016-0549-2
38. Huang P, Wang L, Li Q, et al. Atorvastatin enhances the therapeutic efficacy of mesenchymal stem cells derived exosomes in acute myocardial infarction via up-regulating long non-coding RNA H19. Cardiovasc Res. 2019. doi:10.1093/cvr/cvz139
39. Zaragoza C, Gomez-Guerrero C, Martin-Ventura JL, et al. Animal models of cardiovascular diseases. J Biomed Biotechnol. 2011;2011:497841. doi:10.1155/2011/497841
40. Chen J, Ceholski DK, Liang L, Fish K, Hajjar RJ. Variability in coronary artery anatomy affects consistency of cardiac damage after myocardial infarction in mice. Am J Physiol Heart Circulatory Physiol. 2017;313(2):H275–H282. doi:10.1152/ajpheart.00127.2017
41. Nichtova Z, Novotova M, Kralova E, Stankovicova T. Morphological and functional characteristics of models of experimental myocardial injury induced by isoproterenol. Gen Physiol Biophys. 2012;31(2):141–151. doi:10.4149/gpb_2012_015
42. Lobo Filho HG, Ferreira NL, Sousa RB, Carvalho ER, Lobo PL, Lobo Filho JG. Experimental model of myocardial infarction induced by isoproterenol in rats. Rev Bras Cirurgia Cardiovasc. 2011;26(3):469–476. doi:10.5935/1678-9741.20110024
43. Li M, Song W, Tang Z, et al. Nanoscaled poly(L-glutamic acid)/doxorubicin-amphiphile complex as pH-responsive drug delivery system for effective treatment of nonsmall cell lung cancer. ACS Appl Mater Interfaces. 2013;5(5):1781–1792. doi:10.1021/am303073u
44. Smith RA, Porteous CM, Gane AM, Murphy MP. Delivery of bioactive molecules to mitochondria in vivo. Proc Natl Acad Sci U S A. 2003;100(9):5407–5412. doi:10.1073/pnas.0931245100
45. Goffart S, von Kleist-Retzow JC, Wiesner RJ. Regulation of mitochondrial proliferation in the heart: power-plant failure contributes to cardiac failure in hypertrophy. Cardiovasc Res. 2004;64(2):198–207. doi:10.1016/j.cardiores.2004.06.030
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.