")
Back to Journals » Drug Design, Development and Therapy » Volume 14
Melatonin Attenuates Anoxia/Reoxygenation Injury by Inhibiting Excessive Mitophagy Through the MT2/SIRT3/FoxO3a Signaling Pathway in H9c2 Cells
Authors Wu J, Yang Y, Gao Y, Wang Z, Ma J
Received 13 February 2020
Accepted for publication 24 April 2020
Published 25 May 2020 Volume 2020:14 Pages 2047—2060
DOI https://doi.org/10.2147/DDDT.S248628
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Anastasios Lymperopoulos
Jinjing Wu,* Yanli Yang,* Yafen Gao, Zhaoqi Wang, Jun Ma
Department of Anesthesiology, Beijing Anzhen Hospital, Capital Medical University-Beijing Institute of Heart Lung and Blood Vessel Diseases, Beijing, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Jun Ma
Department of Anesthesiology, Beijing Anzhen Hospital, Capital Medical University-Beijing Institute of Heart Lung and Blood Vessel Diseases, No. 2 Anzhen Road, Chaoyang District, Beijing 100029, People’s Republic of China
Tel +8613370103571
Email [email protected]
Purpose: Autophagy caused by ischemia/reperfusion (I/R) increases the extent of cardiomyocyte damage. Melatonin (Mel) diminishes cardiac injury through regulating autophagy and mitochondrial dynamics. However, illustrating the specific role of mitophagy in the cardioprotective effects of melatonin remains a challenge. The aim of our research was to investigate the impact and underlying mechanisms of melatonin in connection with mitophagy during anoxia/reoxygenation (A/R) injury in H9c2 cells.
Methods: H9c2 cells were pretreated with melatonin with or without the melatonin membrane receptor 2 (MT2) antagonist 4-P-PDOT, the MT2 agonist IIK7 and the sirtuin 3 (SIRT3) inhibitor 3-TYP for 4 hours and then subjected to A/R injury. Cell viability, cellular apoptosis, necrosis levels and oxidative markers were assessed. The expression of SIRT3 and forkhead box O3a (FoxO3a), mitochondrial function and the levels of mitophagy-related proteins were also evaluated.
Results: A/R injury provoked enhanced mitophagy in H9c2 myocytes. In addition, increased mitophagy was correlated with decreased cellular viability, increased oxidative stress and mitochondrial dysfunction in H9c2 cells. However, melatonin pretreatment notably increased cell survival and decreased cell apoptosis and oxidative response after A/R injury, accompanied by restored mitochondrial function. The inhibition of excessive mitophagy is involved in the cardioprotective effects of melatonin, as shown by the decreased expression of the mitophagy-related molecules Parkin, Beclin1, and BCL2-interacting protein 3-like (BNIP3L, best known as NIX) and decreased light chain 3 II/light chain 3 I (LC3 II/LC3 I) ratio and upregulation of p62 expression. Moreover, the decreased expression of SIRT3 and FoxO3a in A/R-injured H9c2 cells was abrogated by melatonin, but these beneficial effects were attenuated by the MT2 antagonist 4-P-PDOT or the SIRT3 inhibitor 3-TYP and enhanced by the MT2 agonist IIK7.
Conclusion: These results indicate that melatonin protects H9c2 cells during A/R injury through suppressing excessive mitophagy by activating the MT2/SIRT3/FoxO3a pathway. Melatonin may be a useful candidate for alleviating myocardial ischemia/reperfusion (MI/R) injury in the future, and the MT2 receptor might become a therapeutic target.
Keywords: melatonin, mitophagy, anoxia/reoxygenation injury, melatonin receptor, SIRT3
Introduction
Acute myocardial infarction (AMI) is a major cause of sudden death worldwide.1 Early reperfusion is the typical therapy for AMI, as it can efficiently reestablish blood flow in ischemic myocardial tissue. However, reperfusion also increases mortality in AMI patients by accelerating and extending cardiac tissue injury, which is known as MI/R injury;2 this process involves excess oxidative products, disturbed mitochondrial dynamics and excessive autophagy.3 Therefore, novel interventional targets and adjuvant therapies to abate reperfusion injury in the heart are urgently required.
Mitochondria are essential organelles that altered in response to metabolic stress.4 Mitophagy, a selective type of autophagy for the specific degradation of impaired mitochondria, is pivotal to maintain mitochondrial function and prevent cell death.5 Damaged mitophagy can result in the accumulation of defective mitochondria and harmful reactive oxygen species (ROS), which can pass across the plasma membrane.6 There is convincing evidence for the crucial roles of mitophagy in the pathogenesis of a number of chronic conditions, including atherosclerosis;7 cancer;8,9 neurodegenerative conditions;10 and cardiovascular,11,12 cerebral13 and liver diseases.14 The heart, which requires a large amount of dynamic energy to maintain normal contractile function, is enriched with mitochondria.15 Mitophagy ensures the regular function of mitochondria in sustaining cardiomyocytes and plays a dual role in the progression of MI/R via diverse signaling pathways.16,17 Thus, it is necessary to explore the specific role of mitophagy in cardiomyocytes, which may further provide theoretical and practical evidence to effectively reduce MI/R injury.
Melatonin, an endogenous circadian indoleamine, is predominantly generated in the pineal gland.18 Several studies have suggested that melatonin can confer significant protection in heart diseases, such as MI/R injury, heart failure, hypertension and atherosclerosis.19 Because of its amphiphilic nature and relatively small size, melatonin can cross all cell membranes and accumulate in subcellular compartments, particularly mitochondria.20 Hence, melatonin is highly concentrated in mitochondria, where it plays a crucial role in mitochondrial processes such as mitophagy. Previous studies with distinct experimental designs in different cell lines have indicated that melatonin regulates mitophagy by enhancing or suppressing mitophagic activity.21–24 The specific mechanisms of this regulation still require deeper exploration. In addition, it has been shown that melatonin membrane receptors are exclusively associated with the myocardial protective effects of melatonin.25,26 However, the specific melatonin membrane receptor that controls the cardioprotective effect of melatonin and the principal mechanisms of this effect remain unclear. Therefore, investigating the role of melatonin membrane receptors in melatonin-induced cardioprotection is constructive for the clinical application of melatonin.
SIRT3, a class III histone deacetylase in mitochondria, modulates the mitochondrial network primarily by regulating lysine acetylation. SIRT3 has been suggested to be an underlying regulator of the beneficial effects of melatonin.27–30 However, the potential interaction between melatonin and SIRT3 has not been completely elucidated. Recent studies have demonstrated that SIRT3 can promote or restrain autophagy to alleviate MI/R injury by stimulating specific downstream targets, including FoxO3a, adenosine monophosphate (AMP)-activated protein kinase (AMPK) and ROS.31 In addition, the relationship between SIRT3 and mitophagy has been previously reported, and the depletion of SIRT3 strongly exacerbated mitophagy inhibition; thus, aged hearts are more inclined to cardiac disorders.32 Moreover, SIRT3 could also exert cardioprotection against diabetic cardiomyopathy (DCM) through initiating Parkin-dependent mitophagy33 and exacerbating mitophagy by the voltage-dependent anion channel 1 (VDAC1)-Parkin interaction.34 Remarkably, SIRT3 has also been demonstrated to interact with FoxO3a and regulate its mitochondrial activity.35
Thus, the present study aimed to inspect whether the cardioprotective effects of melatonin are linked to its capability to suppress excessive mitophagy and to determine the potential mechanism. We hypothesized that melatonin plays a vital role in myocardial protection in A/R-treated H9c2 cells through mitophagy inhibition. To verify this assumption, we developed an A/R model in H9c2 myocytes to simulate MI/R in vivo and pretreated H9c2 cells with melatonin for 4 hours prior to A/R injury. Subsequently, the impacts of melatonin on cellular injury, oxidative stress, apoptosis, mitochondrial function and mitophagy were measured. Furthermore, we investigated whether melatonin could protect H9c2 cells from A/R-induced excessive mitophagy by activating SIRT3/FoxO3a signaling in an MT2-dependent manner.
Materials and Methods
Cell Culture and Treatment
The H9c2 cell line, which was provided by the Shanghai Cell Library of China, was cultivated in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS) (Gibco, Grand Island, NY, USA) and 1% (v/v) penicillin/streptomycin at 37°C in 5% CO2. A/R treatment was administered as follows: after serum starvation, H9c2 cells were first subjected to hypoxic buffer containing (in mM) 12 KCl, 0.9 CaCl2, 137 NaCl, 0.49 MgCl2, 10 deoxyglucose, 0.75 sodium dithionate, 4 HEPES, and 20 lactate (pH 6.5) for 3 hours in a humidified cell culture incubator (95% N2, 5% CO2, 37°C). The hypoxic buffer was pre-gassed and saturated with 95% N2 and 5% CO2 for 30 min in advance.36–38 Subsequently, reoxygenation was performed by resuspending the cells in regular culture medium and incubating them for 4 hours in a humidified cell culture incubator (95% air, 5% CO2, 37°C). To select the appropriate concentration of melatonin, H9c2 myocytes were pretreated with melatonin at a range of concentrations (50, 100, 150 and 200 μM) for 4 hours in normal and A/R-injured H9c2 cells.26,30,39 Cell viability assays suggested that the pro-survival effect of melatonin was most obvious with a concentration of 150 μM. Consequently, a dose of 150 μM was chosen for the subsequent experiments.
Then, H9c2 myocytes were randomly distributed into six groups as follows: the control group, the cells in which were pretreated using serum-free DMEM; the A/R group, the cells in which were exposed to A/R treatment as mentioned above; the A/R + Mel group, the cells in which were pretreated with melatonin (150 μM) for 4 hours and then exposed to A/R damage; the A/R + Mel + 4-P-PDOT group, the cells in which were treated with melatonin (150 μM) and 4-P-PDOT (10 μM) for 4 hours prior to A/R treatment; the A/R + Mel + IIK7 group, the cells in which were treated with melatonin (150 μM) and IIK7 (10 μM) for 4 hours prior to A/R treatment; and the A/R + Mel + 3-TYP group, the cells in which were treated with melatonin (150 μM) and 3-TYP (5 μM) for 4 hours before A/R treatment. The doses of 4P-PDOT, IIK7 and 3-TYP were chosen based on recent studies.24,40,41
Determination of Cell Viability
Cell viability was evaluated using methyl thiazolyl tetrazolium (MTT) assay. Briefly, after the treated cells were washed with phosphate buffer saline (PBS), a mixture of 120 μL of cell culture medium and 20 μL of MTT solution was added to the cells, which were cultured for 4 hours at 37°C. The medium was then discarded, and 150 μL of dimethyl sulfoxide (DMSO) was added to the medium to dissolve the formazan crystals. The absorbance at a wavelength of 570 nm was measured using an ELX-800 microplate reader (BioTek, Winooski, VT, USA). Cell viability was calculated by dividing the optical density of the samples by the optical density of the control group.
Detection of Cell Apoptosis
Cell apoptosis was quantified using an Annexin V-fluorescein isothiocyanate (FITC) and propidium iodide (PI) apoptosis assay kit (Wanleibio, Shenyang, China) by flow cytometry. In brief, H9c2 myocytes were collected after treatment, washed twice with PBS, resuspended and cultured in 5 μL of Annexin V‐FITC and 10 μL of PI for 15 min in the dark. Samples were assessed using flow cytometer (ACEA Bio, San Diego, CA, USA). The results are expressed as the calculated apoptotic index.
Detection of Intracellular Ca2+, Lactate Dehydrogenase (LDH) and Creatine Kinase-MB (CK-MB) Levels
Cellular calcium was examined using the calcium-dependent fluorescent dye Fluo-3 acetoxymethyl ester (AM) (Solarbio, Beijing, China) in accordance with the manufacturer’s protocol, and cellular calcium was subsequently analyzed under a confocal microscope (Olympus FV1000S-SIM/IX81, Tokyo, Japan). The excitation and emission wavelengths were 488 nm and 526 nm, respectively. The average fluorescence intensity of Fluo-3 AM is regarded as an indicator of the cellular calcium concentration. LDH and CK-MB levels were spectrophotometrically (Yoke, Shanghai, China) quantified using an LDH assay kit (Wanleibio, Shenyang, China) and a CK-MB assay kit (USCN, Wuhan, China) in accordance with the manufacturers’ protocols.
Determination of ROS, Malonaldehyde (MDA), Superoxide Dismutase (SOD) and Glutathione Peroxidase (GSH-Px) Levels
The levels of ROS, MDA, SOD and GSH-Px in the incubated cells were measured utilizing commercial kits (Wanleibio, Shenyang, China). All assays were performed in accordance with the manufacturers’ instructions. ROS production was quantified using flow cytometry. In brief, treated cells were gathered and rinsed with 1 mL of 2′,7′-dichlorofluorescein diacetate (DCF-DA) (1:1000, FBS-free medium) at 37°C for 40 min, and the cells were then analyzed using a flow cytometer (ACEA Bio, San Diego, CA, USA). The concentrations of MDA, SOD and GSH-Px were determined using a spectrophotometer (Yoke, Shanghai, China) by measuring the absorbance at 532, 550 and 412 nm, respectively.
Measurement of Mitochondrial Permeability Transition Pore (MPTP) Opening and the Mitochondrial Membrane Potential (MMP)
In the MPTP opening assay, cells were collected and stained with calcein-AM/CoCl2 for 15 min at 37°C in the dark. Then, the cells were washed three times with PBS, and the immunofluorescence intensity of calcein-AM was determined by flow cytometry as a reflection of MPTP opening. The MMP was assessed by flow cytometry after 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethyl- imidacarbocyanine iodide (JC-1) (Beyotime, Beijing, China) staining. In brief, treated H9c2 myocytes were rinsed with a JC-1 probe solution for 20 min at 37°C in the dark. The cells were then washed twice with incubation buffer and collected for subsequent flow cytometry analysis. The results are expressed as a relative red/green fluorescence ratio.
Western Blot Analysis
Briefly, the total protein was collected from H9c2 cells and separated on sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) gels. Subsequently, the proteins were electrotransferred to polyvinylidene difluoride membranes (Millipore, Billerica, MA, USA) and incubated overnight (4°C) with primary antibodies (Table 1). After that, the membranes were washed and incubated with appropriate secondary antibodies for 2 hours (37°C). Protein bands were developed using an enhanced chemiluminescence (ECL) reagent (Wanleibio, Shenyang, China), and the band intensity was quantified using Gel-Pro Analyzer Software (Media Cybernetics, Bethesda, MD, USA). β-actin served as an internal loading control. The relative protein level was determined as the ratio of the gray value of the target band to that of the β-actin band.
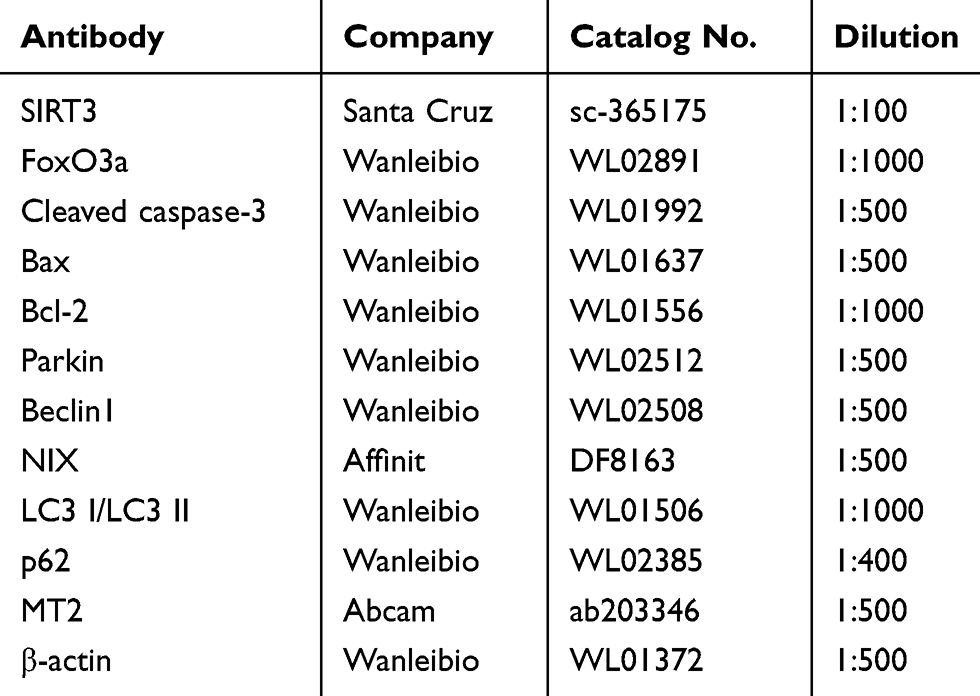 |
Table 1 Primary Antibodies Used in Western Blot |
Confocal Microscopy
H9c2 myocytes were briefly transfected with Ad-GFP-LC3 and Ad-HBmTur-Mito (Hanbio, Shanghai, China) for 6 hours. After the different treatments, yellow puncta in at least 100 cells for each individual experiment were detected with a confocal microscope (Olympus, FV1000S-SIM/IX81, Tokyo, Japan). The Manders’ overlap coefficient was used to assess the extent of colocalization with Image Pro-Plus software.
Coimmunoprecipitation
H9c2 myocytes were treated and lysed in cell lysis buffer. Cell lysates were centrifuged at 12,000 ×g for 10 min at 4°C and prepared for immunoprecipitation. Sample proteins (1 μg/μL) were incubated with 2 μg of primary antibodies targeting MT2 (ab203346, Abcam) or SIRT3 (sc-365175, Santa Cruz) overnight at 4°C, followed by incubation with protein agarose beads (Beyotime, Shanghai, China) for 2 hours at 4°C. The beads were washed three times, and protein complexes were dissolved in 60 μL of 2× SDS sample buffer and probed by SDS‐PAGE.
Statistical Analysis
All the results are shown as the means ± SEM. Statistical analyses were performed with GraphPad Prism 8.0 software (GraphPad Software, Inc., San Diego, CA, USA). Data were evaluated by one-way analysis of variance (ANOVA) followed by Tukey’s post hoc test. A p value < 0.05 indicated a statistically significant difference.
Results
Effects of Melatonin with or Without 4-P-PDOT, IIK7 and 3-TYP on Cell Viability; Apoptosis; and Cellular Ca2+, LDH and CK-MB levels in A/R H9c2 cells
To investigate the effects of melatonin on H9c2 cells after 4 hours of reoxygenation, melatonin at different concentrations (50, 100, 150 and 200 μM) was initially administered to normal and A/R-injured H9c2 cells for 4 hours. Then, cell viability was assessed via MTT assay. As shown in Figure 1A, melatonin alone exerted no substantial effects on the viability of control cells. As shown in Figure 1B, A/R injury triggered a notable reduction in cell viability compared with that in control cells, while melatonin at three concentrations (100, 150 and 200 μM) significantly improved the cell viability of A/R-injured cells; no notable difference between the A/R and A/R+50 μM Mel group was observed. The beneficial effect of melatonin was most evident at a concentration of 150 μM. Therefore, a dose of 150 μM was chosen for subsequent studies. In addition, the effects of 4-P-PDOT, IIK7 and 3-TYP treatments were evaluated in control and A/R-treated cells. As shown in Figure 1C, 4-P-PDOT, IIK7 and 3-TYP treatments exerted no remarkable change in the cell viability of treated cells compared to that of control cells. In A/R-treated cells, 4-P-PDOT and 3-TYP treatment also resulted in no notable difference in cell viability, while IIK7 treatment significantly increased cell viability compared with that in the A/R group, which is in accordance with a previous study.42 Melatonin evidently increased cell viability after A/R injury (Figure 1D). In addition, the apoptotic index was markedly reduced by melatonin pretreatment compared with that in the A/R group (Figure 1E and I). Moreover, cellular Ca2+, LDH, and CK-MB levels were drastically decreased with melatonin treatment compared to those in the A/R group (Figure 1F–H, J). Nevertheless, the beneficial changes due to melatonin observed in the A/R + Mel group were abolished by either 4-P-PDOT or 3-TYP; conversely, these beneficial effects were enhanced by IIK-7. These results provided evidence that melatonin pretreatment alleviated A/R damage by attenuating cell apoptosis and necrosis. MT2 and SIRT3 signaling might participate in this process.
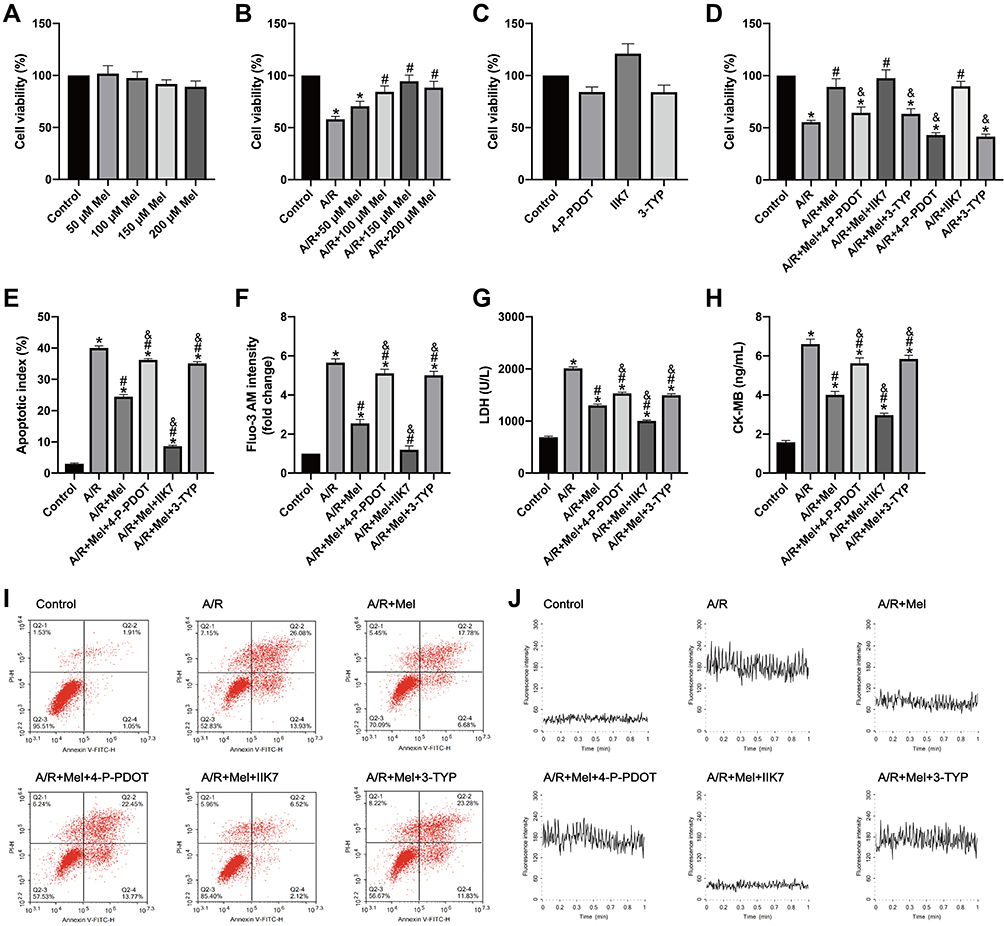 |
Figure 1 Melatonin ameliorated A/R injury in H9c2 cells by increasing cellular viability and reducing the apoptotic index, cellular Ca2+ level, LDH release and CK-MB level, but these effects were attenuated by 4-P-PDOT or 3-TYP and increased by IIK7. (A–D) Cell viability was examined by MTT assay and was calculated by dividing the optical density of samples by that of control group. (E) Apoptotic cells were evaluated by Annexin V/PI staining; the results are expressed as the calculated apoptotic index. (F) The mean fluorescence of Fluo-3 AM-stained cells was assessed using a confocal microscope, and the data were normalized to the control group. (G) LDH levels. (H) CK-MB levels. (I) Representative apoptosis data from flow cytometry. (J) The fluorescence intensity of Fluo-3 AM is representative of the cellular calcium concentration. Data are described as the mean ± SEM (n=6 in each group). *P < 0.05 vs the control group; #P < 0.05 vs the A/R group; &P < 0.05 vs the A/R + Mel group. |
Effects of Melatonin with or Without 4-P-PDOT, IIK7 and 3-TYP on Oxidative Stress, MPTP Opening and MMP in A/R H9c2 Cells
Biomarkers of the level of oxidative stress were further examined. Melatonin treatment effectively reduced cellular ROS generation and MDA levels in H9c2 cells after 4 hours of reperfusion compared to those in the A/R group (Figure 2A and B). Additionally, the decreased activities of SOD and GSH-Px were reversed by melatonin treatment compared to those in the A/R group (Figure 2C and D). Accordingly, these antioxidative effects of melatonin were alleviated by either 4-P-PDOT or 3-TYP but intensified by IIK-7, indicating that melatonin reduced oxidative damage induced by A/R injury. MT2 and SIRT3 might play a role in this process. Moreover, mitochondrial function was also evaluated. MPTP opening was shown to be substantially promoted due to excessive oxidative injury, resulting in MMP repression.43 A/R injury increased MPTP opening and decreased the MMP compared to those in the control group (Figure 2E–H). However, melatonin pretreatment decreased MPTP opening and rescued changes to the MMP. In accordance, these protective effects in the A/R + Mel group were abrogated by either 4-P-PDOT or 3-TYP; in contrast, they were enhanced by IIK-7. Mechanistically, these results suggested that melatonin decreased oxidative stress and MPTP opening and restored the MMP in H9c2 cells, and MT2 and SIRT3 are involved in this process.
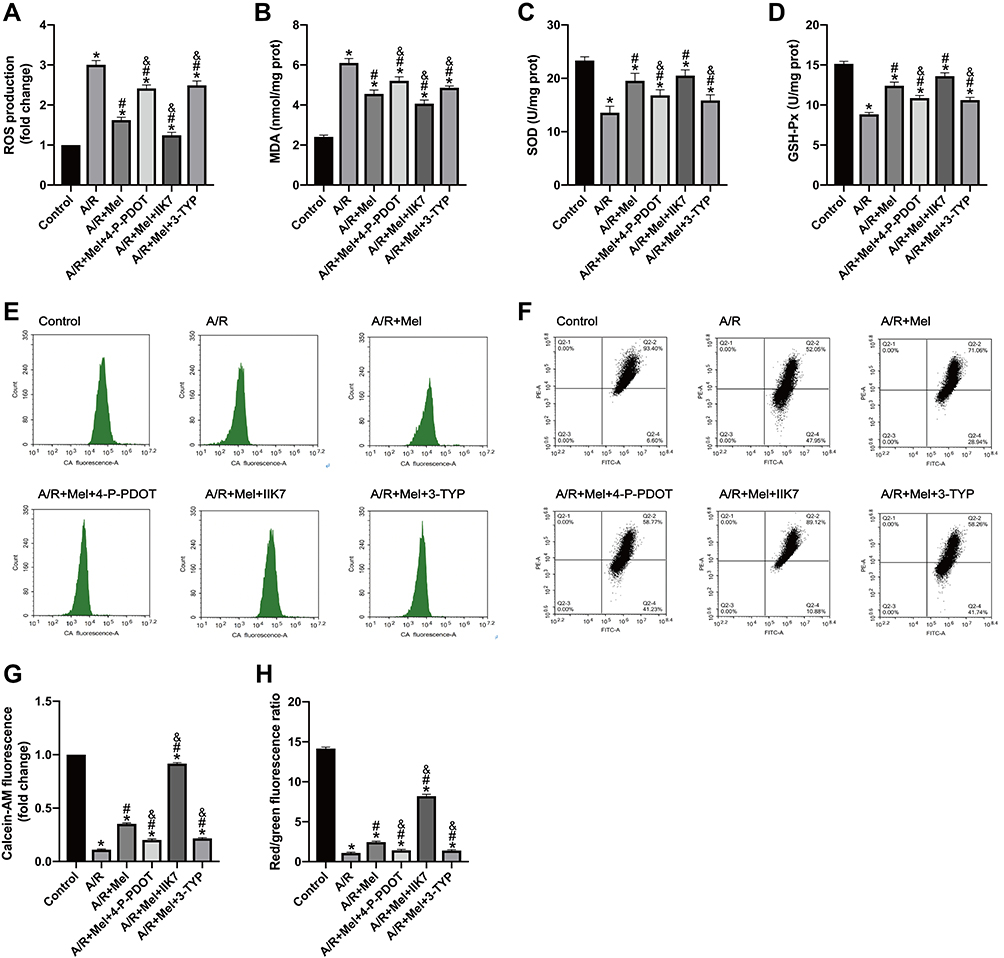 |
Figure 2 Melatonin attenuated A/R injury in H9c2 cells by alleviating myocardial oxidative stress, decreasing MPTP opening and restoring the MMP, but these effects were lessened by 4-P-PDOT or 3-TYP and enhanced by IIK7. (A) ROS production. (B) MDA content. (C) SOD content. (D) GSH-Px level. (E) MPTP opening was determined by calcein-AM flow cytometry assay. (F) The MMP was determined by JC-1 staining. (G) Fold change in calcein-AM fluorescence. (H) The MMP is expressed as the ratio of red/green fluorescence. Data are described as the mean ± SEM (n=6 in each group). *P < 0.05 vs the control group; #P < 0.05 vs the A/R group; &P < 0.05 vs the A/R + Mel group. |
Effects of Melatonin with or Without 4-P-PDOT, IIK7 and 3-TYP on SIRT3 and Apoptotic Signaling in A/R H9c2 Cells
To further explore the protective impact of melatonin on A/R damage in H9c2 cells, SIRT3 and apoptotic signaling were investigated. SIRT3 and FoxO3a expression was significantly reduced in the A/R group compared to that in the control group (Figure 3A and B). However, melatonin counteracted this effect by elevating SIRT3 and FoxO3a expression compared to that in the A/R group, revealing that the positive effects of melatonin might be linked to SIRT3 and FoxO3a. In addition, these protective effects were immediately diminished after the introduction of 4-P-PDOT and the 3-TYP treatment inhibited the FoxO3a expression but not the expression of SIRT3 compared with that in the A/R + Mel group, which is consistent with a previous study.30 In contrast, the protective effects of melatonin were increased by IIK7. In addition, melatonin significantly decreased levels of the apoptosis markers cleaved caspase-3 and Bax in A/R-injured myocytes but upregulated expression of the antiapoptotic protein Bcl-2. Moreover, the presence of 4-P-PDOT and 3-TYP elevated apoptotic signaling by increasing cleaved caspase-3 and Bax expression while decreasing Bcl-2 expression compared with that in the A/R + Mel group (Figure 3C–E), which suggested that melatonin’s protective effects are associated with the MT2 and SIRT3 signaling pathway.
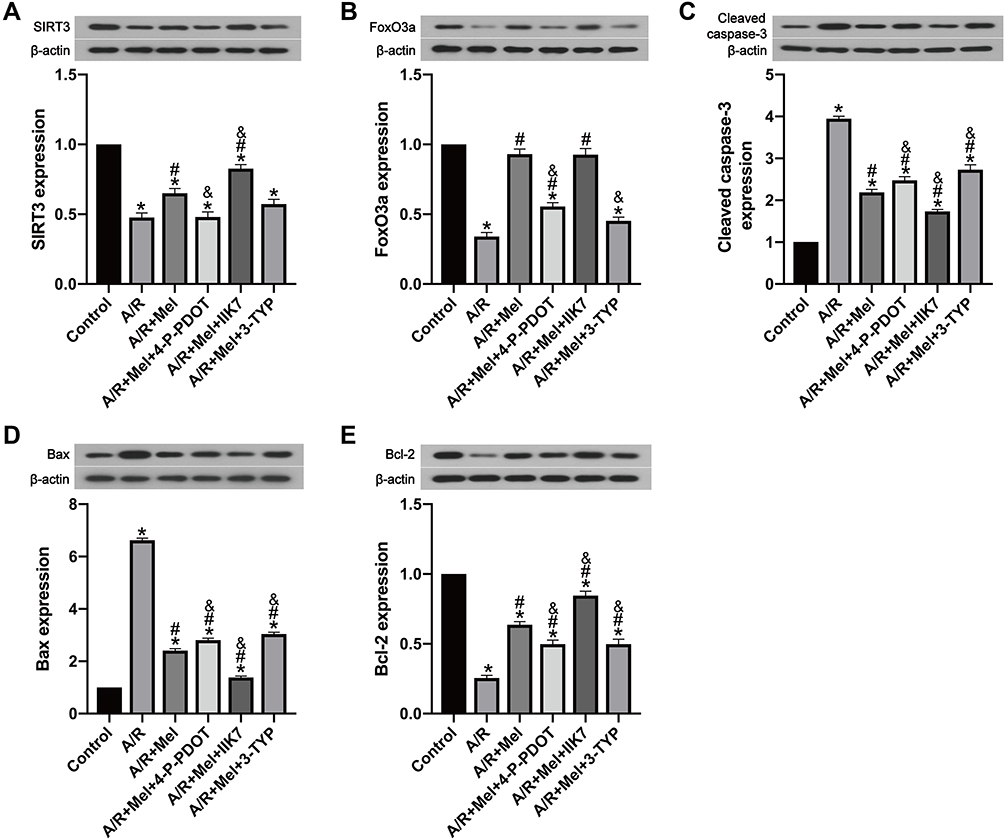 |
Figure 3 Melatonin reduced A/R injury in H9c2 cells by upregulating SIRT3-related signaling and reducing cellular apoptosis, but these effects were suppressed by 4-P-PDOT or 3-TYP and improved by IIK7, while 3-TYP did not decrease the beneficial effect of melatonin towards SIRT3 expression. (A) SIRT3 expression. (B) FoxO3a expression. (C) Cleaved caspase-3 expression. (D) Bax expression. (E) Bcl-2 expression. Data are described as the mean ± SEM (n=6 in each group). *P < 0.05 vs the control group; #P < 0.05 vs the A/R group; &P < 0.05 vs the A/R + Mel group. |
Effects of Melatonin with or Without 4-P-PDOT, IIK7 and 3-TYP on Mitophagy in A/R H9c2 Cells
To reveal whether melatonin restrains mitophagy levels, the expression of molecules involved in mitophagy and autophagy was determined by Western blotting. Parkin, Beclin1 and NIX protein expression in A/R-treated cells was significantly increased, and this increase was clearly attenuated by melatonin (Figure 4A–C). Autophagy index proteins were also investigated after A/R injury, which revealed an elevated LC3 II/LC3 I ratio and decreased p62 level. Melatonin markedly decreased the LC3 II/LC3 I ratio and upregulated the p62 protein level (Figure 4D and E), indicating that melatonin exerted an inhibitory effect on the mitophagic process. These observations were confirmed by measuring the overlap between the GFP-LC3 distribution and MitoTracker-labeled mitochondria via confocal microscopy. Compared with the control cells, A/R-damaged H9c2 cells after melatonin pretreatment displayed a diminished number of autophagic vacuoles engulfing mitochondria (Figure 4F and G). To illustrate the specific role of MT2 and SIRT3 in A/R-induced mitophagy, the specific MT2 antagonist 4-P-PDOT and the SIRT3 inhibitor 3-TYP were employed. 4-P-PDOT suppressed the inhibitory effect of melatonin on mitophagy in H9c2 cells observed in the A/R + Mel group, leading to a significant increase in Parkin, Beclin1 and NIX protein expression. In addition, there was a notable increase in the LC3 II/LC3 I ratio with a decrease in p62 expression compared to those in the A/R + Mel group (Figure 4A–E). The SIRT3 inhibitor 3-TYP had the same effects as shown by an obvious increase in Parkin, Beclin1, and NIX expression and an increased LC3 II/LC3 I ratio, with a decrease in p62 expression. Moreover, the results of microscopic analysis of the degree to which GFP-LC3 colocalized with MitoTracker-labeled mitochondria were consistent with the results of Western blot analysis. Melatonin treatment decreased the degree to which GFP-LC3 and MitoTracker colocalized following A/R; both 4-P-PDOT and 3-TYP abrogated this decrease in GFP-LC3 and MitoTracker colocalization (Figure 4F and G). Overall, the above results suggested the excessive activation of mitophagy in H9c2 cells after A/R treatment and that melatonin can alleviate A/R injury in H9c2 cells by reducing mitophagic activation. Moreover, these inhibitory effects of melatonin on mitophagy might be modulated by MT2 and SIRT3 signaling pathway.
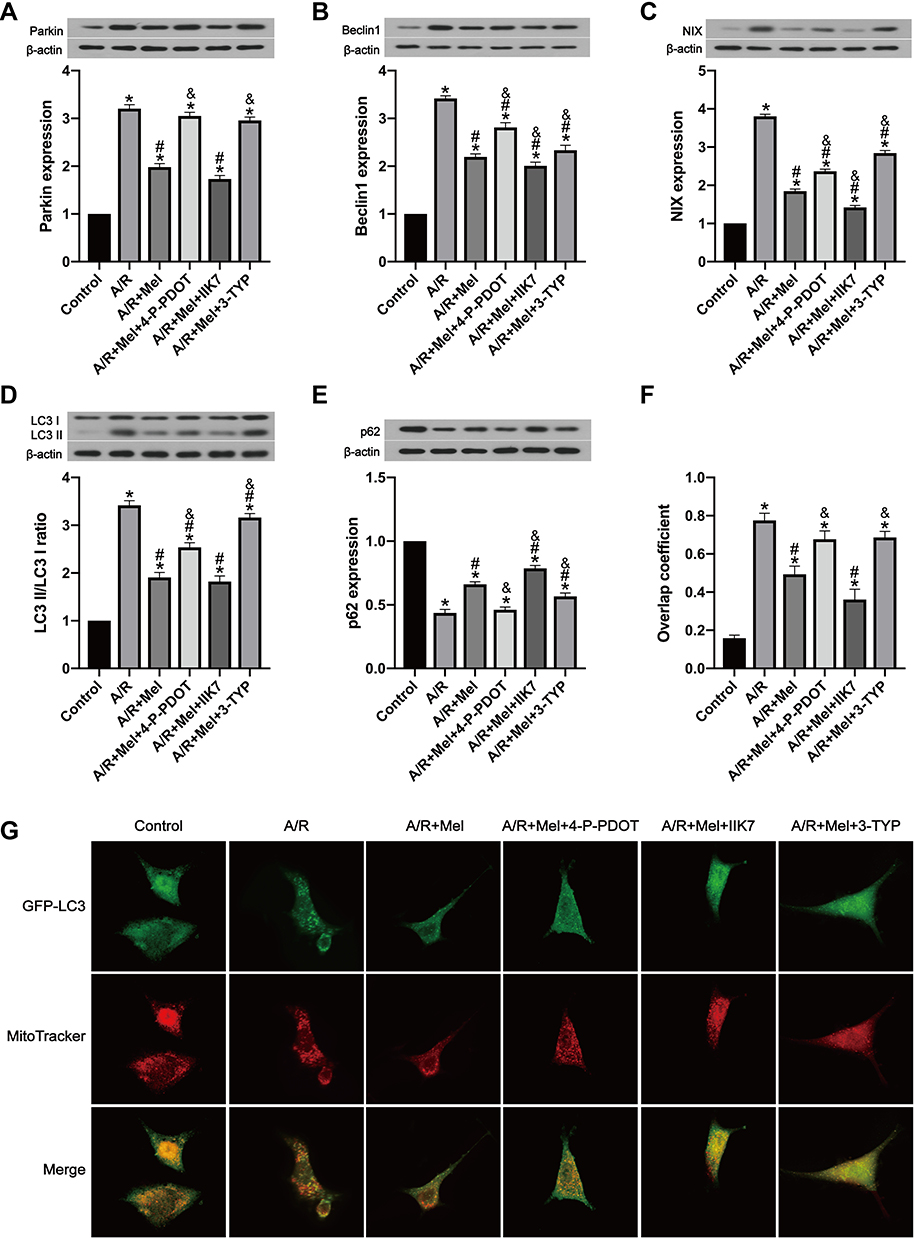 |
Figure 4 Melatonin alleviated A/R injury in H9c2 cells by inhibiting mitophagy, but these effects were inhibited by 4-P-PDOT or 3-TYP and enhanced by IIK7. (A) Parkin expression. (B) Beclin1 expression. (C) NIX expression. (D) LC3 II/LC3 I ratio. (E) p62 expression. (F) Manders’ overlap coefficient for GFP-LC3 and mitochondria. (G) Colocalization of GFP-LC3 and MitoTracker. Fluorescence images were obtained by confocal microscopy. Data are described as the mean ± SEM (n=6 in each group). *P < 0.05 vs the control group; #P < 0.05 vs the A/R group; &P < 0.05 vs the A/R + Mel group. |
Interaction Between SIRT3 and MT2 and Its Impact on the Effects of Melatonin in A/R H9c2 Cells
According to previous research, the SIRT3 signaling pathway plays a critical role in the defensive effects of melatonin against A/R damage,44 but the association between SIRT3 and MT2 is still uncertain. In the present in vitro study, the expression levels of both SIRT3 and MT2 were extensively decreased following A/R. Melatonin treatment restored SIRT3 and MT2 expression after A/R (Figures 3A and 5A). Remarkably, the SIRT3 inhibitor 3-TYP had a significant effect on MT2 expression (Figure 5A), and specific inhibition of the MT2 with 4-P-PDOT also generated a notable decrease in SIRT3 expression (Figure 3A). Moreover, a coimmunoprecipitation assay showed that SIRT3 interacted with the MT2 receptor in both control cells and A/R-injured cells. However, A/R treatment attenuated the interaction between SIRT3 and the MT2 receptor (Figure 5B). Collectively, these data may demonstrate the role of MT2/SIRT3 signaling in the cardioprotective effects of melatonin via the inhibition of mitophagy.
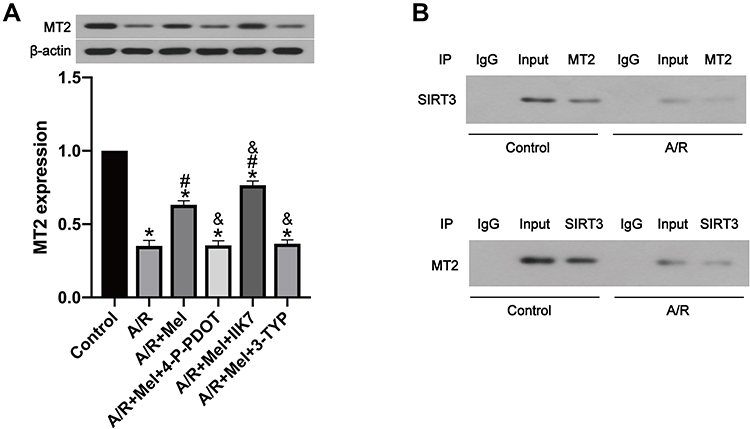 |
Figure 5 The interaction between SIRT3 and MT2 may promote the protective effects of melatonin in A/R H9c2 cells. (A) MT2 expression. (B) A coimmunoprecipitation assay indicated the interaction between SIRT3 and MT2 in control and A/R-injured cells. Data are described as the mean ± SEM (n=6 in each group). *P < 0.05 vs the control group; #P < 0.05 vs the A/R group; &P < 0.05 vs the A/R + Mel group. |
Discussion
In the current study, we revealed that melatonin pretreatment notably alleviated cellular injury, apoptosis and oxidative stress and attenuated mitochondrial dysfunction after A/R-induced injury in vitro. In addition, we demonstrated the underlying mechanism of these effects in the melatonin-induced inhibition of excessive mitophagy. Our results indicated that melatonin significantly suppressed mitophagy by inhibiting expression of the mitophagy-related proteins Parkin, Beclin1, and NIX; decreasing the LC3 II/LC3 I ratio; and consequently protected H9c2 cells from A/R-damaged cellular death. Additional experiments suggested that melatonin pretreatment restored the expression of SIRT3 and FoxO3a and prevented excessive mitophagy in A/R-injured H9c2 cells. However, these effects were notably attenuated by 4-P-PDOT or 3-TYP, revealing that the cardioprotective effect of melatonin due to the inhibition of excessive mitophagy might be mediated, at least in part, by MT2 and SIRT3. In summary, we identified a specific mechanism by which melatonin attenuates A/R injury in H9c2 cells via inhibition of mitophagy through the MT2/SIRT3/FoxO3a signaling pathway. This provides new evidence of melatonin as a prospective drug to alleviate MI/R injury and demonstrates that the combination of melatonin with an MT2 agonist might produce a more promising result.
Mitophagy is essential to homeostasis in both mitochondria and cells. Mitophagy dysfunction might be responsible for the pathogenesis of numerous chronic diseases, including cancer, Parkinson’s disease, and heart and liver disorders.45 The dysregulation of mitophagy leads to decreased cellular resistance to MI/R injury due to energy shortage. Increasing evidence demonstrates that excessive mitophagy and autophagy during the reperfusion period could give rise to irreparable damage in cardiac myocytes and ultimately death.46–48 Our data also indicated that the triggering of excess mitophagy during the reperfusion phase was damaging to H9c2 cells. In this study, the expression of Parkin, Beclin1, and NIX and the LC3 II/LC3 I ratio were significantly increased after A/R treatment, whereas p62 expression was notably decreased, indicating that excessive mitophagy was activated; the activation of excessive mitophagy was also confirmed by increased overlap between the GFP-LC3 distribution and MitoTracker signal measured using confocal microscopy. Furthermore, these changes might be due to an alternative form of mitophagy known as receptor‐mediated mitophagy in which proteins in the outer mitochondrial membrane (OMM) including BNIP3 and NIX, directly bind LC3 through its BH3 domain under hypoxic conditions.49 The results indeed showed excessive mitophagy in myocardial cells exposed to A/R, which was in accordance with the above data. Thus, it is conceivable that inhibiting mitophagy could indirectly benefit cardiomyocytes and be developed into a strategy for MI/R injury mitigation.
Melatonin, the chief hormone in the pineal gland, is highly pleiotropic and regulates a variety of physiological functions in numerous organs through receptor-mediated and receptor-independent mechanisms.50 In the present investigation, we discovered that melatonin pretreatment had cardioprotective effects on an A/R model in vitro, as evidenced by increased cell viability and a decreased oxidative response, cellular apoptosis and necrosis. The role of melatonin during I/R damage was recently discussed, and melatonin was shown to play a dual role by either enhancing or suppressing autophagy during I/R injury in different established models.11,51-53 Moreover, mitophagy, a specific form of autophagy, is also considered to be affected by melatonin in a dual manner. The precise mechanism by which melatonin affects mitophagy is still an enigma. According to recent investigations, melatonin could attenuate MI/R by potentiating mitophagy by initiating different signaling pathways, including the mammalian Ste20-like kinase 1(Mst1) inhibition, AMPK‐optic atrophy 1(OPA1) signaling and uncoupling protein 2 (UCP2) regulatory pathways. In contrast, melatonin could also attenuate I/R injury in the myocardial microvasculature through suppressing the VDAC1-hexokinase 2 (HK2)-mitophagy axis.22,25,54-56 Our results showed that melatonin significantly suppressed mitophagy by decreasing the receptor‐mediated mitophagy proteins Parkin, Beclin1, and NIX and the LC3 II/LC3 I ratio and upregulating p62 expression. These effects of melatonin conferred cardioprotection against A/R treatment, resulting in the improved survival of H9c2 cells. Moreover, melatonin treatment also restored changes to MPTP opening and the MMP in H9c2 myocytes after A/R injury, suggesting its role in defense against mitochondrial dysfunction.
FoxO3a is known to directly modulate the promotion of mitophagy by stimulating the downstream targets Parkin, BNIP3 and sequestosome. Its activity was suggested to be altered by its physical interaction with mitochondrial SIRT3, as SIRT3 elevates the DNA-binding activity of FoxO3a, leading to the upregulation of FoxO3a-dependent genes.35 As a principal member of the mitochondrial sirtuin family, SIRT3 acts as a crucial modulator of cellular metabolic maintenance, the oxidative response and mitophagy in cardiomyocytes. Recent studies demonstrated that SIRT3-FoxO3a signaling could activate mitophagy by promoting the Pink-1/Parkin pathway when I/R occurred.33,57,58 Our data suggested that SIRT3 and FoxO3a were decreased in A/R-injured myocytes, and their expression was accordingly correlated with oxidative damage and mitochondrial dysfunction. Melatonin treatment was also shown to elevate SIRT3 protein level as well as FoxO3a expression in H9c2 cells after A/R injury. By suppressing SIRT3 with the SIRT3 inhibitor 3-TYP, the beneficial effects of melatonin against A/R were demonstrated to involve SIRT3. Moreover, we found that 3-TYP abolished the melatonin-induced upregulation of FoxO3a expression. Consequently, SIRT3 might be a vital upstream molecule that affects the levels of the FoxO3a protein during the mechanism of melatonin-mediated cardioprotection.
Three melatonin membrane receptors have been detected thus far. Melatonin membrane receptor 1 (MT1) and MT2 are expressed in humans, while melatonin membrane receptor 3 (MT3) has been found in hamsters.59 MT1 and MT2 were recently identified in mitochondrial membranes.60 Several studies exploiting the nonspecific melatonin receptor antagonist luzindole have also illustrated that the cardioprotective impact of melatonin is chiefly attributed to melatonin receptors.28,44 However, the specific melatonin membrane receptor that regulates melatonin‐induced cardioprotection remains unclear. It has been reported that MT2 expression was enhanced due to MI/R, and melatonin exerted its cardioprotective activity during ischemic injury through MT2, but not MT1.25 Therefore, this in vitro study was mainly focused on MT2. However, our results showed that the MT2 level was instead decreased after A/R injury, which is consistent with a previous study indicating that traumatic brain injury (TBI) in rats led to the decreased expression of MT1 and MT2.61 This discrepancy may be explained in part by the different stress conditions and cultured cell lines. In addition, melatonin-mediated cardioprotection against A/R damage was considerably suppressed by 4-P-PDOT and conversely enhanced by IIK-7. Taken together, these results suggest that melatonin exerts an important effect by inhibiting mitophagy through an MT2-dependent cardioprotective mechanism.
However, there are some limitations in the present study. First, the MT2 antagonist 4-P-PDOT significantly but not completely abolished the cardioprotective effects of melatonin in A/R-injured cells. As for melatonin treatment, the role of MT1 antagonist should be considered and further explored as well as the combination of IIK7 and 4-P-PDOT. Second, the blockage of FoxO3a, which might be an underlying contributor to the functional work of melatonin, was not examined in the present study. Third, our research was particularly focused on in vitro studies. Further investigations are needed to support the present findings applied to an animal model.
In conclusion, melatonin pretreatment drastically reduced injury after A/R treatment and enhanced the viability of H9c2 cells by inhibiting excessive mitophagy. The protective effects of melatonin against A/R injury were modulated by suppressing excessive mitophagy through regulating the SIRT3/FoxO3a signaling pathway in an MT2-dependent manner. The above results indicated that melatonin might be a candidate for the treatment of MI/R injury in cardiovascular diseases, and the MT2 receptor represents a potentially appealing molecular target as well.
Acknowledgments
This research was supported by grants from the National Natural Science Foundation of China (No. 81871592 and No. 81471902) and Beijing Municipal Administration of Hospitals Clinical Medicine Development of Special Funding Support (ZYLX 201810).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Heusch G, Gersh BJ. The pathophysiology of acute myocardial infarction and strategies of protection beyond reperfusion: a continual challenge. Eur Heart J. 2017;38(11):774–784. doi:10.1093/eurheartj/ehw224
2. Hausenloy DJ, Yellon DM. Targeting myocardial reperfusion injury-the search continues. N Engl J Med. 2015;373(11):1073–1075. doi:10.1056/NEJMe1509718
3. Sadoshima J. The role of autophagy during ischemia/reperfusion. Autophagy. 2008;4(4):402–403. doi:10.4161/auto.5924
4. Han D, Ybanez MD, Johnson HS, et al. Dynamic adaptation of liver mitochondria to chronic alcohol feeding in mice: biogenesis, remodeling, and functional alterations. J Biol Chem. 2012;287(50):42165–42179. doi:10.1074/jbc.M112.377374
5. Williams JA, Ding WX. Mechanisms, pathophysiological roles and methods for analyzing mitophagy-recent insights. Biol Chem. 2018;399(2):147–178. doi:10.1515/hsz-2017-0228
6. Bienert GP, Moller AL, Kristiansen KA, et al. Specific aquaporins facilitate the diffusion of hydrogen peroxide across membranes. J Biol Chem. 2007;282(2):1183–1192. doi:10.1074/jbc.M603761200
7. Ma S, Chen J, Feng J, et al. Melatonin ameliorates the progression of atherosclerosis via mitophagy activation and NLRP3 inflammasome inhibition. Oxid Med Cell Longev. 2018;2018:9286458. doi:10.1155/2018/9286458
8. Shen YQ, Guerra-Librero A, Fernandez-Gil BI, et al. Combination of melatonin and rapamycin for head and neck cancer therapy: suppression of AKT/mTOR pathway activation, and activation of mitophagy and apoptosis via mitochondrial function regulation. J Pineal Res. 2018;64:3. doi:10.1111/jpi.12461
9. Fernandez-Gil BI, Guerra-Librero A, Shen YQ, et al. Melatonin enhances cisplatin and radiation cytotoxicity in head and neck squamous cell carcinoma by stimulating mitochondrial ROS generation, apoptosis, and autophagy. Oxid Med Cell Longev. 2019;2019:7187128. doi:10.1155/2019/7187128
10. Phillipson OT. Alpha-synuclein, epigenetics, mitochondria, metabolism, calcium traffic, & circadian dysfunction in Parkinson’s disease. An integrated strategy for management. Ageing Res Rev. 2017;40:149–167. doi:10.1016/j.arr.2017.09.006
11. Zhang Y, Wang Y, Xu J, et al. Melatonin attenuates myocardial ischemia-reperfusion injury via improving mitochondrial fusion/mitophagy and activating the AMPK-OPA1 signaling pathways. J Pineal Res. 2019;66(2):e12542. doi:10.1111/jpi.12542
12. Zhong J, Tan Y, Lu J, et al. Therapeutic contribution of melatonin to the treatment of septic cardiomyopathy: a novel mechanism linking Ripk3-modified mitochondrial performance and endoplasmic reticulum function. Redox Biol. 2019;26:101287. doi:10.1016/j.redox.2019.101287
13. Liu Y, Yan J, Sun C, et al. Ameliorating mitochondrial dysfunction restores carbon ion-induced cognitive deficits via co-activation of NRF2 and PINK1 signaling pathway. Redox Biol. 2018;17:143–157. doi:10.1016/j.redox.2018.04.012
14. Zhou H, Du W, Li Y, et al. Effects of melatonin on fatty liver disease: the role of NR4A1/DNA-PKcs/p53 pathway, mitochondrial fission, and mitophagy. J Pineal Res. 2018;64:1. doi:10.1111/jpi.12450
15. Dorn GW
16. Li YZ, Wu XD, Liu XH, Li PF. Mitophagy imbalance in cardiomyocyte ischaemia/reperfusion injury. Acta Physiol (Oxf). 2019;225(4):e13228. doi:10.1111/apha.13228
17. Cao S, Sun Y, Wang W, et al. Poly (ADP-ribose) polymerase inhibition protects against myocardial ischaemia/reperfusion injury via suppressing mitophagy. J Cell Mol Med. 2019;23(10):6897–6906. doi:10.1111/jcmm.14573
18. Brzezinski A. Melatonin in humans. N Engl J Med. 1997;336(3):186–195. doi:10.1056/NEJM199701163360306
19. Pandi-Perumal SR, BaHammam AS, Ojike NI, et al. Melatonin and human cardiovascular disease. J Cardiovasc Pharmacol Ther. 2017;22(2):122–132. doi:10.1177/1074248416660622
20. Boga JA, Caballero B, Potes Y, et al. Therapeutic potential of melatonin related to its role as an autophagy regulator: a review. J Pineal Res. 2019;66(1):e12534. doi:10.1111/jpi.12534
21. Lee WJ, Chen LC, Lin JH, et al. Melatonin promotes neuroblastoma cell differentiation by activating hyaluronan synthase 3-induced mitophagy. Cancer Med. 2019;8(10):4821–4835. doi:10.1002/cam4.2389
22. Zhou H, Zhang Y, Hu S, et al. Melatonin protects cardiac microvasculature against ischemia/reperfusion injury via suppression of mitochondrial fission-VDAC1-HK2-mPTP-mitophagy axis. J Pineal Res. 2017;63:1. doi:10.1111/jpi.12413
23. Chen Y, Wu Y, Shi H, et al. Melatonin ameliorates intervertebral disc degeneration via the potential mechanisms of mitophagy induction and apoptosis inhibition. J Cell Mol Med. 2019;23(3):2136–2148. doi:10.1111/jcmm.14125
24. Onphachanh X, Lee HJ, Lim JR, et al. Enhancement of high glucose-induced PINK1 expression by melatonin stimulates neuronal cell survival: involvement of MT2/Akt/NF-kappaB pathway. J Pineal Res. 2017;63:2. doi:10.1111/jpi.12427
25. Han D, Wang Y, Chen J, et al. Activation of melatonin receptor 2 but not melatonin receptor 1 mediates melatonin-conferred cardioprotection against myocardial ischemia/reperfusion injury. J Pineal Res. 2019;67(1):e12571. doi:10.1111/jpi.12571
26. Yu L, Liang H, Lu Z, et al. Membrane receptor-dependent Notch1/Hes1 activation by melatonin protects against myocardial ischemia-reperfusion injury: in vivo and in vitro studies. J Pineal Res. 2015;59(4):420–433. doi:10.1111/jpi.12272
27. Reiter RJ, Tan DX, Rosales-Corral S, Galano A, Jou MJ, Acuna-Castroviejo D. Melatonin mitigates mitochondrial meltdown: interactions with SIRT3. Int J Mol Sci. 2018;19:8. doi:10.3390/ijms19082439
28. Pi H, Xu S, Reiter RJ, et al. SIRT3-SOD2-mROS-dependent autophagy in cadmium-induced hepatotoxicity and salvage by melatonin. Autophagy. 2015;11(7):1037–1051. doi:10.1080/15548627.2015.1052208
29. Mayo JC, Sainz RM, Gonzalez Menendez P, Cepas V, Tan DX, Reiter RJ. Melatonin and sirtuins: a “not-so unexpected” relationship. J Pineal Res. 2017;62:2. doi:10.1111/jpi.12391
30. Zhai M, Li B, Duan W, et al. Melatonin ameliorates myocardial ischemia reperfusion injury through SIRT3-dependent regulation of oxidative stress and apoptosis. J Pineal Res. 2017;63:2. doi:10.1111/jpi.12419
31. Zheng Y, Shi B, Ma M, Wu X, Lin X. The novel relationship between Sirt3 and autophagy in myocardial ischemia-reperfusion. J Cell Physiol. 2019;234(5):5488–5495. doi:10.1002/jcp.27329
32. Li Y, Ma Y, Song L, et al. SIRT3 deficiency exacerbates p53/Parkinmediated mitophagy inhibition and promotes mitochondrial dysfunction: implication for aged hearts. Int J Mol Med. 2018;41(6):3517–3526. doi:10.3892/ijmm.2018.3555
33. Yu W, Gao B, Li N, et al. Sirt3 deficiency exacerbates diabetic cardiac dysfunction: role of Foxo3A-Parkin-mediated mitophagy. Biochim Biophys Acta Mol Basis Dis. 2017;1863(8):1973–1983. doi:10.1016/j.bbadis.2016.10.021
34. Qiao A, Wang K, Yuan Y, et al. Sirt3-mediated mitophagy protects tumor cells against apoptosis under hypoxia. Oncotarget. 2016;7(28):43390–43400. doi:10.18632/oncotarget.9717
35. Jacobs KM, Pennington JD, Bisht KS, et al. SIRT3 interacts with the daf-16 homolog FOXO3a in the mitochondria, as well as increases FOXO3a dependent gene expression. Int J Biol Sci. 2008;4(5):291–299. doi:10.7150/ijbs.4.291
36. Wang Z, Lin D, Zhang L, Liu W, Tan H, Ma J. Penehyclidine hydrochloride prevents anoxia/reoxygenation injury and induces H9c2 cardiomyocyte apoptosis via a mitochondrial pathway. Eur J Pharmacol. 2017;797:115–123. doi:10.1016/j.ejphar.2017.01.012
37. Lin D, Cui B, Ren J, Ma J. Regulation of VDAC1 contributes to the cardioprotective effects of penehyclidine hydrochloride during myocardial ischemia/reperfusion. Exp Cell Res. 2018;367(2):257–263. doi:10.1016/j.yexcr.2018.04.004
38. Lin D, Cui B, Ma J, Ren J. MiR-183-5p protects rat hearts against myocardial ischemia/reperfusion injury through targeting VDAC1. Biofactors. 2020;46(1):83–93. doi:10.1002/biof.1571
39. Yu L, Li B, Zhang M, et al. Melatonin reduces PERK-eIF2alpha-ATF4-mediated endoplasmic reticulum stress during myocardial ischemia-reperfusion injury: role of RISK and SAFE pathways interaction. Apoptosis. 2016;21(7):809–824. doi:10.1007/s10495-016-1246-1
40. He YM, Deng HH, Shi MH, et al. Melatonin modulates the functions of porcine granulosa cells via its membrane receptor MT2 in vitro. Anim Reprod Sci. 2016;172:164–172. doi:10.1016/j.anireprosci.2016.07.015
41. Wang C, Yang Y, Zhang Y, Liu J, Yao Z, Zhang C. Protective effects of metformin against osteoarthritis through upregulation of SIRT3-mediated PINK1/Parkin-dependent mitophagy in primary chondrocytes. Biosci Trends. 2019;12(6):605–612. doi:10.5582/bst.2018.01263
42. Yu J, Wei J, Ji L, Hong X. Exploration on mechanism of a new type of melatonin receptor agonist Neu-p11 in hypoxia-reoxygenation injury of myocardial cells. Cell Biochem Biophys. 2014;70(2):999–1003. doi:10.1007/s12013-014-0009-2
43. Zhao Q, Wang W, Cui J. Melatonin enhances TNF-alpha-mediated cervical cancer HeLa cells death via suppressing CaMKII/Parkin/mitophagy axis. Cancer Cell Int. 2019;19:58. doi:10.1186/s12935-019-0777-2
44. Yu L, Sun Y, Cheng L, et al. Melatonin receptor-mediated protection against myocardial ischemia/reperfusion injury: role of SIRT1. J Pineal Res. 2014;57(2):228–238. doi:10.1111/jpi.12161
45. Redmann M, Dodson M, Boyer-Guittaut M, Darley-Usmar V, Zhang J. Mitophagy mechanisms and role in human diseases. Int J Biochem Cell Biol. 2014;53:127–133. doi:10.1016/j.biocel.2014.05.010
46. Huang Z, Han Z, Ye B, et al. Berberine alleviates cardiac ischemia/reperfusion injury by inhibiting excessive autophagy in cardiomyocytes. Eur J Pharmacol. 2015;762:1–10. doi:10.1016/j.ejphar.2015.05.028
47. Shi X, Zhu H, Zhang Y, Zhou M, Tang D, Zhang H. XuefuZhuyu decoction protected cardiomyocytes against hypoxia/reoxygenation injury by inhibiting autophagy. BMC Complement Altern Med. 2017;17(1):325. doi:10.1186/s12906-017-1822-0
48. Wang Y, Wang Q, Zhang L, et al. Coptisine protects cardiomyocyte against hypoxia/reoxygenation-induced damage via inhibition of autophagy. Biochem Biophys Res Commun. 2017;490(2):231–238. doi:10.1016/j.bbrc.2017.06.027
49. Yoo SM, Jung YK. A molecular approach to mitophagy and mitochondrial dynamics. Mol Cells. 2018;41(1):18–26. doi:10.14348/molcells.2018.2277
50. Reiter RJ, Tan DX, Manchester LC, Pilar Terron M, Flores LJ, Koppisepi S. Medical implications of melatonin: receptor-mediated and receptor-independent actions. Adv Med Sci. 2007;52:11–28.
51. Hu Y, Wang Z, Liu Y, et al. Melatonin reduces hypoxic-ischaemic (HI) induced autophagy and apoptosis: an in vivo and in vitro investigation in experimental models of neonatal HI brain injury. Neurosci Lett. 2017;653:105–112. doi:10.1016/j.neulet.2016.11.050
52. Kang JW, Cho HI, Lee SM. Melatonin inhibits mTOR-dependent autophagy during liver ischemia/reperfusion. Cell Physiol Biochem. 2014;33(1):23–36. doi:10.1159/000356647
53. Chen WR, Liu HB, Chen YD, et al. Melatonin attenuates myocardial ischemia/reperfusion injury by inhibiting autophagy via an AMPK/mTOR signaling pathway. Cell Physiol Biochem. 2018;47(5):2067–2076. doi:10.1159/000491474
54. Zhou H, Li D, Zhu P, et al. Melatonin suppresses platelet activation and function against cardiac ischemia/reperfusion injury via PPARgamma/FUNDC1/mitophagy pathways. J Pineal Res. 2017;63:4. doi:10.1111/jpi.12438
55. Wang S, Zhao Z, Feng X, et al. Melatonin activates Parkin translocation and rescues the impaired mitophagy activity of diabetic cardiomyopathy through Mst1 inhibition. J Cell Mol Med. 2018;22(10):5132–5144. doi:10.1111/jcmm.13802
56. Pan P, Zhang H, Su L, Wang X, Liu D. Melatonin balance the autophagy and apoptosis by regulating UCP2 in the LPS-induced cardiomyopathy. Molecules. 2018;23:3. doi:10.3390/molecules23030675
57. Das S, Mitrovsky G, Vasanthi HR, Das DK. Antiaging properties of a grape-derived antioxidant are regulated by mitochondrial balance of fusion and fission leading to mitophagy triggered by a signaling network of Sirt1-Sirt3-Foxo3-PINK1-PARKIN. Oxid Med Cell Longev. 2014;2014:345105. doi:10.1155/2014/345105
58. Zhang M, Zhao Z, Shen M, et al. Polydatin protects cardiomyocytes against myocardial infarction injury by activating Sirt3. Biochim Biophys Acta Mol Basis Dis. 2017;1863(8):1962–1972. doi:10.1016/j.bbadis.2016.09.003
59. Slominski RM, Reiter RJ, Schlabritz-Loutsevitch N, Ostrom RS, Slominski AT. Melatonin membrane receptors in peripheral tissues: distribution and functions. Mol Cell Endocrinol. 2012;351(2):152–166. doi:10.1016/j.mce.2012.01.004
60. Ahluwalia A, Brzozowska IM, Hoa N, Jones MK, Tarnawski AS. Melatonin signaling in mitochondria extends beyond neurons and neuroprotection: implications for angiogenesis and cardio/gastroprotection. Proc Natl Acad Sci U S A. 2018;115(9):E1942–E1943. (). doi:10.1073/pnas.1722131115
61. Osier ND, Pham L, Pugh BJ, et al. Brain injury results in lower levels of melatonin receptors subtypes MT1 and MT2. Neurosci Lett. 2017;650:18–24. doi:10.1016/j.neulet.2017.03.053
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.