")
Back to Journals » Drug Design, Development and Therapy » Volume 10
Looking for inhibitors of the dengue virus NS5 RNA-dependent RNA-polymerase using a molecular docking approach
Authors Galiano V , Garcia-Valtanen P, Micol V, Encinar JA
Received 16 July 2016
Accepted for publication 17 August 2016
Published 11 October 2016 Volume 2016:10 Pages 3163—3181
DOI https://doi.org/10.2147/DDDT.S117369
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Frank Boeckler
Vicente Galiano,1 Pablo Garcia-Valtanen,2 Vicente Micol,3,4 José Antonio Encinar3
1Physics and Computer Architecture Department, Miguel Hernández University (UMH), Elche, Spain; 2Experimental Therapeutics Laboratory, Hanson and Sansom Institute for Health Research, School of Pharmacy and Medical Science, University of South Australia, Adelaide, Australia; 3Molecular and Cell Biology Institute, Miguel Hernández University (UMH), Elche, Spain; 4CIBER: CB12/03/30038, Physiopathology of the Obesity and Nutrition, CIBERobn, Instituto de Salud Carlos III, Palma de Mallorca, Spain
Abstract: The dengue virus (DENV) nonstructural protein 5 (NS5) contains both an N-terminal methyltransferase domain and a C-terminal RNA-dependent RNA polymerase domain. Polymerase activity is responsible for viral RNA synthesis by a de novo initiation mechanism and represents an attractive target for antiviral therapy. The incidence of DENV has grown rapidly and it is now estimated that half of the human population is at risk of becoming infected with this virus. Despite this, there are no effective drugs to treat DENV infections. The present in silico study aimed at finding new inhibitors of the NS5 RNA-dependent RNA polymerase of the four serotypes of DENV. We used a chemical library comprising 372,792 nonnucleotide compounds (around 325,319 natural compounds) to perform molecular docking experiments against a binding site of the RNA template tunnel of the virus polymerase. Compounds with high negative free energy variation (ΔG <-10.5 kcal/mol) were selected as putative inhibitors. Additional filters for favorable druggability and good absorption, distribution, metabolism, excretion, and toxicity were applied. Finally, after the screening process was completed, we identified 39 compounds as lead DENV polymerase inhibitor candidates. Potentially, these compounds could act as efficient DENV polymerase inhibitors in vitro and in vivo.
Keywords: virtual screening, molecular docking, high-throughput computing, AutoDock/Vina, ADMET, SuperNatural database, inhibitors, NS5 RNA-dependent RNA polymerase
Introduction
Members of the Flaviviridae family cause a large variety of diseases in humans and other animal species. Flaviviruses can be transmitted from animals to humans by arthropod vector species such as ticks and mosquitoes. For example, the mosquito Aedes aegypti can transmit the chikungunya, yellow fever, and Zika viruses.1 Also, humans can be infected by contact with infected blood.2 The family Flaviviridae includes four main genera, Flavivirus, Pestivirus, Hepacivirus, and Pegivirus, as well as some unclassified viruses.3 The genus Flavivirus comprises 67 viruses, several of which infect humans, for instance, dengue virus (DENV), Japanese encephalitis virus, yellow fever virus, West Nile virus, and tick-borne encephalitis virus.2 DENV poses a major risk for human health. It is estimated that half of the population of the world is at risk of becoming infected with one serotype of DENV or another.4 Recent data indicated that in 2013, DENV caused 40–58 million symptomatic infections, including 13,586 fatal cases, with an associated financial cost of US$ 8.9 billion.5 DENV infection in humans develops differently in each case. Globally, in 2013, 18% of DENV-infected patients were admitted to hospital, 48% received medical attention or advise outside a hospital, and 34% did not need, find, or seek medical attention.5 Clinical manifestations of DENV disease range from mild dengue fever to severe dengue hemorrhagic fever and dengue shock syndrome. In late 2015 and early 2016, the first dengue vaccine, Dengvaxia (CYD-TDV), manufactured by Sanofi Pasteur (Lyon cedex, France), was registered in several countries for its use in individuals aged between 9 and 45 and living in DENV-endemic areas.6 However, despite its huge impact on public health around the world, effective antiviral therapies against DENV and other flaviviruses have not been developed yet.
The DENV virus genome is made up of a single strand of positive-sense (ie, it is directly translated into protein) RNA, which is copied in endoplasmic reticulum membrane-associated replication complexes in the host cell. Such complexes contain the DENV nonstructural proteins 3 and 5 (NS3 and NS5) along with host proteins.7 The genomic RNA contains a 5′ untranslated region, a single open reading frame, and another 3′ untranslated region. The open reading frame encodes a polyprotein that is processed by viral and host proteases generating ten mature viral proteins: three structural proteins (capsid [C], membrane [M], and envelope [E]) and seven nonstructural proteins (NS1, NS2A, NS2B, NS3, NS4A, NS4B, and NS5).8
NS5 (900 amino acids) exerts three different enzymatic activities.9 The MTase activity protects viral mRNA from degradation by 5′-exoribonucleases and ensures their recognition by eukaryotic translation initiation factor.10 The N-terminal MTase domain (residues 1–262, guanosine-5′-triphosphate-binding site, S-adenosylmethionine binding site, and NS3 protease interacting site) is connected through a ten-residue linker to the C-terminal RNA-dependent RNA polymerase (RdRp) domain formed by residues 273–900.11 In turn, the RdRp domain is formed by three subdomains called fingers (273–315, 416–496, and 543–600), palm (497–542, 601–705), and thumb (706–900).12 The active center of the enzyme is defined by the conserved GDD protein motif located in the palm domain. The amino acid sequence between residues 320 and 405 includes two nuclear localization sequences that are recognized by cellular factors, allowing protein transport to the nucleus. In conjunction with other viral proteins and host cell proteins, the NS5 polymerase domain promotes the formation of double-stranded RNA intermediates, both positive and negative sense. Negative-sense strands serve as templates for the DENV RdRp to synthesize new positive-sense genomic RNA.13,14
RNA viruses, such as DENV, are the only organisms that possess enzymes with RdRp activity that is essential for their replication. The lack of RdRps in human cells as wells as in other animal species makes these polymerases an attractive antiviral target.15 Current polymerase inhibitors belong to one of two types of molecules: first, nucleoside/nucleotide analogs (NIs) that function as RNA chain terminators16 and second, nonnucleoside inhibitors which bind to different sites of the catalytic binding site of the enzyme, blocking the conformational switch from polymerase initiation to elongation or impeding the processivity of polymerase elongation.17 In the literature, we can find examples of shortfalls and side effects of some of the NIs against the DENV polymerase.18 Sometimes due to the lack of selectivity, NIs may have off-target effects on other polymerases such as mitochondrial DNA polymerase-γ, resulting in mitochondrial toxicity.19,20 The first reported allosteric inhibitor of DENV RdRp (NITD-1 compound, IC50 =7.2 μM) was identified by means of high-throughput screening using a primer extension-based assay; the second was modified at the N-sulfonylanthranilic acid lead (NITD-2 compound, IC50 =0.7 μM)21 to selectively inhibit DENV-2 polymerase. NITD-29, a photoreactive derivative of NITD-2 (IC50 =1.5 μM), has been used to map the compound-binding site on the protein and, therefore, define its regulatory allosteric site.21 Mass spectrometry of the ultraviolet cross-linked NITD-29/NS5 complex and molecular docking simulations have helped to further define the allosteric binding pocket near the residues Arg737, Thr413, and Met343 located at the RNA template tunnel. Thus, in light of these findings, it could be assumed that the inhibitory effect of these compounds is due to their ability to compete with the RNA template in the RNA template tunnel.21 Unfortunately, these three compounds are inactive in cell culture.21
In this context, we performed an in silico study to find new potential NS5 RdRp efficient agonists from a chemical library built with molecules sharing ≥70% structural similarity with NITD-1, NITD-2, and NITD-29 (~47,473 compounds recorded in the PubChem database22) together with another chemical library of natural products (~325,319 compounds recorded in the SuperNatural II database23). The best scoring compounds were compared with NITD inhibitors based on their ability to bind to DENV NS5 RdRp at the RNA template tunnel. In addition to this, compounds that showed the best binding scores were also evaluated for their pharmacodynamics, pharmacokinetics, and toxicity properties. At the end of the study, 39 compounds passed the selection process. These compounds scored better than NITD-1, NITD-2, and NITD-29, which suggests that these 39 compounds could overcome some of the problems experienced with NITD molecules, such as their inability to inhibit NS5 RdRp in live cell systems. Our in silico approach provides the basis for subsequent in vitro and in vivo studies to test these newly identified DENV NS5 RdRp inhibitor candidates.
Materials and methods
Protein structures for Dengue NS5 RdRp and chemical libraries
To date, several crystal structures of DENV-3 NS5 RdRd protein have been solved and deposited in the Protein Data Bank (4V0Q,11 4V0R,11 5DTO,24 2J7U,12 2J7W,12 3VWS,25 4C11,26 4HHJ25). However, in all structures, several residues were missing. Herein, the three-dimensional (3D) atomic coordinates for these amino acid sequences were obtained by constructing a model, using a homology modeling approach, for the RdRps of each of the four serotypes of DENV. Three 3D theoretical models of each UniProt dengue NS5 RdRp were built at Swiss-Model server27 using the crystallographic structures of 4C11, 3VWS, 4V0Q, and 5DTO (Table 1) as templates. Arg737, Thr413, and Met343 or equivalent positions in the different protein sequences have been used to define the ligand-binding site.21 For each model, water molecules, ions, or inhibitors were removed.
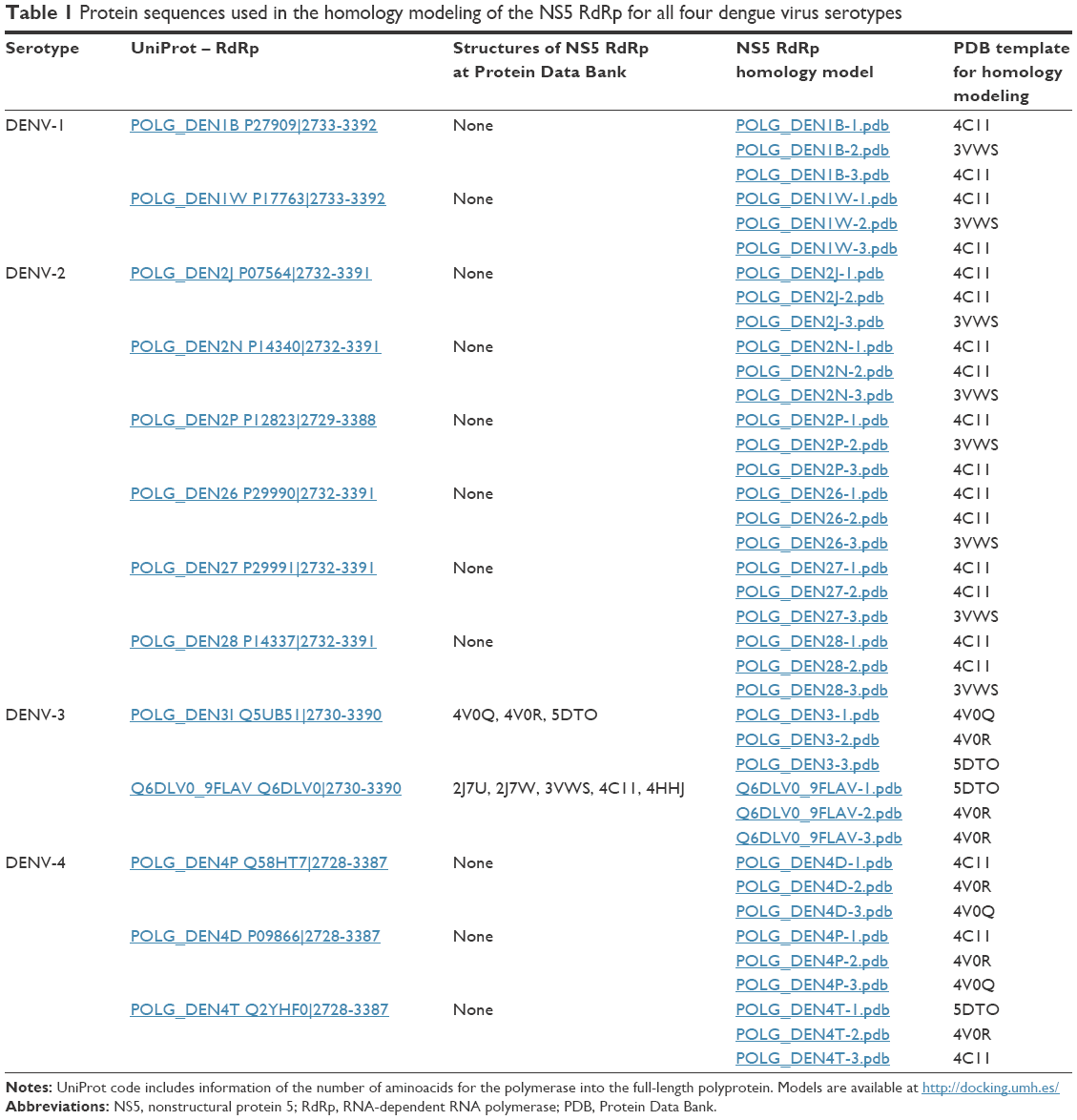 | Table 1 Protein sequences used in the homology modeling of the NS5 RdRp for all four dengue virus serotypes |
The two-dimensional (2D) structures of 47,473 compounds from the PubChem database22 and 325,319 molecules of natural compounds from the SuperNatural II database23 were downloaded in spatial data file format. To handle the large number of ligand structures, we used a script28 to convert 2D spatial data files into individual 3D structures in mol2 format using Marvin Suite 6.0 from ChemAxon (http://www.chemaxon.com). Mol2 files were converted into pdbqt format using the Python script “prepare_ligand4.py” included in the AutoDockTools-1.5.7.rc1.29
Docking procedure
Prior to initiating the docking procedure, the protein (receptor) and ligand structures should be prepared.28 All of the NS5 RdRp modeled protein structures were then subjected to geometry optimization using the repair function of the FoldX algorithm.30 We perform docking with AutoDock/Vina31 using a grid with dimensions of 24×24×24 points centered to residues Arg737, Thr413, and Met343 or equivalent positions in the different protein sequences. AutoDock/Vina was set up on a Linux cluster at calendula.fcsc.es (Linux cluster ROCKS 6.1 distribution) (Castilla y Leon Supercomputing Center), lusitania2.cenits.es Linux cluster (Research, Technological Innovation and Supercomputing Center of Extremadura [CenitS]), and login-hpc.ceta-ciemat.es Linux cluster (Extremadura Research Centre for Advanced Technologies [CETA-CIEMAT]). Compounds with the lowest calculated free energy variations (ie, the best theoretical binding energy) were selected as putative modulators.
In silico analysis of pharmacokinetic parameters and toxicity potential properties
Molecular descriptors, such as the topological polar surface area (TPSA), molecular weight (MW), the estimated logarithm (base 10) of the solubility measured in mol/L (logS), the estimated logarithm (base 10) of the partition coefficient between n-octanol and water (logP), the number of hydrogen bond acceptors, the number of hydrogen bond donors, the violations of Lipinski’s rule of five32 (Ro5 violations), the drug score (DrugScore), and the fragment-based druglikeness (Druglikeness), were calculated using DataWarrior v4.2.2 software (Allschwil, Switzerland).33 The in silico absorption, distribution, metabolism, excretion, and toxicity (ADMET) properties of all compounds were calculated using admetSAR34 and DataWarrior v.4.2.2 software (mutagenicity, tumorigenicity, irritancy, and reproductive effects).
Results and discussion
Homology modeling of NS5 RdRp for all DENV serotypes
The DENV-3 NS5 RdRp X-ray crystallographic structure is the only available in Protein Data Bank (Table 1). Three structures exist for the sequence POLG_DEN3I (UniProt code Q5UB51, sequence 2,730–3,390), that is, 4V0Q, 4V0R, and 5DTO, and five for Q6DLV0_9FLAV (UniProt code Q6DLV0, 2,730–3,390), that is, 2J7U, 2J7W, 3VWS, 4C11, and 4HHJ. None of these structures include all of the residues in RdRp domain. For example, 4V0Q does not include residues 405–415; 453–467, 4V0R residues 404–415; 453–466, and 5DTO residues 405–414; 453–468. Likewise, 2J7U does not contain residues 309–314; 404–416; 449–467, 2J7W residues 308–314; 406–417; 449–469, 3VWS residues 312–315; 342–348; 454–462, 4C11 residues 405–415; 453–470, and 4HHJ residues 405–417; 455–466. In all cases, the unresolved residues are located in the RNA template tunnel, that is, the target site of our molecular docking experiments. Therefore, we generated the structural models for the NS5 RdRp protein sequences of DENV-3 as well as for the other three serotypes DENV-1, -2, and -4 (Table 1).
The protein sequences of NS5 RdRp of DENV-1 to -4 are highly conserved, with a minimum identity of 75% (Figure 1). The sequences close to the amino acids that define the allosteric binding proteins (Arg737, Thr413, and Met343) are especially conserved, except near the DENV-3 Thr413 site. The results of Niyomrattanakit et al strongly indicate that NITD-29 was cross-linked to Met343 within the RdRp domain of NS5 and the sequence alignment showed that this position is absolutely conserved among the four serotypes of DENV. Docking and molecular simulation suggest that the compound binds to the RNA template formed between the finger and thumb subdomains.21 Arg737 is conserved among mosquito-borne flavivirus NS5. NITD-29 compound inhibits RdRp activity through competing with RNA template in the RNA template tunnel.21 With these sequences, three 3D models were generated (Table 1; Figure 1) by homology modeling (in automated mode27) as the first step to perform molecular docking experiments.
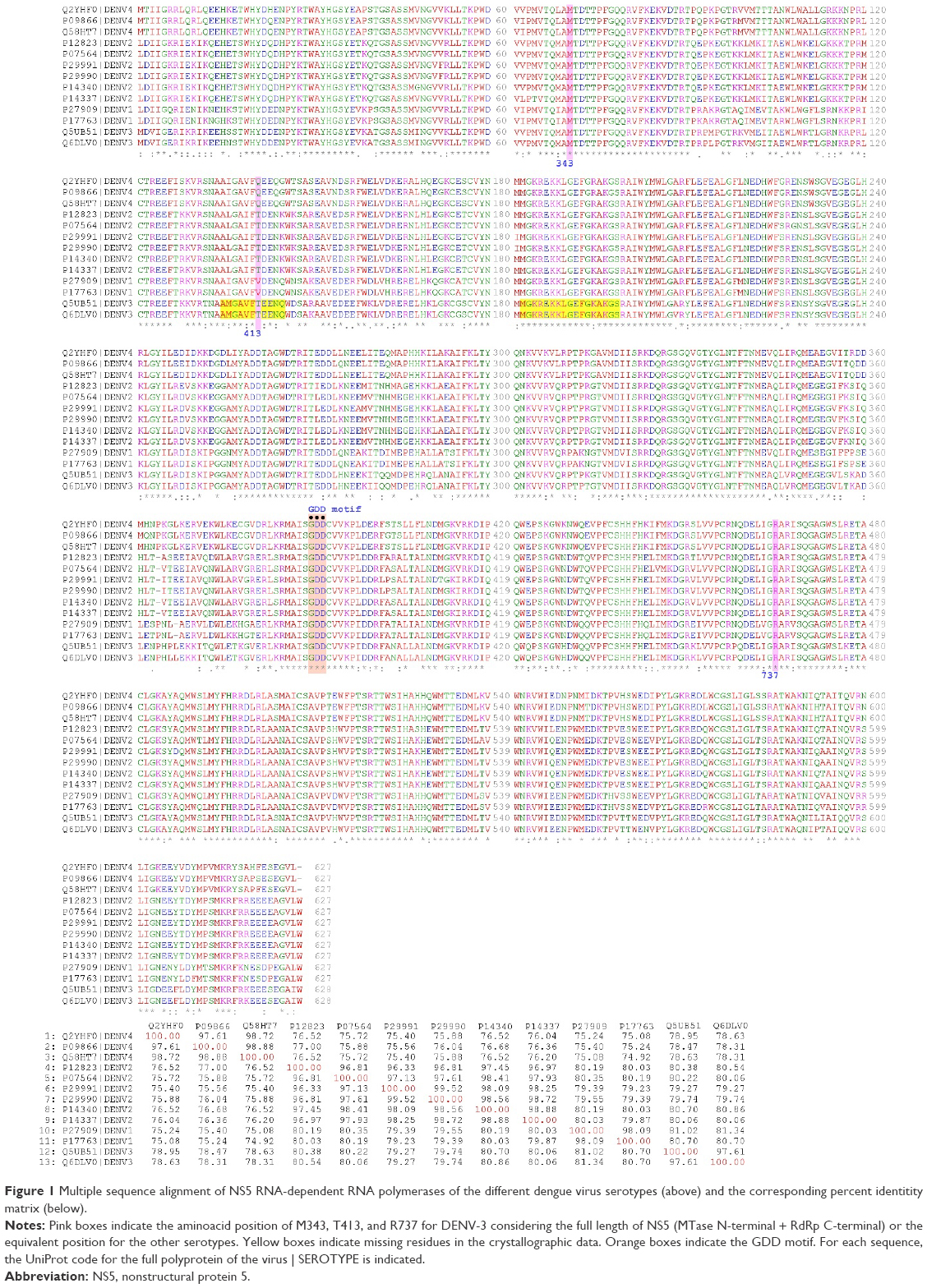 | Figure 1 Multiple sequence alignment of NS5 RNA-dependent RNA polymerases of the different dengue virus serotypes (above) and the corresponding percent identitity matrix (below). |
NS5 RdRp–ligand molecular docking analysis
The “induced-fit” Koshland theory states that the active site of the protein is continually reshaped by interactions with the ligands, as the ligands interact with the protein. Therefore, ligand and receptor should be treated as flexible during molecular docking.35 However, due to limitations in the capacity of computational calculation, molecular docking is performed considering a flexible ligand and a rigid receptor. The aim of molecular docking is to predict the structure of the ligand–receptor complex using computational methods. AutoDock/Vina incorporates Monte Carlo simulated annealing, evolutionary, genetic, and Lamarckian genetic algorithm methods to model the ligand flexibility while keeping the receptor rigid.35 Also, AutoDock Vina uses in its scoring function the AMBER force field, which computes the terms of the contributions of van der Waals interactions, hydrogen bonding, electrostatic interactions, conformational entropy, and desolvation.36 In the current study, 3D models of NS5 RpRd of the four serotypes of DENV were docked with two sets of compounds: molecules sharing ≥70% structural similarity with NITD-1, NITD-2, and NITD-2921 and 37,840 compounds from the SuperNatural II23 database with optimum pharmacokinetic and toxicological profiles.
Tables 2 and 3 show the free energy variation (ΔG) calculated using AutoDocK/Vina for the best docking scores, obtained for the NITD-related compounds and the SuperNatural II database compounds, respectively. In addition, hydrogen bonds and direct contacts based on van der Waals radii for compounds with the lowest free energy variations (ΔG ≤−10.5 kcal/mol) are available at http://docking.umh.es/. The calculated KD (KD = expΔG/RT) for compounds with ΔG ≤−10.5 kcal/mol is in the nanomolar or subnanomolar range that was used as a threshold to filter the docking results.28 Therefore, ΔG values represent the first filter in the selection of putative inhibitors among the compounds studied. Additional filters were applied before proposing the final candidates. The free energy variation is a representative value of the number and intensity of the atomic interactions between the receptor (protein) and the ligand, and can thus be considered a baseline comparison for the selection of lead compounds in the process of drug design.28 Several compounds have ΔG values in the range of −10.5 to −10.0 kcal/mol (see full list at http://docking.umh.es/). Interestingly, the calculated ΔG values for all of the tested compounds against 3D models of DENV-3 NS5 RdRp are greater than −10 kcal/mol. Calculated ΔG values for NITD-1, -2, and -29 against all DENV serotypes are worse than those of the selected compounds in Tables 2 and 3. On the other hand, we must remember that these compounds are often inactive in cell culture.21 Thus, additional filters were applied on these selected compounds (Tables 2 and 3) to further characterize them in terms of pharmacokinetics and toxicity.
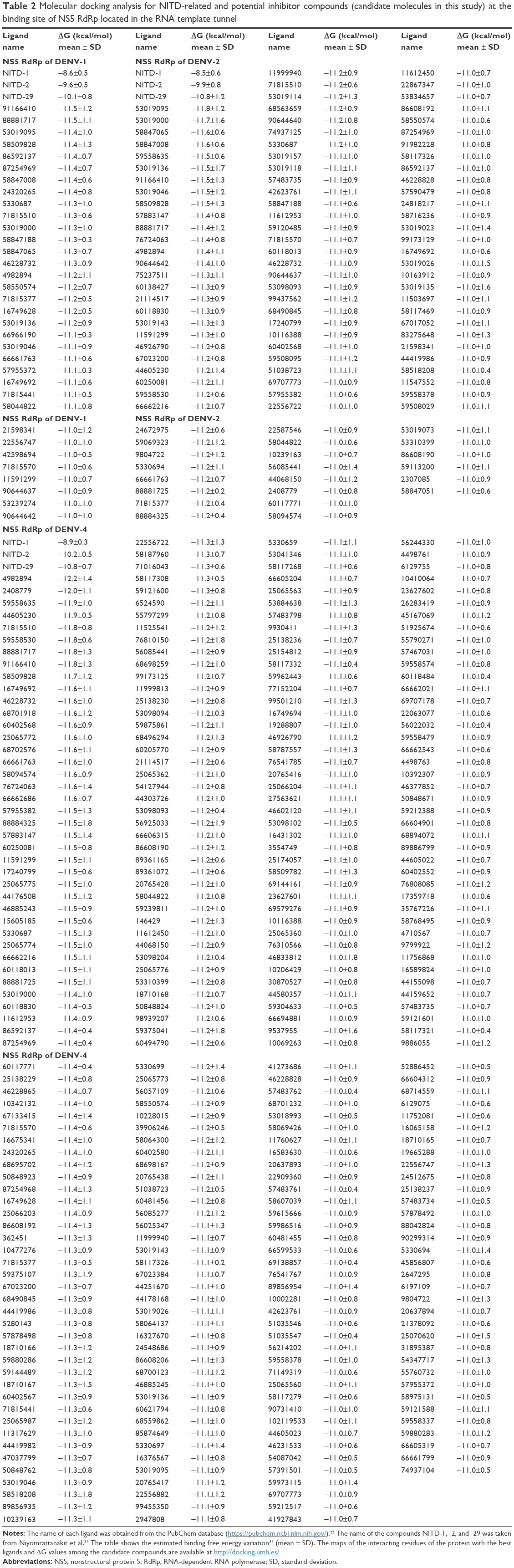 | Table 2 Molecular docking analysis for NITD-related and potential inhibitor compounds (candidate molecules in this study) at the binding site of NS5 RdRp located in the RNA template tunnel |
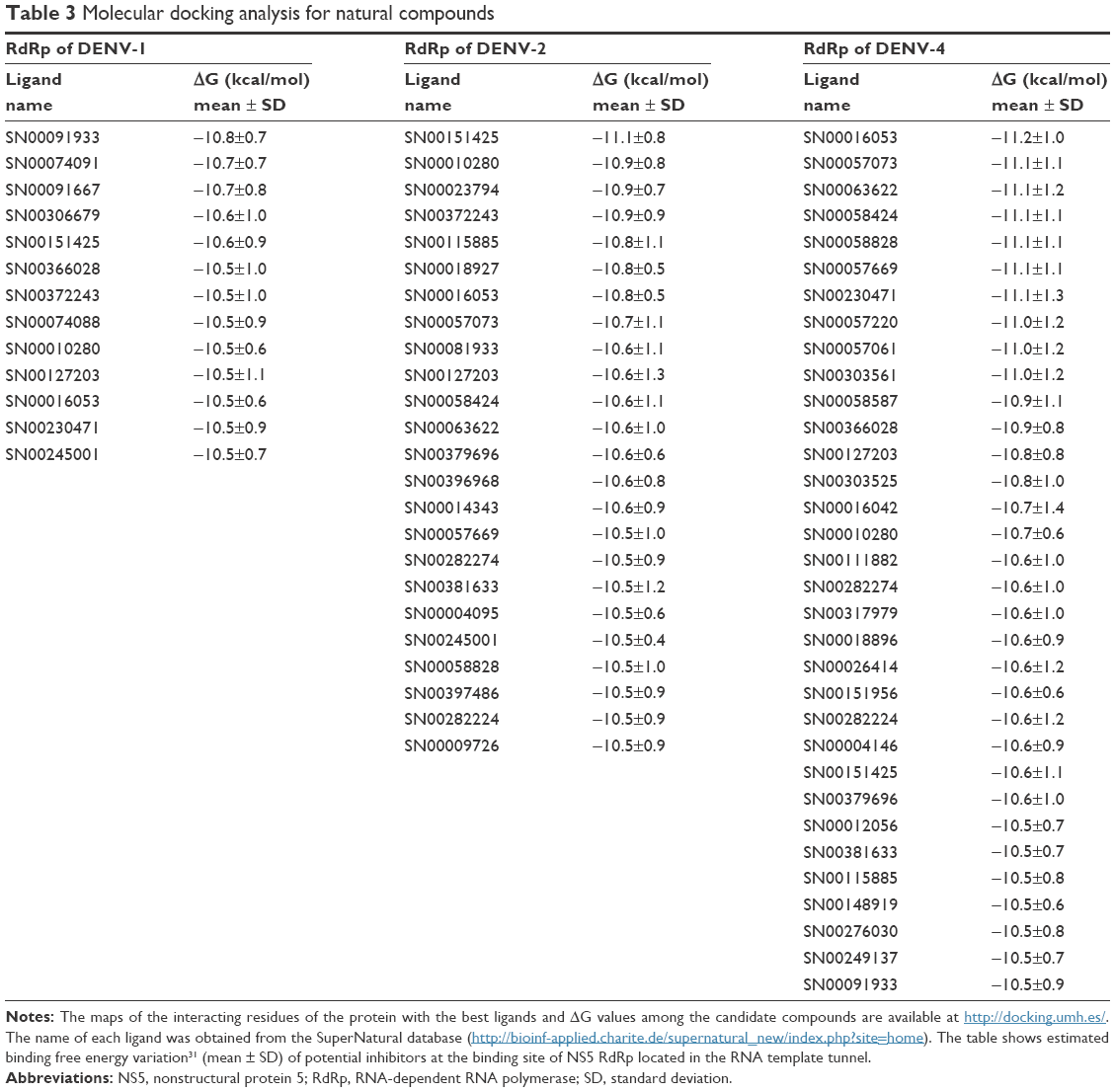 | Table 3 Molecular docking analysis for natural compounds |
Prediction of ADMET profiles
In addition to its efficiency to inhibit the target protein, other factors such as absorption, biodistribution, rate at which it is metabolized, excretion, and toxicity (ie, ADMET profile) upon its administration contributes to the overall success of a drug candidate.37 Moreover, it is recognized that employing computational ADMET, in combination with in vivo and in vitro predictions in early stage in the drug discovery process, helps to reduce the number of potential safety problems.38 In the next selection step, different paramaters were calculated to predict which compounds from Tables 2 and 3 would exhibit optimal ADMET profiles. The logarithm of partition coefficient between n-octanol and water is a well-established measure of the compound’s hydrophilicity. It has been shown that for compounds to have a reasonable probability of being well absorbed, their calculated logP value must not be greater than 5.0.33 Low solubility of a compound negatively impacts its absorption and, consequently, its biodistribution. Therefore, compounds with a calculated logS (logarithm base 10 of the solubility measured in mol/L) less than −4.0 were discarded.33 Compounds with fragment-based druglikeness ≤0 and a drugscore (calculated as a combination of druglikeness, calculated logP, calculated logS, MW, and toxicity risks) ≤0.5 were also discarded. Most orally administered drugs have an MW of ≤500, a calculated logP ≤5, five or fewer hydrogen bond donor sites, and ten or fewer hydrogen bond acceptor sites (Lipinski’s rule of five).32 Molecules violating more than one of these rules were ruled out. Also, mutagenicity, tumorigenicity, irritancy, and reproductive effects calculated with DataWarrior software33 were used to further identify compounds with low tendency for toxicity. Table 4 shows the compounds which satisfied all of the criteria described above, which reduced the number of putative NS5 RdRp inhibitors to 39 compounds, which is 0.010% of all compounds in the two databases screened in this study. Figure 2 shows the calculated ΔG and molecular formula for the 39 selected compounds against each of the four serotypes of DENV. The calculated ΔG for the selected compounds is lower (ie, potentially better candidates) for DENV-2 and -4 and higher for DENV-3. Each compound interacts with multiple residues of the protein (Figure 3) after docking to the binding site of the RNA template tunnel. The interaction maps for the best compounds (ΔG <−10.5 kcal/mol) were calculated and are available at http://docking.umh.es/. The TPSA has been correlated with passive molecular transport through membranes; therefore, TPSA is able to predict transport properties of drugs. Usually, compounds passively absorbed and with TPSA <140 Å2 are likely to exhibit high oral bioavailability. According to Zhao et al,39 the percentage of absorption can be estimated using the lineal equation: % of absorption =109–0.345× TPSA. Using this formula, the calculated percentages of absorption for selected compounds ranged between 70.86% and 92.85% (Table 4). What is the behavior of NITD-1, -2, and -29 compounds in relation to the above parameters? None of these three compounds satisfied all of the criteria imposed in this study to select potential DENV NS5 RdRp inhibitors. NITD-1 and -29 have low percentages of absorption (66% and 56%, respectively) (Table 4). Also, their calculated drug score value is less than 0.5 and their druglikeness is less than 0 (Table 4). The MW of NITD-29 is >500 Da, a violation of Lipinski’s rule of five. NITD-2 presents a percentage of absorption of 72%; however, its logS value is less than −4. The low aqueous solubility of drug candidates is a significant hurdle in many drug development projects.
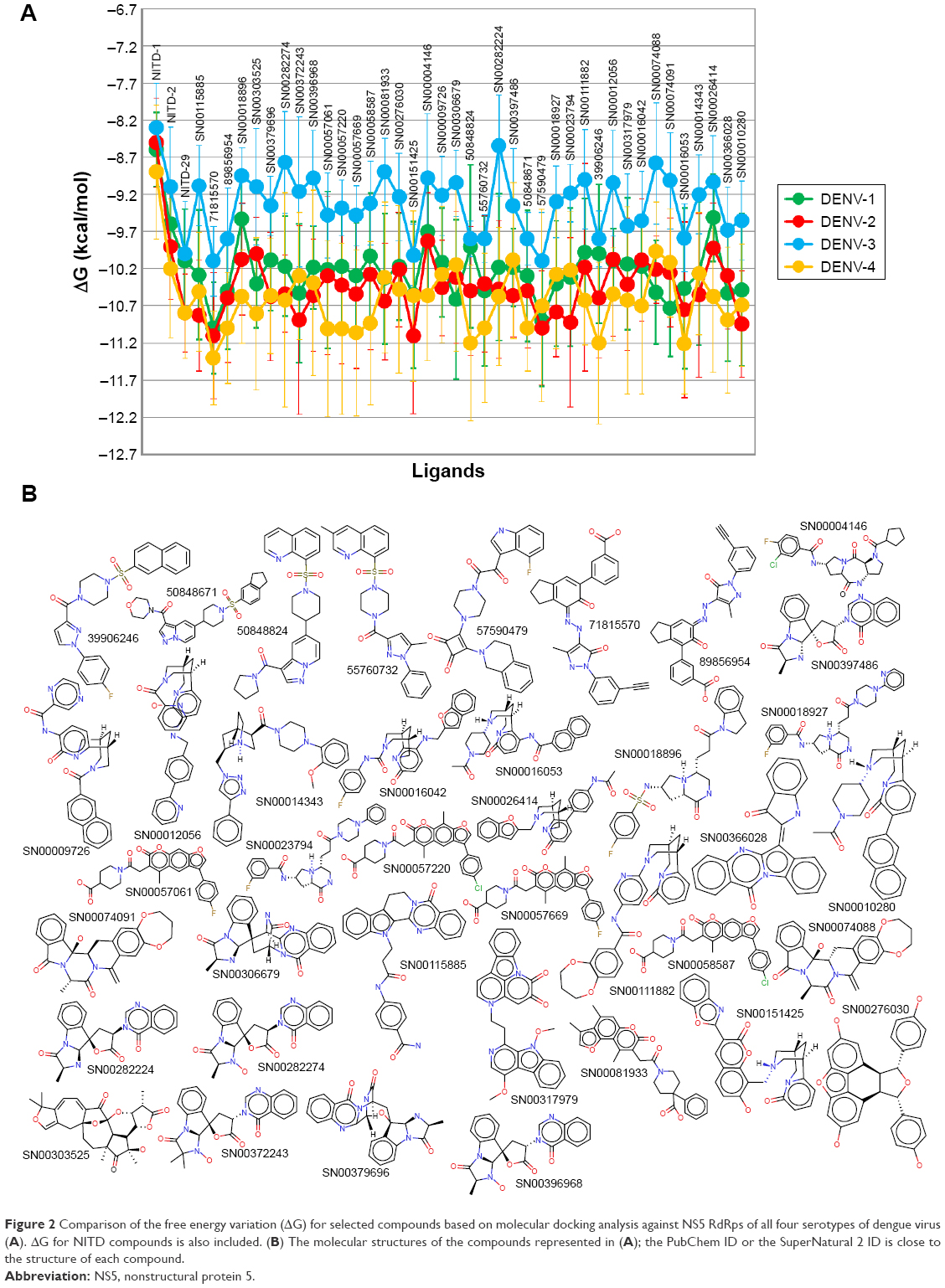 | Figure 2 Comparison of the free energy variation (ΔG) for selected compounds based on molecular docking analysis against NS5 RdRps of all four serotypes of dengue virus (A). ΔG for NITD compounds is also included. (B) The molecular structures of the compounds represented in (A); the PubChem ID or the SuperNatural 2 ID is close to the structure of each compound. |
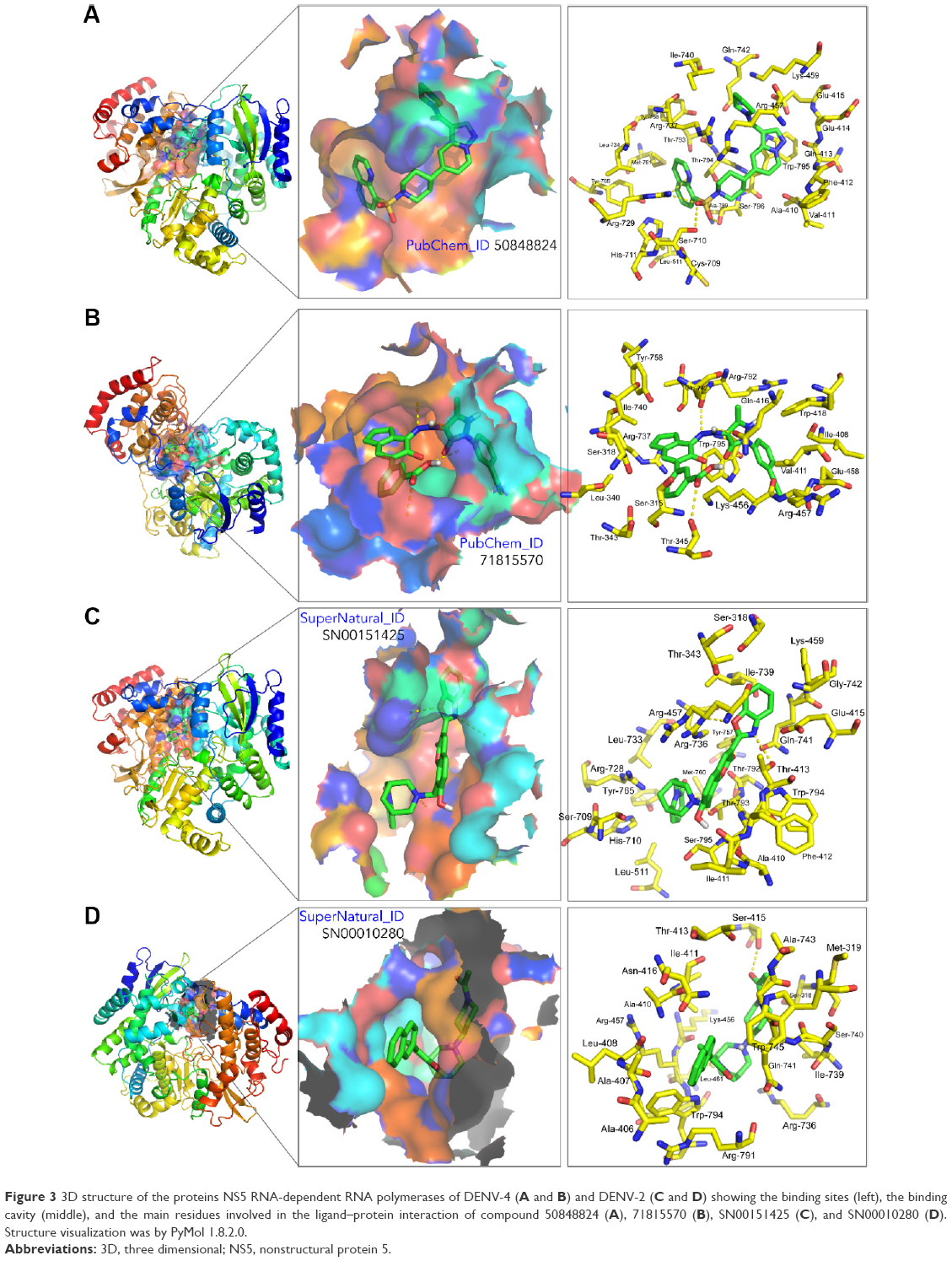 | Figure 3 3D structure of the proteins NS5 RNA-dependent RNA polymerases of DENV-4 (A and B) and DENV-2 (C and D) showing the binding sites (left), the binding cavity (middle), and the main residues involved in the ligand–protein interaction of compound 50848824 (A), 71815570 (B), SN00151425 (C), and SN00010280 (D). Structure visualization was by PyMol 1.8.2.0. |
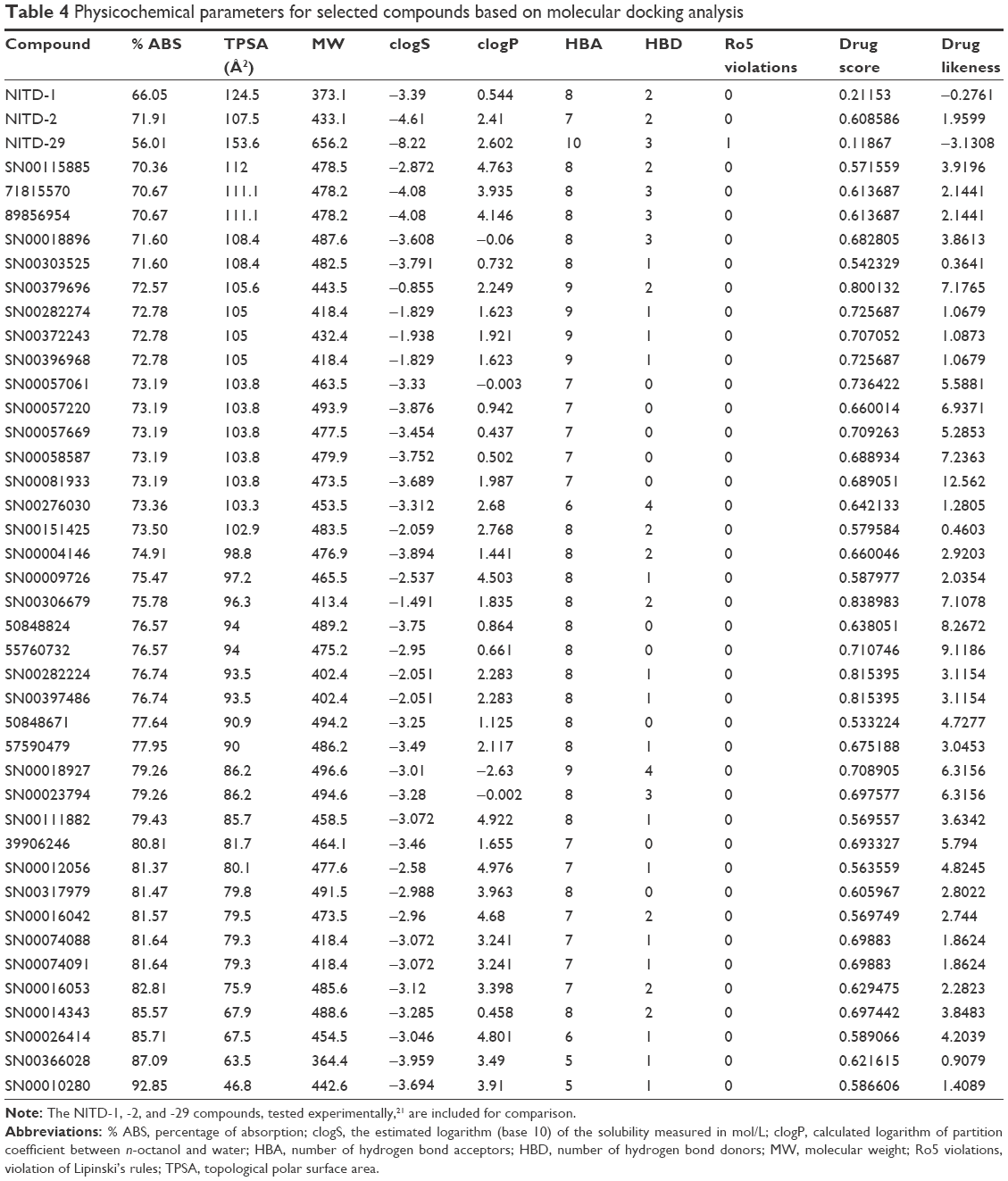 | Table 4 Physicochemical parameters for selected compounds based on molecular docking analysis |
We calculated additional pharmacokinetic properties following Cheng et al34 (Table 5). Almost all of the selected compounds showed optimum results for predicted human intestinal absorption and some compounds did so for transportation through blood–brain barrier and Caco-2. Also, the selected compounds showed no inhibitory side effects in terms of renal cation transport. The cytochrome P450 protein family catalyzes the oxidation of drugs and xenobiotics, generating more soluble compounds that are more readly removable from the body.40 These enzymes are expressed in the liver, small intestine (reducing drug bioavailability), lungs, placenta, and kidneys. Most of the selected putative inhibitors (Table 5) did not serve as substrates for cytochrome 2C9 and 2D6, whereas almost all of them were found to act as substrates for cytochrome P450 3A4. Inhibitors of cytochrome P450 decrease the enzymatic activity of these enzymes in a dose-dependent manner and promote the accumulation of drugs to toxic levels; therefore, it is desirable that the selected compounds do not serve as inhibitors of cytochrome P450. Most of the compounds included in Table 5 satisfy this condition.
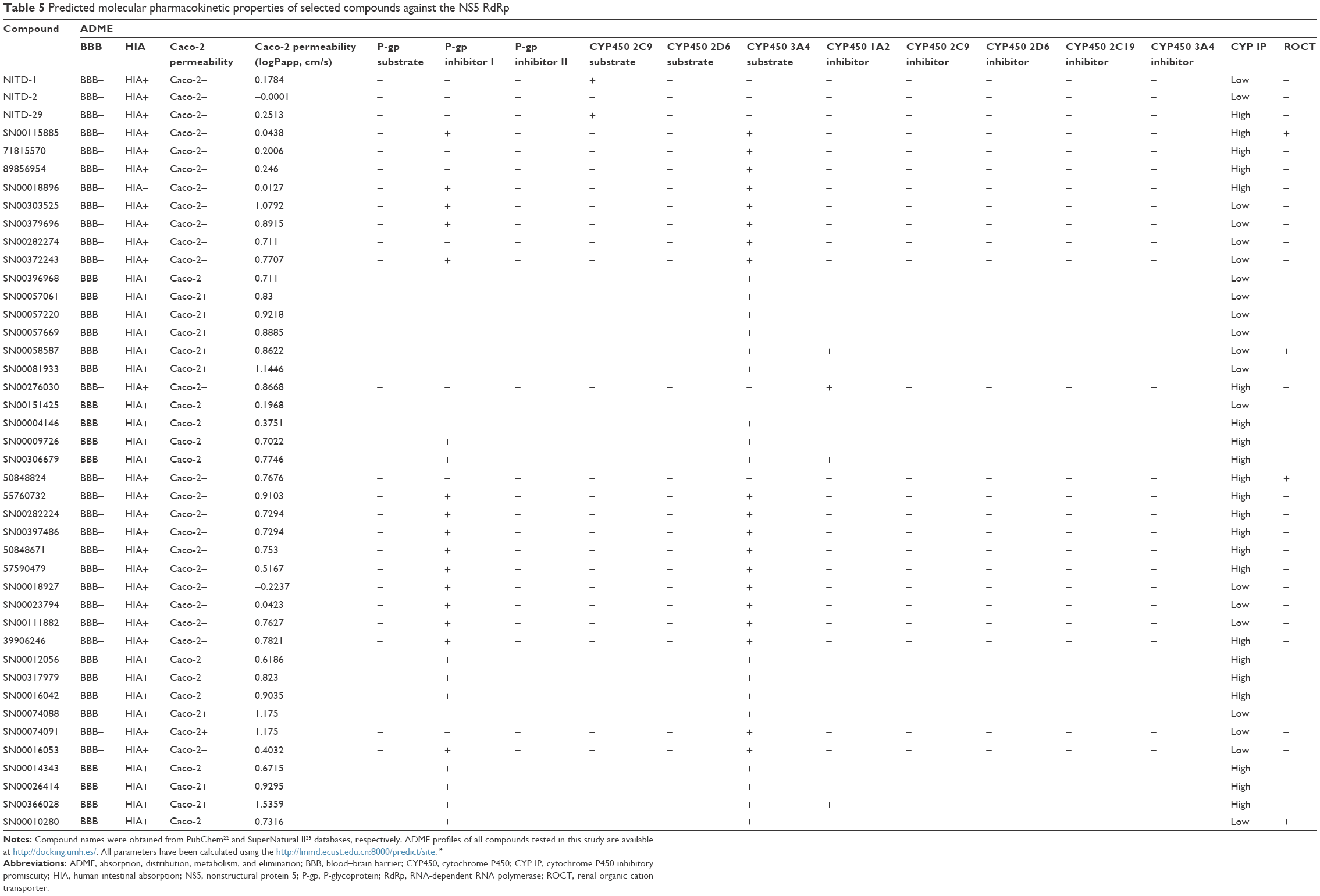 | Table 5 Predicted molecular pharmacokinetic properties of selected compounds against the NS5 RdRp |
Today it is very useful to predict the risk of toxicity of drug candidates with bioinformatics tools.37 The toxicity risk predictor software identifies functional group similarity of the query molecule with the in vitro- and the in vivo-validated molecules included in software’s built-in database; thus, similarity indicates a potential risk of toxicity. Table 6 presents the toxicity screening results for the selected compounds against DENV NS5 RdRp. None of the compounds presented potentially risks of tumorigenicity, mutagenicity, for reproductive function, or irritation.33 Similarly, the selected compounds were negative for AMES toxicity, did not potentially inhibit human ether-a-go-go-related genes, or exhibited no properties that posed significant toxicity risk for humans. The human ether-a-go-go-related gene channel is a voltage-gated potassium channel in cardiac cells, and is essential for cardiac repolarization. With the inhibition of this channel, the electrical depolarization and repolarization of the heart ventricles can be extended, leading to potentially fatal cardiac malfunction.41
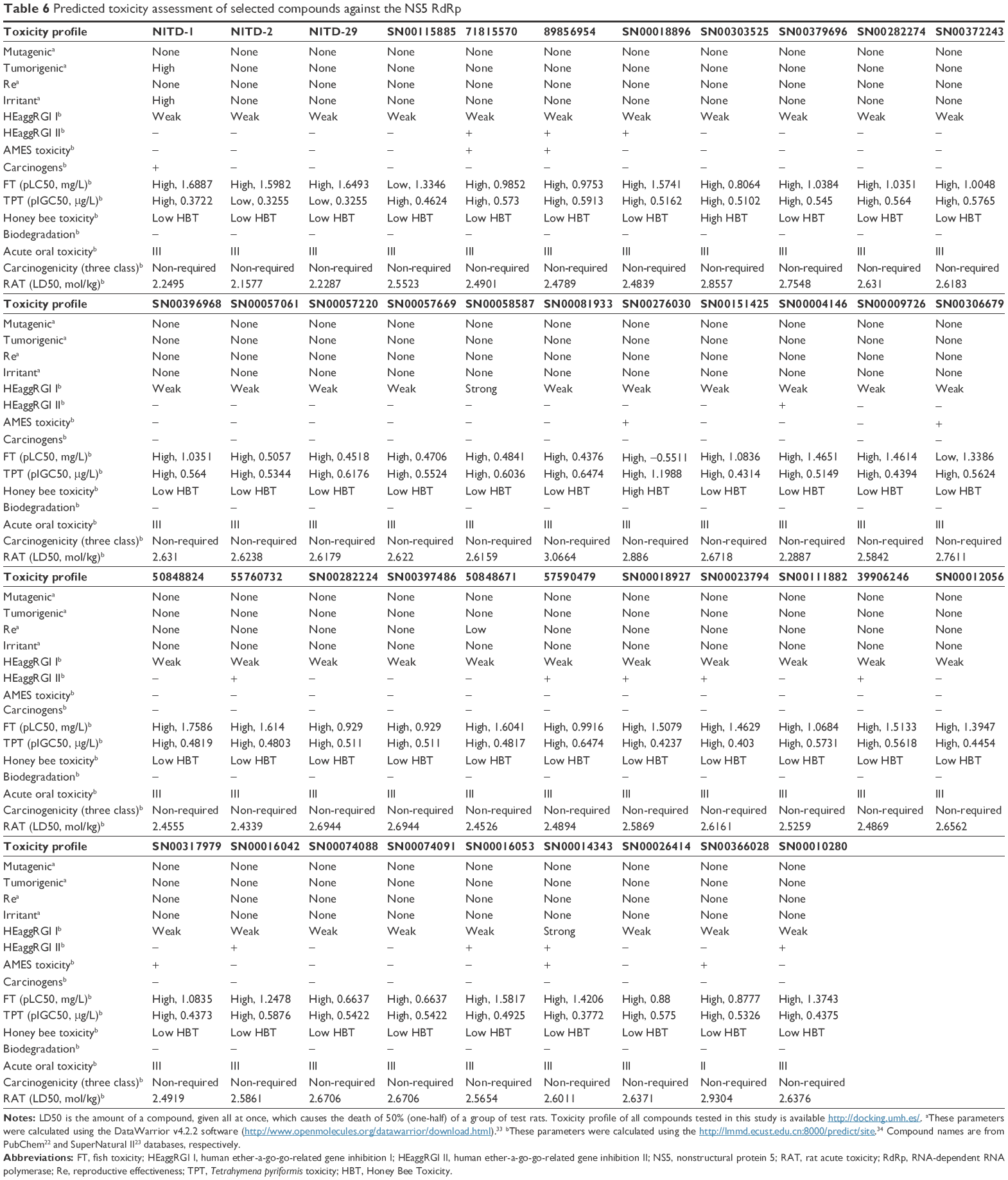 | Table 6 Predicted toxicity assessment of selected compounds against the NS5 RdRp |
Conclusion
The selected putative DENV NS5 RdRp inhibitors identified and characterized in this in silico study showed strong theoretical binding affinity (high negative free energy variation, ΔG), as determined by molecular docking against the binding site at the RNA template tunnel for the four DENV serotypes. This, to a great extent, is explained by the multiple sites of interaction of these compounds (Figure 3) with DENV NS5 RdRp. The selected compounds bind to the allosteric site located near the amino acids Met343, Arg737, and Thr413 in a similar way to NITD-29.21 Also, most of the selected compounds presented favorable druggability and optimum ADMET profiles, which suggests that as drug candidates, they will exhibit favourable traits, such as optimal absorption and biodistribution, compound stability, or low toxicity, all of which are critical for the success of a drug candidate. Further in vitro and in vivo studies will be required to confirm whether the in silico predicted ability of the selected compounds to inhibit DENV NS5 RdRp can be used to reduce DENV in live biological systems and strictly select those with the best potential to be used in real case scenarios. We believe that the information presented here will be useful for other laboratories interested in developing inhibitors against the NS5 RdRp of DENV.
Acknowledgments
We thank the “Fundación del Centro de Supercomputación de Castilla y León (FCSCL)” for allowing us to use their facilities and resources on the Linux cluster calendula.fcsc.es. We are grateful to Research, Technological Innovation and Supercomputing Center of Extremadura (CenitS) and COMPUTAEX for allowing us to use the supercomputing facilities (LUSITANIA II) and for the support provided to us. Also, we thank the computing center of the Extremadura Research Centre for Advanced Technologies (CETA-CIEMAT), funded by the European Regional Development Fund. CETA-CIEMAT belongs to CIEMAT and the Government of Spain. We thank the editor and three anonymous reviewers for their constructive comments, which helped us to improve the manuscript. This work was partly supported by grants AGL2015-67995-C3-1-R and AGL2014-51773-C3-1-R from the Spanish MICINN, and PROMETEO/2016/006 grant from Generalitat Valenciana.
Disclosure
The authors report no conflicts of interest in this work.
References
Weissenbock H, Hubalek Z, Bakonyi T, Nowotny N. Zoonotic mosquito-borne flaviviruses: worldwide presence of agents with proven pathogenicity and potential candidates of future emerging diseases. Vet Microbiol. 2010;140(3–4):271–280. | ||
Papageorgiou L, Loukatou S, Sofia K, Maroulis D, Vlachakis D. An updated evolutionary study of Flaviviridae NS3 helicase and NS5 RNA-dependent RNA polymerase reveals novel invariable motifs as potential pharmacological targets. Mol Biosyst. 2016;2(7):2080–2093. | ||
International Committee on Taxonomy of Viruses, King AMQ. Virus Taxonomy: Classification and Nomenclature of Viruses Ninth Report of the International Committee on Taxonomy of Viruses. London: Academic Press; 2012. | ||
Brady OJ, Gething PW, Bhatt S, et al. Refining the global spatial limits of dengue virus transmission by evidence-based consensus. PLoS Negl Trop Dis. 2012;6(8):e1760. | ||
Shepard DS, Undurraga EA, Halasa YA, Stanaway JD. The global economic burden of dengue: a systematic analysis. Lancet Infect Dis. 2016;16(8):935–941. | ||
Alagarasu K. Introducing dengue vaccine: implications for diagnosis in dengue vaccinated subjects. Vaccine. 2016;34(25):2759–2761. | ||
Mackenzie J. Wrapping things up about virus RNA replication. Traffic. 2005;6(11):967–977. | ||
Paul D, Bartenschlager R. Flaviviridae replication organelles: oh, what a tangled web we weave. Annu Rev Virol. 2015;2(1):289–310. | ||
Issur M, Geiss BJ, Bougie I, et al. The flavivirus NS5 protein is a true RNA guanylyltransferase that catalyzes a two-step reaction to form the RNA cap structure. RNA. 2009;15(12):2340–2350. | ||
Decroly E, Ferron F, Lescar J, Canard B. Conventional and unconventional mechanisms for capping viral mRNA. Nat Rev Microbiol. 2012;10(1):51–65. | ||
Zhao Y, Soh TS, Zheng J, et al. A crystal structure of the Dengue virus NS5 protein reveals a novel inter-domain interface essential for protein flexibility and virus replication. PLoS Pathog. 2015;11(3):e1004682. | ||
Yap TL, Xu T, Chen YL, et al. Crystal structure of the dengue virus RNA-dependent RNA polymerase catalytic domain at 1.85-angstrom resolution. J Virol. 2007;81(9):4753–4765. | ||
Bartholomeusz A, Thompson P. Flaviviridae polymerase and RNA replication. J Viral Hepat. 1999;6(4):261–270. | ||
You S, Falgout B, Markoff L, Padmanabhan R. In vitro RNA synthesis from exogenous dengue viral RNA templates requires long range interactions between 5′- and 3′-terminal regions that influence RNA structure. J Biol Chem. 2001;276(19):15581–15591. | ||
Malet H, Masse N, Selisko B, et al. The flavivirus polymerase as a target for drug discovery. Antiviral Res. 2008;80(1):23–35. | ||
De Clercq E, Neyts J. Antiviral agents acting as DNA or RNA chain terminators. Handb Exp Pharmacol. 2009(189):53–84. | ||
De Francesco R, Tomei L, Altamura S, Summa V, Migliaccio G. Approaching a new era for hepatitis C virus therapy: inhibitors of the NS3-4A serine protease and the NS5B RNA-dependent RNA polymerase. Antiviral Res. 2003;58(1):1–16. | ||
Yin Z, Chen YL, Schul W, et al. An adenosine nucleoside inhibitor of dengue virus. Proc Natl Acad Sci U S A. 2009;106(48):20435–20439. | ||
Kohler JJ, Lewis W. A brief overview of mechanisms of mitochondrial toxicity from NRTIs. Environ Mol Mutagen. 2007;48(3–4):166–172. | ||
Behnam MA, Nitsche C, Boldescu V, Klein CD. The medicinal chemistry of dengue virus. J Med Chem. 2016;59(12):5622–5649. | ||
Niyomrattanakit P, Chen YL, Dong H, et al. Inhibition of dengue virus polymerase by blocking of the RNA tunnel. J Virol. 2010;84(11):5678–5686. | ||
Wang Y, Xiao J, Suzek TO, Zhang J, Wang J, Bryant SH. PubChem: a public information system for analyzing bioactivities of small molecules. Nucleic Acids Res. 2009;37(Web Server issue):W623–W633. | ||
Banerjee P, Erehman J, Gohlke BO, Wilhelm T, Preissner R, Dunkel M. Super Natural II – a database of natural products. Nucleic Acids Res. 2015;43(Database issue):D935–D939. | ||
Zhao Y, Soh TS, Lim SP, et al. Molecular basis for specific viral RNA recognition and 2′-O-ribose methylation by the dengue virus nonstructural protein 5 (NS5). Proc Natl Acad Sci U S A. 2015;112(48):14834–14839. | ||
Noble CG, Lim SP, Chen YL, et al. Conformational flexibility of the Dengue virus RNA-dependent RNA polymerase revealed by a complex with an inhibitor. J Virol. 2013;87(9):5291–5295. | ||
Lim SP, Koh JH, Seh CC, et al. A crystal structure of the dengue virus non-structural protein 5 (NS5) polymerase delineates interdomain amino acid residues that enhance its thermostability and de novo initiation activities. J Biol Chem. 2013;288(43):31105–31114. | ||
Biasini M, Bienert S, Waterhouse A, et al. SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 2014;42(Web Server issue):W252–W258. | ||
Encinar JA, Fernandez-Ballester G, Galiano-Ibarra V, Micol V. In silico approach for the discovery of new PPARgamma modulators among plant-derived polyphenols. Drug Des Devel Ther. 2015;9:5877–5895. | ||
Morris GM, Huey R, Lindstrom W, et al. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J Comput Chem. 2009;30(16):2785–2791. | ||
Schymkowitz J, Borg J, Stricher F, Nys R, Rousseau F, Serrano L. The FoldX web server: an online force field. Nucleic Acids Res. 2005;33(Web Server issue):W382–W388. | ||
Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31(2):455–461. | ||
Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 2001;46(1–3):3–26. | ||
Sander T, Freyss J, von Korff M, Rufener C. DataWarrior: an open-source program for chemistry aware data visualization and analysis. J Chem Inf Model. 2015;55(2):460–473. | ||
Cheng F, Li W, Zhou Y, et al. admetSAR: a comprehensive source and free tool for assessment of chemical ADMET properties. J Chem Inf Model. 2012;52(11):3099–3105. | ||
Meng XY, Zhang HX, Mezei M, Cui M. Molecular docking: a powerful approach for structure-based drug discovery. Curr Comput Aided Drug Des. 2011;7(2):146–157. | ||
Jaghoori MM, Bleijlevens B, Olabarriaga SD. 1001 Ways to run AutoDock Vina for virtual screening. J Comput Aided Mol Des. 2016;30(3):237–249. | ||
Moroy G, Martiny VY, Vayer P, Villoutreix BO, Miteva MA. Toward in silico structure-based ADMET prediction in drug discovery. Drug Discov Today. 2012;17(1–2):44–55. | ||
Merlot C. Computational toxicology – a tool for early safety evaluation. Drug Discov Today. 2010;15(1–2):16–22. | ||
Zhao YH, Abraham MH, Le J, et al. Rate-limited steps of human oral absorption and QSAR studies. Pharm Res. 2002;19(10):1446–1457. | ||
Anzenbacher P, Anzenbacherova E. Cytochromes P450 and metabolism of xenobiotics. Cell Mol Life Sci. 2001;58(5–6):737–747. | ||
Roche O, Trube G, Zuegge J, Pflimlin P, Alanine A, Schneider G. A virtual screening method for prediction of the HERG potassium channel liability of compound libraries. Chembiochem. 2002;3(5): 455–459. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.