")
Back to Journals » Drug Design, Development and Therapy » Volume 14
Metabolite Profiling in Anticancer Drug Development: A Systematic Review
Authors Muhamad N , Na-Bangchang K
Received 1 July 2019
Accepted for publication 20 March 2020
Published 9 April 2020 Volume 2020:14 Pages 1401—1444
DOI https://doi.org/10.2147/DDDT.S221518
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Qiongyu Guo
Nadda Muhamad,1 Kesara Na-Bangchang1– 3
1Chulabhorn International College of Medicine, Thammasat University, Pathum Thani 12120, Thailand; 2Center of Excellence in Pharmacology and Molecular Biology of Malaria and Cholangiocarcinoma, Chulabhorn International College of Medicine, Thammasat University, Pathum Thani 12120, Thailand; 3Drug Discovery and Development Center, Office of Advanced Sciences and Technology, Thammasat University, Pathum Thani 12120, Thailand
Correspondence: Kesara Na-Bangchang
Chulabhorn International College of Medicine, Thammasat University (Rangsit Center), 99 Moo 18, Paholyothin Road, Klongnung, Klongluang, Pathum Thani 12120, Thailand
Tel +66 2 564 4400 ext 1800
Fax +66 2 564 4398
Email [email protected]
Abstract: Drug metabolism is one of the most important pharmacokinetic processes and plays an important role during the stage of drug development. The metabolite profile investigation is important as the metabolites generated could be beneficial for therapy or leading to serious toxicity. This systematic review aims to summarize the research articles relating to the metabolite profile investigation of conventional drugs and herb-derived compounds for cancer chemotherapy, to examine factors influencing metabolite profiling of these drugs/compounds, and to determine the relationship between therapeutic efficacy and toxicity of their metabolites. The literature search was performed through PubMed and ScienceDirect databases up to January 2019. Out of 830 published articles, 78 articles were included in the analysis based on pre-defined inclusion and exclusion criteria. Both phase I and II enzymes metabolize the anticancer agents/herb-derived compounds . The major phase I reactions include oxidation/hydroxylation and hydrolysis, while the major phase II reactions are glucuronidation, methylation, and sulfation. Four main factors were found to influence metabolite formation, including species, gender, and route and dose of drug administration. Some metabolites were identified as active or toxic metabolites. This information is critical for cancer chemotherapy and anticancer drug development.
Keywords: metabolism, metabolite profile, anticancer, herbal medicine, cancer
Introduction
Cancer remains the major cause of death globally. In 2018, approximately 18 million new cases and 9 million deaths from cancer were estimated to occur worldwide.1 Several chemotherapeutic agents have been developed for treatment and prevention of cancer, either chemically synthetic drugs or herb-derived compounds.2–6 As herb-derived anticancer drugs are considered to be less toxic compared with synthetic drugs, attentions to developing new drugs originating from herbal products have substantially been paid worldwide. These include leelamine, the natural active compound from the bark of pine tree,7 atractylodin and β-eudesmol, the natural active compounds from rhizomes of Atractylodes lancea (Thunb) DC,8 and alantolactone, an active sesquiterpene from Inula helenium L.9
Drug metabolism and pharmacokinetic (DMPK) studies play an important role in all steps of drug discovery and development, including anticancer drugs.10 Metabolism is the process of which xenobiotics or endogenous substances in the body are biotransformed to the metabolic products that facilitate their elimination.11 Drug metabolism involves two main phases, i.e., phase I and phase II metabolism. The primary enzyme system involved in phase I metabolism is cytochrome P450 (CYP), and the major enzymes involved in phase II are UDP-glucuronosyltransferase (UGT), sulfotransferase (SULT), glutathione-S-transferase (GST), N-acetyltransferase (NAT), and methyltransferase (MT).12 Drug metabolism plays a crucial role in determining the efficiency and safety of drugs. Drugs undergo metabolism to form numerous stable metabolites, most of which are pharmacologically inactive. On the other hand, in some cases, metabolism may lead to reactive metabolites that can induce adverse effects. Various types of drug metabolism studies have been incorporated during the process of drug discovery and development to generate new chemical entities (NCE) with acceptable safety profiles. This is particularly important for cancer chemotherapeutic drugs with a narrow therapeutic window. Metabolite profiling and identification studies of these compounds and currently used drugs are therefore essential. The main aim of metabolic profiling studies is to identify metabolic pathways and metabolites generated from the biotransformation process. The information obtained from the studies would help to optimize lead compounds for optimal pharmacokinetic and pharmacodynamic properties. Besides, it will help to identify new chemical entities based on the metabolites generated to minimize potential safety liabilities due to the formation of reactive or toxic metabolites. Comparison of information obtained from preclinical studies in animals and humans would also ensure potential adequate coverage of human metabolites in animals and for supporting human prediction.
This systematic review aimed to summarize the research articles relating to the metabolite profiling studies of anticancer drugs (conventional chemical synthetic drugs and targeted small-molecules) and candidate compounds from herbal sources. Factors influencing metabolite profiles (metabolic pathways and types of metabolites generated) and their relationship with anticancer activity and toxicity in vitro, in vivo (animals), and human were also investigated.
Materials and Methods
Study Selection and Inclusion and Exclusion Criteria
This systematic review was performed through the search from PubMed (via Endnote) and ScienceDirect databases up to January 2019. The following keywords were used: “anticancer drug”, “anticancer agent”, “chemotherapy”, “chemotherapeutic drug”, “traditional medicine”, herbal medicine, “natural compound”, “metabolism”, “metabolite profile”, “metabolite identification”, “metabolite characterization”, and “cancer”. No other search conditions were applied. All articles obtained from the two databases were checked for duplication. The remaining articles were initially screened as per the inclusion criteria based on the content of the abstract section. The inclusion criteria for article selection were 1) articles in full-texts and written in English; 2) articles with the investigation (in vitro, in vivo, or clinical studies) of metabolite profiles of conventional chemotherapeutic drugs, small molecules targeted therapy, candidate synthetic anticancer agents, natural products-derived anticancer compounds or drug candidates. The duplicates, review articles, short communications, case reports, articles with the investigation of other types of drug metabolism studies, or those with insufficient information of metabolite(s) or metabolic pathway(s) were excluded from data analysis.
Data Extraction and Collection
Two independent researchers performed data extraction from all articles. When conflicting opinions arose, the decision was sought from higher professional level personnel, and the decision was considered final. The title and abstract of each article search from PubMed (via Endnote) and ScienceDirect databases using the keywords mentioned above were initially screened for relevant original articles based on the inclusion and exclusion criteria. The full-text articles were carefully examined to confirm their compliance with the defined eligibility criteria. The studies of metabolite profile of chemotherapeutic drugs, targeted small molecules, candidate synthesized anticancer agents, traditional or herbal medicines, and natural compounds for cancer were classify according to types of anticancer agents. The information extracted included: name of anticancer drug or compound/herb, type of studies (in vitro, in vivo, and clinical studies), gender and species of animals used, route and dose of administration of the investigated drugs/compounds, biochemical tools used (liver, prostate, intestine, or kidney microsomes, subcellular fractions, hepatocytes, recombinant enzymes, and whole blood), type of biological samples (plasma, urine, bile, feces, and tumor), and study conclusion.
Results
Study Description
Three hundred and twenty-one out of 830 articles were duplicated or review articles and were initially excluded from the analysis. The title and abstract were further screened based on eligible criteria, and 411 articles were further excluded from the analysis. Finally, 78 out of 98 articles were included in the analysis; 20 excluded articles were case reports, short communications, and articles with insufficient information. The flow diagram of the search process is presented in Figure 1. Information on metabolite profiles including biochemical tools/biological samples used in the studies of conventional anticancer drugs, synthetic anticancer candidates, small molecules targeted therapy, herb-derived compounds with anticancer activities are summarized in Tables 1–3, respectively and the anticancer activities of each compound are presented in Table 4.
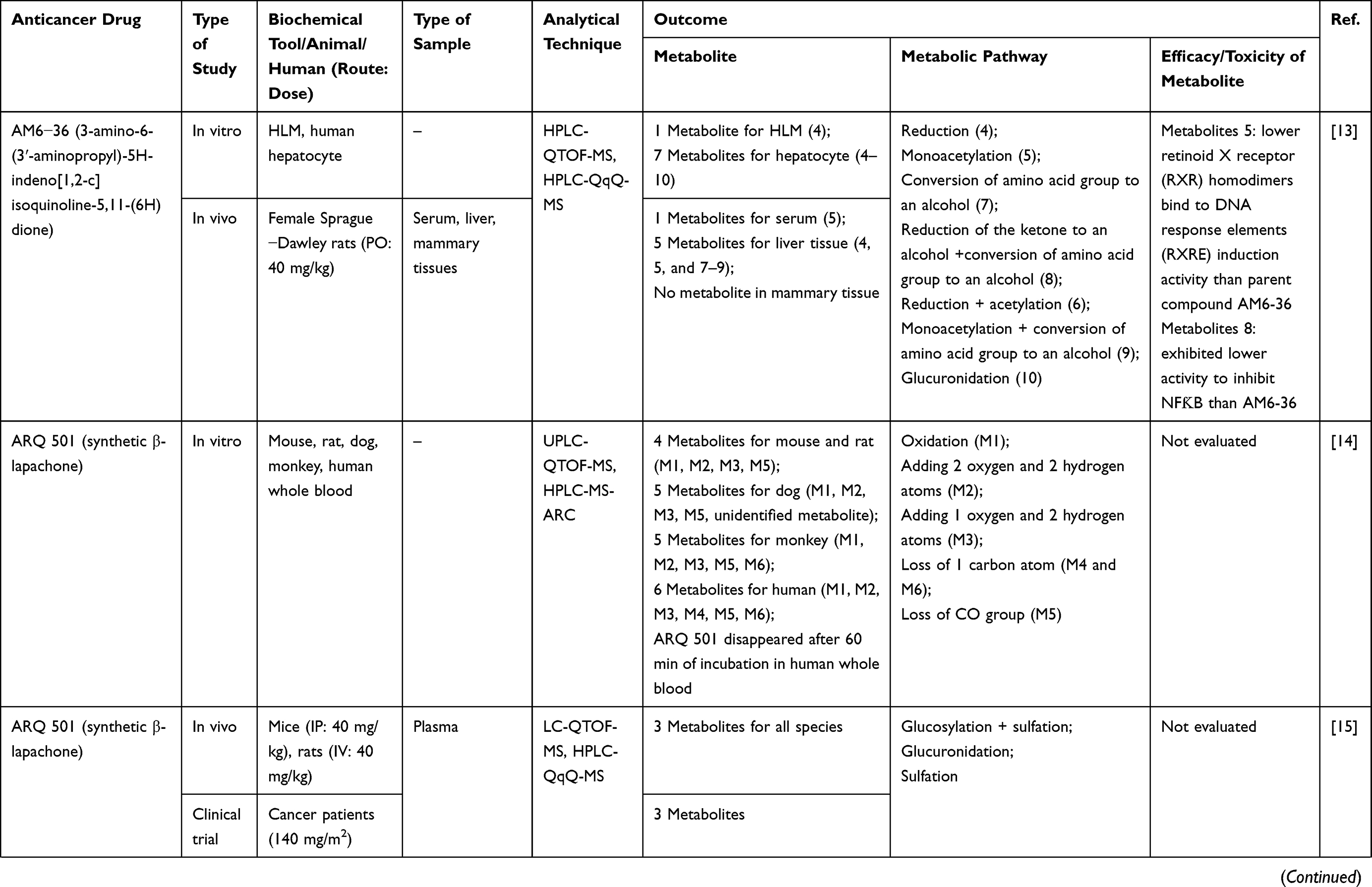 | | 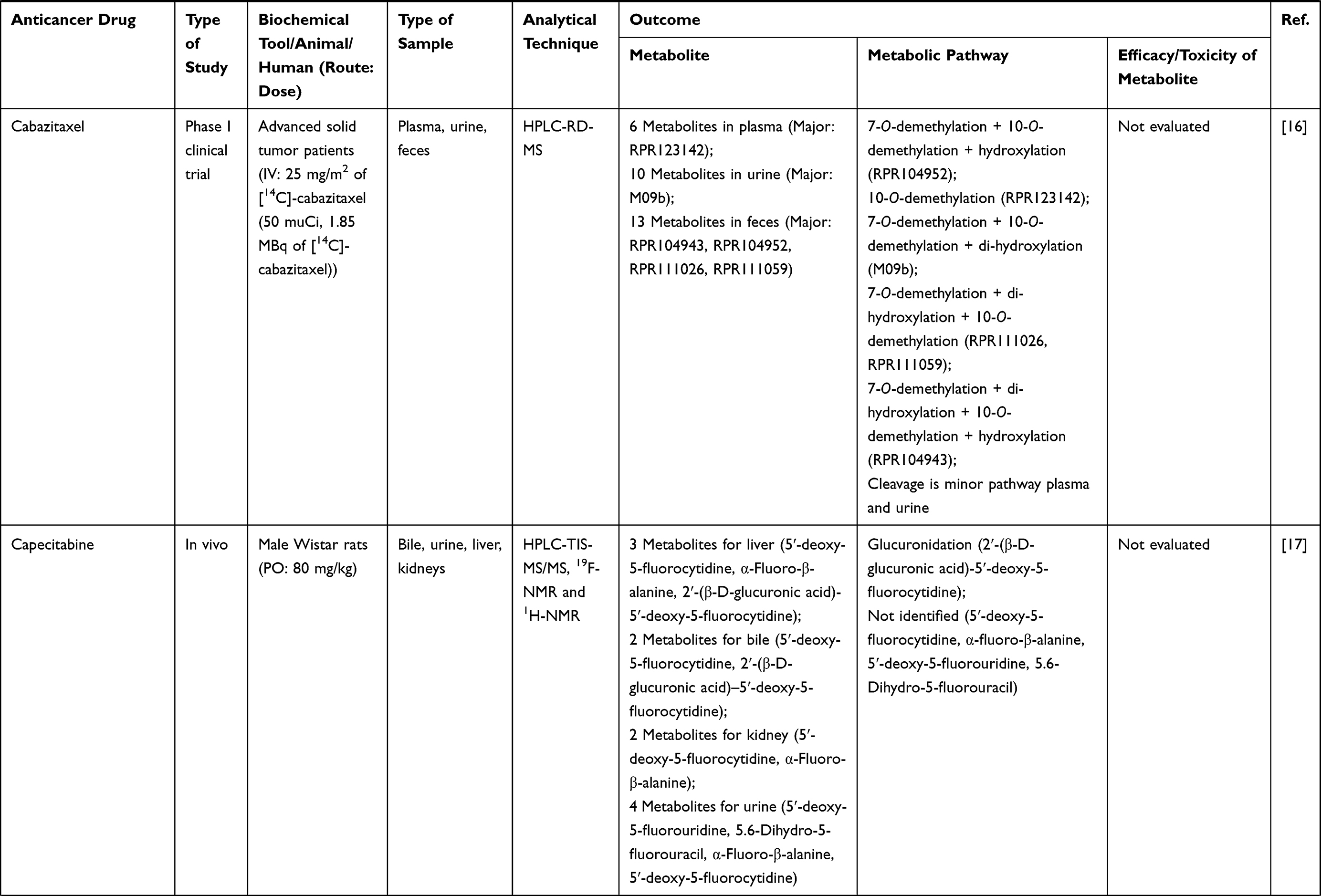 | 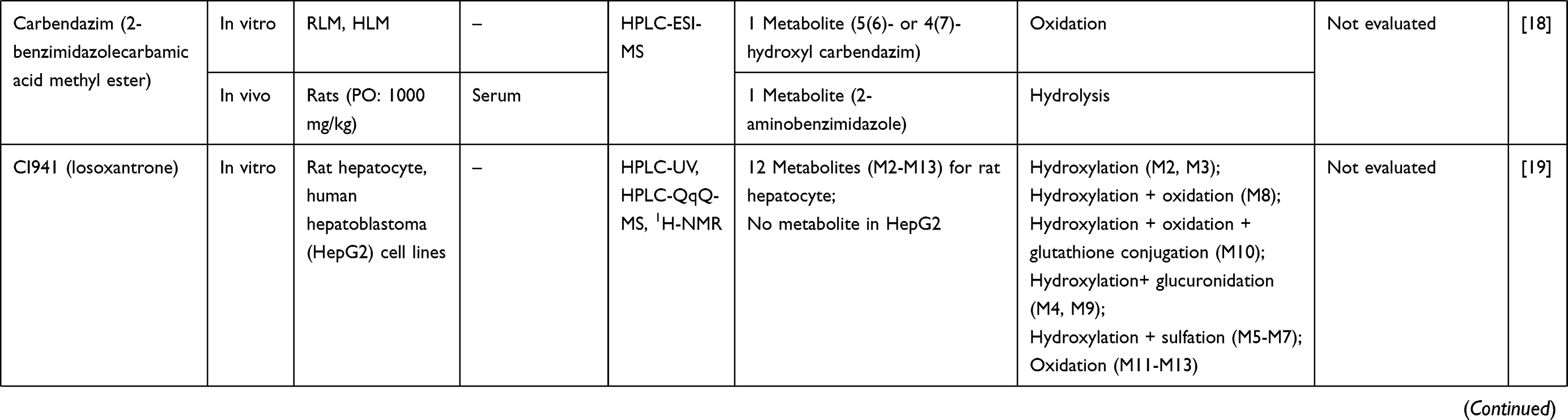 | 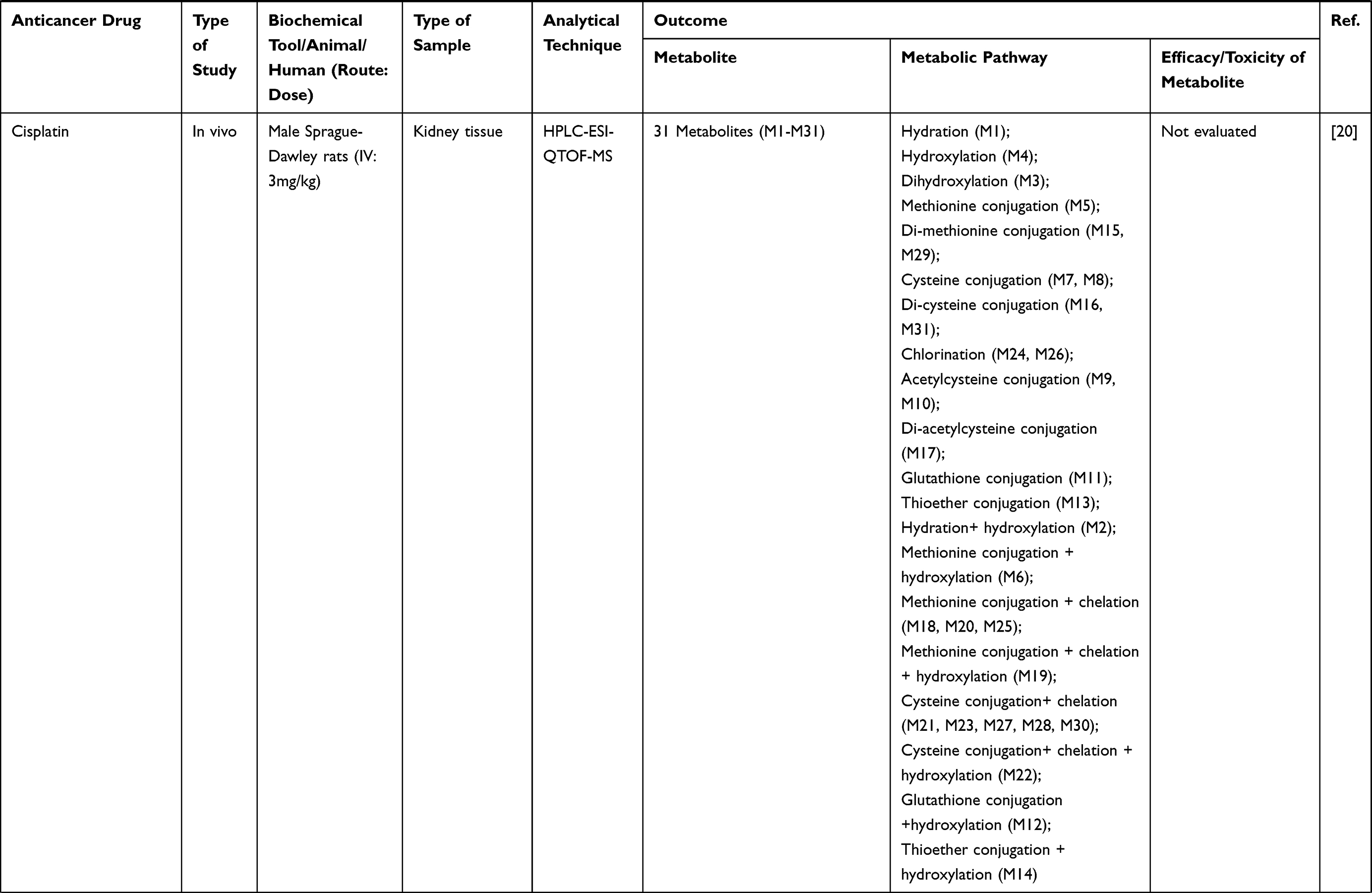 |  |  |  |  | 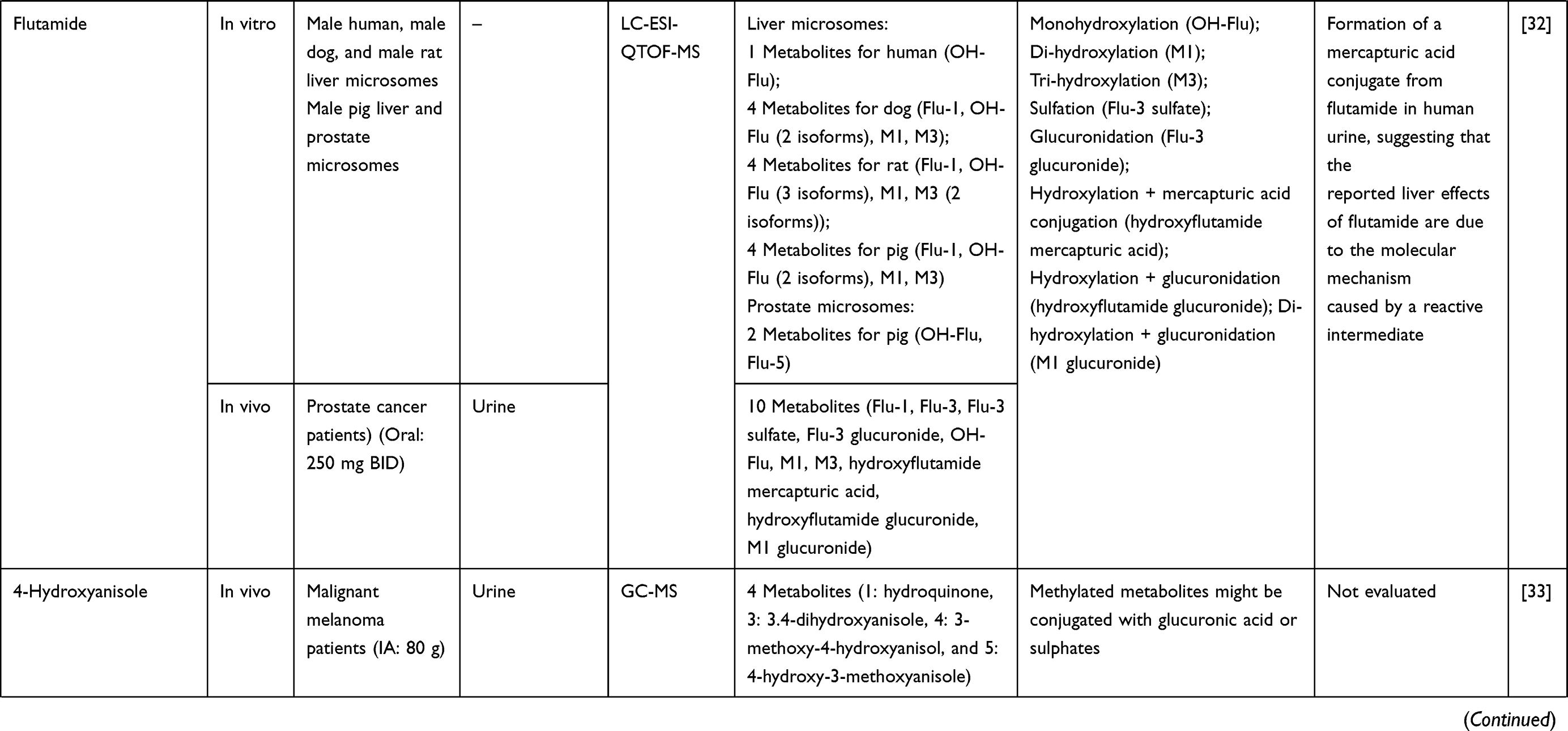 | 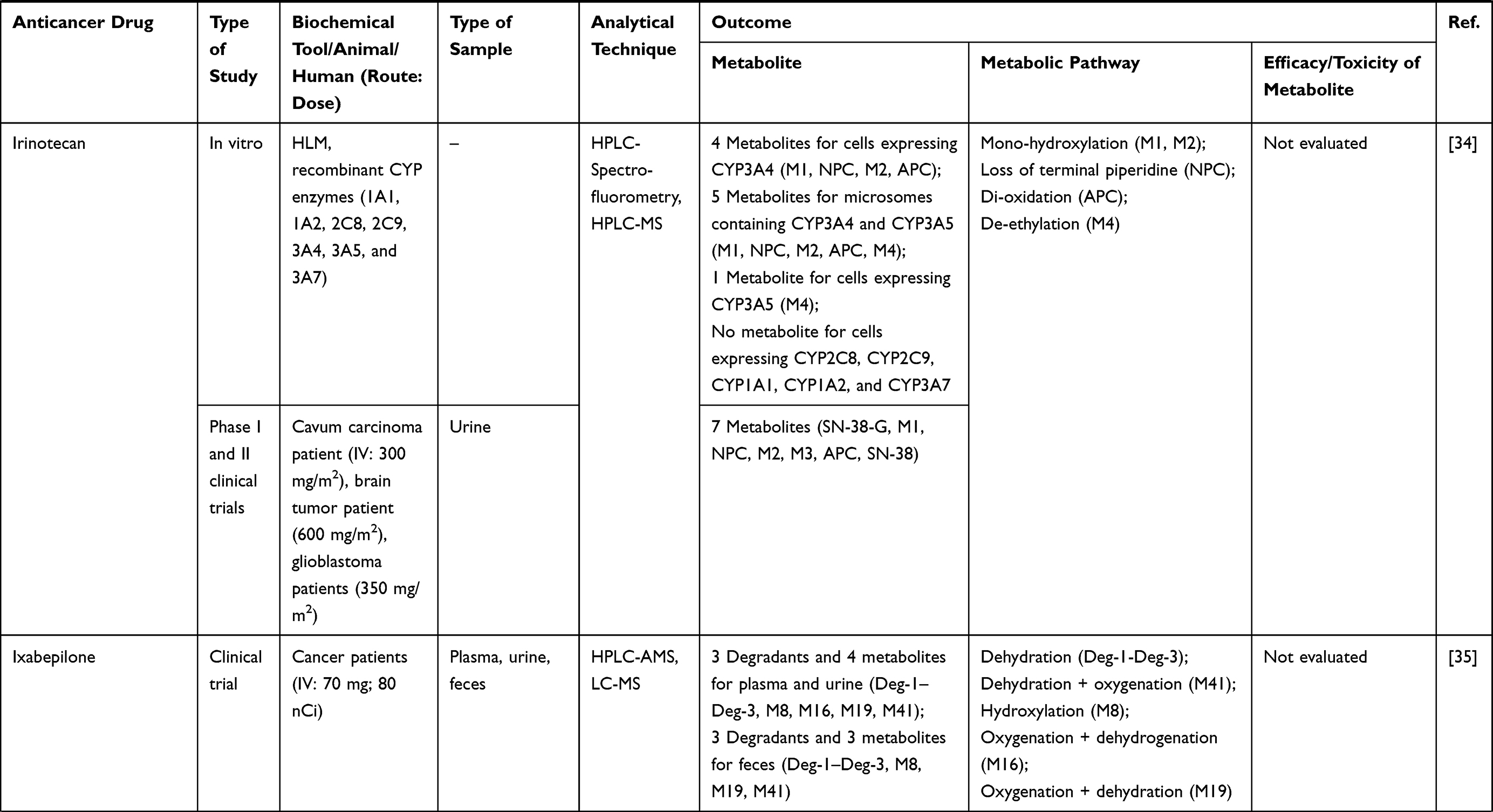 | 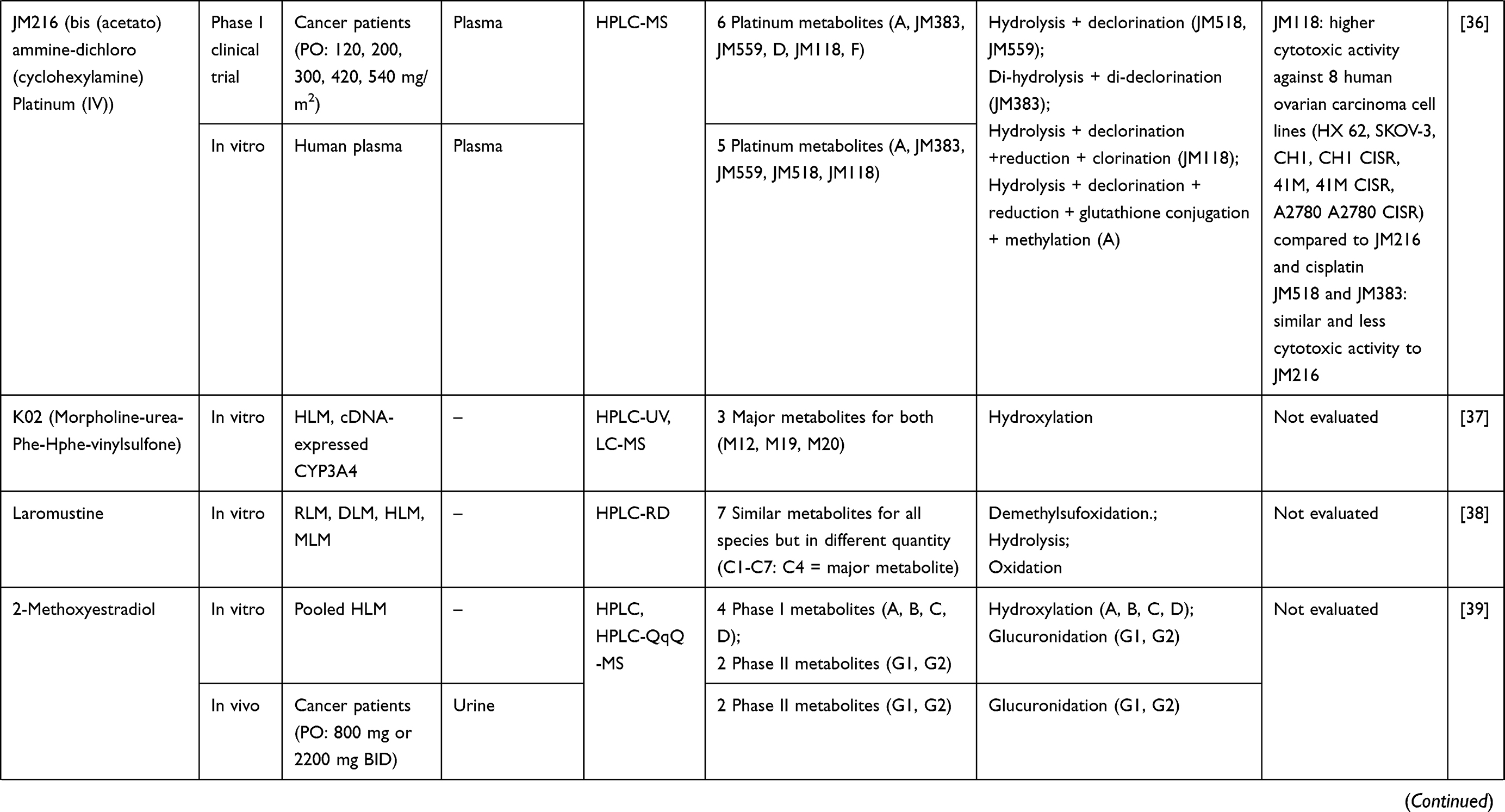 | 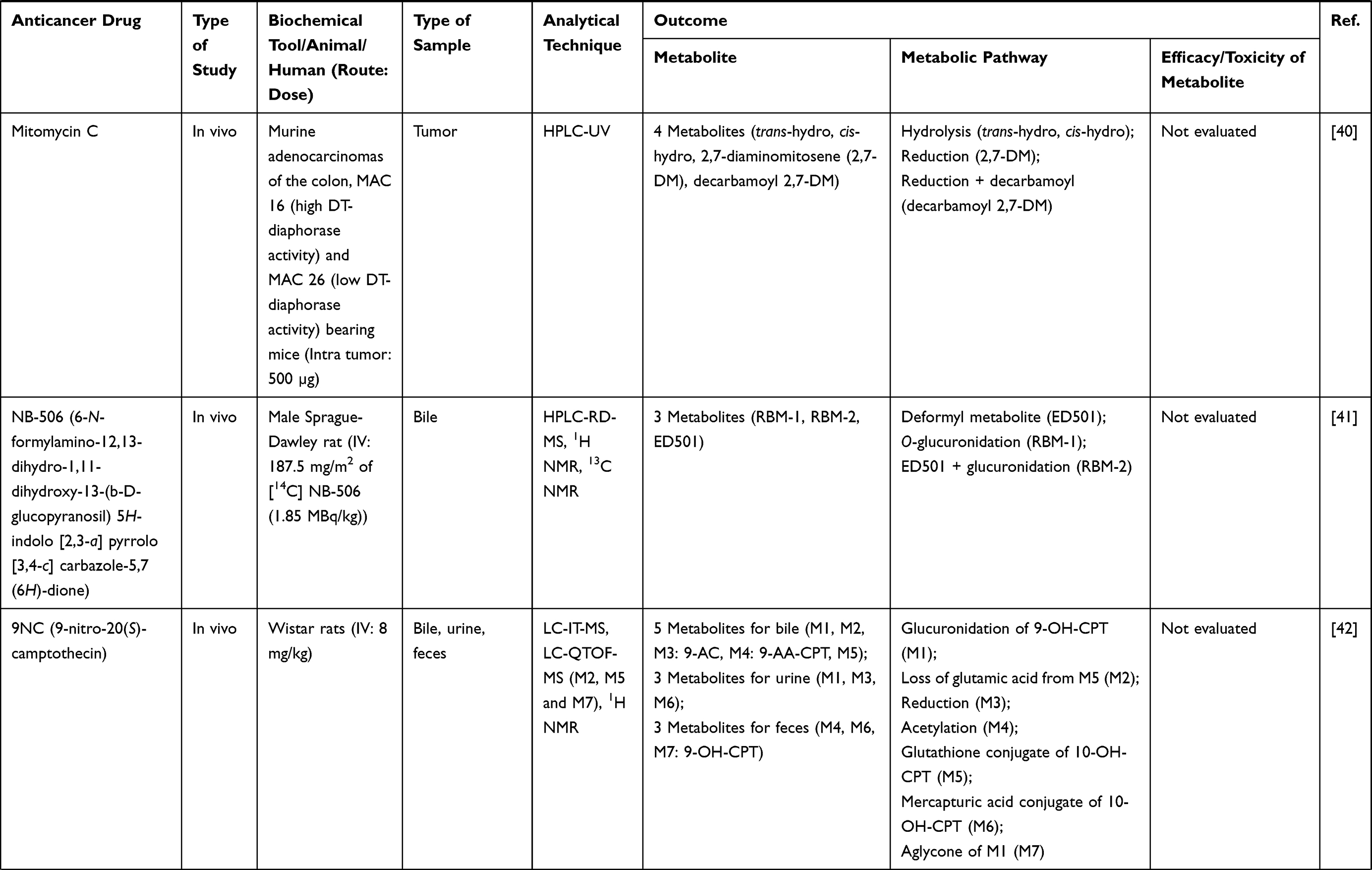 | 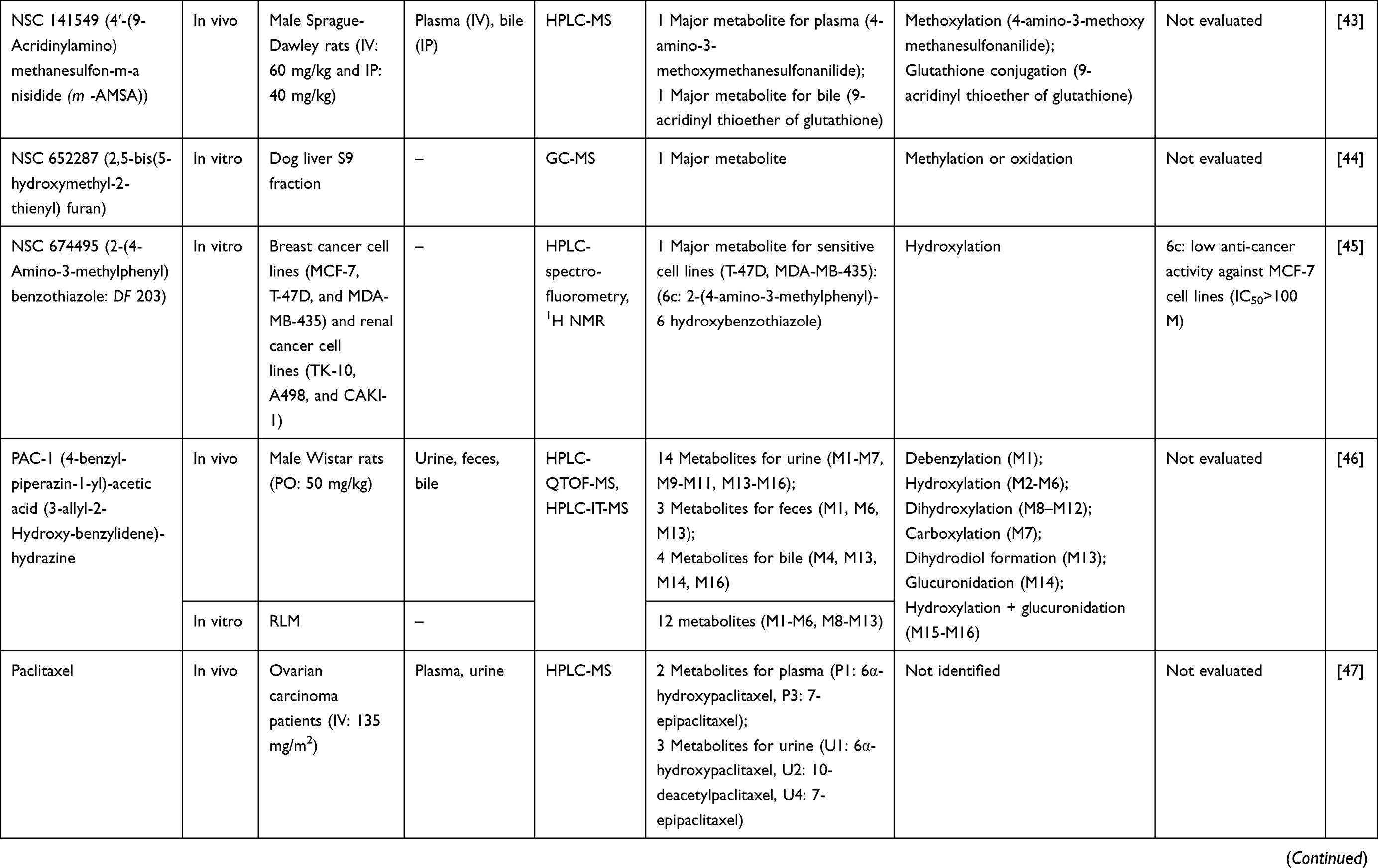 | 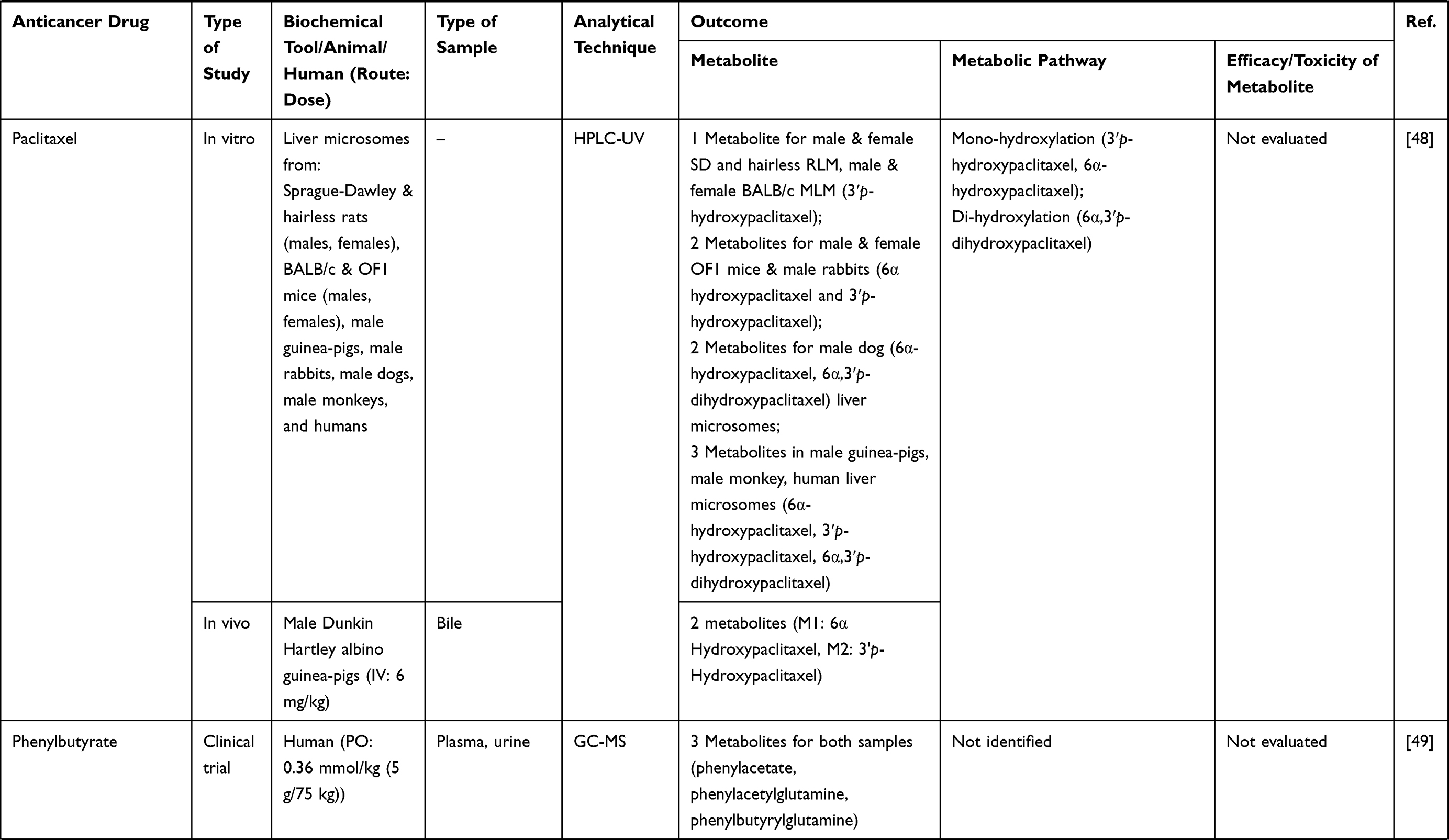 | 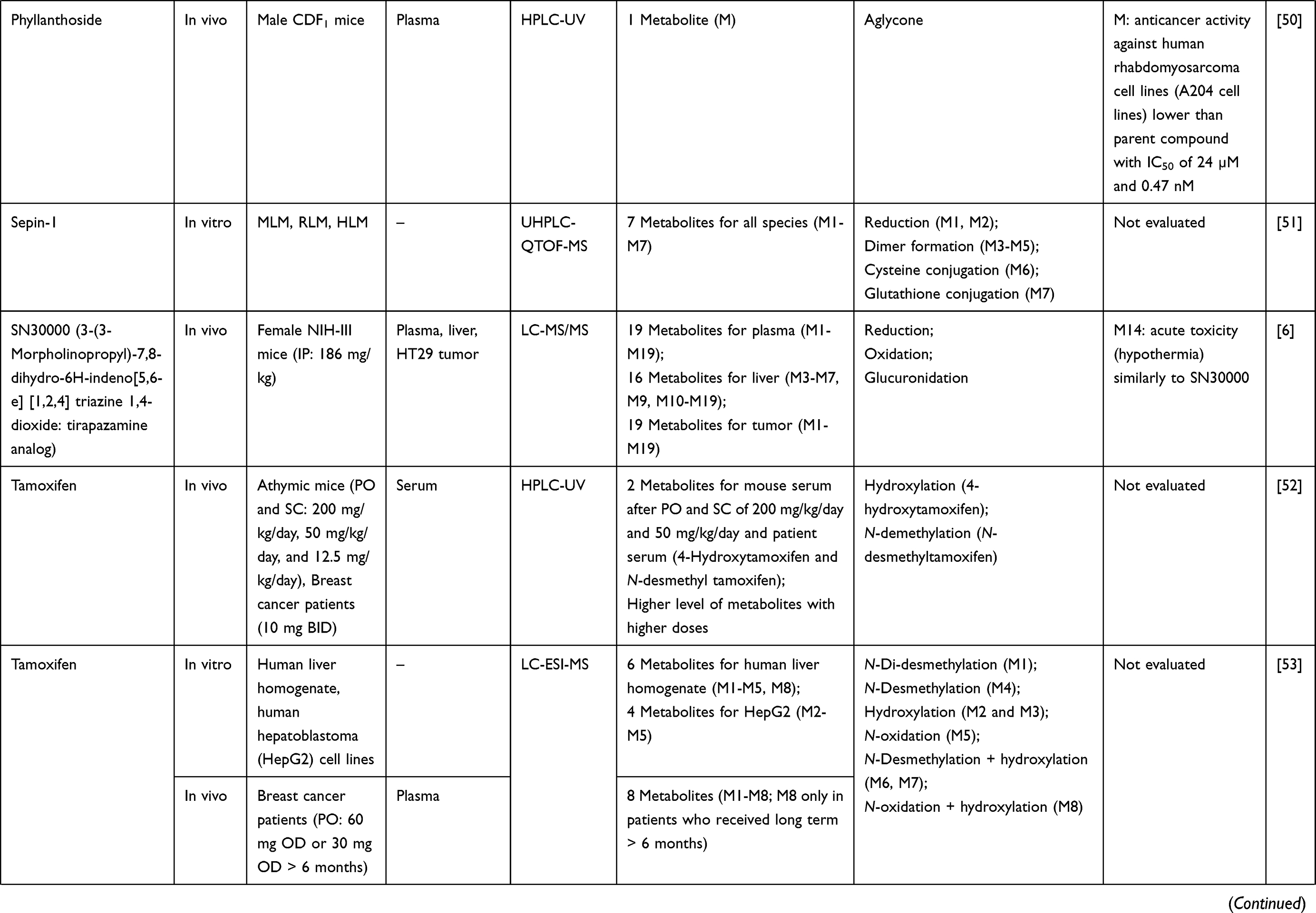 | 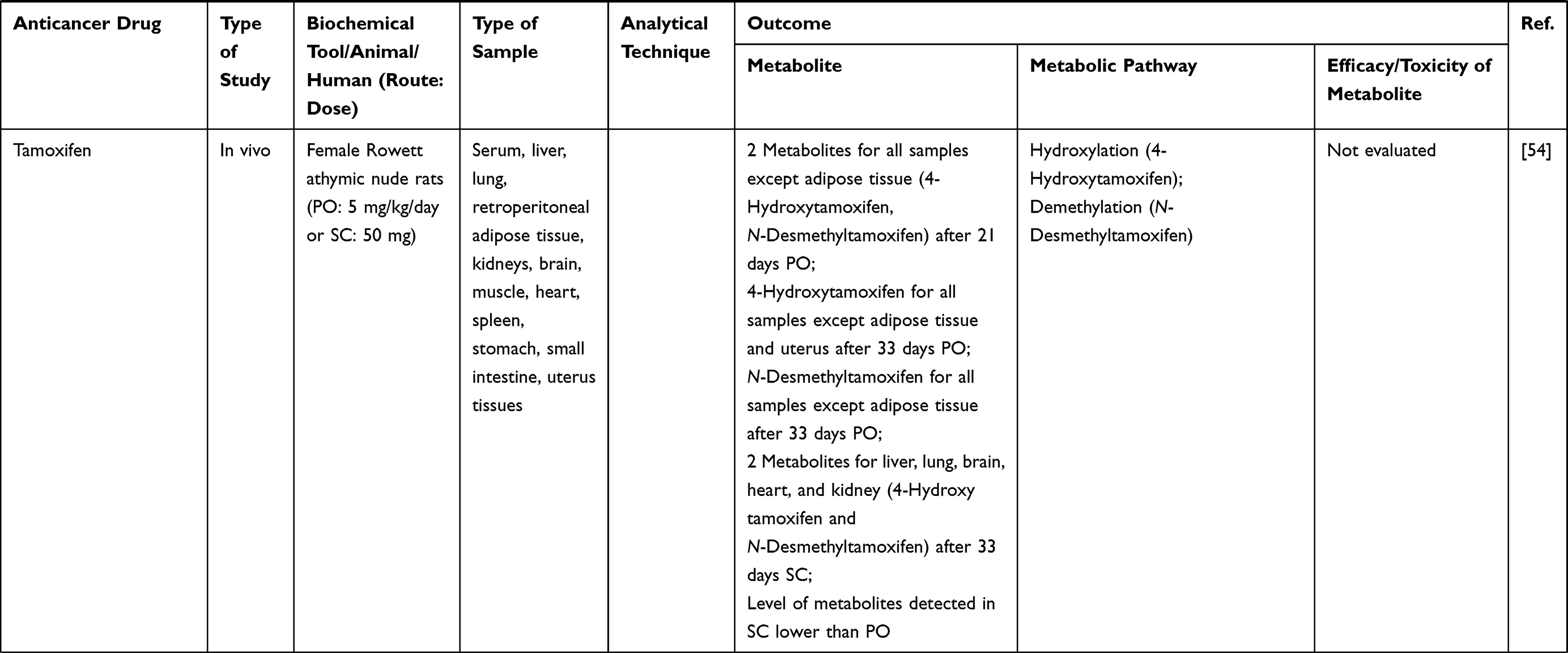 | 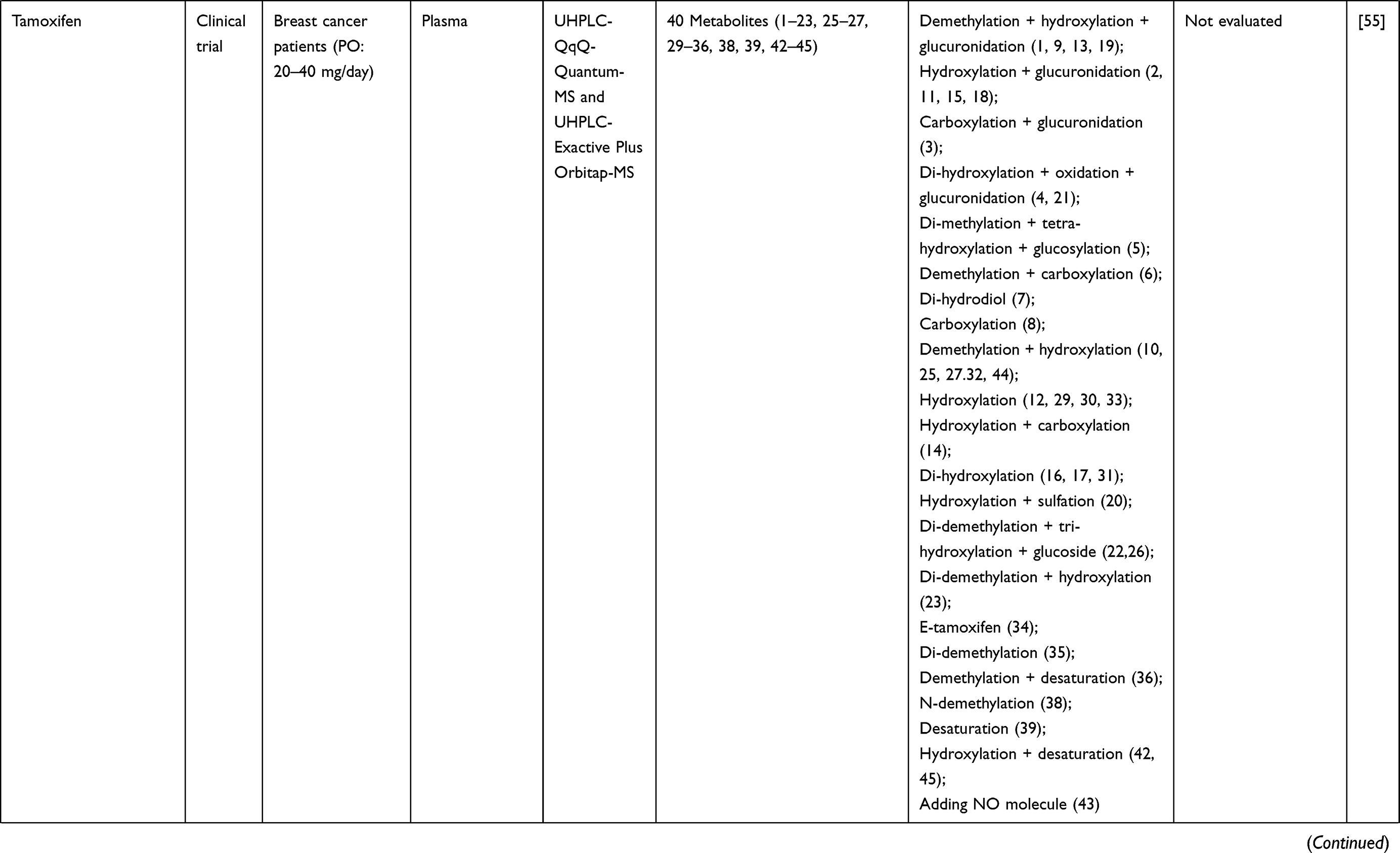 | 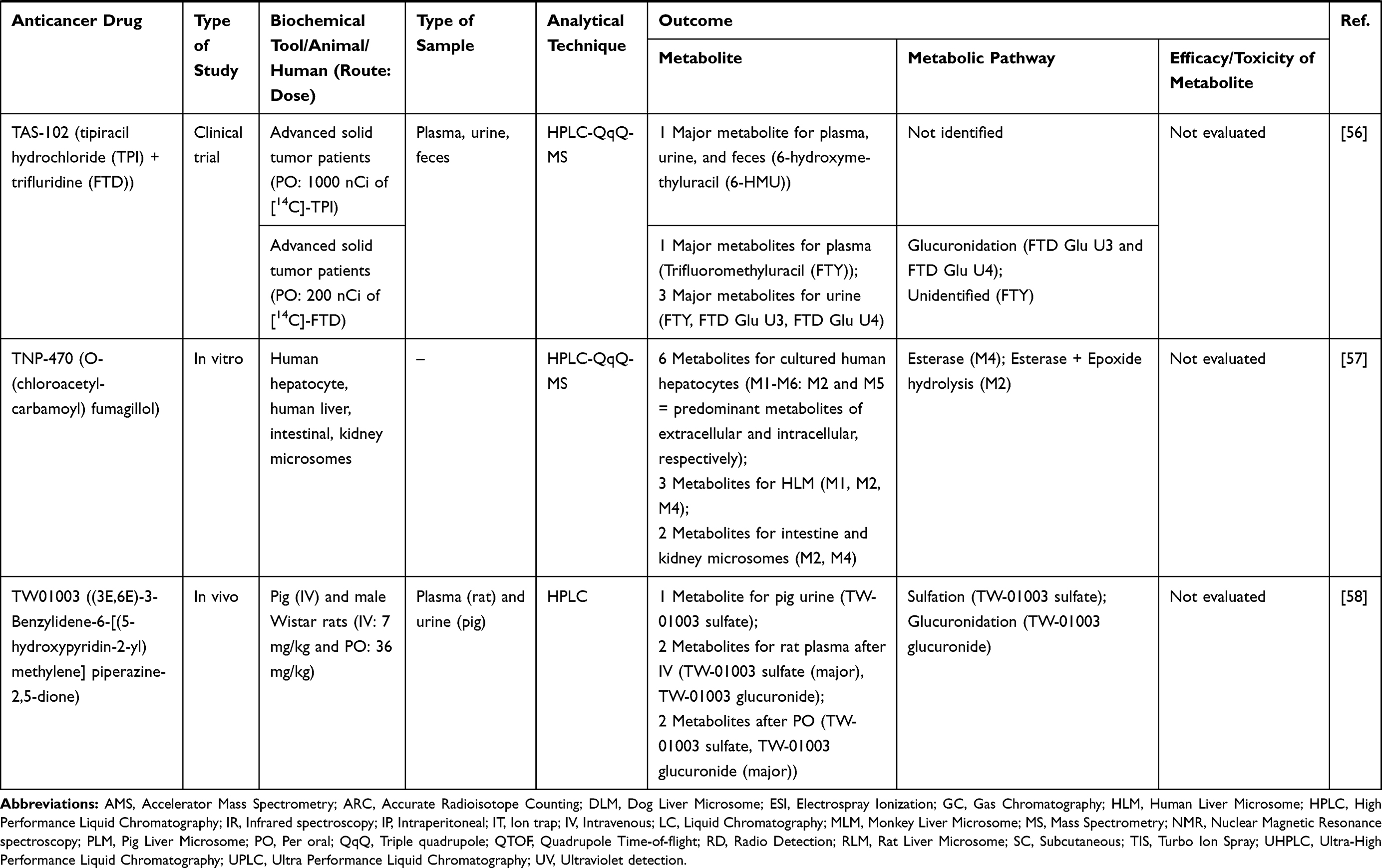 |
Table 1 Metabolism Studies (Metabolite Profiling) of Conventional Synthetic Anticancer Drugs |
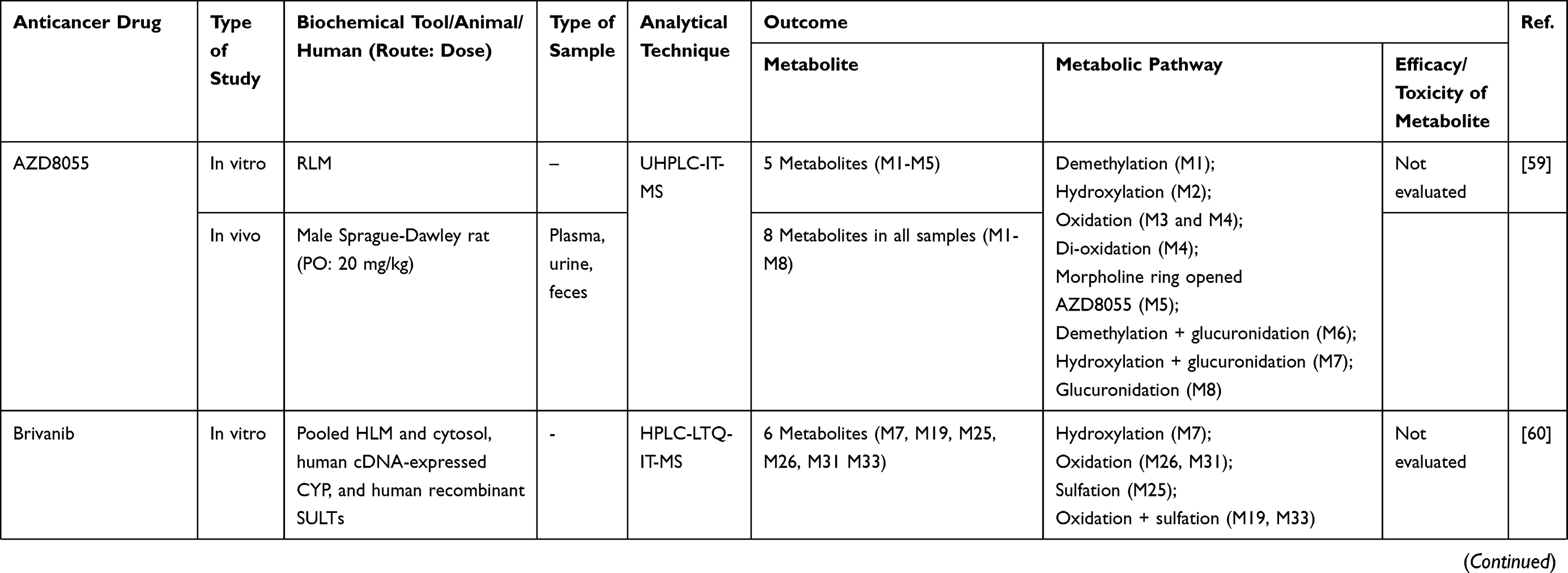 |  | 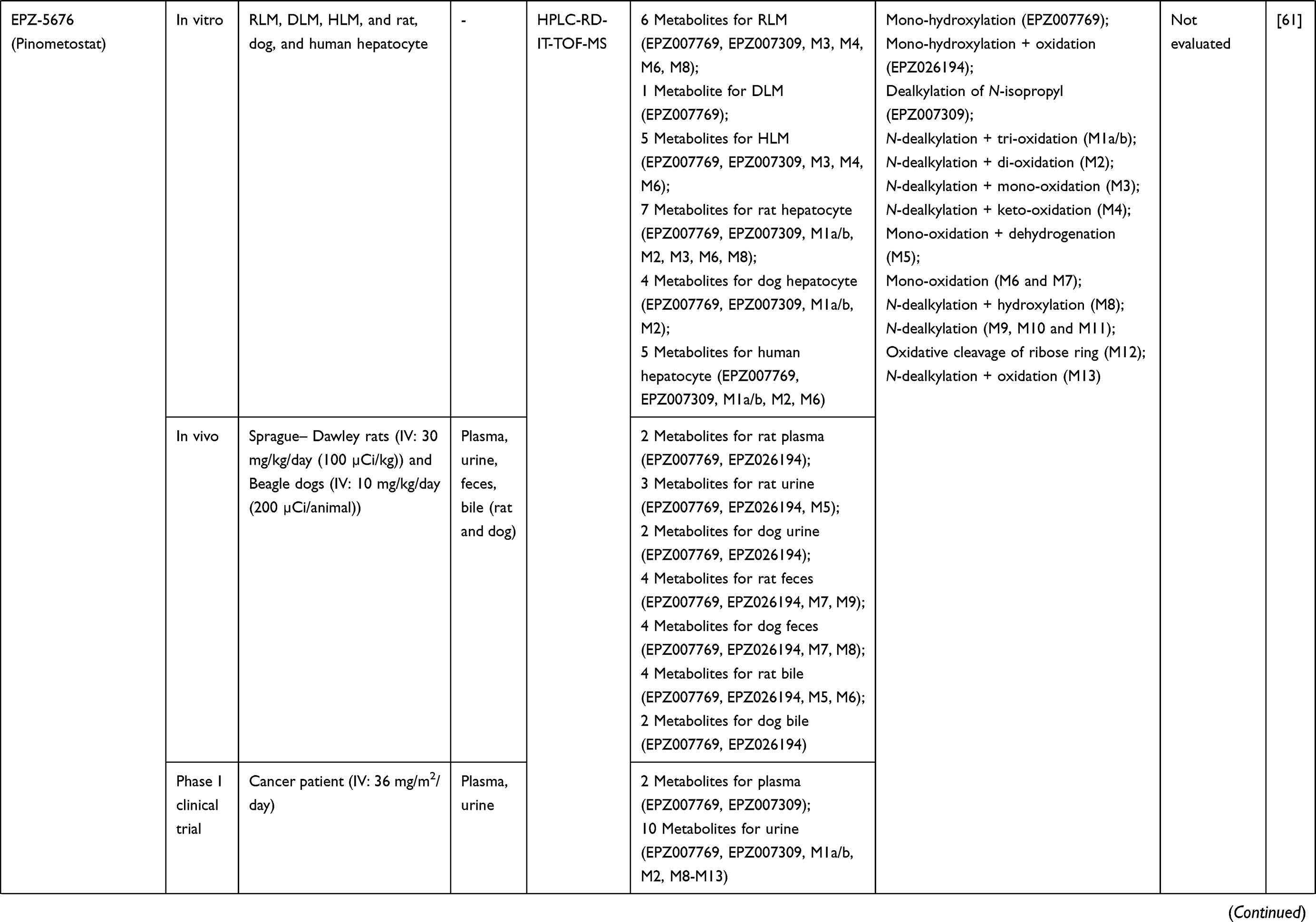 | 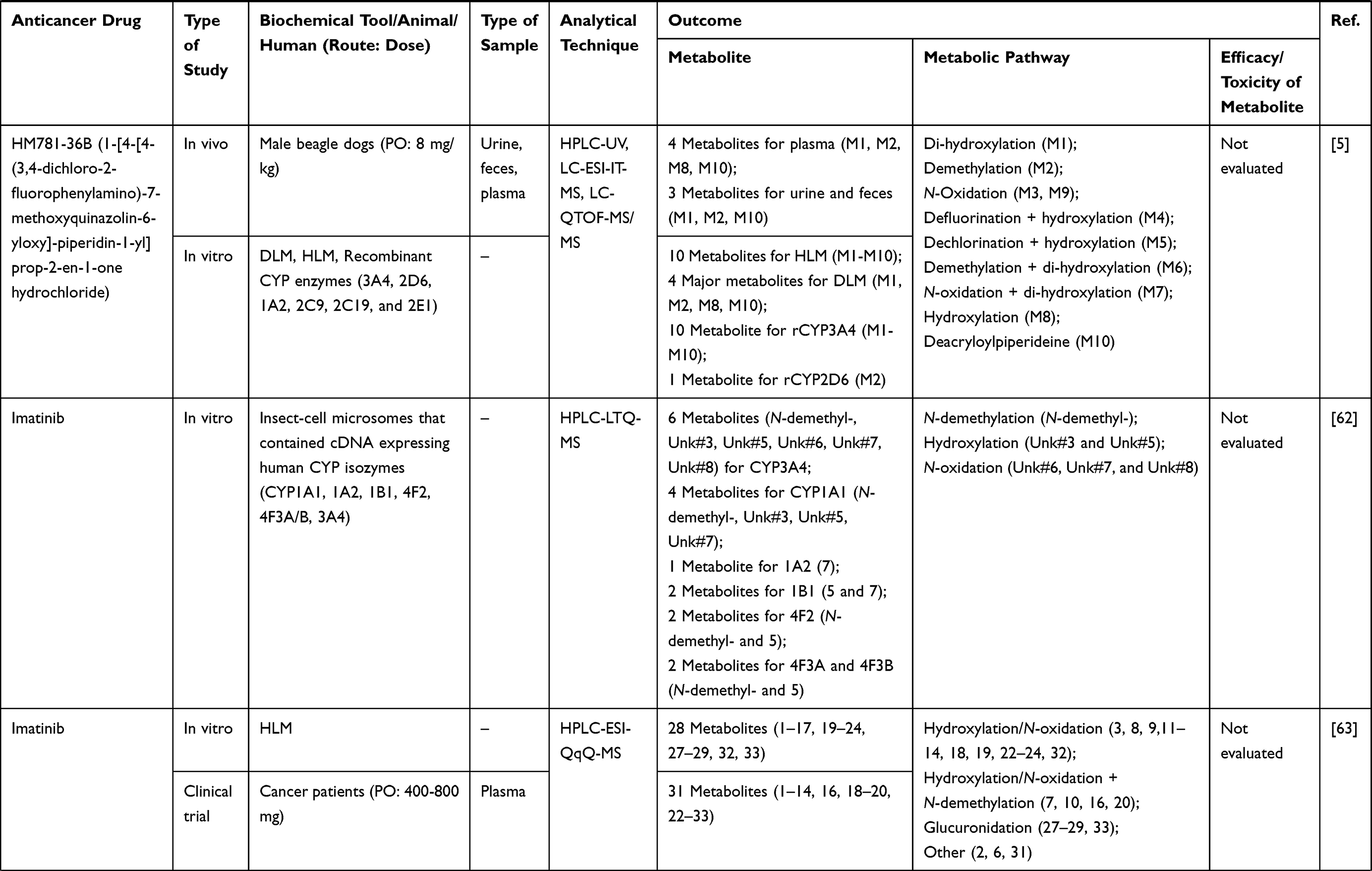 | 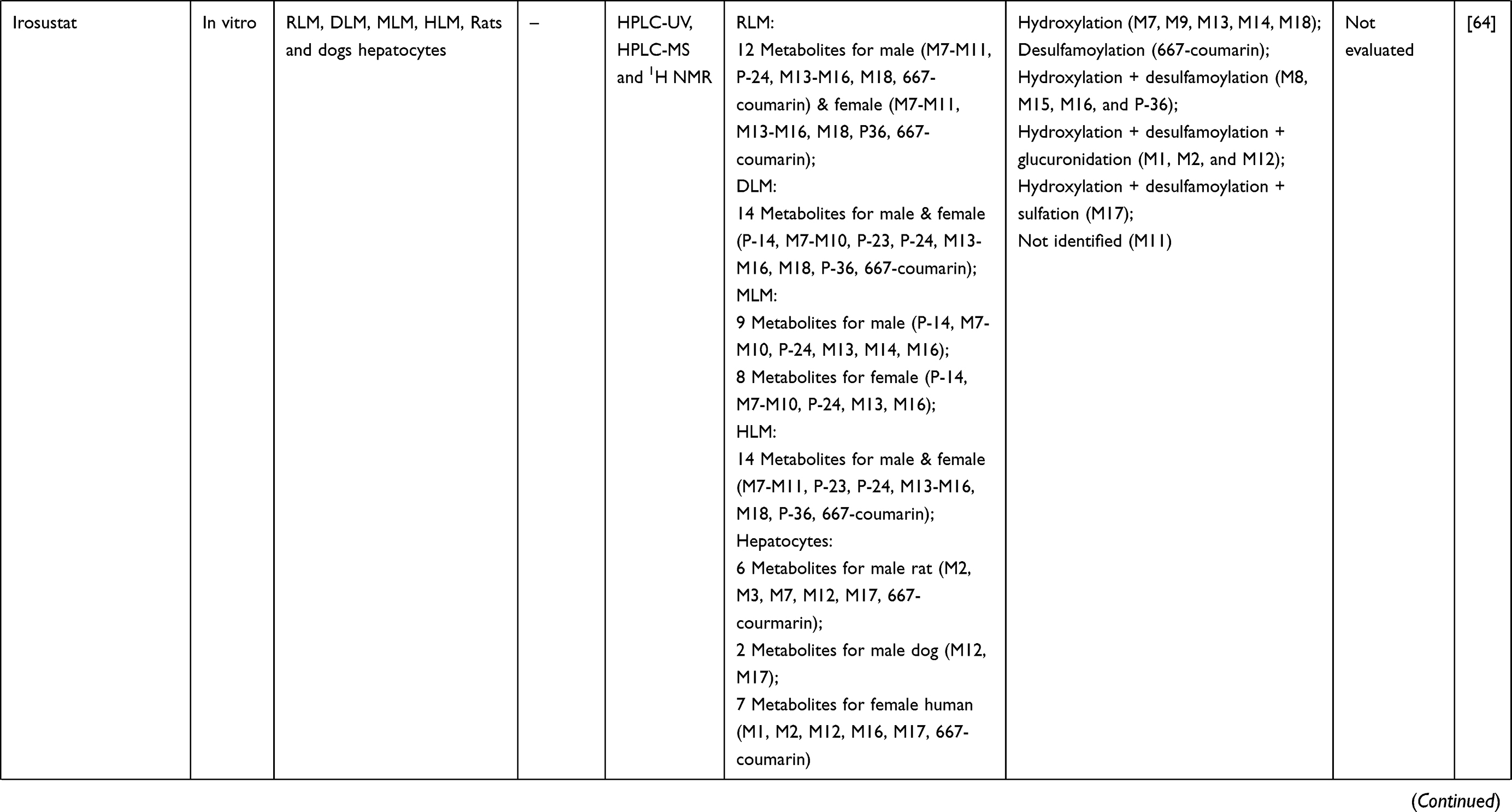 | 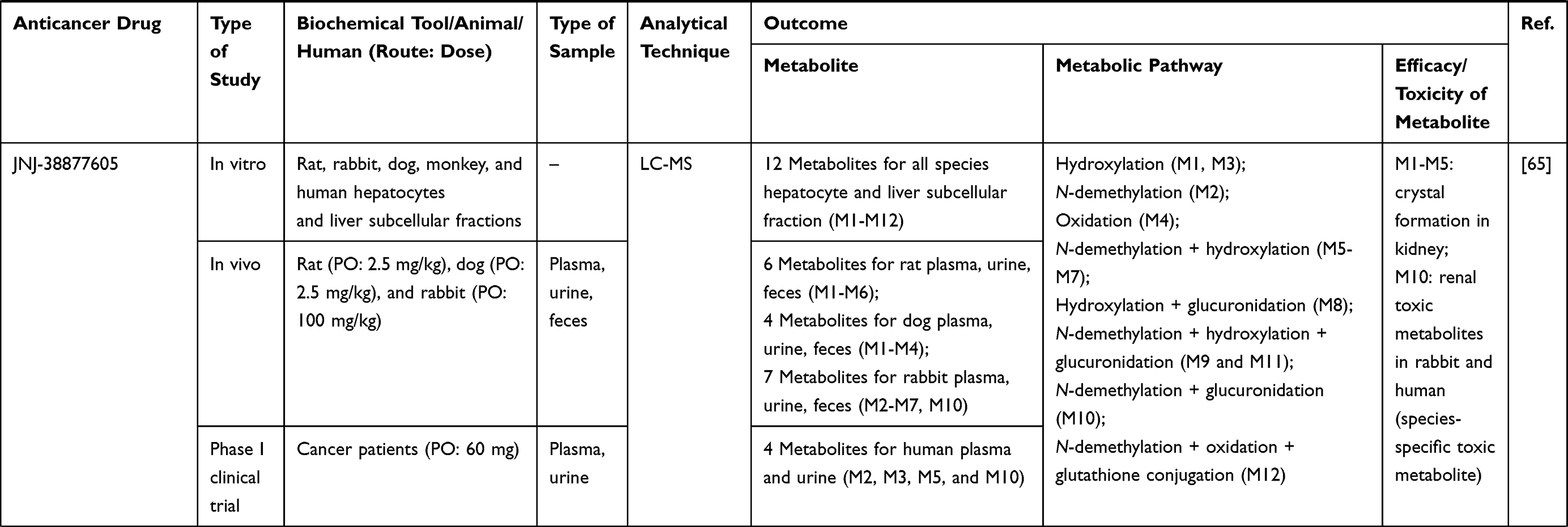 | 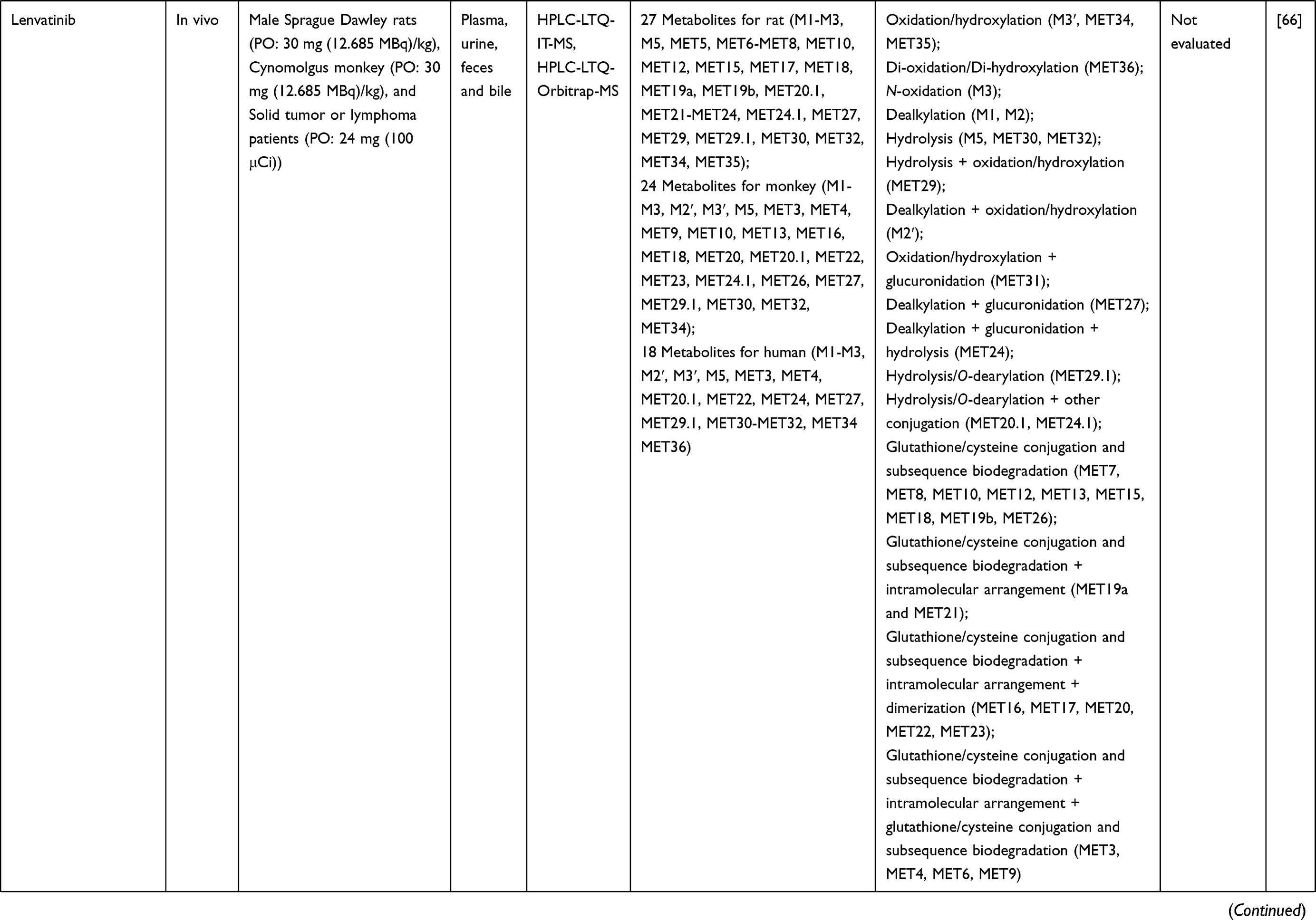 |
Table 2 Metabolism Studies (Metabolite Profiling) of Small Molecules-Targeted Anticancer Drugs |
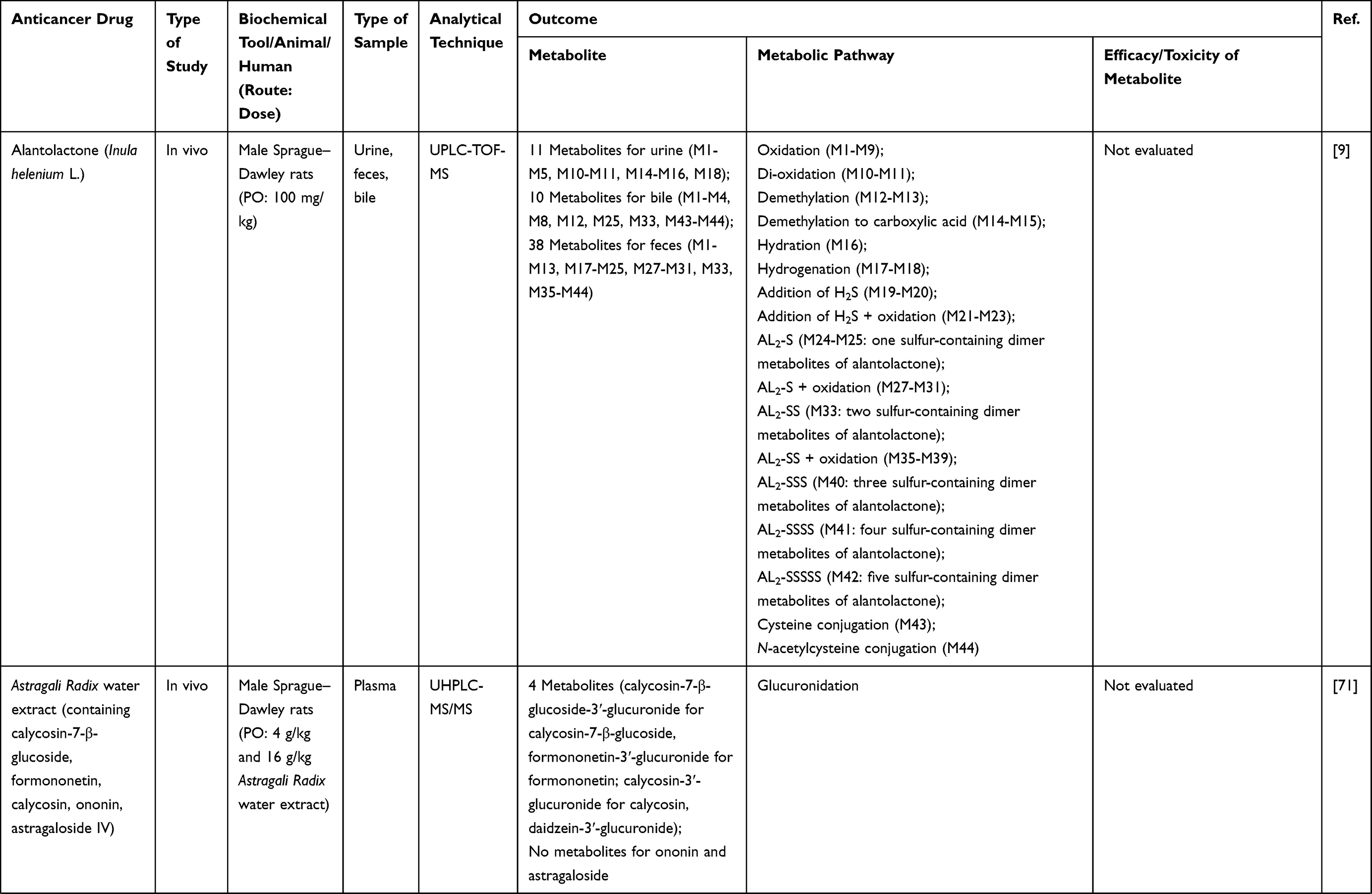 | 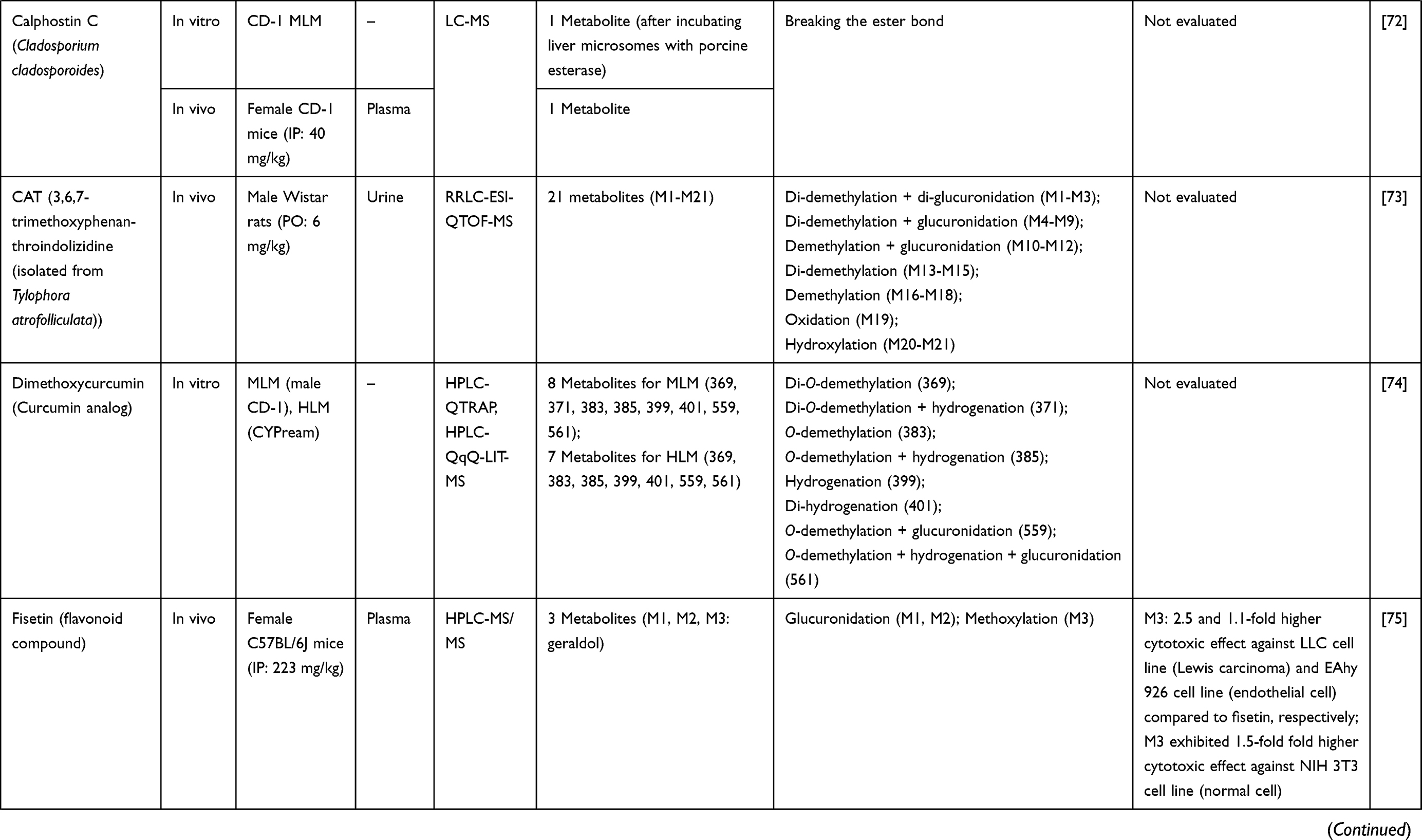 | 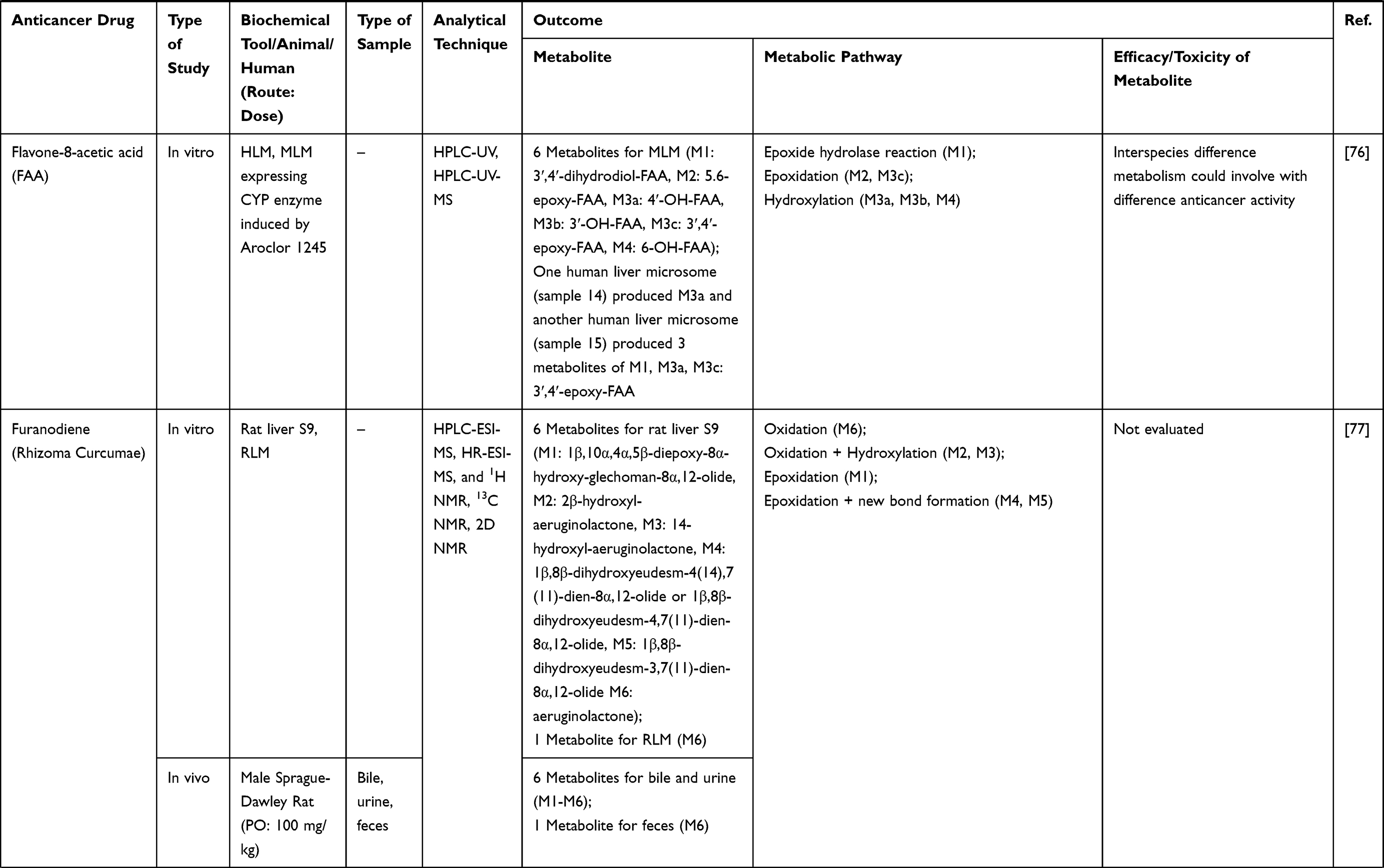 | 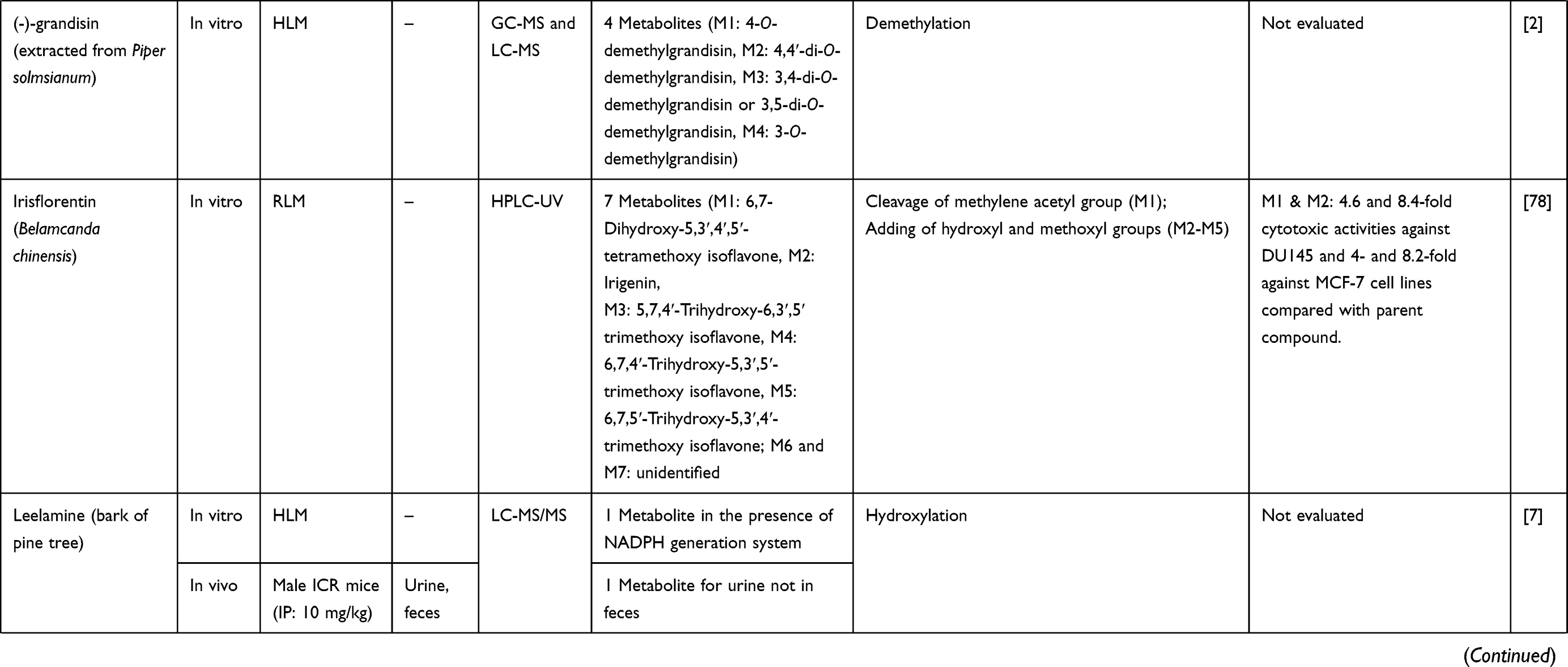 | 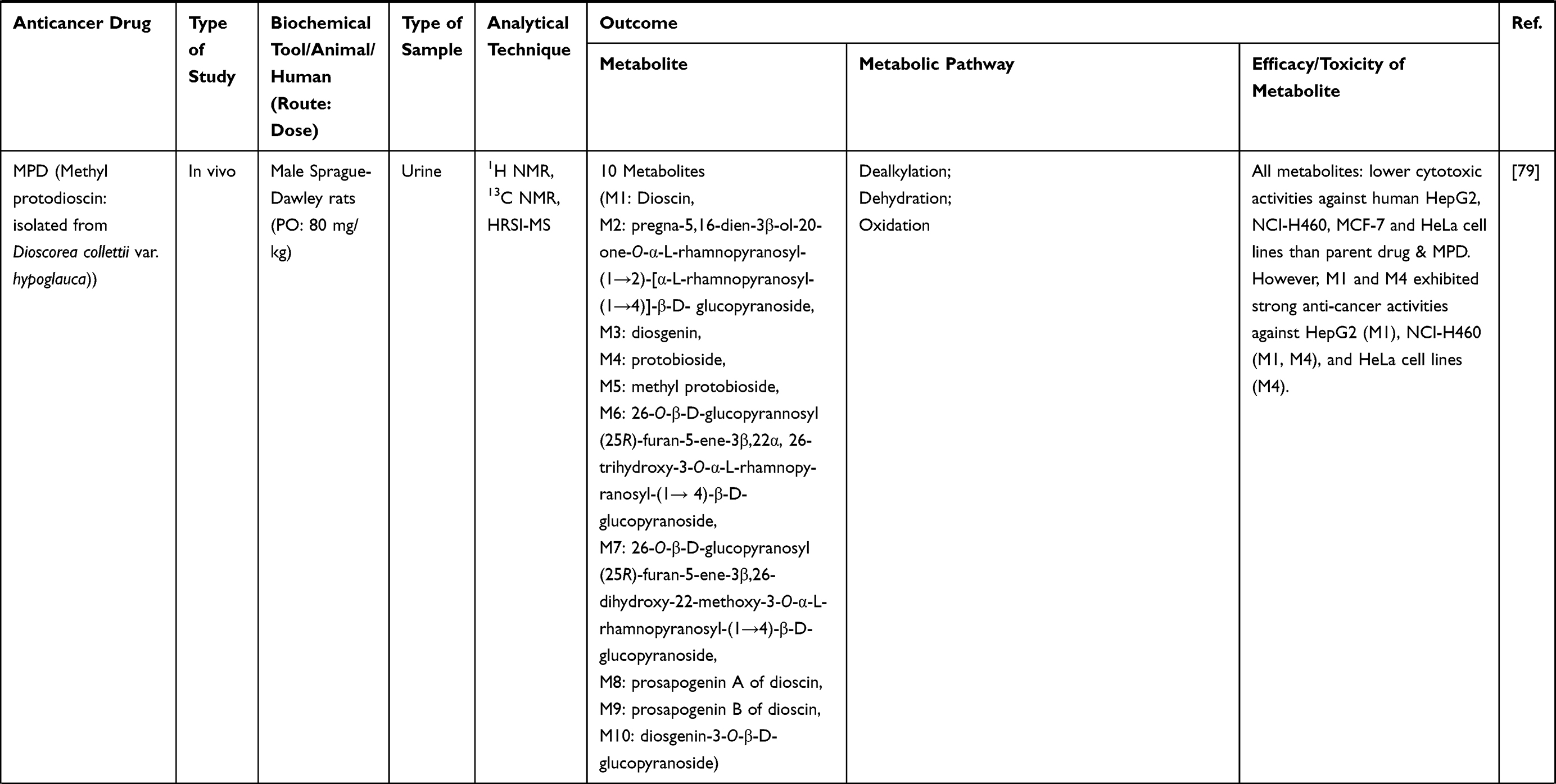 | 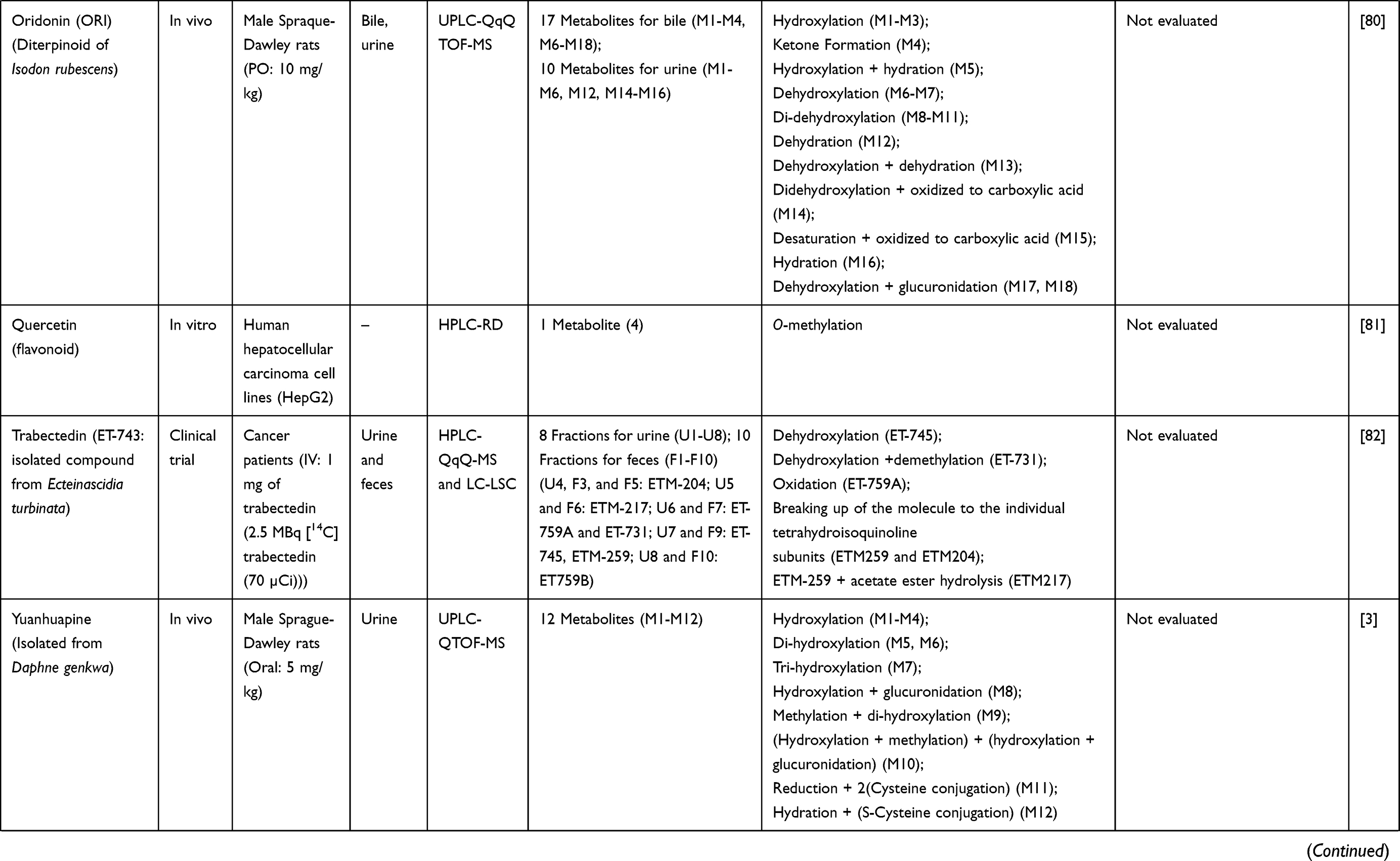 | 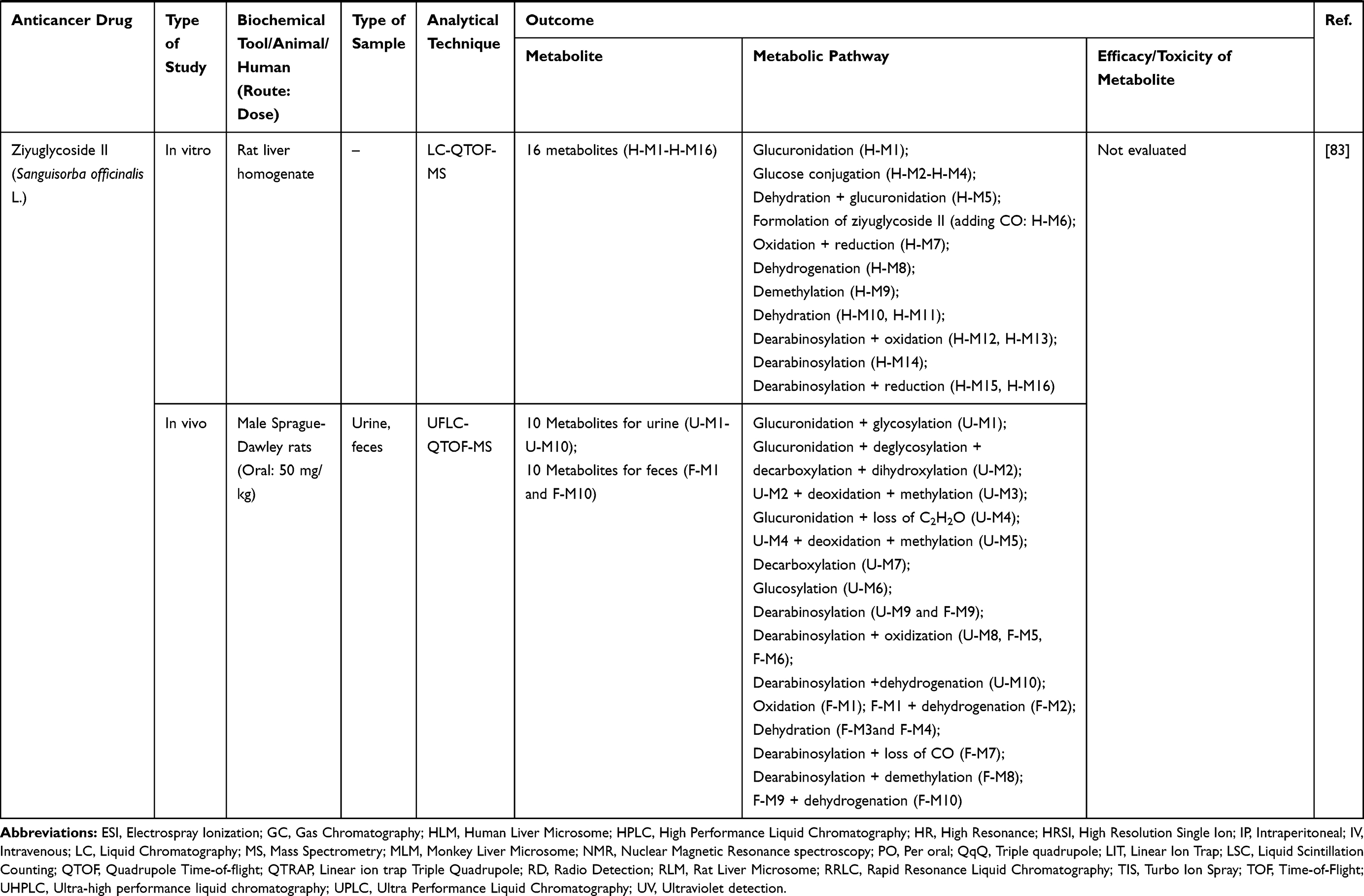 |
Table 3 Metabolism Studies (Metabolite Profiling) of Herb-Derived Compounds with Anticancer Activities |
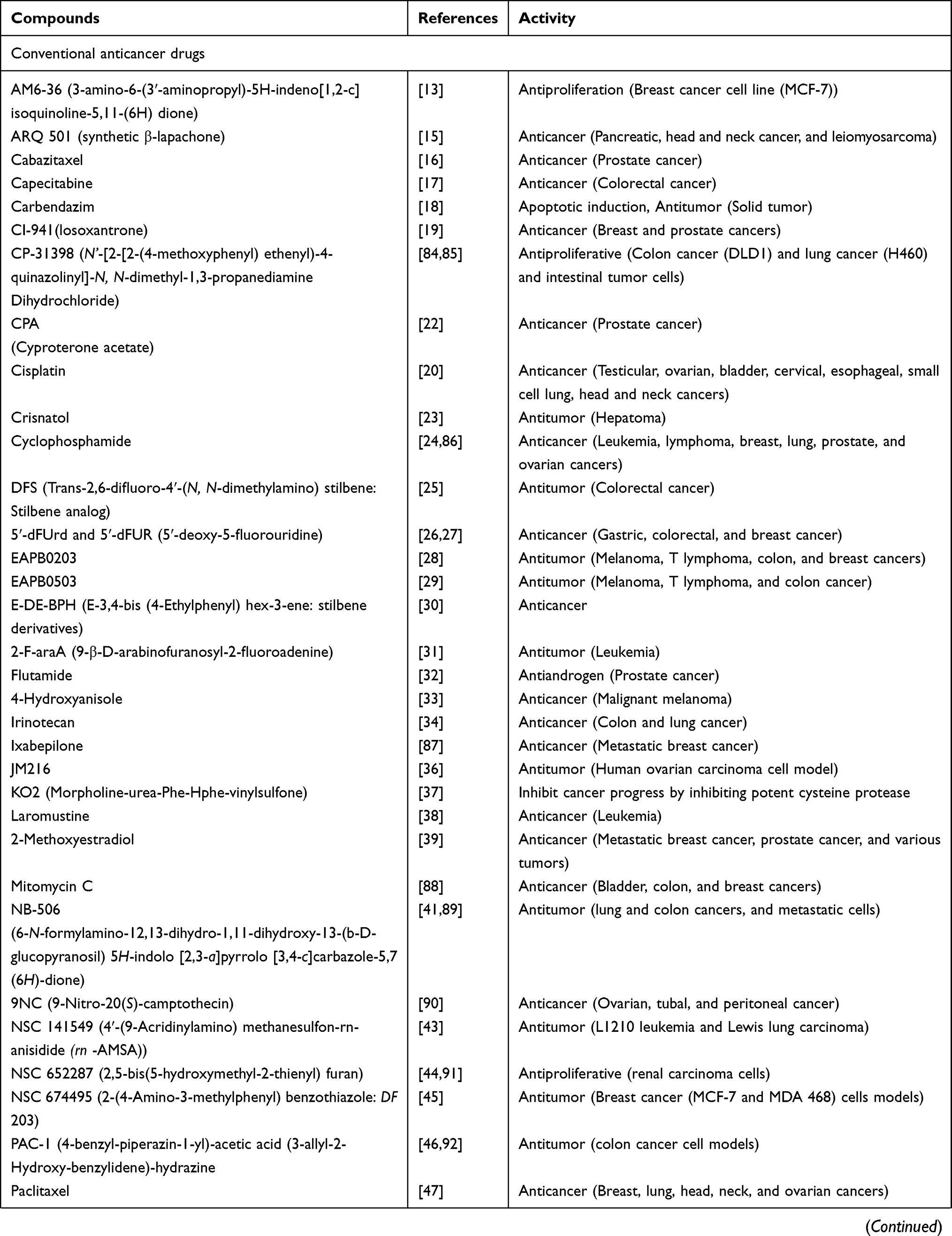 | 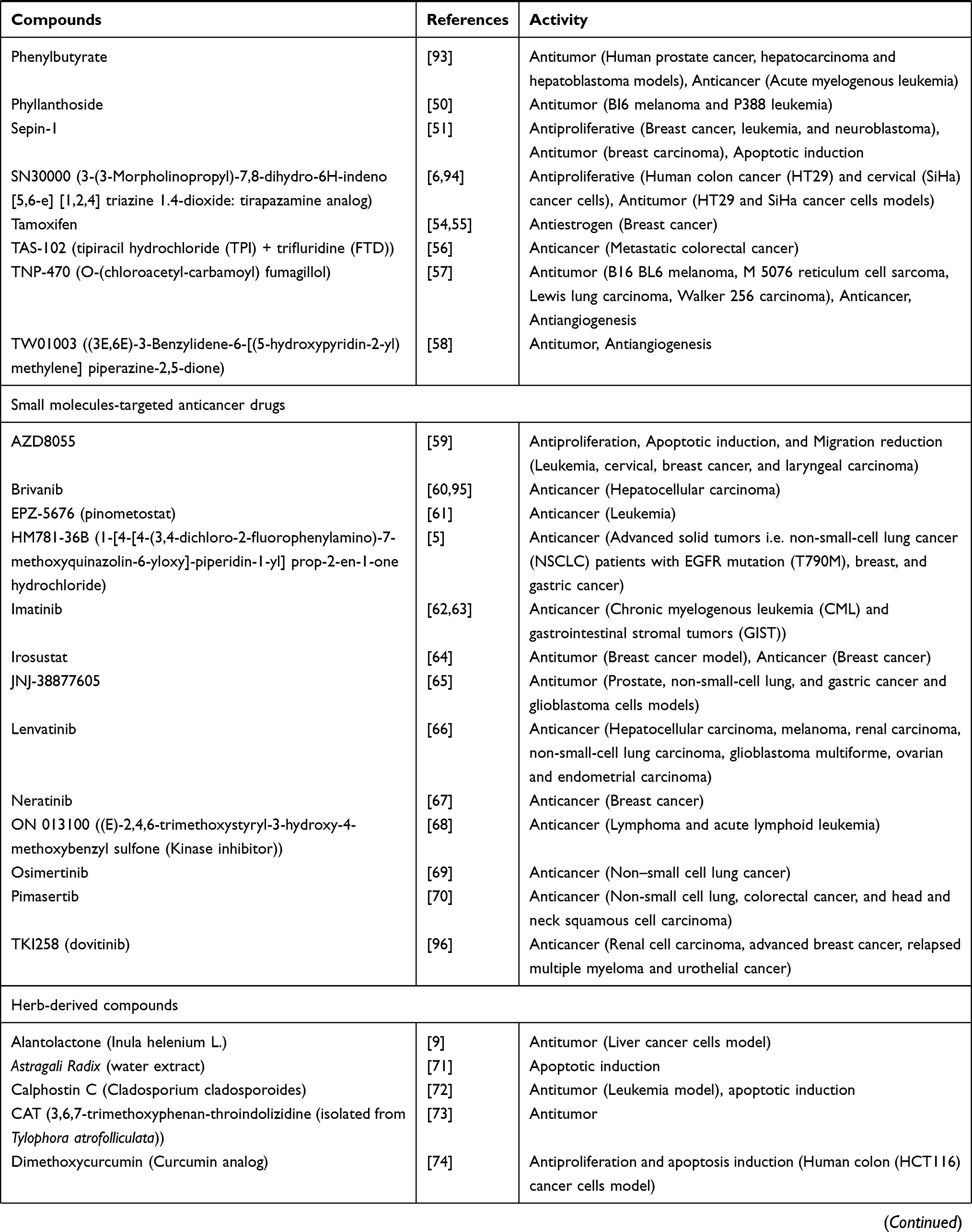 | 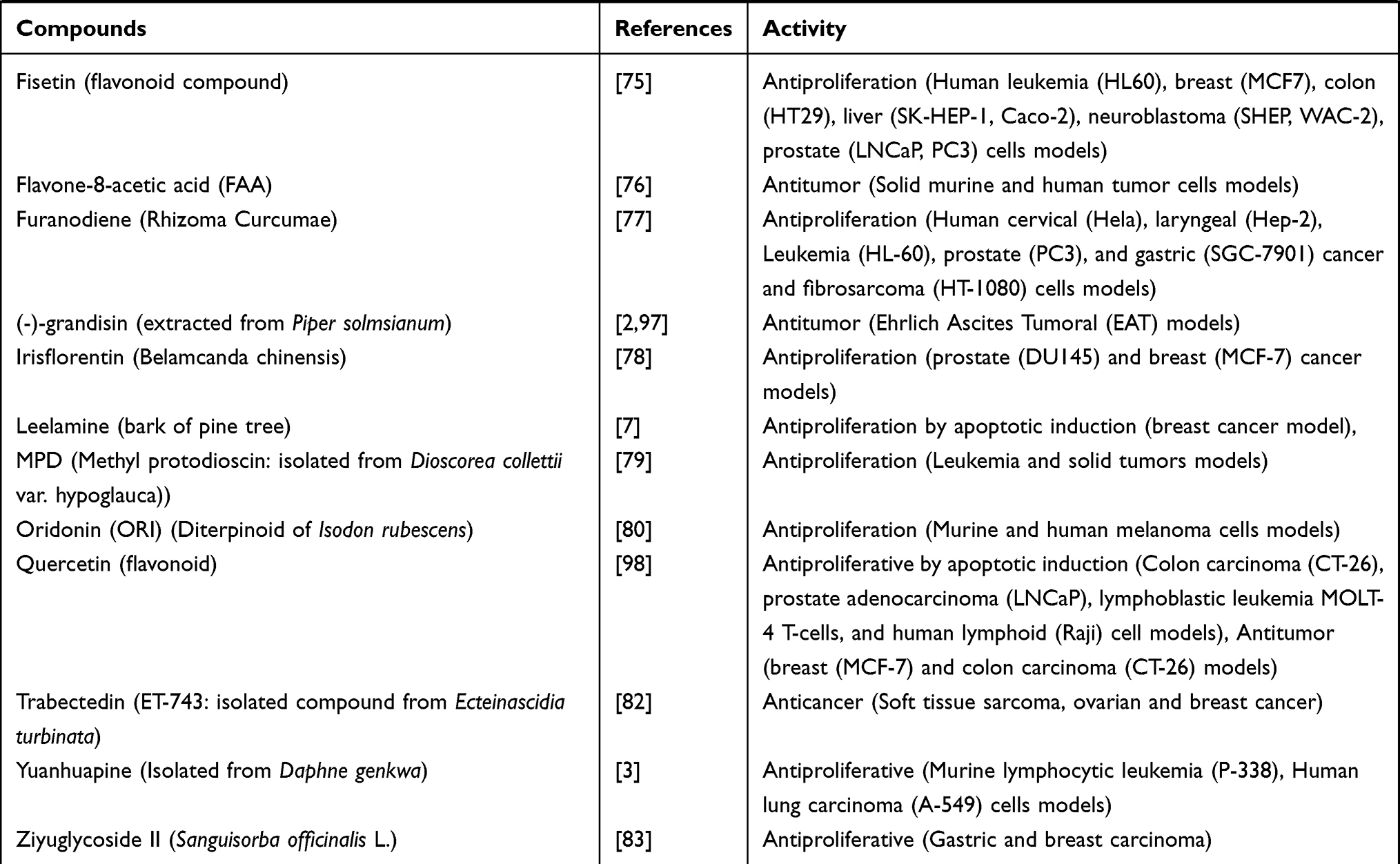 |
Table 4 Activities of Conventional Anticancer Drugs, Synthetic Anticancer Candidates, Small Molecules Targeted Therapy, Herb-Derived Compounds |
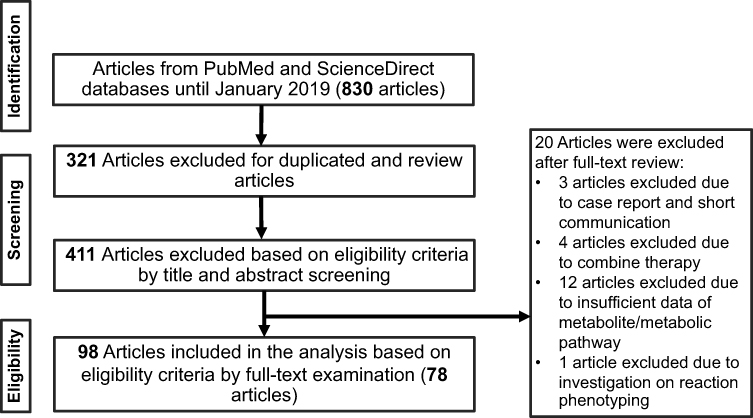 |
Figure 1 Flow diagram summarizing steps for exclusion and inclusion of the research articles included in the analysis. |
Out of 78 articles included in the analysis (42 in vitro, 42 in vivo, and 16 clinical studies), 47 (60.3%), 14 (17.9%) and 17 (21.8%) articles respectively, investigated metabolite profiles of conventional anticancer drugs/synthetic anticancer candidates, small-molecules targeted therapy, and herb-derived compounds with anticancer activities. These studies involved a total of 57 (57.0%) conventional anticancer drugs/synthetic anticancer candidates,6,13–58 22 (22.0%) studies for small-molecules targeted therapy,4,5,59–70 and 21 (21.0%) studies for herb-derived compounds with anticancer activities2,3,7,9,71–83 (Tables 1–3). Anticancer drugs or candidate compounds are metabolized by either Phase I, or phase II metabolizing enzyme alone, or both phase I and phase II metabolizing enzymes. The major metabolic pathways of phase I reaction include oxidation/hydroxylation and hydrolysis. The major metabolic pathways of phase II reaction are glucuronidation, methylation, and sulfation (Tables 1–3).
Several factors were found to influence the metabolite profiles of these drugs/compounds, including species, gender, and route and dose of administration. Relationship between the generated metabolites and anticancer activity and/or toxicity were reported in 10 (17.5%) studies for conventional anticancer drugs/synthetic anticancer candidates,6,13,22,31,32,36,45,50 1 (4.5%) studies for small-molecules targeted therapy,65 and 4 (19.0%) studies for herb-derived compounds with anticancer activities.75,76,78,79
Discussion
The goals of conducting drug metabolism studies are to identify and characterize all major metabolites of the investigated drugs and specific enzymes responsible for their metabolism; to evaluate the impacts of the metabolites on safety and efficacy of the drug, and to utilize the drug metabolism information to maximize their intellectual property. The identity of metabolites present in any matrix of animal or human provides essential information about the biotransformation pathways involved in the clearance of a drug. Technological advances during the past decade have greatly improved analytical capabilities to detect, identify, and characterize metabolites at previously unattainable levels. Chromatography and electrophoresis are usually methods of choice. Electrospray ionization (ESI) and atmospheric pressure chemical ionization (APCI) liquid chromatography-mass spectrometry (LC/MS) have become ideal and widely used methods in the identification, structure characterization, and quantitative analysis of drug metabolites. This is due to its superior specificity, sensitivity, and efficiency over other methods such as radioimmunoassay (RIA), gas chromatography/mass spectrometry (GC/MS),44 and liquid chromatography (LC) with ultraviolet detection (UV),19,76 fluorescence,34,45 radioactivity38,81 and mass spectrometry (MS)13,18,64 detection. Most of the works in metabolite analysis were carried out using triple-quadrupole mass spectrometers.28,39,55,57,63,82 The main advantages include its superior quantitative capabilities in the multiple reaction monitoring (MRM) mode and the fact that a family of metabolites can easily be identified using neutral-loss and precursor ion scans.
Factors Influencing Metabolite Formation
Several factors were shown to influence metabolite formation of the parent drugs/compounds with anticancer activities. These included species, gender, and route and dose of administration of the parent drugs/compounds.
Species
Metabolic pathways and metabolite profiles of anticancer drugs/candidate compounds varied with animal species investigated. This is explained by the difference in the expression of metabolizing enzymes. For example, CYP1A1 and 1A2 enzymes are presented in mouse, rat, dog, monkey, and human, whereas CYP2C9, 2C19, 2D6, and 3A4 enzymes are only presented in human.99 In a study of flutamide (nonsteroidal antiandrogen used primarily for prostate cancer) metabolism, only one metabolite (designated OH-flu) was detected in human after incubation with human liver microsomes. On the other hand, four metabolites (Flu-1, OH-flu, M1, and M3) were detected after incubation of the compound with liver microsomes from rat, dog, and pig.32 Moreover, similar metabolite (OH-flu) was detected in rat and pig, but this metabolite was detected as 3 and 2 isoforms in rat and pig, respectively.32 For the synthetic β-lapachone, ARQ 501, similar phase I metabolites (M1-M3, and M5) were detected after incubation with whole blood of mouse, rat, dog, monkey, and human. On the other hand, only one phase I metabolite M4 was detected in human and M6 metabolite was detected in both human and monkey.14
Gender
Gender is another factor that influences the metabolite profiles of anticancer drugs/candidate compounds due to difference in the expression levels of metabolizing enzymes between males and females. For example, in human, the levels of CYP2E1 and 1A2 are found to be higher in males than females, while the level of CYP3A4 is higher in females.100 In rat liver, the expression level of CYP2C12 is higher in females than males, while the level of CYP2C11 is higher in males than females.101 Ventura et al investigated the metabolic profiles of irosustat (inhibitor of steroid sulfatase under development for hormone-sensitive cancers such as breast and prostate cancer) using liver microsomes from male and female rats, dogs, monkeys, and humans. In rats, 11 metabolites (designated M7-M11, M13-M16, M18 and 667-coumarin) were detected in both genders, while 1 different metabolite each was detected in males (P-24) and females (P-36).64 Eight similar metabolites (P-14, M7-M10, M13, M16, and P-24) were detected in both genders of monkeys, whereas only M14 metabolite was detected in only male monkeys. In dogs and humans, similar 14 metabolites were detected in both genders.64 These results suggest that the metabolites generated from the parent anticancer drugs/candidate compounds may vary depending on the expression levels of the responsible metabolizing enzymes in each gender.
Route of Administration
Route of administration was found to be one contributing factor that influences the metabolite profiles of anticancer drugs/candidate compounds. Following intravenous administration of TW-01003 (a piperazinedione derivative designed as an antimitotic agent), the major metabolite (TW-01003 sulfate) was detected in rat plasma. Following oral administration, on the other hand, TW-01003 glucuronide became major metabolite.58 This might be due to the enterohepatic recirculation, which increases the level of glucuronide conjugates in the systemic circulation.58 In another metabolism study of tamoxifen (selective estrogen-receptor modulator for breast cancer), however, two similar metabolites (4-hydroxytamoxifen and N-desmethyltamoxifen) were detected in mouse serum following both oral and subcutaneous routes of administration.52
Dose
The dose level of anticancer drugs/candidate compounds was not found to be the major factor that influenced their metabolite profiles, but the metabolite levels. In the study of tamoxifen metabolism in mice, higher levels of metabolites were reported after an oral dose of 200 compared with 50 mg/kg/day.52 In the study of JM 216 or satraplatin, a platinum drug, the number of metabolites detected was similar among patients receiving different doses of the compound (120, 200, 300, 420, and 540 mg/m2). Plasma levels of each metabolite were not significantly different in patients who received different doses of the drug. For example, metabolite A was detected at 20–58.9% for 120 mg/m2, 10.2–50% for 200 mg/m2, 8.7–76.2% for 300 mg/m2, 8.2–85.9% for 420 mg/m2, and 5.1–44.4% for 540 mg/m2.36 Such broad range of metabolite levels in plasma observed in patients receiving the same dose might be due to inter-individual variability metabolizing enzyme(s) responsible for JM216 metabolism in each patient. These results suggest that the metabolite profiles of anticancer drugs/candidate compounds are not completely affected by the dose of administration but may be influenced by the dose level and duration of administration.
Contribution of Metabolites to Anticancer Activity and Toxicity of Anticancer Drugs/Candidate Compounds
The metabolites generated from the parent anticancer drugs/candidate compounds can be both active and inactive metabolites. This information is essential for cancer chemotherapy with a narrow therapeutic window, particularly in individuals with increased or decreased drug-metabolizing enzyme activities influenced by genetic and nongenetic factors. Several studies showed that biotransformation could significantly influence the activity and toxicity of the anticancer drugs/candidate compounds. The metabolites of some active drugs/compounds exhibited more potent anticancer activity than their parent compounds. For example, the cytotoxic activity of the M3 metabolite of fisetin, a plant polyphenol, against Lewis carcinoma (LLC) cell line was shown to be about 2-fold of fisetin itself (IC50 or 50% inhibitory concentrations of 24 vs. 59 µM). However, both M3 and fisetin exhibited similar cytotoxic activities against the endothelial EAhy 926 cell line (IC50 72 vs. 76 µM). It was noted however, that the cytotoxic activity of this metabolite against the normal cell line, NIH 3T3, was relatively higher than fisetin (IC50 128 vs. 195 µM).75 The M1 and M2 metabolites of the synthetic compound irisflorentin were shown to exhibit more potent cytotoxic activity against human prostate cancer (DU145) and breast cancer (MCF-7) cell lines compared with the parent drug (IC50 65.12 vs. 35.71 vs.>300 µM and 74.17 vs. 36.30 vs.>300 µM, respectively).78 The metabolite of a potent antileukemic compound, phyllanthoside, also exhibited lower cytotoxic activity against human rhabdomyosarcoma A204 cell line compared with the parent compound (IC50 of 24 µM and 0.47 nM, respectively).50 Some metabolites showed low or no cytotoxic activity. For example, The metabolite 6c of NSC 674495, an antitumor 2-(4-Aminophenyl) benzothiazoles, exhibited low cytotoxic activity against MCF-7 cell line (IC50>100 M) and was not shown to be an active metabolite.45 The anticancer activities of the metabolites of these compounds should be further investigated in animals and humans to confirm their activities in vivo. Some metabolites may be pharmacologically inactive, while some may exhibit toxicity in animal models. For examples, 2-F-araH, the metabolite of 2-F-araA and the antitumor 9-β-D-arabinofuranosyl-2-fluoroadenine exhibited no antitumor activity in BDF1 mice bearing L1210 leukemia cells following 200 mg/kg intraperitoneal dosing for nine days.31 The M1 to M5 metabolites of JNJ-38877605 (a potent and selective Met receptor tyrosine kinase inhibitor) were shown to be associated with crystal formation in the kidney. The M10 metabolite, on the other hand, was shown to be the toxic metabolite found in humans (at sub-therapeutic dose 60 mg OD) and rats and was also noted as a species-specific toxic metabolite.65 Since renal toxicity could be observed even at a subtherapeutic dose, this toxicity is also likely to be observed at standard dose. The M14 metabolite of the tirapazamine analog SN30000 was found to be the cause of acute toxicity (hypothermia) similarly to the parent drug after the administration at the maximum tolerated dose (MTD: 186 mg/kg SN30000).6 However, hypothermia was also observed at 50% of MTD. Thus, this toxicity can be observed at the therapeutic dosage (75% of MTD).94
Conclusion
Drug metabolism plays a critical role in determining the pharmacological and toxicological effects of a drug in human. Metabolite profiling study is essential in determining therapeutic efficacy and toxicity of anticancer drugs/compounds from both chemical synthesis or natural sources. The generated metabolite profiles, either the types and amounts can be significantly influenced by factors such as species, gender, and route and dose of administration. Moreover, the metabolites can be both active and toxic metabolites, and some metabolites are inactive. The information of metabolite profile wills provides beneficial knowledge in anticancer agent development to improve anticancer activity and safety profiles of anticancer drugs or drug candidates.
Acknowledgments
The authors would like to thank the Center of Excellence in Pharmacology and Molecular Biology of Malaria and Cholangiocarcinoma (Thammasat University), Chulabhorn International College of Medicine (Thammasat University), and Drug Discovery and Development Center (Thammasat University) for providing all the necessary support in conducting this systematic review.
Author Contributions
Nadda Muhamad: Conception and design, data analysis and interpretation, drafting the article, final approval, agreement to be accountable for the accuracy or integrity of the work.
Kesara Na-Bangchang: Conception and design, data analysis and interpretation, revising the article, final approval, agreement to be accountable for the accuracy or integrity of the work.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. doi:10.3322/caac.21492
2. Barth T, Habenschus MD, Lima Moreira F, Ferreira Lde S, Lopes NP, Moraes de Oliveira AR. In vitro metabolism of the lignan (-)-grandisin, an anticancer drug candidate, by human liver microsomes. Drug Test Anal. 2015;7(9):780–786. doi:10.1002/dta.1743
3. Chen Y, Guo J, Tang Y, et al. Pharmacokinetic profile and metabolite identification of yuanhuapine, a bioactive component in Daphne genkwa by ultra-high performance liquid chromatography coupled with tandem mass spectrometry. J Pharm Biomed Anal. 2015;112:60–69. doi:10.1016/j.jpba.2015.04.023
4. Dubbelman AC, Upthagrove A, Beijnen JH, et al. Disposition and metabolism of 14C-dovitinib (TKI258), an inhibitor of FGFR and VEGFR, after oral administration in patients with advanced solid tumors. Cancer Chemother Pharmacol. 2012;70(5):653–663. doi:10.1007/s00280-012-1947-2
5. Kim E, Kim H, Suh K, et al. Metabolite identification of a new tyrosine kinase inhibitor, HM781-36B, and a pharmacokinetic study by liquid chromatography/tandem mass spectrometry. Rapid Commun Mass Spectrom. 2013;27(11):1183–1195. doi:10.1002/rcm.6559
6. Gu Y, Chang TT, Wang J, et al. Reductive metabolism influences the toxicity and pharmacokinetics of the hypoxia-targeted benzotriazine di-oxide anticancer agent SN30000 in mice. Front Pharmacol. 2017;8:531. doi:10.3389/fphar.2017.00531
7. Shrestha R, Jo JJ, Lee D, Lee T, Lee S. Characterization of in vitro and in vivo metabolism of leelamine using liquid chromatography-tandem mass spectrometry. Xenobiotica. 2019;49(5):577–583.
8. Na-Bangchang K, Plengsuriyakarn T, Karbwang J. Research and development of Atractylodes lancea (Thunb) DC. as a promising candidate for cholangiocarcinoma chemotherapeutics. J Evid Based Complementary Altern Med. 2017;2017:16.
9. Yao D, Li Z, Huo C, et al. Identification of in vitro and in vivo metabolites of alantolactone by UPLC-TOF-MS/MS. J Chromatogr B Analyt Technol Biomed Life Sci. 2016;1033–1034:250–260. doi:10.1016/j.jchromb.2016.08.034
10. Elhassan G. Drug development: stages of drug development. J Pharmacovigil. 2015;3:3.
11. Gunaratna C. Drug metabolism and pharmacokinetics in drug discovery: a primer for bioanalytical chemists, part I. Curr Sep. 2000;19(1):17–23.
12. Gonzalez FJ, Tukey RH. Drug Metabolism.
13. Chen L, Conda-Sheridan M, Reddy PV, et al. Identification, synthesis, and biological evaluation of the metabolites of 3-amino-6-(3ʹ-aminopropyl)-5H-indeno[1,2-c]isoquinoline-5,11-(6H)dione (AM6-36), a promising rexinoid lead compound for the development of cancer chemotherapeutic and chemopreventive agents. J Med Chem. 2012;55(12):5965–5981.
14. Miao XS, Song P, Savage RE, et al. Identification of the in vitro metabolites of 3,4-dihydro-2,2-dimethyl-2H-naphthol[1,2-b]pyran-5,6-dione (ARQ 501; beta-lapachone) in whole blood. Drug Metab Dispos. 2008;36(4):641–648. doi:10.1124/dmd.107.018572
15. Savage RE, Tyler AN, Miao XS, Chan TC. Identification of a novel glucosylsulfate conjugate as a metabolite of 3,4-dihydro-2,2-dimethyl-2H-naphtho[1,2-b]pyran-5,6-dione (ARQ 501, beta-lapachone) in mammals. Drug Metab Dispos. 2008;36(4):753–758. doi:10.1124/dmd.107.018655
16. Ridoux L, Semiond DR, Vincent C, et al. A phase I open-label study investigating the disposition of [14C]-cabazitaxel in patients with advanced solid tumors. Anticancer Drugs. 2015;26(3):350–358. doi:10.1097/CAD.0000000000000185
17. Desmoulin F, Gilard V, Martino R, Malet-Martino M. Isolation of an unknown metabolite of capecitabine, an oral 5-fluorouracil prodrug, and its identification by nuclear magnetic resonance and liquid chromatography-tandem mass spectrometry as a glucuroconjugate of 5ʹ-deoxy-5-fluorocytidine, namely 2ʹ-(beta-D-glucuronic acid)-5ʹ-deoxy-5-fluorocytidine. J Chromatogr B Analyt Technol Biomed Life Sci. 2003;792(2):323–332. doi:10.1016/s1570-0232(03)00319-2
18. Jia L, Wong H, Wang Y, Garza M, Weitman SD. Carbendazim: disposition, cellular permeability, metabolite identification, and pharmacokinetic comparison with its nanoparticle. J Pharm Sci. 2003;92(1):161–172. doi:10.1002/jps.10272
19. Renner UD, Piperopoulos G, Gebhardt R, Ehninger G, Zeller KP. The oxidative biotransformation of losoxantrone (CI-941). Drug Metab Dispos. 2002;30(4):464–478. doi:10.1124/dmd.30.4.464
20. Bandu R, Ahn HS, Lee JW, et al. Liquid chromatography electrospray ionization tandem mass spectrometric (LC/ESI-MS/MS) study for the identification and characterization of in vivo metabolites of cisplatin in rat kidney cancer tissues: online hydrogen/deuterium (H/D) exchange study. PLoS One. 2015;10(8):e0134027. doi:10.1371/journal.pone.0134027
21. Johnson WD, Muzzio M, Detrisac CJ, Kapetanovic IM, Kopelovich L, McCormick DL. Subchronic oral toxicity and metabolite profiling of the p53 stabilizing agent, CP-31398, in rats and dogs. Toxicology. 2011;289(2):141–150. doi:10.1016/j.tox.2011.08.009
22. Kerdar RS, Baumann A, Brudny-Kloppel M, Biere H, Blode H, Kuhnz W. Identification of 3 alpha-hydroxy-cyproterone acetate as a metabolite of cyproterone acetate in the bile of female rats and the potential of this and other already known or putative metabolites to form DNA adducts in vitro. Carcinogenesis. 1995;16(8):1835–1841. doi:10.1093/carcin/16.8.1835
23. Patel DK, Shockcor JP, Chang SY, Sigel CW, Huber BE. Metabolism of a novel antitumor agent, crisnatol, by a human hepatoma cell line, Hep G2, and hepatic microsomes. Characterization of metabolites. Biochem Pharmacol. 1991;42(2):337–346. doi:10.1016/0006-2952(91)90721-G
24. Fenselau C, Kan MN, Billets S, Colvin M. Identification of phosphorodiamidic acid mustard as a human metabolite of cyclophosphamide. Cancer Res. 1975;35(6):1453–1457.
25. Yeo SC, Sviripa VM, Huang M, et al. Analysis of trans-2,6-difluoro-4ʹ-(N,N-dimethylamino)stilbene (DFS) in biological samples by liquid chromatography-tandem mass spectrometry: metabolite identification and pharmacokinetics. Anal Bioanal Chem. 2015;407(24):7319–7332. doi:10.1007/s00216-015-8893-x
26. Malet-Martino MC, Martino R, Lopez A, et al. New approach to metabolism of 5ʹ-deoxy-5-fluorouridine in humans with fluorine-19 NMR. Cancer Chemother Pharmacol. 1984;13(1):31–35. doi:10.1007/BF00401443
27. Zambonin CG, Palmisano F. Gas chromatography-mass spectrometry identification of a novel N3-methylated metabolite of 5ʹ-deoxy-5-fluorouridine in plasma of cancer patients undergoing chemotherapy. J Pharm Biomed Anal. 1996;14(11):1521–1528. doi:10.1016/0731-7085(96)01798-0
28. Lafaille F, Banaigs B, Inguimbert N, et al. Characterization of a new anticancer agent, EAPB0203, and its main metabolites: nuclear magnetic resonance and liquid chromatography-mass spectrometry studies. Anal Chem. 2012;84(22):9865–9872. doi:10.1021/ac3021483
29. Lafaille F, Solassol I, Enjalbal C, et al. Structural characterization of in vitro metabolites of the new anticancer agent EAPB0503 by liquid chromatography-tandem mass spectrometry. J Pharm Biomed Anal. 2014;88:429–440. doi:10.1016/j.jpba.2013.09.015
30. Fabian EJ, Metzler M. Selective metabolism of E-3,4-bis(4-ethylphenyl)hex-3-ene in rat liver microsomes. Arch Toxicol. 2006;80(1):17–26. doi:10.1007/s00204-005-0007-7
31. Struck RF, Shortnacy AT, Kirk MC, et al. Identification of metabolites of 9-beta-D-arabinofuranosyl-2-fluoroadenine, an antitumor and antiviral agent. Biochem Pharmacol. 1982;31(11):1975–1978. doi:10.1016/0006-2952(82)90407-5
32. Tevell A, Lennernas H, Jonsson M, et al. Flutamide metabolism in four different species in vitro and identification of flutamide metabolites in human patient urine by high performance liquid chromatography/tandem mass spectrometry. Drug Metab Dispos. 2006;34(6):984–992. doi:10.1124/dmd.105.008516
33. Pavel S, Holden JL, Riley PA. Metabolism of 4-hydroxyanisole: identification of major urinary excretory products. Pigment Cell Res. 1989;2(5):421–426. doi:10.1111/pcr.1989.2.issue-5
34. Santos A, Zanetta S, Cresteil T, et al. Metabolism of irinotecan (CPT-11) by CYP3A4 and CYP3A5 in humans. Clin Cancer Res. 2000;6(5):2012–2020.
35. Nilgün Çömezoğlu S, Ly VT, Zhang D, et al. Biotransformation profiling of [14C]Ixabepilone in human plasma, urine and feces samples using accelerator mass spectrometry (AMS). Drug Metab Pharmacokinet. 2009;24(6):511–522. doi:10.2133/dmpk.24.511
36. Raynaud FI, Mistry P, Donaghue A, et al. Biotransformation of the platinum drug JM216 following oral administration to cancer patients. Cancer Chemother Pharmacol. 1996;38(2):155–162. doi:10.1007/s002800050464
37. Zhang Y, Guo X, Lin ET, Benet LZ. Overlapping substrate specificities of cytochrome P450 3A and P-glycoprotein for a novel cysteine protease inhibitor. Drug Metab Dispos. 1998;26(4):360–366.
38. Nassar AE, King I, Paris BL, et al. An in vitro evaluation of the victim and perpetrator potential of the anticancer agent laromustine (VNP40101M), based on reaction phenotyping and inhibition and induction of cytochrome P450 enzymes. Drug Metab Dispos. 2009;37(9):1922–1930. doi:10.1124/dmd.109.027516
39. Lakhani NJ, Sparreboom A, Xu X, et al. Characterization of in vitro and in vivo metabolic pathways of the investigational anticancer agent, 2-methoxyestradiol. J Pharm Sci. 2007;96(7):1821–1831. doi:10.1002/jps.20837
40. Spanswick VJ, Cummings J, Ritchie AA, Smyth JF. Pharmacological determinants of the antitumour activity of mitomycin C. Biochem Pharmacol. 1998;56(11):1497–1503. doi:10.1016/S0006-2952(98)00164-6
41. Takenaga N, Ishii M, Nakajima S, et al. In vivo metabolism of a new anticancer agent, 6-N-formylamino-12, 13-dihydro-1,11-dihydroxy-13-(beta-D-glucopyranosil)5H-indolo [2,3-a]pyrrolo [3,4-c]carbazole-5,7(6H)-dione (NB-506) in rats and dogs: pharmacokinetics, isolation, identification, and quantification of metabolites. Drug Metab Dispos. 1999;27(2):205–212.
42. Li K, Chen X, Zhong D, Li Y. Identification of the metabolites of 9-nitro-20(S)-camptothecin in rats. Drug Metab Dispos. 2003;31(6):792–797. doi:10.1124/dmd.31.6.792
43. Przybylski M, Cysyk RL, Shoemaker D, Adamson RH. Identification of conjugation and cleavage products in the thiolytic metabolism of the anticancer drug 4ʹ-(9-acridinylamino)methanesulfon-m-anisidide. Biomed Mass Spectrom. 1981;8(10):485–491. doi:10.1002/bms.1200081004
44. Phillip LR, Jorden JL, Rivera MI, Wolfe TL, Upadhyayb K, Stinson SF. Identification of the major metabolite of 2,5-bis(5-hydroxymethyl-2-thienyl)furan (NSC 652287), an antitumor agent, in the S9 subcellular fraction of dog liver cells. J Chromatogr B Analyt Technol Biomed Life Sci. 2002;767(1):27–33. doi:10.1016/S0378-4347(01)00530-8
45. Kashiyama E, Hutchinson I, Chua MS, et al. Antitumor benzothiazoles. 8. Synthesis, metabolic formation, and biological properties of the C- and N-oxidation products of antitumor 2-(4-aminophenyl)benzothiazoles. J Med Chem. 1999;42(20):4172–4184. doi:10.1021/jm990104o
46. Ren L, Bi K, Gong P, et al. Characterization of the in vivo and in vitro metabolic profile of PAC-1 using liquid chromatography-mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2008;876(1):47–53. doi:10.1016/j.jchromb.2008.10.006
47. Royer I, Alvinerie P, Armand JP, Ho LK, Wright M, Monsarrat B. Paclitaxel metabolites in human plasma and urine: identification of 6 alpha-hydroxytaxol, 7-epitaxol and taxol hydrolysis products using liquid chromatography/atmospheric-pressure chemical ionization mass spectrometry. Rapid Commun Mass Spectrom. 1995;9(6):495–502. doi:10.1002/rcm.1290090605
48. Bun SS, Giacometti S, Fanciullino R, Ciccolini J, Bun H, Aubert C. Effect of several compounds on biliary excretion of paclitaxel and its metabolites in guinea-pigs. Anticancer Drugs. 2005;16(6):675–682. doi:10.1097/00001813-200507000-00013
49. Comte B, Kasumov T, Pierce BA, et al. Identification of phenylbutyrylglutamine, a new metabolite of phenylbutyrate metabolism in humans. J Mass Spectrom. 2002;37(6):581–590. doi:10.1002/jms.316
50. Chapman DE, Moore DJ, Melder DC, Breau A, Powis G. Isolation, identification and biological activity of a phyllanthoside metabolite produced in vitro by mouse plasma. Cancer Chemother Pharmacol. 1989;25(3):184–188. doi:10.1007/BF00689580
51. Li F, Zhang N, Gorantla S, Gilbertson SR, Pati D. The metabolism of separase inhibitor sepin-1 in human, mouse, and rat liver microsomes. Front Pharmacol. 2018;9:313. doi:10.3389/fphar.2018.00313
52. Robinson SP, Langan-Fahey SM, Jordan VC. Implications of tamoxifen metabolism in the athymic mouse for the study of antitumor effects upon human breast cancer xenografts. Eur J Cancer Clin Oncol. 1989;25(12):1769–1776. doi:10.1016/0277-5379(89)90347-7
53. Poon GK, Walter B, Lonning PE, Horton MN, McCague R. Identification of tamoxifen metabolites in human Hep G2 cell line, human liver homogenate, and patients on long-term therapy for breast cancer. Drug Metab Dispos. 1995;23(3):377–382.
54. Kisanga ER, Moi LLH, Gjerde J, Mellgren G, Lien EA. Induction of hepatic drug-metabolising enzymes and tamoxifen metabolite profile in relation to administration route during low-dose treatment in nude rats. J Steroid Biochem Mol Biol. 2005;94(5):489–498. doi:10.1016/j.jsbmb.2004.12.037
55. Dahmane E, Boccard J, Csajka C, et al. Quantitative monitoring of tamoxifen in human plasma extended to 40 metabolites using liquid-chromatography high-resolution mass spectrometry: new investigation capabilities for clinical pharmacology. Anal Bioanal Chem. 2014;406(11):2627–2640. doi:10.1007/s00216-014-7682-2
56. Lee JJ, Seraj J, Yoshida K, et al. Human mass balance study of TAS-102 using (14)C analyzed by accelerator mass spectrometry. Cancer Chemother Pharmacol. 2016;77(3):515–526. doi:10.1007/s00280-016-2965-2
57. Placidi L, Cretton-Scott E, de Sousa G, Rahmani R, Placidi M, Sommadossi JP. Disposition and metabolism of the angiogenic moderator O-(chloroacetyl-carbamoyl) fumagillol (TNP-470; AGM-1470) in human hepatocytes and tissue microsomes. Cancer Res. 1995;55(14):3036–3042.
58. Wang CL, Chen CK, Wang YH, Cheng YW. In search of the active metabolites of an anticancer piperazinedione, TW01003, in rats. Biomed Res Int. 2014;2014:793504.
59. Rashid MM, Oh HA, Lee H, Jung BH. Metabolite identification of AZD8055 in Sprague-Dawley rats after a single oral administration using ultra-performance liquid chromatography and mass spectrometry. J Pharm Biomed Anal. 2017;145:473–481. doi:10.1016/j.jpba.2017.06.059
60. Gong J, Gan J, Iyer RA. Identification of the oxidative and conjugative enzymes involved in the biotransformation of brivanib. Drug Metab Dispos. 2012;40(1):219–226. doi:10.1124/dmd.111.042457
61. Waters NJ, Smith SA, Olhava EJ, et al. Metabolism and disposition of the DOT1L inhibitor, pinometostat (EPZ-5676), in rat, dog and human. Cancer Chemother Pharmacol. 2016;77(1):43–62. doi:10.1007/s00280-015-2929-y
62. Marull M, Rochat B. Fragmentation study of imatinib and characterization of new imatinib metabolites by liquid chromatography-triple-quadrupole and linear ion trap mass spectrometers. J Mass Spectrom. 2006;41(3):390–404. doi:10.1002/jms.1002
63. Rochat B, Fayet A, Widmer N, et al. Imatinib metabolite profiling in parallel to imatinib quantification in plasma of treated patients using liquid chromatography-mass spectrometry. J Mass Spectrom. 2008;43(6):736–752. doi:10.1002/jms.1369
64. Ventura V, Sola J, Celma C, Peraire C, Obach R. In vitro metabolism of irosustat, a novel steroid sulfatase inhibitor: interspecies comparison, metabolite identification, and metabolic enzyme identification. Drug Metab Dispos. 2011;39(7):1235–1246. doi:10.1124/dmd.111.038315
65. Lolkema MP, Bohets HH, Arkenau HT, et al. The c-Met tyrosine kinase inhibitor JNJ-38877605 causes renal toxicity through species-specific insoluble metabolite formation. Clin Cancer Res. 2015;21(10):2297–2304. doi:10.1158/1078-0432.CCR-14-3258
66. Dubbelman AC, Nijenhuis CM, Jansen RS, et al. Metabolite profiling of the multiple tyrosine kinase inhibitor lenvatinib: a cross-species comparison. Invest New Drugs. 2016;34(3):300–318. doi:10.1007/s10637-016-0342-y
67. Liu W, Li S, Wu Y, et al. Metabolic profiles of neratinib in rat by using ultra-high-performance liquid chromatography coupled with diode array detector and Q-exactive orbitrap tandem mass spectrometry. Biomed Chromatogr. 2018:e4272. doi:10.1002/bmc.4272
68. Cho SY, Cosenza SC, Pallela V, et al. Determination of the glucuronide metabolite of ON 013100, a benzylstyrylsulfone antineoplastic drug, in colon cancer cells using LC/MS/MS. J Pharm Biomed Anal. 2013;75:138–144. doi:10.1016/j.jpba.2012.11.022
69. MacLeod AK, Lin D, Huang JT, McLaughlin LA, Henderson CJ, Wolf CR. Identification of novel pathways of osimertinib disposition and potential implications for the outcome of lung cancer therapy. Clin Cancer Res. 2018;24(9):2138–2147. doi:10.1158/1078-0432.CCR-17-3555
70. von Richter O, Massimini G, Scheible H, Udvaros I, Johne A. Pimasertib, a selective oral MEK1/2 inhibitor: absolute bioavailability, mass balance, elimination route, and metabolite profile in cancer patients. Br J Clin Pharmacol. 2016;82(6):1498–1508. doi:10.1111/bcp.13078
71. Shi J, Zheng L, Lin Z, et al. Study of pharmacokinetic profiles and characteristics of active components and their metabolites in rat plasma following oral administration of the water extract of Astragali radix using UPLC–MS/MS. J Ethnopharmacol. 2015;169:183–194. doi:10.1016/j.jep.2015.04.019
72. Chen CL, Tai HL, Zhu DM, Uckun FM. Pharmacokinetic features and metabolism of calphostin C, a naturally occurring perylenequinone with antileukemic activity. Pharm Res. 1999;16(7):1003–1009. doi:10.1023/A:1018923430094
73. Tian Y, He J, Zhang R, et al. Integrated rapid resolution liquid chromatography-tandem mass spectrometric approach for screening and identification of metabolites of the potential anticancer agent 3,6,7-trimethoxyphenanthroindolizidine in rat urine. Anal Chim Acta. 2012;731:60–67. doi:10.1016/j.aca.2012.04.024
74. Tamvakopoulos C, Dimas K, Sofianos ZD, et al. Metabolism and anticancer activity of the curcumin analogue, dimethoxycurcumin. Clin Cancer Res. 2007;13(4):1269–1277. doi:10.1158/1078-0432.CCR-06-1839
75. Touil YS, Auzeil N, Boulinguez F, et al. Fisetin disposition and metabolism in mice: identification of geraldol as an active metabolite. Biochem Pharmacol. 2011;82(11):1731–1739. doi:10.1016/j.bcp.2011.07.097
76. Pham MH, Auzeil N, Regazzetti A, et al. Identification of new flavone-8-acetic acid metabolites using mouse microsomes and comparison with human microsomes. Drug Metab Dispos. 2007;35(11):2023–2034. doi:10.1124/dmd.107.017012
77. Chen M, Lou Y, Wu Y, et al. Characterization of in vivo and in vitro metabolites of furanodiene in rats by high performance liquid chromatography-electrospray ionization mass spectrometry and nuclear magnetic resonance spectra. J Pharm Biomed Anal. 2013;86:161–168. doi:10.1016/j.jpba.2013.08.008
78. Jia YW, Zeng ZQ, Shi HL, et al. Characterization of in vitro metabolites of irisflorentin by rat liver microsomes using high-performance liquid chromatography coupled with tandem mass spectrometry. Biomed Chromatogr. 2016;30(9):1363–1370. doi:10.1002/bmc.3692
79. He X, Qiao A, Wang X, et al. Structural identification of methyl protodioscin metabolites in rats’ urine and their antiproliferative activities against human tumor cell lines. Steroids. 2006;71(9):828–833. doi:10.1016/j.steroids.2006.05.013
80. Tian T, Jin Y, Ma Y, et al. Identification of metabolites of oridonin in rats with a single run on UPLC-triple-TOF-MS/MS system based on multiple mass defect filter data acquisition and multiple data processing techniques. J Chromatogr B Analyt Technol Biomed Life Sci. 2015;1006:80–92. doi:10.1016/j.jchromb.2015.10.006
81. Boulton DW, Walle UK, Walle T. Fate of the flavonoid quercetin in human cell lines: chemical instability and metabolism. J Pharm Pharmacol. 1999;51(3):353–359. doi:10.1211/0022357991772367
82. Beumer JH, Rademaker-Lakhai JM, Rosing H, et al. Metabolism of trabectedin (ET-743, Yondelis) in patients with advanced cancer. Cancer Chemother Pharmacol. 2007;59(6):825–837. doi:10.1007/s00280-006-0342-2
83. Xiao J, Chen H, Fu H, et al. Development of a novel sectional multiple filtering scheme for rapid screening and classifying metabolites of ziyuglycoside II in rat liver and excreta specimen based on high-resolution mass spectrometry. J Pharm Biomed Anal. 2016;129:310–319. doi:10.1016/j.jpba.2016.06.053
84. Takimoto R, Wang W, Dicker DT, Rastinejad F, Lyssikatos J, el-Deiry WS. The Mutant p53-Conformation Modifying Drug, CP-31398, Can Induce Apoptosis. Cancer Biol Ther. 2002;1(1):47–55. doi:10.4161/cbt.1.1.41
85. Rao CV, Swamy MV, Patlolla JMR, Kopelovich L. Suppression of familial adenomatous polyposis by CP-31398, a TP53 modulator, in APCmin/+ mice. Cancer Res. 2008;68(18):7670–7675. doi:10.1158/0008-5472.CAN-08-1610
86. Nicol BM, Prasad S. The effects of cyclophosphamide alone and in combination with ascorbic acid against murine ascites Dalton’s lymphoma. Indian J Pharmacol. 2006;38(4):260–265. doi:10.4103/0253-7613.27022
87. Cobham MV, Donovan D. Ixabepilone: a new treatment option for the management of taxane-resistant metastatic breast cancer. Cancer Manag Res. 2009;1:69–77. doi:10.2147/CMAR.S5723
88. Snodgrass RG, Collier AC, Coon AE, Pritsos CA. Mitomycin C inhibits ribosomal RNA: a novel cytotoxic mechanism for bioreductive drugs. J Biol Chem. 2010;285(25):19068–19075. doi:10.1074/jbc.M109.040477
89. Ren J, Bailly C, Chaires JB. NB-506, an indolocarbazole topoisomerase I inhibitor, binds preferentially to triplex DNA. FEBS Lett. 2000;470(3):355–359. doi:10.1016/S0014-5793(00)01335-1
90. Verschraegen CF, Gupta E, Loyer E, et al. A phase II clinical and pharmacological study of oral 9-nitrocamptothecin in patients with refractory epithelial ovarian, tubal or peritoneal cancer. Anticancer Drugs. 1999;10(4):375–383. doi:10.1097/00001813-199904000-00005
91. Rivera MI, Stinson SF, Vistica DT, Jorden JL, Kenney S, Sausville EA. Selective toxicity of the tricyclic thiophene NSC 652287 in renal carcinoma cell lines: differential accumulation and metabolism. Biochem Pharmacol. 1999;57(11):1283–1295. doi:10.1016/S0006-2952(99)00046-5
92. Putt KS, Chen GW, Pearson JM, et al. Small-molecule activation of procaspase-3 to caspase-3 as a personalized anticancer strategy. Nat Chem Biol. 2006;2(10):543–550. doi:10.1038/nchembio814
93. Al-Keilani MS, Al-Sawalha NA. Potential of phenylbutyrate as adjuvant chemotherapy: an overview of cellular and molecular anticancer mechanisms. Chem Res Toxicol. 2017;30(10):1767–1777. doi:10.1021/acs.chemrestox.7b00149
94. Hicks KO, Siim BG, Jaiswal JK, et al. Pharmacokinetic/pharmacodynamic modeling identifies SN30000 and SN29751 as tirapazamine analogues with improved tissue penetration and hypoxic cell killing in tumors. Clin Cancer Res. 2010;16(20):4946. doi:10.1158/1078-0432.CCR-10-1439
95. Park JW, Finn RS, Kim JS, et al. Phase II, open-label study of brivanib as first-line therapy in patients with advanced hepatocellular carcinoma. Clin Cancer Res. 2011;17(7):1973–1983. doi:10.1158/1078-0432.CCR-10-2011
96. Hasinoff BB, Wu X, Nitiss JL, Kanagasabai R, Yalowich JC. The anticancer multi-kinase inhibitor dovitinib also targets topoisomerase I and topoisomerase II. Biochem Pharmacol. 2012;84(12):1617–1626. doi:10.1016/j.bcp.2012.09.023
97. Cortez AP, Menezes EGP, Benfica PL, et al. Grandisin induces apoptosis in leukemic K562 cells. Braz J Pharm Sci. 2017;53(1).
98. Hashemzaei M, Delarami Far A, Yari A, et al. Anticancer and apoptosis-inducing effects of quercetin in vitro and in vivo. Oncol Rep. 2017;38(2):819–828. doi:10.3892/or.2017.5766
99. Martignoni M. Species and Strain Differences in Drug Metabolism in Liver and Intestine. s.n.; 2006:136.
100. Sakuma T, Kawasaki Y, Jarukamjorn K, Nemoto N. Sex differences of drug-metabolizing enzyme: female predominant expression of human and mouse cytochrome P450 3A isoforms. J Drug Metab Toxicol. 2012;3(3):325–337.
101. Mugford CA, Kedderis GL. Sex-dependent metabolism of xenobiotics. Drug Metab Rev. 1998;30(3):441–498. doi:10.3109/03602539808996322
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.