")
Back to Journals » Drug Design, Development and Therapy » Volume 12
MEK inhibitors for the treatment of NRAS mutant melanoma
Authors Sarkisian S, Davar D
Received 3 April 2018
Accepted for publication 15 June 2018
Published 20 August 2018 Volume 2018:12 Pages 2553—2565
DOI https://doi.org/10.2147/DDDT.S131721
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Anastasios Lymperopoulos
Saro Sarkisian,1 Diwakar Davar2
1Division of General Internal Medicine, University of Pittsburgh, Pittsburgh, PA, USA; 2Division of Hematology-Oncology, Hillman Cancer Center and University of Pittsburgh, Pittsburgh, PA, USA
Abstract: Melanoma is increasing rapidly in incidence and prevalence, especially in younger females and older males. Treatment options have expanded beyond high-dose interleukin 2 and adoptive T-cell therapy to include inhibitors of immune checkpoints programmed death 1 (PD-1) and cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) and small molecular inhibitors of pathways activated in melanoma, in particular the mitogen-activated protein kinase (MAPK) pathway. PD-1/CTLA-4 inhibitors and inhibitors of MAPK such as BRAF/MEK inhibitors have significantly improved survival in both the metastatic and, more recently, adjuvant settings. In this review, we discuss the preclinical data, clinical development, and potential use of novel MEK inhibitor binemetinib, particularly in the setting of NRAS mutant melanoma.
Keywords: advanced, metastatic, melanoma, BRAF, MEK, NRAS, binimetinib, MAPK, ERK
Introduction
Surveillance, Epidemiology, and End Results data estimated that in 2017 alone, 87,110 new cases of cutaneous malignant melanoma (CMM) were reported along with 9,730 deaths.1,2 The incidence of CMM has been increasing by 1.4% annually on average over the last 10 years – in stark comparison to the incidence of other common malignancies like female breast cancer (stable incidence), prostate cancer (declining by 5.8%), colorectal cancer (CRC; declining by 2.7%), and lung cancer (declining by 2.0%).1,2 This increase in incidence is disproportionally seen in young women and older men.1,2
Compelling epidemiologic data suggest that primary risk factor for CMM is exposure to ultraviolet (UV) light. These include the significantly greater incidence of cutaneous melanoma in fair-skinned compared to dark-skinned individuals3 and the greater incidence of melanoma in countries further away from the equator such as the United States,1,2 Australia,4 and New Zealand.5 However, several factors including the occurrence of melanoma in patients with intermittent sun exposure and on body sites typically not exposed to the sun such as the trunk suggests that the relationship between melanoma tumorigenesis and UV exposure is not a simple model in which risk correlated directly with exposure.6,7
This led Whiteman et al to propose the divergent pathway hypothesis: an understanding that “melanomas are a consequence of unchecked melanocytic proliferation, and that the degree of proliferation is a trait that is variably distributed in the population”.8–10 This model predicted that patients with “greater inherent predisposition” toward melanoma would only require modest levels of sun exposure to trigger melanoma and that these would typically occur at sites such as the trunk and/or extremities. Conversely, patients with “lower inherent predisposition” toward melanoma would require much greater levels of sun exposure to drive melanoma tumorigenesis, and hence, these tumors would typically occur at sun-exposed areas such as the head and neck. The differential incidence in truncal melanoma (peaking in middle-age) compared to head and neck melanoma (increasingly common with older age) supports this hypothesis.11 Genetic studies supported the divergent pathway hypothesis – mutations in BRAF were primarily seen in younger patients with tumors arising in areas only intermittently exposed to the sun. Correspondingly, BRAF mutations were less commonly identified in tumors from older patients in chronically sun-exposed areas and were particularly unusual in acral skin or mucosal sites.12
The critical breakthrough in our understanding of the mutational landscape of CMM, and, in particular, the link between UV exposure and its effects on preexisting nevi and melanoma came through large-scale collaborative sequencing efforts from groups including The Cancer Genome Atlas (TCGA). Beyond clarifying links between UV exposure and melanoma tumorigenesis, these data provided critical insight and led to the pursuit of inhibitors of genetic drivers as a potential therapeutic option in melanoma. In this review, we dissect the genetic diversity of CMM with particular attention to the mitogen-activated protein kinase (MAPK) pathway and the role of BRAF/MEK inhibitors in the treatment of this disease. We describe the recent development of the selective MEK inhibitor binemetinib in the treatment of metastatic melanoma, and focus on its clinical development and describe its place in the melanoma therapeutic armamentarium.
Genetic diversity of cutaneous melanoma
Seminal studies by Curtin et al using an array-based comparative genomic hybridization approach clarified the mutational landscape of cutaneous melanoma particularly in relation to UV exposure and chronic sun damage (CSD).13 Evaluating acral, mucosal, and melanoma associated with CSD skin and non-CSD skin, they discovered that the majority of non-CSD melanomas exhibited mutations in BRAF or NRAS while the majority of CSD melanomas were BRAF wild type (WT). Although BRAF mutations occasionally occurred with PTEN loss, BRAF and NRAS mutations appeared to be mutually exclusive suggesting that each was a distinct driver. Acral and mucosal melanomas were not strongly enriched for either BRAF and/or NRAS mutations.
Subsequently, advances in next-generation sequencing permitted large-scale sequencing of hundreds of specimens by TCGA, which provided a framework for the genomic classification of CMM: BRAF mutant, NRAS mutant, NF1 mutant, and WT. Collectively, BRAF (37%–50%), NRAS (13%–25%), and NF1 (12%) mutations account for the majority of mutations found in cutaneous melanoma. CSD melanomas typically arise in older (>55 years) patients on sun-exposed areas (head/neck, dorsal surfaces of distal extremities) and are associated with specific driver mutations (BRAFnonV600E, NRAS, NF1, or KIT) and an increased incidence of cytidine to thymidine (C>T) transitions that is characteristic of an ultraviolet (UV) light-induced mutational signature genetic signatures.14–17 Conversely, non-CSD melanomas typically arise in younger (<55 years) patients on intermittently sun-exposed areas (trunk, proximal extremities), and are associated with BRAFV600E driver mutations while lacking genetic signature of UV mutagenesis.14–17
NRAS mutations are found in 15%–20% of CMM in either exon 2 (codons 12, 13) or exon 3 (codon 61).13,18,19 Unlike BRAF mutations, which are typically seen in non-CSD skin, NRAS mutations occur in both CSD and non-CSD skin although a predisposition for the upper extremities has been observed in multiple series.13,20 NRAS mutant primary melanomas are typically thicker with higher Breslow depth and Clark level although they are less likely to have mitoses and/or ulceration20 and lower tumor-infiltrating lymphocyte (TIL) grade.21 Also, while BRAF mutations are typically present in most melanocytic nevi, NRAS mutations are conspicuously absent in nevi other than congenital nevi and the associated congenital nevus-derived melanoma.22 NRAS mutant melanomas (NEMOs) have been linked with increased risk of visceral and central nervous system metastases23 compared to NRAS WT tumors. Although some reports have linked NRAS mutant metastatic melanoma to improved outcomes with immunotherapies including high-dose interleukin 224 and programmed death 1 (PD-1) or cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) checkpoint inhibition,25 the converse has been observed in other series26 and has not been conclusively validated in prospective series.
Eighty percent of NRAS mutations generally occur in codon 61 and typically comprise amino acid substitutions at position 61 from glutamine (Q) to arginine (R – 38%), lysine (K – 34%), leucine (L – 10%), and less commonly to glutamate (E), histidine (H), or proline (P). These mutations are typically in the switch II region of the G domain. Conversely, NRAS codon 12 or 13 mutations comprise 20% of all NRAS mutations and generally occur within the P-loop region of the G domain secondary to an amino acid substitution from glycine (G) to most commonly aspartic acid (D). Codon 61 mutations are associated with locking of the Ras protein into its activated conformation and impaired GTPase activity.27 Codon 12/13 mutations render Ras insensitive to normal physiologic inactivation by Ras-GTPase-activating proteins.27 Both codon 61 or codon 12/13 mutations result in constitutively activated RAS signaling – which drives cellular transformation through a network of signal transduction pathways involved in growth, motility, and survival, thereby enhancing tumor growth.28–31
Roles of BRAF and MEK inhibition in melanoma
Activation of MAPK pathway signaling starts with initiation of receptor tyrosine kinase (RTK) on the cell surface through growth factors, cytokines, and extracellular mitogens.32 In the quiescent state, inactive Ras-GDP is plasma membrane-bound while inactive Raf, MEK, and ERK are cytoplasmic. Ligand binding activates RTK auto-phosphorylation, which generates binding sites for the SHC and GRB2 adaptor molecules, which in turn recruit cytoplasmic SOS (Ras GTPase exchange factor [RasGEF]) to the plasma membrane.29,30,33,34 SOS/RasGEF catalyzes exchange of GDP for GTP and Ras-GTP and then recruits Raf to the membrane where it is activated.29,35–37 Raf activates MEK1/2 and MEK1/2 activates ERK1/2 through activation loop phosphorylation, while ERK negatively regulates SOS to negatively regulate the pathway.32,37–40 Terminal targets of phosphorylated ERK1/2 include cytoplasmic signaling proteins such as RSK (p90 ribosomal S6 kinase) and multiple transcription factors including ternary complex factor (TCF) transcription factors.38,39 TCF transcription factors are key to the regulation of immediate early gene expression, products of which include c-Fos and c-Myc.41,42 Thus, physiologic activation of MEK1/2 and ERK1/2 is required for multiple normal cellular survival, proliferation, motility, and metabolism, while overactivation of the pathway leads to constitutive activation and malignant transformation. Hence, inhibition of overactive signaling in MAPK pathway components such as BRAF and MEK represents an attractive strategy in controlling BRAF and NRAS mutant malignancies.
As discussed, somatic mutations in BRAF and NRAS are seen in 37%–50% and 13%–25% of CMM, where they tend to be nonoverlapping with other oncogenic mutations found in melanoma (mutations in NF1 and KIT). BRAF inhibitors inhibit downstream signaling in BRAF mutant tumors, and consequently, BRAF inhibitors vemurafenib (Zelboraf®; Genentech, San Francisco, CA, USA) and dabrafenib (Tafinlar®; Novartis, Basel, Switzerland) have both demonstrated profound reductions in tumor burden (~50% objective responses) that lasted 6–7 months in randomized Phase III trials of BRAFV600E/K mutant melanoma.43,44 Responses to single-agent BRAF inhibition are fleeting secondary to acquired resistance. Multiple mechanisms of acquired resistance to single-agent BRAF inhibition have been described including MAPK pathway-independent and MAPK pathway-dependent methods such as acquired RAS mutations, mutant BRAF amplification, and alternative splicing, of which the MAPK pathway-dependent methods are more common and account for 70% of acquired resistance.37,45–58 Although the respective frequencies of each of these mechanisms of acquired resistance are not yet known, most resistance appears to functionally result in reactivation of MAPK pathway signaling. Hence, combined BRAF and MEK inhibitor therapy is an attractive strategy to delay or prevent resistance. Three pivotal Phase III studies established the superiority of combination BRAF and MEK inhibition over BRAF inhibition alone: COMBI-d (dabrafenib/trametinib vs dabrafenib/placebo), COMBI-v (dabrafenib/trametinib vs vemurafenib), and coBRIM (vemurafenib/cobimetinib vs vemurafenib/placebo). As expected, BRAF/MEK inhibitor combinations produced greater rates of response (64%–68%) and median durations of response (9.9–11.5 months) compared to BRAF inhibitor monotherapy.59–61 Interestingly, the rate of cutaneous squamous-cell carcinomas was much lower with combination therapy reflecting the more profound degree of MAPK pathway inhibition achieved with combined BRAF and MEK inhibition. Based on these results, US Food and Drug Administration (FDA) approval was granted for both dabrafenib/trametinib and vemurafenib/cobimetinib combinations. Separately, combined BRAF and MEK inhibition is effective after failure of BRAF inhibitor monotherapy.62–64 One clear role for MEK inhibitors is in the combination with BRAF inhibitors to potentiate the response of these agents in BRAF mutant melanoma.
Recently, a body of work has emerged that suggests that oncogenic activation of the Ras/MAPK pathway interacts with and affects tumor-associated immunity. In the BRAFV600E/PTENnull melanoma mouse model, MAPK pathway activation mediates T-cell exclusion, while BRAF/MEK inhibition enhances CD8+ T-cell infiltration into tumor microenvironment (TME) and synergizes with PD-1 blockade.65 Within TME, while BRAF inhibition alone increases tumor-associated macrophages and T regulatory cell infiltration, combined BRAF/MEK inhibition reverses both these trends while increasing major histocompatibility complex expression and inflammatory immune-related gene upregulation – arguing for potential synergies between BRAF/MEK inhibition and PD-1 blockade.66
The adverse effects of MAPK pathway activation on systemic immunity are supported by clinical data from triple-negative breast cancer patients in whom MAPK pathway activation is associated with significantly decreased levels of TILs and poorer survival.67–69 Programmed death ligand 1 (PD-L1) expression appears to correlate with the presence of KRAS mutations70–73 although this observation has to be considered with regard to both the diversity of the methods used to evaluate PD-1/PD-L1 expression and the heterogeneity of the KRAS mutant lung cancer population. The immune effects of Ras signaling may be better understood given the recently reported role of tristetraprolin (TTP) in stabilizing PD-L1 mRNA. In instances where Ras signaling is constitutively enhanced, the activity of AU-rich element-binding protein TTP is decreased resulting in stabilization of PD-L1 mRNA and increased PD-L1 expression.74 Approximately 40%–50% of KRAS mutant lung cancers are concurrently TP53 or STK11 mutant – a subgroup characterized by greater immune infiltrate and somatic mutation burden.75,76 However, whether this can be used as a predictive biomarker for PD-1/PD-L1 inhibition is unknown and will need to wait till this has been assessed in a prospective study. Overall, the available data suggest that RAS activation represents a non-cell autonomous mechanism that mediates tumor development and modulates efficacy of antitumor immunity and argues for another possible role for MEK inhibitors in HRAS/KRAS/NRAS mutant tumors in combination with PD-1 blockade. Although multiple clinical trials are testing the efficacy of the PD-1/MEK inhibitor combination in multiple malignancies including tumors known to harbor NRAS mutations (melanoma – NCT01940809; CRC – NCT03374254; advanced malignancies – NCT03182673 and NCT02586987), other than a single study in KRAS mutant lung cancer (NCT03004105), these studies are not focused on malignancies in which Ras signaling is overactive through one or more mechanisms.
MEK inhibitors: clinical development
The MEK protein is a typical kinase with an adenosine triphosphate (ATP) binding pocket that toggles between catalytically active (phosphorylated) and catalytically inactive (unphosphorylated) states. Three types of MEK inhibitors have been developed: ATP-competitive (type I) and ATP noncompetitive (types II and III).77,78 Type I inhibitors occupy the ATP-binding pocket, while type II inhibitors bind to a binding site that includes the ATP-binding pocket and an adjacent less-conserved allosteric site. However, both type I and type II inhibitors are equally nonselective. Type III inhibitors occupy highly specific allosteric sites and are more selective.
PD098059
The first MEK inhibitor to be disclosed was PD098059. Unlike earlier agents that had variably inhibited both RAF and MEK, PD098059 was a non-ATP competitive MEK inhibitor that inhibited MEK in vitro at micromolar concentrations (half-maximal inhibitory concentration [IC50] 2 μM) without inhibiting RAF.79 In vitro testing clearly established that PD098059 inhibited MAPK activation, inhibited cell growth, and reversed phenotype of ras-transformed cells.80,81 However, the evaluation of PD098059 in Raf/MEK signaling cancer was truncated by data that showed that PD098059 while effectively inhibiting MEK signaling paradoxically promoted ERK1/2 phosphorylation through an Raf-1-independent/MEK-dependent mechanism mediated by reactive oxygen species.82 This suggested that PD098059 may not completely ablate MEK activity and may have differential activity dependent on the redox state of the target cell, and resulted in the truncation of clinical development.
Selumetinib (AZD6244, ARRY-142886)
Selumetinib (AZD6244) is a potent, highly selective inhibitor of MEK1/2 (IC50 10–14 nM) and ERK1/2 phosphorylation (IC50 10 nM). Preclinical data demonstrated activity of AZD6244 in both in vitro and human tumor mouse xenograft models of non-small-cell lung cancer (NSCLC) and melanoma.32,83,84 Dose-finding studies studied two separate formulations including an oral free-base suspension and a hydrogen sulfate (Hyd-Sulfate) salt formulation.85,86 Both studies identified separate maximal tolerated doses (MTD): 100 mg twice daily (oral free-base suspension) and 75 mg twice daily (Hyd-Sulfate). Dose-finding studies also evaluated the combination of selumetinib with cetuximab,87 erlotinib,88 sorafenib,89 cisplatin/gemcitabine,90 docetaxel,91,92 dacarbazine,93,94 and platinum-based chemotherapy95 in a variety of tumors.
Subsequent Phase II studies across a variety of histologies were largely negative except in BRAF mutated cutaneous melanoma and uveal melanoma. In a Phase II study of uveal melanoma patients who were randomized to either selumetinib or chemotherapy, selumetinib modestly improved progression-free survival (PFS) although overall survival (OS) was not improved and selumetinib was associated with considerable toxicity.96 In BRAF mutant melanoma, responses observed were restricted to patients with tumors that expressed low levels of phosphorylated AKT (pAKT),97 confirming the effect of selumetinib and vemurafenib observed in low pAKT cell lines.98 Signaling through the phosphatidylinositol 3-kinase (PI3K)/AKT/mammalian target of rapamycin (mTOR) pathway has been implicated in intrinsic resistance to MEK (and BRAF) inhibitor therapy,39,99 and co-blockade of MEK and AKT was reported to be additive in KRAS mutant tumor cell lines and in vivo model systems100 – providing a rationale to target both MEK and PI3K/AKT in tumors driven by oncogenic RAS signaling. Unfortunately, studies testing selumetinib in combination with AKT inhibitor MK-2206 in KRAS mutant cancers,101 KRAS mutant CRC,102 and KRAS mutant pancreatic cancer103 yielded negative results – likely due to inability to dose-escalate selumetinib and MK-2206 to doses capable of eliciting sufficient pERK/pAKT inhibition.
More recently, selumetinib has been studied in nonmalignant conditions characterized by elevated MAPK signaling such as neurofibromatosis type 1-related plexiform neurofibromas. MTD of 25 mg/m2 was ~60% of the adult dose with promising efficacy and relatively tolerable toxicity profile. Separately, based on observations linking MAPK and PI3K-AKT-mTOR pathway activity to lower tumor immune infiltrates,65 a study of selumetinib in combination with PD-1 and CTLA-4 immune checkpoint inhibitors has been launched (NCT02586987).
Trametinib (GSK1120212)
Trametinib is an allosteric, non-ATP-competitive inhibitor of both MEK1 (IC50 0.7 nM) and MEK2 (IC50 0.9 nM) kinases with excellent efficacy and high activity in preclinical studies.104–106 Following initial dose-finding studies, the clinical evaluation of trametinib centered around BRAF mutant tumors and melanoma in particular.
In melanoma, trametinib monotherapy was initially compared to chemotherapy (dacarbazine or paclitaxel) in the Phase III METRIC study of BRAFV600E/K mutant melanoma that accrued 322 patients in a 2:1 ratio.107 Trametinib monotherapy resulted in an overall response rate (ORR) of 22% and improved PFS and OS (summarized in Table 1). These data resulted in the FDA approval of trametinib to treat BRAFV600E/K mutant unresectable or metastatic melanoma in May 2013.
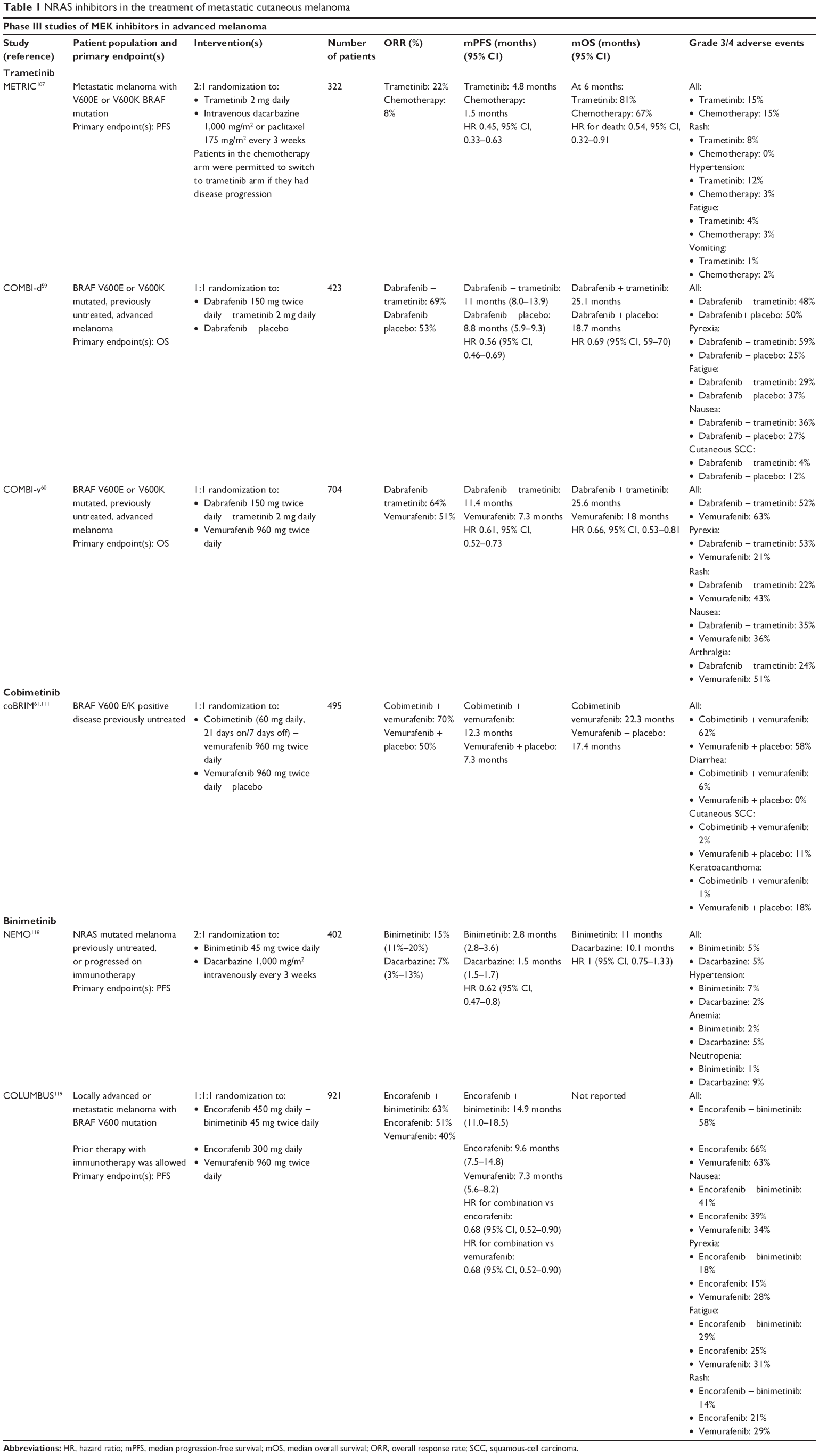 | Table 1 NRAS inhibitors in the treatment of metastatic cutaneous melanoma |
Following a successful open-label Phase III trial against dacarbazine in BRAFV600E/K mutant melanoma, dabrafenib independently garnered FDA approval in May 2013.44 The dabrafenib/trametinib combination was studied in two separate placebo-controlled Phase III trials against dabrafenib (COMBI-d) and vemurafenib (COMBI-v) (summarized in Table 1). In both studies, dabrafenib/trametinib combination had greater ORR (COMBI-d: 67%; COMBI-v: 76%) and median PFS (COMBI-d: 9.3 months; COMBI-v: 11.4 months) with improved OS compared to BRAF inhibitor monotherapy.59,60 The significant improvement observed resulted in FDA approval for the dabrafenib/trametinib combination to treat BRAFV600E/K mutant melanoma in January 2014.
Given the success of dabrafenib/trametinib in treating metastatic patients, its use in the high-risk resected setting has been explored in stage III patients.108 In a blinded Phase III trial of dabrafenib/trametinib against matched placebo in stage III A–C BRAFV600E/K mutant melanoma (COMBI-AD), risk of relapse was 53% lower in the treatment group (hazard ratio for relapse or death, 0.47). Relapse-free survival rates were similarly greater in the treatment group: 1-year 88% (vs 56%), 2-year 67% (vs 44%), and 3-year 58% (vs 39%). The combination was granted breakthrough therapy status.
Given possible synergies between BRAF/MEK inhibition with PD-1 inhibitors, the triple combination of dabrafenib, trametinib, and pembrolizumab has been studied in BRAFV600E/K mutant melanoma. A Phase I/II study of the triple combination (KEYNOTE-022) reported ORR of 67% with no toxicity signals.109 A Phase III trial (COMBI-i) is comparing the triple combination of dabrafenib, trametinib, and investigational PD-1 inhibitor PDR001 to dabrafenib, trametinib, and placebo (NCT02967692).
Cobimetinib (GDC-0973, XL518)
Cobimetinib is a potent and highly selective inhibitor of MEK1 (IC50 0.9 nM) with greater than 100-fold selectivity for MEK1 over MEK2 (IC50 199 nM). Preclinical data demonstrated broad in vivo efficacy in xenograft models utilizing BRAF or KRAS mutant cell lines.110 Unlike trametinib, cobimetinib was not evaluated as a single agent but rather in combination with vemurafenib in BRAF mutant melanoma.
In melanoma, cobimetinib was initially evaluated in the Phase III coBRIM study that compared vemurafenib/cobimetinib combination with vemurafenib/placebo in 495 BRAFV600 mutant melanoma patients (summarized in Table 1). As expected, combination therapy resulted in greater ORR (68% vs 45%), PFS (9.9 months vs 6.2 months), and OS (22.3 months vs 17.4 months) compared to vemurafenib monotherapy,61,111 resulting in FDA approval for the combination in November 2015.
Concordantly with the development of dabrafenib/trametinib, cobimetinib has also been evaluated alongside PD-1 inhibitors across a variety of histologies. Separate Phase Ib studies evaluated the cobimetinib/atezolizumab combination in microsatellite stable (MSS) CRC and BRAF WT melanoma with ORR of 17% (MSS CRC) and 45% (melanoma).112 Further evaluation in MSS CRC is ongoing in the randomized Phase II ATELIER study (NCT03340558) that is comparing the cobimetinib/atezolizumab combination with atezolizumab monotherapy in CRC with liver metastases. In melanoma, the triple combination of vemurafenib, cobimetinib, and atezolizumab was initially studied in a Phase Ib study in BRAFV600 mutant melanoma with encouraging results (85% ORR). These results will be formally assessed in the Phase III TRILOGY trial of the triple combination compared with vemurafenib/cobimetinib/placebo (NCT02908672).
Binimetinib (MEK162, ARRY-162, ARRY-438162)
Binimetinib is an orally bioavailable, highly selective, ATP noncompetitive allosteric MEK inhibitor that was initially discovered by Array BioPharm., and subsequently codeveloped by Array BioPharm. and Novartis. Binimetinib inhibits MEK1/2 at nanomolar concentrations (IC50 12 nM) with significant antitumor activity in preclinical studies involving cell lines and murine xenograft models.113,114
In a Phase I study of 93 patients, 19 patients with a variety of solid tumors were enrolled during dose escalation with a further 74 patients during dose expansion.115 The expansion cohort primarily enrolled patients with gastrointestinal malignancies including biliary cancer (28 patients) and CRC (31 KRAS mutant and 15 BRAF mutant patients). MTD was 60 mg twice daily although after greater than expected incidence of ocular toxicities was observed at MTD during dose-expansion phase, 45 mg twice daily was selected as the recommended Phase II dose (RP2D). Common adverse events (AEs) across all dose levels included rash (81%), nausea (56%), vomiting (52%), diarrhea (51%), peripheral edema (46%), and fatigue (43%), with the most common grade 3 events including anemia (11%), abdominal pain (4%), and dehydration (4%). The incidence of grade 4 AEs was low at 6% with no grade 5 AE reported. AE-related treatment discontinuation occurred in 15% (60 mg twice daily) to 25% (80 mg twice daily) of patients, most commonly due to ocular events. Of the 26 patients with evaluable biliary cancer, one complete response and one partial response were observed. A separate Phase I study in Japanese patients also identified 45 mg twice daily as RP2D.116
Binimetinib at 45 mg twice daily monotherapy was subsequently evaluated in a Phase II study of 71 patients with NRAS (30 patients) or BRAFV600E/K (41 patients) mutant melanoma.117 Most patients had previously been treated with either chemotherapy or cytokine-based immunotherapy, but relatively few had received prior immune checkpoint inhibitors (30% of NRAS mutant; 15% of BRAF mutant).117 Although no complete responses were observed, ORR was 11% in NRAS mutant patients and 5% in BRAF mutant patients. Latter was substantially lower than the 22% ORR observed in the METRIC study with trametinib in BRAF mutant melanoma although 17% of these had received prior BRAF inhibitor therapy.107,117 Toxicity profile was relatively favorable with relatively low incidence of grade 3/4 AEs except for serum creatine phosphokinase elevations (23% of NRAS mutant; 17% of BRAF mutant). The incidence of ocular events was low (27% of NRAS mutant; 12% of BRAF mutant) with no grade 3/4 events.
Following Phase II data, separate Phase III studies in metastatic NEMO and BRAFV600E/K mutant melanoma (COLUMBUS) were launched (data summarized in Table 1). In NEMO, binimetinib monotherapy was compared with dacarbazine.118 Binimetinib resulted in ORR 15% with median PFS 2.8 months that compared favorably to dacarbazine (ORR 7%, median PFS 1.5 months). Although statistically significant, these results have to be interpreted within the context of a relatively good-risk patient population (69% normal lactose dehydrogenase) with minimal prior exposure to effective immune checkpoint inhibitor therapy (prior ipilimumab 13%; prior PD-1/PD-L1 6%). Although a new drug application (NDA) for binimetinib was filed for this indication, this was subsequently withdrawn based on feedback from the FDA during a preplanned review meeting.
In COLUMBUS, binimetinib combined with BRAF inhibitor encorafenib was compared with both vemurafenib and encorafenib separately.119 Encorafenib/binimetinib combination was unsurprisingly better than either encorafenib or vemurafenib monotherapy with 63% ORR (encorafenib 51%; vemurafenib 40%) and median PFS of 14.9 months (encorafenib 9.6 months; vemurafenib 7.3 months). Although the ORR in the combination arm was similar to the dabrafenib/trametinib and vemurafenib/cobimetinib combinations, the median PFS of encorafenib/binimetinib was greater than what was observed with the BRAF/MEK inhibitor combination arms in COMBI-d, COMBI-v, and coBRIM.59–61 Incidence of grade 3/4 AEs was lower in the encorafenib/binimetinib group (58%) than in either encorafenib (66%) or vemurafenib (63%) group. Concordantly, the treatment discontinuation rate of the combination was lower than in either monotherapy arm. An NDA for the use of encorafenib/binimetinib in this indication has been submitted to the FDA, results of which are pending.
The further development of binimetinib is along a similar trajectory to that described for trametinib and cobimetinib. Combinations with PD-1/CTLA-4 immune checkpoint inhibitors (NCT03271047 and NCT03374254 – MSS CRC); CDK4/6 inhibitor palbociclib in KRAS mutant NSCLC (NCT03170206); MET inhibitor PF-02341066 in RAS mutant/WT CRC with aberrant C-MET signaling (NCT02510001); chemotherapy in KRAS mutant NSCLC (NCT02185690), CRC (NCT02928224, NCT02613650), and biliary tract cancer (NCT02773459); erlotinib in KRAS/EGFR mutant NSCLC (NCT01859026); imatinib in gastrointestinal stromal tumors (NCT01991379); and as monotherapy in NF1 plexiform neurofibromas (NCT03231306).
Conclusion
Following the initial approval of trametinib to treat BRAF mutant melanoma in MEK inhibitors in May 2013, the clinical development of MEK inhibitors has proceeded rapidly. Initial success in BRAF mutant melanoma in combination with BRAF inhibitors has been parlayed into other indications where BRAF is an oncogenic driver including NSCLC120 and papillary/anaplastic thyroid cancer.121
Separately, preclinical studies have demonstrated that MEK inhibitors either singly or in combination with BRAF inhibitors exert immunomodulatory effects on the tumor and the TME and may enhance immune activation.65,66 These observations are buttressed by clinical trials demonstrating efficacy of the PD-1/MEK inhibitor combination in CRC and BRAF WT melanoma while the PD-1/MEK/BRAF inhibitor triple combination has produced startling response rates in early-phase studies, although confirmatory studies are ongoing.
Finally, data regarding the clinical benefit of dose-adjusted selumetinib in treating children and adults with highly symptomatic plexiform neurofibromas associated with neurofibromatosis type I, a common genetic tumor syndrome caused by germline mutations in NF1, are encouraging. These reports argue for expanding the role of NRAS inhibitors beyond traditional oncologic indications to RASopathies and other conditions associated with dysregulation of the RAS/MAPK pathway.
Acknowledgment
The authors have not received any funding or benefits from the industry to conduct this review.
Disclosure
Diwakar Davar has received the following funding: Checkmate Pharmaceuticals, Merck, Bristol-Myers Squibb (research support). The other author reports no conflicts of interest in this work.
References
Melanoma of the Skin – SEER Stat Fact Sheets. Available from: http://seer.cancer.gov/statfacts/html/melan.html. Accessed May 10, 2016. | ||
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66(1):7–30. | ||
Armstrong BK, Kricker A. How much melanoma is caused by sun exposure? Melanoma Res. 1993;3(6):395–402. | ||
Jelfs PL, Giles G, Shugg D, et al. Cutaneous malignant melanoma in Australia, 1989. Med J Aust. 1994;161(3):182–187. | ||
Bulliard J-L. Site-specific risk of cutaneous malignant melanoma and pattern of sun exposure in New Zealand. Int J Cancer. 2000;85(5):627–632. | ||
Beral V, Robinson N. The relationship of malignant melanoma, basal and squamous skin cancers to indoor and outdoor work. Br J Cancer. 1981;44(6):886–891. | ||
Green A, Maclennan R, Youl P, Martin N. Site distribution of cutaneous melanoma in queensland. Int J Cancer. 1993;53(2):232–236. | ||
Whiteman DC, Parsons PG, Green AC. p53 expression and risk factors for cutaneous melanoma: a case-control study. Int J Cancer. 1998;77(6):843–848. | ||
Whiteman DC, Watt P, Purdie DM, Hughes MC, Hayward NK, Green AC. Melanocytic nevi, solar keratoses, and divergent pathways to cutaneous melanoma. J Natl Cancer Inst. 2003;95(11):806–812. | ||
Whiteman DC. Testing the divergent pathway hypothesis for melanoma: recent findings and future challenges. Expert Rev Anticancer Ther. 2010;10(5):615–618. | ||
Lachiewicz AM, Berwick M, Wiggins CL, Thomas NE. Epidemiologic support for melanoma heterogeneity using the Surveillance, Epidemiology, and End Results program. J Invest Dermatol. 2008;128(5):1340–1342. | ||
Maldonado JL, Fridlyand J, Patel H, et al. Determinants of BRAF mutations in primary melanomas. J Natl Cancer Inst. 2003;95(24):1878–1890. | ||
Curtin JA, Fridlyand J, Kageshita T, et al. Distinct sets of genetic alterations in melanoma. N Engl J Med Overseas Ed. 2005;353(20):2135–2147. | ||
Curtin JA, Busam K, Pinkel D, Bastian BC. Somatic activation of KIT in distinct subtypes of melanoma. J Clin Oncol. 2006;24(26):4340–4346. | ||
Hodis E, Watson IR, Kryukov GV, et al. A landscape of driver mutations in melanoma. Cell. 2012;150(2):251–263. | ||
Cancer Genome Atlas Network. Genomic classification of cutaneous melanoma. Cell. 2015;161(7):1681–1696. | ||
Kappes UP, Luo D, Potter M, Schulmeister K, Rünger TM, et al. Short- and long-wave UV light (UVB and UVA) induce similar mutations in human skin cells. J Invest Dermatol. 2006;126(3):667–675. | ||
Ball NJ, Yohn JJ, Morelli JG, et al. RAS mutations in human melanoma: a marker of malignant progression. J Invest Dermatol. 1994;102(3):285–290. | ||
van’t Veer LJ, Burgering BM, Versteeg R, et al. N-ras mutations in human cutaneous melanoma from sun-exposed body sites. Mol Cell Biol. 1989;9(7):3114–3116. | ||
Ellerhorst JA, Greene VR, Ekmekcioglu S, et al. Clinical correlates of NRAS and BRAF mutations in primary human melanoma. Clin Cancer Res. 2011;17(2):229–235. | ||
Thomas NE, Edmiston SN, Alexander A, et al. Association between NRAS and BRAF mutational status and melanoma-specific survival among patients with higher-risk primary melanoma. JAMA Oncol. 2015;1(3):359–368. | ||
Lu C, Zhang J, Nagahawatte P, et al. The genomic landscape of childhood and adolescent melanoma. J Invest Dermatol. 2015;135(3):816–823. | ||
Adler NR, Wolfe R, Kelly JW, et al. Tumour mutation status and sites of metastasis in patients with cutaneous melanoma. Br J Cancer. 2017;117(7):1026–1035. | ||
Joseph RW, Sullivan RJ, Harrell R, et al. Correlation of NRAS mutations with clinical response to high-dose IL-2 in patients with advanced melanoma. J Immunother. 2012;35(1):66–72. | ||
Johnson DB, Lovly CM, Flavin M, et al. Impact of NRAS mutations for patients with advanced melanoma treated with immune therapies. Cancer Immunol Res. 2015;3(3):288–295. | ||
Kirchberger MC, Ugurel S, Mangana J, et al. NRAS-mutated melanoma patients have similar response rates to therapy with checkpoint inhibitors as other cohorts. J Clin Oncol. 2017;35:e21035. | ||
Fedorenko IV, Gibney GT, Smalley KSM. NRAS mutant melanoma: biological behavior and future strategies for therapeutic management. Oncogene. 2013;32(25):3009–3018. | ||
Bos JL. Ras oncogenes in human cancer: a review. Cancer Res. 1989;49(17):4682–4689. | ||
Buday L, Downward J. Epidermal growth factor regulates p21ras through the formation of a complex of receptor, Grb2 adapter protein, and Sos nucleotide exchange factor. Cell. 1993;73(3):611–620. | ||
Colicelli J. Human RAS superfamily proteins and related GTPases. Sci Signal. 2004;2004(250):RE13. | ||
Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417(6892):949–954. | ||
Haass NK, Sproesser K, Nguyen TK, et al. The mitogen-activated protein/extracellular signal-regulated kinase kinase inhibitor AZD6244 (ARRY-142886) induces growth arrest in melanoma cells and tumor regression when combined with docetaxel. Clin Cancer Res. 2008;14(1):230–239. | ||
Marcus K, Mattos C. Direct attack on RAS: intramolecular communication and mutation-specific effects. Clin Cancer Res. 2015;21(8):1810–1818. | ||
Cox AD, der CJ, Philips MR. Targeting RAS membrane association: back to the future for anti-RAS drug discovery? Clin Cancer Res. 2015;21(8):1819–1827. | ||
Hatzivassiliou G, Song K, Yen I, et al. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature. 2010;464(7287):431–435. | ||
Poulikakos PI, Zhang C, Bollag G, Shokat KM, Rosen N, et al. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature. 2010;464(7287):427–430. | ||
Nissan MH, Pratilas CA, Jones AM, et al. Loss of NF1 in cutaneous melanoma is associated with RAS activation and MEK dependence. Cancer Res. 2014;74(8):2340–2350. | ||
Kono M, Dunn IS, Durda PJ, et al. Role of the mitogen-activated protein kinase signaling pathway in the regulation of human melanocytic antigen expression. Mol Cancer Res. 2006;4(10):779–792. | ||
Wagle N, van Allen EM, Treacy DJ, et al. MAP kinase pathway alterations in BRAF-mutant melanoma patients with acquired resistance to combined RAF/MEK inhibition. Cancer Discov. 2014;4(1):61–68. | ||
Marusiak AA, Edwards ZC, Hugo W, et al. Mixed lineage kinases activate MEK independently of RAF to mediate resistance to RAF inhibitors. Nat Commun. 2014;5(1):3901. | ||
Zullo JN, Faller DV. P21 v-ras inhibits induction of c-myc and c-fos expression by platelet-derived growth factor. Mol Cell Biol. 1988;8(12):5080–5085. | ||
Tabib A, Bachrach U. Activation of the proto-oncogene c-myc and c-fos by c-ras: involvement of polyamines. Biochem Biophys Res Commun. 1994;202(2):720–727. | ||
Chapman PB, Hauschild A, Robert C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med Overseas Ed. 2011;364(26):2507–2516. | ||
Hauschild A, Grob J-J, Demidov LV, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet. 2012;380(9839):358–365. | ||
Poulikakos PI, Persaud Y, Janakiraman M, et al. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E). Nature. 2011;480(7377):387–390. | ||
Shi H, Moriceau G, Kong X, et al. Melanoma whole-exome sequencing identifies V600E B-RAF amplification-mediated acquired B-RAF inhibitor resistance. Nat Commun. 2012;3(1):724. | ||
Johannessen CM, Boehm JS, Kim SY, et al. COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature. 2010;468(7326):968–972. | ||
Straussman R, Morikawa T, Shee K, et al. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature. 2012;487(7408):500–504. | ||
Wilson TR, Fridlyand J, Yan Y, et al. Widespread potential for growth-factor-driven resistance to anticancer kinase inhibitors. Nature. 2012;487(7408):505–509. | ||
Villanueva J, Vultur A, Lee JT, et al. Acquired resistance to BRAF inhibitors mediated by a RAF kinase switch in melanoma can be overcome by cotargeting MEK and IGF-1R/PI3K. Cancer Cell. 2010;18(6):683–695. | ||
Emery CM, Vijayendran KG, Zipser MC, et al. MEK1 mutations confer resistance to MEK and B-RAF inhibition. Proc Natl Acad Sci U S A. 2009;106(48):20411–20416. | ||
Wagle N, Emery C, Berger MF, et al. Dissecting therapeutic resistance to RAF inhibition in melanoma by tumor genomic profiling. J Clin Oncol. 2011;29(22):3085–3096. | ||
Maertens O, Johnson B, Hollstein P, et al. Elucidating distinct roles for NF1 in melanomagenesis. Cancer Discov. 2013;3(3):338–349. | ||
Whittaker SR, Theurillat J-P, van Allen E, et al. A genome-scale RNA interference screen implicates NF1 loss in resistance to RAF inhibition. Cancer Discov. 2013;3(3):350–362. | ||
Nazarian R, Shi H, Wang Q, et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature. 2010;468(7326):973–977. | ||
Shi H, Hugo W, Kong X, et al. Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer Discov. 2014;4(1):80–93. | ||
van Allen EM, Wagle N, Sucker A, et al. The genetic landscape of clinical resistance to RAF inhibition in metastatic melanoma. Cancer Discov. 2014;4(1):94–109. | ||
Paraiso KHT, Xiang Y, Rebecca VW, et al. PTEN loss confers BRAF inhibitor resistance to melanoma cells through the suppression of BIM expression. Cancer Res. 2011;71(7):2750–2760. | ||
Long GV, Stroyakovskiy D, Gogas H, et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N Engl J Med Overseas Ed. 2014;371(20):1877–1888. | ||
Robert C, Karaszewska B, Schachter J, et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med Overseas Ed. 2015;372(1):30–39. | ||
Larkin J, Ascierto PA, Dréno B, et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N Engl J Med Overseas Ed. 2014;371(20):1867–1876. | ||
Johnson DB, Flaherty KT, Weber JS, et al. Combined BRAF (Dabrafenib) and MEK inhibition (Trametinib) in patients with BRAFV600-mutant melanoma experiencing progression with single-agent BRAF inhibitor. J Clin Oncol. 2014;32(33):3697–3704. | ||
Ribas A, Gonzalez R, Pavlick A, et al. Combination of vemurafenib and cobimetinib in patients with advanced BRAFV600-mutated melanoma: a phase 1b study. Lancet Oncol. 2014;15(9):954–965. | ||
Schreuer M, Jansen Y, Planken S, et al. Combination of dabrafenib plus trametinib for BRAF and MEK inhibitor pretreated patients with advanced BRAF V600-mutant melanoma: an open-label, single arm, dual-centre, phase 2 clinical trial. Lancet Oncol. 2017;18(4):464–472. | ||
Deken MA, Gadiot J, Jordanova ES, et al. Targeting the MAPK and PI3K pathways in combination with PD1 blockade in melanoma. Oncoimmunology. 2016;5(12):e1238557. | ||
Hu-Lieskovan S, Mok S, Homet Moreno B, et al. Improved antitumor activity of immunotherapy with BRAF and MEK inhibitors in BRAF (V600E) melanoma. Sci Transl Med. 2015;7(279):ra41. | ||
Balko JM, Schwarz LJ, Bhola NE, et al. Activation of MAPK pathways due to DUSP4 loss promotes cancer stem cell-like phenotypes in basal-like breast cancer. Cancer Res. 2013;73(20):6346–6358. | ||
Balko JM, Giltnane JM, Wang K, et al. Molecular profiling of the residual disease of triple-negative breast cancers after neoadjuvant chemotherapy identifies actionable therapeutic targets. Cancer Discov. 2014;4(2):232–245. | ||
Loi S, Dushyanthen S, Beavis PA, et al. RAS/MAPK activation is associated with reduced tumor-infiltrating lymphocytes in triple-negative breast cancer: therapeutic cooperation between MEK and PD-1/PD-L1 immune checkpoint inhibitors. Clin Cancer Res. 2016;22(6):1499–1509. | ||
D’incecco A, Andreozzi M, Ludovini V, et al. PD-1 and PD-L1 expression in molecularly selected non-small-cell lung cancer patients. Br J Cancer. 2015;112(1):95–102. | ||
Garon EB, Rizvi NA, Hui R, et al. Pembrolizumab for the treatment of non–small-cell lung cancer. N Engl J Med. 2015;372(21):2018–2028. | ||
Skoulidis F, Hellmann MD, Awad MM, et al. STK11/LKB1 co-mutations to predict for de novo resistance to PD-1/PD-L1 axis blockade in KRAS-mutant lung adenocarcinoma. J Clin Oncol. 2017;35:9016. | ||
Dhar M, Wong J, Che J, et al. Evaluation of PD-L1 expression on vortex-isolated circulating tumor cells in metastatic lung cancer. Sci Rep. 2018;8(1):2592. | ||
Coelho MA, de Carné Trécesson S, Rana S, et al. Oncogenic RAS signaling promotes tumor immunoresistance by stabilizing PD-L1 mRNA. Immunity. 2017;47(6):1083–1099. | ||
Skoulidis F, Byers LA, Diao L, et al. Co-occurring genomic alterations define major subsets of KRAS-mutant lung adenocarcinoma with distinct biology, immune profiles, and therapeutic vulnerabilities. Cancer Discov. 2015;5(8):860–877. | ||
Schabath MB, Welsh EA, Fulp WJ, et al. Differential association of STK11 and TP53 with KRAS mutation-associated gene expression, proliferation and immune surveillance in lung adenocarcinoma. Oncogene. 2016;35(24):3209–3216. | ||
Roskoski R. Allosteric MEK1/2 inhibitors including cobimetanib and trametinib in the treatment of cutaneous melanomas. Pharmacol Res. 2017;117:20–31. | ||
Kooistra AJ, Kanev GK, vanLinden OPJ, et al. KLIFS: a structural kinase-ligand interaction database. Nucleic Acids Res. 2016;44(D1):D365–D371. | ||
Alessi DR, Cuenda A, Cohen P, Dudley DT, Saltiel AR. PD 098059 is a specific inhibitor of the activation of mitogen-activated protein kinase kinase in vitro and in vivo. J Biol Chem. 1995;270(46):27489–27494. | ||
Dudley DT, Pang L, Decker SJ, Bridges AJ, Saltiel AR. A synthetic inhibitor of the mitogen-activated protein kinase cascade. Proc Natl Acad Sci U S A. 1995;92(17):7686–7689. | ||
Pang L, Sawada T, Decker SJ, Saltiel AR. Inhibition of MAP kinase kinase blocks the differentiation of PC-12 cells induced by nerve growth factor. J Biol Chem. 1995;270(23):13585–13588. | ||
Cerioni L, Palomba L, Cantoni O. The Raf/MEK inhibitor PD98059 enhances ERK1/2 phosphorylation mediated by peroxynitrite via enforced mitochondrial formation of reactive oxygen species. FEBS Lett. 2003;547(1–3):92–96. | ||
Davies BR, Logie A, Mckay JS, et al. AZD6244 (ARRY-142886), a potent inhibitor of mitogen-activated protein kinase/extracellular signal-regulated kinase kinase 1/2 kinases: mechanism of action in vivo, pharmacokinetic/pharmacodynamic relationship, and potential for combination in preclinical models. Mol Cancer Ther. 2007;6(8):2209–2219. | ||
Yeh TC, Marsh V, Bernat BA, et al. Biological characterization of ARRY-142886 (AZD6244), a potent, highly selective mitogen-activated protein kinase kinase 1/2 inhibitor. Clin Cancer Res. 2007;13(5):1576–1583. | ||
Banerji U, Camidge DR, Verheul HM, et al. The first-in-human study of the hydrogen sulfate (Hyd-sulfate) capsule of the MEK1/2 inhibitor AZD6244 (ARRY-142886): a phase I open-label multicenter trial in patients with advanced cancer. Clin Cancer Res. 2010;16(5):1613–1623. | ||
Adjei AA, Christian M, Ivy P. Novel designs and end points for phase II clinical trials. Clin Cancer Res. 2009;15(6):1866–1872. | ||
Deming DA, Cavalcante LL, Lubner SJ, et al. A phase I study of selumetinib (AZD6244/ARRY-142866), a MEK1/2 inhibitor, in combination with cetuximab in refractory solid tumors and KRAS mutant colorectal cancer. Invest New Drugs. 2016;34(2):168–175. | ||
Ah K, Bekaii-Saab T, van Ziffle J, et al. A multicenter, open-label phase II clinical trial of combined MEK plus EGFR inhibition for chemotherapy-refractory advanced pancreatic adenocarcinoma. Clin Cancer Res. 2016;22(1):61–68. | ||
Tai WM, Yong WP, Lim C, et al. A phase Ib study of selumetinib (AZD6244, ARRY-142886) in combination with sorafenib in advanced hepatocellular carcinoma (HCC). Ann Oncol. 2016;27(12):2210–2215. | ||
Bridgewater J, Lopes A, Beare S, et al. A phase 1b study of Selumetinib in combination with Cisplatin and Gemcitabine in advanced or metastatic biliary tract cancer: the ABC-04 study. BMC Cancer. 2016;16(1):153. | ||
Lorusso PM, Infante JR, Kim KB, et al. A phase I dose-escalation study of selumetinib in combination with docetaxel or dacarbazine in patients with advanced solid tumors. BMC Cancer. 2017;17(1):173. | ||
Jänne PA, Smith I, Mcwalter G, et al. Impact of KRAS codon subtypes from a randomised phase II trial of selumetinib plus docetaxel in KRAS mutant advanced non-small-cell lung cancer. Br J Cancer. 2015;113(2):199–203. | ||
Carvajal RD, Schwartz GK, Mann H, Smith I, Nathan PD. Study design and rationale for a randomised, placebo-controlled, double-blind study to assess the efficacy of selumetinib (AZD6244; ARRY-142886) in combination with dacarbazine in patients with metastatic uveal melanoma (SUMIT). BMC Cancer. 2015;15(1):467. | ||
Patel SP, Lazar AJ, Papadopoulos NE, et al. Clinical responses to selumetinib (AZD6244; ARRY-142886)-based combination therapy stratified by gene mutations in patients with metastatic melanoma. Cancer. 2013;119(4):799–805. | ||
Greystoke A, Steele N, Arkenau H-T, et al. SELECT-3: a phase I study of selumetinib in combination with platinum-doublet chemotherapy for advanced NSCLC in the first-line setting. Br J Cancer. 2017;117(7):938–946. | ||
Carvajal RD, Sosman JA, Quevedo JF, et al. Effect of selumetinib vs chemotherapy on progression-free survival in uveal melanoma. JAMA. 2014;311(23):2397–2405. | ||
Catalanotti F, Solit DB, Pulitzer MP, et al. Phase II trial of MEK inhibitor selumetinib (AZD6244, ARRY-142886) in patients with BRAFV600E/K-mutated melanoma. Clin Cancer Res. 2013;19(8):2257–2264. | ||
Sweetlove M, Wrightson E, Kolekar S, et al. Inhibitors of pan-PI3K signaling synergize with BRAF or MEK inhibitors to prevent BRAF-mutant melanoma cell growth. Front Oncol. 2015;5:135. | ||
Janku F, Lee JJ, Tsimberidou AM, et al. PIK3CA mutations frequently coexist with RAS and BRAF mutations in patients with advanced cancers. PLoS One. 2011;6(7):e22769. | ||
Wee S, Jagani Z, Xiang KX, et al. PI3K pathway activation mediates resistance to MEK inhibitors in KRAS mutant cancers. Cancer Res. 2009;69(10):4286–4293. | ||
Tolcher AW, Khan K, Ong M, et al. Antitumor activity in RAS-driven tumors by blocking AKT and MEK. Clin Cancer Res. 2015;21(4):739–748. | ||
Do K, Speranza G, Bishop R, et al. Biomarker-driven phase 2 study of MK-2206 and selumetinib (AZD6244, ARRY-142886) in patients with colorectal cancer. Invest New Drugs. 2015;33(3):720–728. | ||
Chung V, Mcdonough S, Philip PA, et al. Effect of selumetinib and MK-2206 vs oxaliplatin and fluorouracil in patients with metastatic pancreatic cancer after prior therapy: SWOG S1115 study randomized clinical trial. JAMA Oncol. 2017;3(4):516–522. | ||
Gilmartin AG, Bleam MR, Groy A, et al. GSK1120212 (JTP-74057) is an inhibitor of MEK activity and activation with favorable pharmacokinetic properties for sustained in vivo pathway inhibition. Clin Cancer Res. 2011;17(5):989–1000. | ||
Abe H, Kikuchi S, Hayakawa K, et al. Discovery of a highly potent and selective MEK inhibitor: GSK1120212 (JTP-74057 DMSO Solvate). ACS Med Chem Lett. 2011;2(4):320–324. | ||
Yamaguchi T, Kakefuda R, Tajima N, Sowa Y, Sakai T. Antitumor activities of JTP-74057 (GSK1120212), a novel MEK1/2 inhibitor, on colorectal cancer cell lines in vitro and in vivo. Int J Oncol. 2011;39(1):23–31. | ||
Flaherty KT, Robert C, Hersey P, et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med. 2012;367(2):107–114. | ||
Long GV, Hauschild A, Santinami M, et al. Adjuvant dabrafenib plus trametinib in stage III BRAF-mutated melanoma. N Engl J Med. 2017;377(19):1813–1823. | ||
Ribas A, Hodi FS, Lawrence D. 1216O – KEYNOTE-022 update: phase 1 study of first-line pembrolizumab (pembro) plus dabrafenib (D) and trametinib (T) for BRAF-mutant advanced melanoma. Ann Oncol. 2017;28(Suppl 5):V428–V448. | ||
Choo EF, Ng CM, Berry L, et al. PK-PD modeling of combination efficacy effect from administration of the MEK inhibitor GDC-0973 and PI3K inhibitor GDC-0941 in A2058 xenografts. Cancer Chemother Pharmacol. 2013;71(1):133–143. | ||
Ascierto PA, Mcarthur GA, Dréno B, et al. Cobimetinib combined with vemurafenib in advanced BRAF(V600)-mutant melanoma (coBRIM): updated efficacy results from a randomised, double-blind, phase 3 trial. Lancet Oncol. 2016;17(9):1248–1260. | ||
Bendell JC, Kim TW, Goh BC, et al. Clinical activity and safety of cobimetinib (cobi) and atezolizumab in colorectal cancer (CRC). J Clin Oncol. 2016;34(Suppl). Abstract 3502. | ||
Woessner R, Winski S, Rana S, et al. ARRY-162, a potent and selective MEK 1/2 inhibitor, shows enhanced efficacy in combination with other targeted kinase inhibitors and with chemotherapy. Cancer Res. 2010;70(Suppl 8). Abstract 2514. | ||
Lee PA, Wallace E, Marlow A, et al. preclinical development of ARRY-162, a potent and selective MEK 1/2 inhibitor. Cancer Res. 2010;70(Suppl 8). Abstract 2515. | ||
Bendell JC, Javle M, Bekaii-Saab TS, et al. A phase 1 dose-escalation and expansion study of binimetinib (MEK162), a potent and selective oral MEK1/2 inhibitor. Br J Cancer. 2017;116(5):575–583. | ||
Watanabe K, Otsu S, Hirashima Y, et al. A phase I study of binimetinib (MEK162) in Japanese patients with advanced solid tumors. Cancer Chemother Pharmacol. 2016;77(6):1157–1164. | ||
Ascierto PA, Schadendorf D, Berking C, et al. MEK162 for patients with advanced melanoma harbouring NRAS or Val600 BRAF mutations: a non-randomised, open-label phase 2 study. Lancet Oncol. 2013;14(3):249–256. | ||
Dummer R, Schadendorf D, Ascierto PA, et al. Binimetinib versus dacarbazine in patients with advanced NRAS-mutant melanoma (NEMO): a multicentre, open-label, randomised, phase 3 trial. Lancet Oncol. 2017;18(4):435–445. | ||
Dummer R, Ascierto PA, Gogas HJ, et al. Encorafenib plus binimetinib versus vemurafenib or encorafenib in patients with BRAF-mutant melanoma (COLUMBUS): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2018;19(5):603–615. | ||
Planchard D, Smit EF, Groen HJM, et al. Dabrafenib plus trametinib in patients with previously untreated BRAFV600E-mutant metastatic non-small-cell lung cancer: an open-label, phase 2 trial. Lancet Oncol. 2017;18(10):1307–1316. | ||
Falchook GS, Millward M, Hong D, et al. BRAF inhibitor dabrafenib in patients with metastatic BRAF-mutant thyroid cancer. Thyroid. 2015;25(1):71–77. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.