")
Back to Archived Journals » Virus Adaptation and Treatment » Volume 9
Lujo virus: current concepts
Authors Sewlall NH, Paweska JT
Received 19 May 2017
Accepted for publication 20 September 2017
Published 7 December 2017 Volume 2017:9 Pages 41—47
DOI https://doi.org/10.2147/VAAT.S113593
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Jonathan Dinman
Nivesh H Sewlall,1,2 Janusz T Paweska2,3
1Department of Medicine, Morningside MediClinic, Johannesburg, South Africa; 2Department of Medicine, University of the Witwatersrand, Johannesburg, South Africa; 3Department of Medicine, National Institute for Communicable Diseases of the National Health Laboratory Service, Johannesburg, South Africa
Abstract: Lujo virus (LUJV), a novel Old World arenavirus, was found to cause a fulminant viral hemorrhagic fever syndrome in an outbreak in 2008. The primary patient was from Lusaka, Zambia, and subsequent nosocomial transmission occurred to four other patients in Johannesburg, South Africa, hence the name Lujo virus. Like all arenaviruses, LUJV is a segmented, single-stranded, negative-sense RNA virus. Genomic sequencing confirmed that LUJV G1 glycoprotein was novel, diverse and genetically equidistant from other arenaviruses. A clinical syndrome resembling severe, fulminant Lassa virus infection was responsible for a high case fatality rate of 80% (4/5 cases). This review describes briefly the clinical course of the disease, laboratory findings and diagnosis. Recent studies address the current aspects of epidemiology, pathophysiology and treatments, specifically comparing with Lassa virus.
Keywords: Lujo virus, viral hemorrhagic fever, outbreak, epidemiology, pathophysiology, treatment
Introduction
Viral hemorrhagic fever (VHF) is an acute illness characterized by fever and nonspecific symptoms, usually progressing to bleeding, shock and multiorgan failure.
Viruses causing VHF belong to four taxonomic families: Arenaviridae, Filoviridae, Bunyaviridae and Flaviviridae. Arenaviruses comprise over 22 recognized species. These are divided into New World (NW) or Tacaribe Complex and Old World (OW) or Lassa-lymphocytic choriomeningitis complex.1 Various members of both families are known to cause VHF in South America and Africa. Lassa fever is caused by the Lassa virus (LASV), which was the only OW arenavirus known to cause VHF in West Africa.2,38
Lujo virus (LUJV), a novel OW arenavirus, was isolated during an outbreak that occurred in Johannesburg, South Africa, in 2008, after an index patient was airlifted from Lusaka, Zambia, hence the taxonomic “Lujo virus”.3 This is the second pathogenic arenavirus to be identified in Africa.
Outbreak
Clinical descriptions of the outbreak have been extensively reported (Table 1).4 The primary patient (Patient 1) was a 36-year-old white female from Lusaka, Zambia. She lived in an agricultural smallholding in the outskirts of the capital, with horses, dogs, cats and evidence of rodents found in the stables.3
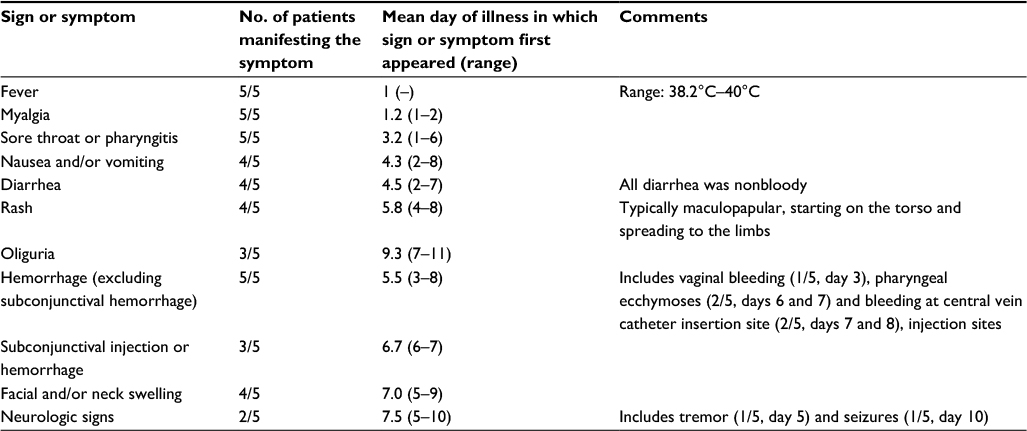 | Table 1 Clinical signs and symptoms of five patients with Lujo hemorrhagic fever Notes: Signs and symptoms are listed in order of appearance during the course of infection. Only the manifestations noted during the first 2 weeks of illness are shown. Adapted from Sewlall NH, Richards G, Duse A, et al. Clinical features and patient management of lujo hemorrhagic fever. PLoS Negl Trop Dis. 2014;8(11):e3233.3 |
She fell ill on September 2, Illness Day 1 (ID-1), with an abrupt onset of fever, myalgia, sore throat and severe headache. This was followed a day later with non-bloody diarrhea and vomiting. A fine, erythematous, macular rash appeared on ID-5 on her torso. Fever was intermittent with spikes up to 39°C and was accompanied by retrosternal chest pain and worsening sore throat. She presented to a local hospital on ID-8, where she received fluids and broad-spectrum antibiotics. She continued to deteriorate with spreading of the rash and facial swelling. She was then intubated with difficulty and prepared for evacuation to a private tertiary hospital in Johannesburg on ID-11.
On admission to the hospital in Johannesburg, she was found to be completely comatose with extensive cerebral edema noted on computed tomography scan (Glasgow Coma Score 3/10). A fine, diffuse macular rash was observed with generalized edema (worse over the face and neck) evident. An eschar resembling a tick bite was visible on her right foot. No overt bleeding was noted. Progressive organ failure ensued despite aggressive supportive care which included mechanical ventilation, continuous hemodialysis and broad-spectrum antimicrobials. Blood tests indicated leukocytosis (27×109/L), thrombocytopenia (42×109/L), elevated hepatic transaminases (aspartate transaminase 1029 IU/L, alanine transaminase 386 IU/L) and a mildly elevated C-reactive protein (27 mg/L). Rapid hemodynamic collapse and death occurred on ID-13.
Patient 2 was a 33-year-old white paramedic who had accompanied Patient 1 on the medical evacuation from Zambia to South Africa. He had participated in her intubation and wore only disposable gloves. He could not recall specific exposure to blood or other bodily fluids. On September 21 (ID-1), which was 9 days after his last contact with the primary case, he developed an abrupt onset fever, headache and myalgias. On ID-4, he was admitted to a local clinic in Lusaka where he was getting treatment for a respiratory tract infection. He subsequently developed a diffuse, erythematous rash, sore throat and myalgias. He was transferred to the same Johannesburg hospital as Patient 1 on ID-7.
He was fully awake initially with a diffuse, maculopapular rash and severe subconjunctival hemorrhage. Facial and neck swelling followed with palatal ecchymoses. Laboratory examination revealed leukopenia (2×109/L), thrombocytopenia (52×109/L), hepatic transaminitis and positive d-dimer (>10 mg/L). Investigations proved negative for malaria, rickettsiae and salmonella. Following a presumptive diagnosis of thrombotic thrombocytopenic purpura, plasmapheresis was initiated.
He was seen on ID-9 by the Intensive Care Unit (ICU) physician who had cared for Patient 1, and an epidemiologic link was made. Precautions for VHF were immediately implemented, and given the history, a possible filovirus was suspected. Contact tracing began by the hospital infection control team. The patient died on ID-12.
Patient 3 was a 34-year-old black nurse who was involved in the care of Patient 1. She became ill on September 25 (ID-1), 9 days after caring for Patient 1. She was involved in turning and cleaning the patient. This included washing the corpse and removing the dialysis catheter. No breaches in protective equipment (gown, mask, latex gloves and plastic visor) were reported. She was found ill by the contact tracing team at a hospital west of Johannesburg close to her home. The patient had a similar clinical sequence and was given initial doses of oral ribavirin. She demised on ID-10.
Patient 4 was a 38-year-old black female with a history of advanced human immunodeficiency virus infection (CD4 count 250/μL), who worked as a cleaner involved in the terminal disinfection of the hospital room of Patient 1. Cleaning was performed with a scrub gown, surgical mask, latex gloves and a plastic visor. No exposure to body fluids was reported. She fell ill on September 27 (ID-1), 13 days after cleaning the room. A similar clinical picture ensued, and she was admitted to a local hospital on ID-6. Empiric treatment for tuberculosis was started, but she continued to deteriorate. Contact tracing located her on ID-9, and she was transferred to a tertiary academic hospital. She deteriorated and died on ID-10.
Patient 5 was a 47-year-old white female who worked as an ICU nurse caring for Patient 2 from September 27 to 29. She was exposed to vomitus and bloody dressings. She reported wearing plastic aprons, masks and disposable gloves, although potential lapses in the consistent use of these were possible. She was placed on twice daily temperature monitoring, as were all contacts. On October 10 (ID-1), she developed fever (38.4°C) with headache and nausea. Initial blood tests demonstrated thrombocytopenia (91×109/L), leukopenia (1.3×109/L) and elevated D-dimer (2.84 mg/L). A diagnosis of probable VHF was made (2 days prior to an etiologic agent being identified). Oral ribavirin was commenced (2 g loading; 1 g q 6 hours). At that time, intravenous (IV) ribavirin was not available. Concurrent treatment included atorvastatin (80 mg qd) and N-acetylcysteine, primarily for their immunomodulatory and anti-inflammatory properties.5,6
Her clinical scenario was similar to the other patients described. Persistent fever, myalgias, facial swelling and pharyngitis were prominent. Thrombocytopenia and hepatic transaminitis worsened. IV recombinant factor VIIa was given (1.2 mg q 6 hours) and N-acetylcysteine was switched to IV administration (1 g q 8 hours). She continued to deteriorate hemodynamically and developed respiratory failure. Following a difficult intubation due to airway swelling, mechanical ventilation continued. A supply of IV ribavirin was obtained and commenced on ID-8 (20 mg/kg q 6 hours), which was being switched for the oral drug. The course was completed on ID-18.
Subconjunctival hemorrhages, as well as the distinctive rash, appeared with slow improvement noted clinically. Weaning from mechanical ventilation occurred on ID-15. Subsequent issues were related to cardiac failure (presumed myocarditis), development of hepatosplenomegaly (ID-15) and distal neuropathic weakness (ID-13).
Slow recovery was noted with hospital discharge on ID-42. Long-term problems seemed predominantly neurologic, which included anxiety, mood fluctuation, peripheral neuropathy, myopathy and tremors. Complete, non-scarring alopecia developed from ID-83 and resolved over a 4-month period.
In summary, the five patients’ ages ranged from 33 to 47 years. They were two white females, two black females and one white male. The incubation periods ranged from 9 to 13 days. Four deaths occurred from the five cases (case fatality 80%).
Signs and symptoms
The illness began with an abrupt onset of nonspecific symptoms including fever, malaise, headache and myalgias. Sore throat, nausea, vomiting and non-bloody diarrhea followed with variable retrosternal or epigastric pain. A blanching, erythematous, maculopapular rash appeared toward the end of the first week which spread from the torso to the limbs, but sparing the palms and soles. In the survivor, this rash coalesced and then disappeared by ID-14.
Facial swelling with subconjunctival hemorrhage appeared soon after the rash. Neurologic signs tended to occur later in the disease with tremors, seizures and cerebral edema noted on computed tomography scan. Hepatosplenomegaly developed in the survivor between ID-15 and ID-40. No major hemorrhage was noted, but subconjunctival hemorrhages, palatal ecchymoses and injection site bleeding were noted. Rapid deterioration with multiorgan failure developed between ID-7 and ID-10. Death occurred at a mean of 9 days (range: 6–12) in the fatalities. Convalescence was prolonged in the survivor.
Laboratory findings
Typical features included leukopenia with lymphopenia. Leukocytosis occurred later with thrombocytopenia. Hepatic transaminases were elevated (aspartate transaminase 2–3 times greater than alanine transaminase), with an elevated lactate dehydrogenase. Elevated D-dimer levels and prolonged partial thromboplastin time were noted in three patients, suggestive of disseminated intravascular coagulopathy. Renal function was remarkably stable initially with normal (or marginally elevated) C-reactive protein and procalcitonin.
Diagnosis
Blood samples from Patient 2 (September 29) and Patient 3 (October 3) were screened at the National Institute for Communicable Disease of the National Health Laboratory Service (NICD-NHLS) in Johannesburg by reverse transcription-polymerase chain reaction, antigen and antibody detection enzyme-linked immunosorbent assays for known agents of African VHF, including Lassa Fever Virus, and all tested negative. These included primers to detect all known OW arenaviruses.7,8
Samples of liver biopsy and skin pinch biopsy were taken on October 9 in a biosafety level four facility at NICD from Patients 2 and 3. While laboratory tests on collected biopsies continued in South Africa, specimens were sent concurrently to the Centers for Disease Control and Prevention (CDC, Atlanta, GA, USA) and Columbia University (New York, NY, USA).4 On October 10, the Department of Anatomical Pathology at the University of the Witwatersrand described hepatocyte necrosis and skin vasculitic changes in keeping with VHF histopathology. On October 11, the Infectious Disease Pathology Branch, CDC, detected antigen in the liver and skin using immunohistochemical stains with monoclonal antibody broadly cross-reactive with OW arenaviruses. As a result, generic reverse transcription polymerase chain reaction procedures for detecting OW arenaviruses were applied to the liver samples, and they provided positive results simultaneously at special pathogens unit NICD/NHLS and Special Pathogens Branch, CDC. Phylogenetic analyses at special pathogens unit NICD/NHLS indicated that the agent was not LASV, but likely a novel, previously unreported OW arenavirus. Specimens sent to Columbia University underwent 454-pyrosequencing and produced full-length genome sequences of the arenavirus. This demonstrated that the LUJV G1 glycoprotein was novel, highly diverse and genetically equidistant from other OW and NW arenaviruses.9,33
Epidemiology
Arenaviruses are segmented, single-stranded, negative-sense RNA viruses. Most of these cause chronic infection in rodents with viral excretion in urine. Contamination of food or household items leads to human infection.10 Transmission can also occur via inoculation with infected body fluids.11 Several NW arenaviruses in South America can cause VHF, including Junin, Machupo, Sabia, Chapare and Guanarito. OW arenaviruses include the prototypic lymphocytic choriomeningitis virus (LCMV), which is associated with pet rodents. Infection ranges from asymptomatic to severe meningoencephalitis in humans. Dandenong virus was described in Australia from organ transplant recipients of an index donor and looks closely related to LCMV.12
The clinical disease of LUJV appears strikingly similar to Lassa fever.1 Lassa is usually associated with mild disease with a mortality estimated to be around 5% when considering community infection. Up to 20%–30% of hospitalized cases are severe or fatal.36,37 Nosocomial outbreaks have indicated up to 50% mortality.13 The known reservoir is the mouse Mastomys natalensis and is seen to cause large numbers of infection in West Africa.
OW arenaviruses not known to cause human infection include Ippy, Mobala and Morogoro.14 Two studies have investigated the natural host of LUJV in Zambia.15,16 Among the captured animals, 24 arenavirus-positive rodents were identified, most of which were captured in periurban environments. LUJV was not identified; however, two other novel arenaviruses were detected: Lusaka-Namwala Virus and Lusaka-New Kasama Virus. The pathogenic propensity to human infection is not known.
In essence, the distribution and prevalence of LUJV in humans and rodents is unknown, as are the ecology, distribution, and mode of transmission from reservoir host to humans. Of note was a tick bite-like eschar on the foot of the primary patient, which may or may not have been significant. The nosocomial spread and presumed high viremic load of body fluids may have impacted the high case fatality rate. Typical rates of LASV transmissibility seem to be low, but higher rates are noted in outbreaks where barrier nursing may not have been maintained.11,17,35
Pathogenesis
Our understanding of the molecular biology of arenavirus assembly, budding, life cycle and cellular interaction is evolving and is the subject of ongoing research.18
Arenaviruses are enveloped with a bisegmented negative- strand RNA genome. The S (small) segment encodes the viral nucleoprotein and glycoprotein precursor (GPC). The L (large) segment encodes the viral matrix protein (Z) and protein L, an RNA-dependent RNA polymerase. The host cell surface protein α-dystroglycan is widely expressed. This is the main receptor for the OW arenaviruses, LASV and LCMV.19 In contrast, human transferrin receptor 1 is the primary receptor for pathogenic NW arenaviruses.20 Following binding to the respective receptor, endocytosis of the virions into the infected cell cytoplasm occurs. The release of viral ribonucleoprotein intracellularly triggers viral ribonucleoprotein-associated RNP polymerase activity, which in turn induces gene transcription and viral RNA replication.
A recent study described the development of pseudotype viruses (vesicular stomatitis virus) bearing arenavirus envelope proteins as a surrogate virus system. It was demonstrated that pseudotype vesicular stomatitis virus bearing LUJV envelope protein induced infection independently of α-dystroglycan and human transferrin receptor 1, which indicates that LUJV utilizes an unidentified host cell receptor.21
Further study of the mechanisms of viral release indicated that the process is driven by matrix protein (Z) and GPC. It was found that the L domain, which is a short amino acid motif in Z, interacts with cellular factors to mediate viral particle budding. However, the cellular factors required for LUJV budding were different from those required for LASV. Furthermore, GPC was found to be cleaved by site 1 protease (S1P). As a follow-on experiment, treatment with an S1P inhibitor (PF-429242) seemed to reduce LUJV production.22
Treatment
Organized, randomized trials of treatments of VHF are scarce. Treatment protocols usually represent a composite view of expert opinion. For the most part, supportive therapy is the fundamental basis of treatment.23 It is presumed that the pathophysiologic basis of VHF shares similarity with multiorgan failure associated with severe sepsis/systemic inflammatory response. This is in part mediated by various proinflammatory cytokines, mediators and nitric oxide dysregulation.24
Ribavirin is a purine nucleoside analog, licensed as an antiviral agent in chronic hepatitis C and respiratory syncytial virus infections. Its mechanism of action broadly includes inhibition of RNA polymerase and inhibition of initiation/elongation of RNA fragments. It is widely considered the most efficacious drug in arenavirus infections, including LASV and other NW arenaviruses.25–28 The mechanism of action in arenavirus infections is not entirely clear, but it seemingly targets different steps in the life cycle.29
In the LUJV outbreak, oral ribavirin was briefly attempted in Patient 3, and then early in the illness of Patient 5 (ID-1; 2 g loading and 1 g q 6 hours thereafter). The oral bioavailability of ribavirin is reported at ~60% and is subject to significant first-pass metabolism. It has also not been shown to be definitively effective in arenavirus infection. Patient 5 continued to deteriorate on oral therapy, with IV doses commencing on ID-8 (20 mg/kg q 6 hours). At this point, it would be remiss of us to completely disregard the efficacy of ribavirin for treating LUJV infection.
A project has described the mutagenic potential of ribavirin to LCMV, the premise being that an increase in the rate of mutation would increase viral extinction (lethal mutagenesis). This has been demonstrated at subinhibitory concentrations of ribavirin.30 This would suggest potential for combination or adjunctive therapies.
Various adjunctive therapies were attempted (mainly in Patient 5) with demonstrable or theoretic efficacy in severe sepsis or inflammation. These included statins (HMG-CoA reductase inhibitors), N-acetylcysteine and recombinant factor VIIa.4
Further supportive measures included mechanical ventilation, hemodialysis and plasmapheresis (Patient 2). These are invasive procedures, requiring access to sophisticated ICUs. They also represent significant exposure to blood/body fluids, and we are suspicious of this being a potential cause of the high secondary attack rate in this outbreak.
Ongoing projects defining cellular mechanisms will provide potential targets of therapy, as shown in inhibition of S1P with PF-42924222 and U18666A, a cholesterol transport inhibitor that inhibited LUJV envelope protein infectivity.21
Discussion
VHF has typically been relegated to footnotes in medical texts, with sporadic, deadly outbreaks being identified since the 1960s. The high case fatality rates were seen as a prime reason for rapid “burnout” of disease outbreaks. However, the recent Ebola virus disease (EVD) epidemic in 2013–2016 had far-reaching consequences, with EVD being declared a public health emergency of international concern in West Africa by the World Health Organization. Ebola virus was previously identified to cause outbreaks of EVD only in central equatorial Africa but emerged in West Africa in late 2013 in Guinea and spread into Liberia, Sierra Leone, Nigeria and Senegal. For many months, EVD cases in Guinea were misdiagnosed due to the similarity of symptoms with other more common disease conditions and lack of laboratory capacity to establish a specific diagnosis. Cases in the USA and Spain encouraged heightened mainstream media coverage. Of significance was the initial sluggish, disorganized and poorly funded global response.31
The key determinant in the diagnosis of VHF (and subsequent outbreaks) remains a high level of clinical suspicion. This is especially so, given the nonspecific nature of initial complaints. The primary case was from wealthy suburban Lusaka, where no prior arenavirus (or any other VHF virus) had been identified. The differential diagnosis of a febrile illness with rash is also quite diverse. Further confusion ensued when after suspicion of VHF was raised, a negative screen for VHF was documented. Nevertheless, a persistently high index of suspicion was maintained, which ensured that contact tracing and infection control measures remained in place.
The clinical features of suspected VHF require a wide range of differential diagnosis concerning both infectious (parasitic, viral, bacterial) and noninfectious disease conditions, including malaria, rickettsial infection, typhoid, Q fever, tularemia, leptospirosis, brucellosis, viral hepatitis, bacterial sepsis, other VHF, autoimmune vasculitis and hematologic disasters such as acute leukemia. Entertaining all these diagnoses is clinically cumbersome and with inherent risks concerning infection control. Although superfluous, it bears mentioning that detailed history taking and clinical examination remains the cornerstone of medicine. Basic infection control practices which should be routine for nosocomial prevention would suffice for VHF patients. The narrowing of the differential diagnosis would depend on the progress of disease, specific laboratory tests and epidemiologic data.34
The complexity of identifying a novel agent has been described,9 and this delay between suspicion and confirmation certainly would have had greater impact on secondary transmission. Even with a high index of suspicion, three cases of nosocomial transmission occurred despite measures being implemented to ostensibly prevent VHF transmission. Despite exhaustive enquiry, no specific exposure to infected fluids or blood was noted (splashes/needle stick injury). This prompts reassessment of adherence not only to strict barrier practices and wearing of personal protective equipment which includes gloves, gown and mask, but also to effective and safe removal of the personal protective equipment.
Potential outbreak scenarios are also hampered by poor preparation of logistic protocols. Auxiliary staff (including cleaning, catering, laboratory, radiology and transport personnel) had to be hastily considered and organized, with no clear guideline protocols in place. Contact tracing included them in the surveillance, which rapidly increased the total number of contacts to 94.4
Surveillance studies of rodents in Lusaka did not identify a reservoir of LUJV, but the identification of two novel arenaviruses illustrates a worrying possibility.15,16 Rapidly expanding urbanization, travel and accessibility of previously remote areas increase the possibility of further cases of novel agents being found.
It is hoped that increasing awareness among health care systems globally and implementation of broad-based organized structures would impact earlier detection and containment of future outbreaks.
Further studies into the molecular and cellular mechanisms of viral pathogenicity would hopefully elucidate further therapeutic targets. In this vein, therapeutic trials in animal models (such as 13/N strain guinea pigs) may be of value.32
Acknowledgment
The authors thank Dr Daniel Bausch for his invaluable earlier contributions and critical review of the manuscript.
Disclosure
The authors report no conflicts of interest in this work.
References
Enria DA, Mills JN, Bausch D, Shieh W, Peters CJ. Arenavirus Infections. In: Guerrant RL, Walker DH, Weller PF, editors. Tropical Infectious Diseases:Pprinciples, Pathogens, and Practice. 3rd ed. Philadelphia, PA: Churchill Livingstone; 2011:449–461. | ||
Buckley SM, Casals J. Lassa fever, a new virus disease of man from West Africa. 3. Isolation and characterization of the virus. Am J Trop Med Hyg. 1970;19(4):680–691. | ||
Sewlall NH, Richards G, Duse A, et al. Clinical features and patient management of lujo hemorrhagic fever. PLoS Negl Trop Dis. 2014;8(11): e3233. | ||
Paweska JT, Sewlall NH, Ksiazek TG, et al; Outbreak Control and Investigation Teams. Nosocomial outbreak of novel arenavirus infection, Southern Africa. Emerg Infect Dis. 2009;15(10):1598–1602. | ||
Merx MW, Liehn EA, Janssens U, et al. HMG-CoA reductase inhibitor simvastatin profoundly improves survival in a murine model of sepsis. Circulation. 2004;109(21):2560–2565. | ||
Hackman DG, Mamdani M, Li P, Redelmeier DA. Statins and sepsis in patients with cardiovascular disease: a population based cohort analysis. Lancet. 2006;367(9508):413–418. | ||
Clergy JC, Wilson SM, Oram JD. Nucleotide sequence of the S RNA of Lassa virus (Nigerian strain) and comparative analysis of arenavirus gene products. Virus Res. 1991;18(2–3):151–164. | ||
Demby AH, Chamberlain J, Brown DW, Clegg CS. Early diagnosis of lassa fever by reverse transcriptase-PCR. J Clin Microbiol. 1994;32(12):2898–2903. | ||
Briese T, Paweska JT, McMullan LK, et al. Genetic detection and characterization of lujo virus, a new hemorrhagic fever-associated arenavirus from Southern Africa. PLoS Pathog. 2009;5(5):e1000455. | ||
Buchmeier MJ, de la Torre JC, Peters CJ. Arenaviridae: the viruses and their replication. In: Knipe DM, Howley PM, editors. Fields Virology. 5th ed. Philadelphia, PA: Wolters Kluwer Health/Lippincott Williams & Wilkins; 2007:1791–1827. | ||
Fischer-Hoch SP, Tomori O, Nasidi A, et al. Review of cases of nosocomial lassa fever in Nigeria: the high price of poor medical practice. BMJ. 1995;311(7009):857–859. | ||
Palacios G, Druce J, Du L, et al. A new arenavirus in a cluster of fatal transplant-associated diseases. N Eng J Med. 2008;358(10):991–998. | ||
Frame JD, Baldwin JM Jr, Gocke DJ, Troup JM. Lassa fever, a new virus disease of man from West Africa. 1. Clinical description and pathological findings. Am J Trop Med Hyg. 1970;19(4):670–676. | ||
Charred RN, de Lamballerie X, Emonet S. Phylogeny of the genus Arenavirus. Curr Opin Microbiol. 2008;11(4):362–368. | ||
Ishii A, Thomas Y, Moonga L, et al. Novel Arenavirus, Zambia. Emerg Infect Dis. 2011;17(10):1921–1924. | ||
Ishii A, Thomas Y, Moonga L, et al. Molecular surveillance and phylogenetic analysis of Old World arenaviruses in Zambia. J Gen Virol. 2012;93(Pt 10):2247–2251. | ||
Troup JM, White HA, Fom AL, Carey DE. An outbreak of Lassa fever on Jos Plateau, Nigeria, in January – February 1970. A preliminary report. Am J Trop Med Hyg. 1970;19(4):695–696. | ||
Urata S, Yasuda J. Molecular mechanisms of arenavirus assembly and budding. Viruses. 2012;4(10):2049–2079. | ||
Cao W, Henry MD, Borrow P, et al. Identification of alpha-dystroglycan as a receptor for lymphocytic choriomeningitis virus and lassa fever virus. Science. 1998;282(5396):2079–2081. | ||
Radoshitzky SR, Abraham J, Spiropoulou CF, et al. Transferrin receptor 1 is a cellular receptor for New World haemorrhagic fever arenaviruses. Nature. 2007;446(7131):92–96. | ||
Tani H, Iha K, Shimojima M, et al. Analysis of Lujo virus cell entry using pseudotype vesicular stomatitis virus. J Virol. 2014;88(13):7317–7330. | ||
Urata S, Weber J, Storm N, et al. Analysis of assembly ad budding of lujo virus. J Virol. 2015;90(6):3257–3261. | ||
Bausch DG. Viral hemorrhagic fevers. In: Goldman’s Cecil Medicine. Goldman L, Schafer A, editors. 24th ed. Philadelphia, PA: Elsevier Saunders; 2011:2704 | ||
Bausch DG, Sprecher AG, Jeffs B, Boumandouki P. Treatment of marburg and ebola hemorrhagic fevers: a strategy for testing new drugs and vaccines under outbreak conditions. Antiviral Res. 2008;78(1):150–161. | ||
McCormick JB, King IJ, Webb PA, et al. Lassa fever. Effective therapy with ribavirin. N Engl J Med. 1986;314(1):20–26. | ||
Kilgore PE, Ksiazek TG, Rollin PE, et al. Treatment of bolivian hemorrhagic fever with intravenous ribavirin. Clin Infect Dis. 1997;24(4):718–722. | ||
Barry M, Russi M, Armstrong L, et al. Brief report: treatment of a laboratory-acquired Sabia virus infection. N Engl J Med. 1995; 333(5):294–296. | ||
Enria DA, Briggiler AM, Sanchez Z. Treatment of Argentine hemorrhagic fever. Antiviral Res. 2008;78(1):132–139. | ||
Parker WB. Metabolism and antiviral activity of ribavirin. Virus Res. 2005;107(2):165–171. | ||
Moreno H, Gallego I, Sevilla N, de la Torre JC, Domingo E, Martín V. Ribavirin can be mutagenic for arenaviruses. J Virol. 2011;85(14):7246–7255. | ||
Kalra S, Kelkar D, Galwankar SC, et al. The emergence of ebola as a global health security threat: from “lessons learned” to coordinated multilateral containment efforts. J Glob Infect Dis. 2014;6(4):164–177. | ||
Bird BH, Dodd KA, Erickson BR, et al. Severe hemorrhagic fever in strain 13/N guinea pigs infected with Lujo virus. PLoS Negl Trop Dis. 2012;6(8):e1801. | ||
Paweska JT, Jansen van Vuuren P, Weyer J. Lujo Virus Hemorrhagic Fever. In: Viral Hemorrhagic Fevers. Singh SK, Ruzek D, editors. 1st ed. CRC Press London; 2013:287–305. | ||
Paweska JT. Lujo virus hemorrhagic fever. In: Clinical Case Study of Emerging Infectious Diseases. O Ergonul, F Can, M Akova, L Madoff, editors. Elsevier/Academic Press London; 2014:95–110. | ||
Anderson KG, Shapiro BJ, Matranga CB, et al. Clinical sequencing uncovers origins and evolution of lassa virus. Cell. 2015;162(4):738–750. | ||
McCormick JB, King IJ, Webb PA, et al. A case-control study of the clinical diagnosis and course of Lassa fever. J Infect Dis. 1987;155(3):445–455. | ||
McCormick JB, Webb PA, Krebs JW, Johnson KM, Smith ES. A prospective study of the epidemiology and ecology of Lassa fever. J Infect Dis. 1987;155(3):437–444. | ||
Ogbu O, Ajuluchukwa E, Uneke CJ. Lassa fever in West-African sub-region: an overview. J Vector Borne Dis. 2007;44(1):1–11. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.