")
Back to Journals » Drug Design, Development and Therapy » Volume 9
Lefty-1 alleviates TGF-β1-induced fibroblast–myofibroblast transdifferentiation in NRK-49F cells
Authors Zhang L, Zhang J, Xu C, Zhou X, Wang W, Zheng R, Hu W, Wu P
Received 17 April 2015
Accepted for publication 23 June 2015
Published 14 August 2015 Volume 2015:9 Pages 4669—4678
DOI https://doi.org/10.2147/DDDT.S86770
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Shu-Feng Zhou
Lijun Zhang, Jie Zhang, Changgeng Xu, Xiangjun Zhou, Wei Wang, Renping Zheng, Wei Hu, Pin Wu
Department of Urology, Renmin Hospital of Wuhan University, Wuhan, Hubei Province, People’s Republic of China
Abstract: Fibroblast activation and proliferation are important for fibroblast–myofibroblast transdifferentiation, a crucial process in the pathological changes that define renal interstitial fibrosis. The left–right determination factor (Lefty) is an important cytokine of the transforming growth factor (TGF)-β family, with two variants, Lefty-1 and Lefty-2, in mice. Lefty has diverse functions, such as the regulation of embryonic development, the inhibition of TGF-β1 signaling, and the suppression of tumor activity. However, whether Lefty-1 influences fibroblast activation and proliferation, and consequently prevents fibroblast–myofibroblast transdifferentiation, remains unclear. This study aimed to investigate whether Lefty-1 can attenuate TGF-β1-induced fibroblast–myofibroblast transdifferentiation in normal rat kidney interstitial fibroblast cells (NRK-49F), as well as the mechanisms underlying any effects. Results showed that the typical fibroblast cell morphology of NRK-49F cells was altered after TGF-β1 treatment and that Lefty-1 significantly prevented this change in a dose-dependent manner. Further analyses demonstrated decreased proliferating cell nuclear antigen, cyclin D1, collagen I(A1), alpha-smooth muscle actin, and fibronectin expression. Lefty-1 further induced remarkable reductions in TGF-β1-induced Smad3 and mitogen-activated protein kinase-10/c-Jun N-terminal kinase (JNK-3) signaling, and enhanced expression of the antifibrotic factor bone morphogenetic protein (BMP)-5. However, without TGF-β1, Lefty-1 had no effect on Smad3, JNK-3, and BMP-5 activation and fibroblast–myofibroblast transdifferentiation. Taken together, these findings indicate that Lefty-1 can alleviate TGF-β1-mediated activation and the proliferation of fibroblasts. Furthermore, Lefty-1 may prevent fibroblast–myofibroblast transdifferentiation in part via modulations of Smad3, JNK-3, and BMP-5 activities in the TGF-β/BMP signaling pathway.
Keywords: Lefty-1, NRK-49F, fibroblast, myofibroblast, transdifferentiation
Introduction
Chronic kidney disease (CKD) was caused by a variety of etiology, including inflammation, immunity, and obstructive, metabolic, or other systemic diseases. With the gradual decline in renal function as its main pathological feature, the majority of CKD inevitably progresses to end-stage renal disease. CKD is an ongoing global medical problem and lacks timely and effective intervention measures, because of fairly complex underlying mechanisms. Renal interstitial fibrosis is considered to be a common mechanism of renal failure during the end-stage of CKD. Many types of cell interactions are involved in this process, especially myofibroblasts that express alpha-smooth muscle actin (α-SMA) may be responsible for deposition of the extracellular matrix (ECM), which replaces the normal tissue structure during fibrosis.1 Many studies have proposed mechanisms to explain how myofibroblasts accumulate in the injured renal interstitium, including transdifferentiation of resident fibroblasts, recruitment of circulating fibrocytes, and epithelial to mesenchymal transition.2
The activation and proliferation of fibroblasts are important in fibroblast–myofibroblast transdifferentiation, a crucial process of renal interstitial fibrosis. However, as the steps involved in transdifferentiation of fibroblasts to myofibroblasts are not well understood, the molecular mechanisms by which this transdifferentiation occurs in vivo are also unknown so far.3 As a kind of mesenchymal cell, spindle-shaped fibroblasts are widely present in the whole body organs and tissues. Tubulointerstitial myofibroblasts are terminally differentiated cells under nonpathological conditions and are rarely found. These cells can give rise to the synthesis and accumulation of renal interstitial ECM components. However, interstitial fibroblasts are stimulated upon sustained injury, resulting in changing patterns of hydrodynamic and chemical, inflammatory, or other mediators to undergo differentiation into the myofibroblast phenotype. Historically, myofibroblasts have been assumed to originate from resident fibroblasts after renal injury. Although this concept has been challenged, the idea that the majority of myofibroblasts can originate from renal interstitial resident fibroblast activation and proliferation in injured kidneys requires further verification.4 These activated fibroblast cells and myofibroblasts produce large amounts of ECM that accumulate in the tubulointerstitial space; together with renal tubular atrophy, this accumulation causes renal interstitial fibrosis.5 Therefore, fibroblast–myofibroblast transdifferentiation is an important step in renal fibrosis.
In renal fibrosis, transforming growth factor (TGF)-β is considered a key mediator that significantly induces renal scarring by activating its downstream Smad and non-Smad or other signaling pathways. A recent report on the functional roles of the TGF-β signaling pathways has improved our understanding of renal interstitial fibrosis and the molecular mechanisms of CKD inflammation.6 TGF-β1 stimulates ECM production leading to progressive renal fibrosis while inhibiting renal degradation. It also mediates renal fibrosis by inducing the transformation of tubular epithelial cells to myofibroblasts.7 Evidence from in vitro and in vivo studies utilizing neutralizing TGF-β1 antibodies, decorin, and antisense oligonucleotides indicate that blocking TGF-β1 can prevent or improve renal fibrosis.8 Thus, the TGF-β signaling pathway may present a major strategy for preventing activation and transduction during renal fibrosis.
The left–right determination factor (Lefty) is an important cytokine of the TGF-β superfamily. Lefty presents two variants in mice: Lefty-1 and Lefty-2, which are orthologs of LeftyA and LeftyB in humans. These proteins control differentiation of stem cells and regulation of embryonic development.9,10 In previous studies, Lefty was found to inhibit TGF-β1 signaling by limiting Smad2/3 phosphorylation and activating the TGF-β receptor.11 Lefty further suppresses downstream events including R-Smads (Smad2/3) phosphorylation and the formation of the R-Smad/Smad4 complex.12 Recent studies have shown that Lefty may have tumor-suppressive activity in human liver stem cells13 and that LeftyA inhibits the TGF-β signaling pathway in human renal tubular epithelial cells.14,15 These findings have unraveled some of the molecular properties of Lefty and drawn more interest into its biological functions. Despite the availability of information on its beneficial properties, however, the antagonistic roles of Lefty-1 in fibrosis remain incompletely understood. Therefore, this study aims to determine whether Lefty-1 attenuates TGF-β1-induced activation and proliferation of NRK-49F cells.
Materials and methods
Reagents
Dulbecco’s Modified Eagle’s Medium (DMEM; high glucose), trypsin/EDTA solution, and fetal bovine serum (FBS) were purchased from the Gibco® (Thermo Fisher Scientific, Waltham, MA, USA). Recombinant mouse Lefty-1 and TGF-β1 were obtained from R&D Systems, Inc. (Minneapolis, MN, USA). Quantitative real-time polymerase chain reaction (PCR) kit was obtained from Hoffman-La Roche Ltd. (Basel, Switzerland), and Cell Counting Kit-8 (CCK)-8 and the cell cycle detection kit were acquired from MultiSciences Biotech Co., Ltd. (Hangzhou, Zhejiang, People’s Republic of China). Antibodies to fibronectin, collagen I(A1), glyceraldehyde-3-phosphate dehydrogenase (GAPDH), bone morphogenetic protein (BMP)-5, and proliferating cell nuclear antigen (PCNA) were obtained from Santa Cruz Biotechnology, Inc. (Dallas, TX, USA). α-SMA and cyclin D1 were obtained from Abcam plc (Cambridge, UK). All other antibodies used in this study were purchased from Cell Signaling Technology, Inc. (Danvers, MA, USA).
Cell culture and treatment
The NRK-49F cells were purchased from the American Type Culture Collection (Manassas, VA, USA), cultured in DMEM/high-glucose medium, and supplemented with 0.5% penicillin, streptomycin, and 10% FBS in a humidified incubator containing 95% air and 5% CO2 at 37°C. To determine the function of Lefty-1 on TGF-β1-induced activation and proliferation of NRK-49F cells, the cells were treated first in serum-free medium to synchronize for 24 hours. The cells were subsequently induced with TGF-β1 (10 ng/mL), treated with different Lefty-1 concentrations (0 ng/mL, 10 ng/mL, 20 ng/mL, and 50 ng/mL), and then incubated for 48 hours. Then, expressions of related proteins and genes were determined. All experiments were conducted in serum-free conditions and at least repeated thrice.
Cell viability and cell cycle assays
Cells treated with or without Lefty-1 were seeded into 96-well plates at a density of 2,000 cells per well and then incubated for 48 hours. Cell viability was measured using the CCK-8 method, in which 10 μL of the CCK-8 reagent was added and the cells were incubated at 37°C for 4 hours for proliferation measurements. Optical density values represent cell viability, thus detecting the proliferation of NRK-49F cells. Viability was detected using a PerkinElmer Victor3 1420 Multilabel Counter (PerkinElmer, Inc., Waltham, MA, USA). Cells were seeded into six-well plates with 2×105 cells per well for 24 hours of culture for cell cycle analysis. After synchronization and treatment for another 48 hours, the cells were harvested and then fixed with precooled 70% ethanol at 4°C overnight. Fixed cells were washed with phosphate buffered saline (PBS) and then stained with propidium iodide (MultiSciences Biotech Co., Ltd.) for 30 minutes at 4°C. Stained cells were determined using a FACSCalibur system (BD, Franklin Lakes, NJ, USA) to examine cell cycle distribution, followed using the MODFIT software for data analysis.
BrdU incorporation assay
The effect of Lefty-1 on NRK-49F cell proliferation was also evaluated using the 5-Bromo-2-deoxyuridine (BrdU) incorporation assay. Briefly, cells were seeded on coverslips at a density of 1.0×106 cells/well in six-well plates. Cells were incubated for 48 hours, as previously described, and then pulsed with BrdU (10 μg/mL) for 24 hours. Cells were fixed with 4% paraformaldehyde for 15 minutes, and the DNA was denatured with 2 N of HCL at 37°C for 30 minutes, following by washing with PBS. Endogenous peroxidase activity was eliminated using 3% H2O2 in PBS for 20 minutes. After washing with PBS three times, cells were permeabilized with 0.3% Triton X-100 buffer at room temperature for 30 minutes, and nonspecific binding was blocked with 1% goat serum at 37°C for 1 hour. Incorporated BrdU was detected using a rabbit monoclonal anti-BrdU antibody overnight, and then the cells were stained with CY3-conjugated secondary antibody. Finally, nuclei were counterstained with 4,6-diamidino-2-phenylindole (DAPI). The immunofluorescent images were visualized by an Olympus-BX51 fluorescence upright microscope (Olympus Corporation, Tokyo, Japan). The percentage of BrdU-positive nuclei was determined by the proportion of the number of BrdU-positive cells to total NRK-49F cells.
Immunofluorescence
Indirect immunofluorescence was used on control and Lefty-1-treated NRK-49F cells on coverslips. The cells were washed with PBS at 37°C and fixed in 4% paraformaldehyde for 10 minutes, and then permeabilized using 0.5% Triton X-100 buffer for 5 minutes at room temperature. Afterward, the cells were extensively washed thrice with PBS. These slides were blocked with 1% bovine serum albumin (BSA) in PBS for 20 minutes at 4°C and then incubated with the primary antibodies as described above. The following polyclonal antibodies were used: antifibronectin (1:100), anticollagen I (1:100), anti-α-SMA (1:100), and anti-PCNA (1:100). To visualize the primary antibodies, the cells were stained with fluorescein isothiocyanate or cyanine dye-labeled secondary antibodies (1:100). Stained cells were mounted using the antiquenching fluorescence mounting medium (Wuhan Good Biotechnology CO., LTD, Wuhan, Hubei, People’s Republic of China) and viewed using an Olympus-BX51 fluorescence upright microscope (Olympus Corporation). Quantitation of immunofluorescence staining was carried out on coded cell coverslips as the integrated option density value.
Western blot
NRK-49F cells were lysed and harvested in radioimmunoprecipitation assay lysis buffer containing 50 mM of Tris, pH 7.4, 1% TritonX-100, 150 mM of NaCl, 0.1% sodium dodecyl sulfate (SDS), 1% sodium deoxycholate, 2 mM of sodium pyrophosphate, 1 mM of EDTA, 25 mM of β-glycerophosphate, 0.5 μg/mL of leupeptin, and 1 mM of Na3VO4. To measure protein concentrations, a BCA assay kit (Wuhan Boster Bio-Engineering Limited Company, Wuhan, Hubei, People’s Republic of China) was used according to the manufacturer’s instructions. After adding protein loading buffer (200 mM of DTT, 40 mM of Tris/HCl, 40% glycerol, 4% SDS; pH 6.8, 0.032% Bromophenol blue) and denaturing at 95°C for 5 minutes, 30 μg of the total protein was separated from these samples by 10% SDS–polyacrylamide gel electrophoresis (PAGE), and then transferred onto activated polyvinylidene fluoride membranes. After blocking in 5% BSA at 37°C for 1 hour, the membranes were blotted with appropriate primary antibodies at 4°C overnight, followed by fluorescence-labeled secondary antibodies (IRDye700 and IRDye800, goat antimouse/rabbit) for 1 hour at 37°C. Signals were detected with an Odyssey infrared imaging system (LI-COR Biosciences, Lincoln, NE, USA).
RNA isolation and quantitative real-time PCR
Total RNA was extracted using the TRIzol reagent (Thermo Fisher Scientific), and first-strand complementary (c)DNA was synthesized using a Revert Aid First Strand cDNA Synthesis Kit (Thermo Fisher Scientific). Quantitative real-time PCR was conducted using a FastStart Universal SYBR Green Master (Rox) system (Hoffman-La Roche Ltd.). SYBR green real-time PCR mix was used with 7.5 μM each of forward and reverse primers, which are listed in Table 1, for PCR. The reaction conditions were as follows: predenaturation at 95°C for 10 minutes; 40 cycles of 95°C for 15 seconds; 60°C for 60 seconds; 72°C for 20 seconds; and a final extension at 60°C for 5 minutes. GAPDH was used as the reference gene. Relative quantification of gene expression for both target and reference genes was performed by the 2−ΔΔCt method and based on Ct values. Real-time PCR analysis results are presented as the mean ± standard deviation (SD) of fold-change in expression.
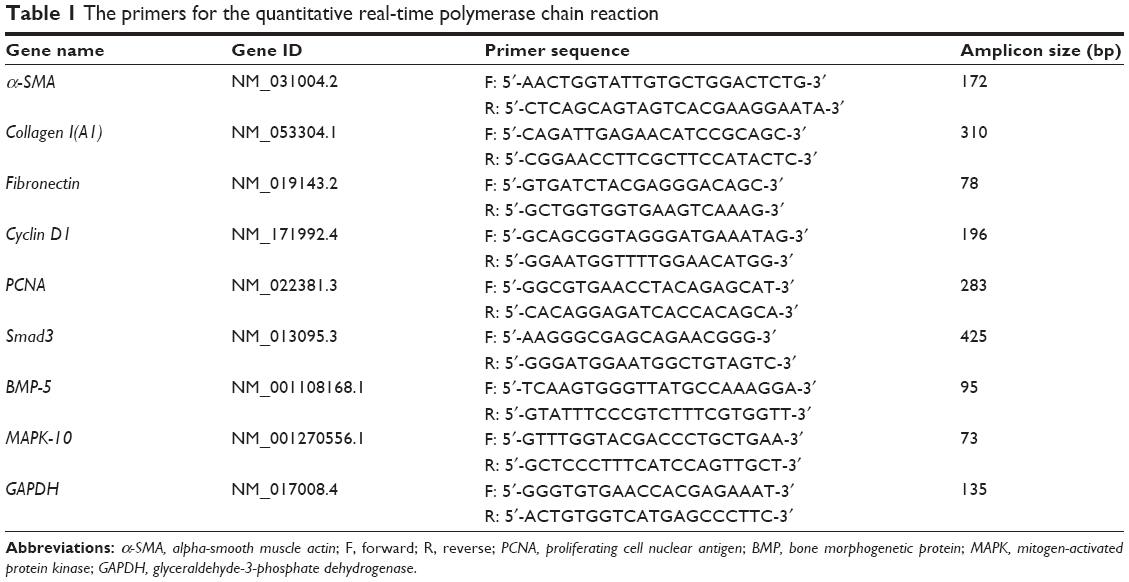 | Table 1 The primers for the quantitative real-time polymerase chain reaction |
Gene microarray
TGF-β/BMP signal transduction PCR array profiles consisting of 88 key genes were used (CTB-PA21; CT Biosciences, Changzhou, Jiangsu, People’s Republic of China). The real-time PCR allowed for the easy and reliable analysis of the expression of genes related to the TGF-β signal transduction pathways to determine the effect of Lefty-1 on TGF-β1-induced activation and proliferation of NRK-49F cells.
Statistical analyses
All statistical analyses were performed with SPSS 19.0 (IBM Corporation, Armonk, NY, USA), and data are presented here as the mean ± SD. The data were further subjected to analysis of variance. Differences between two groups were determined using Student’s t-test, and multiple means were compared by Tukey’s test. P-values <0.05 were considered statistically significant.
Results
Lefty-1 prevents TGF-β1-induced morphological changes in NRK-49F cells
After induction with TGF-β1 (10 ng/mL) for 48 hours, NRK-49F cells underwent a series of morphological changes. Following induction by TGF-β1, the cytoskeleton of NRK-49F cells exhibited bundles of internal microfilaments with gradual thickening and elongation, as well as a loss of spindle or stellate-shaped fibroblast appearance. Lefty-1 significantly prevented these TGF-β1-induced phenotypic changes with significant effects observed at 50 ng/mL. No significant morphological changes were observed in cells treated only with Lefty-1 (Figure 1).
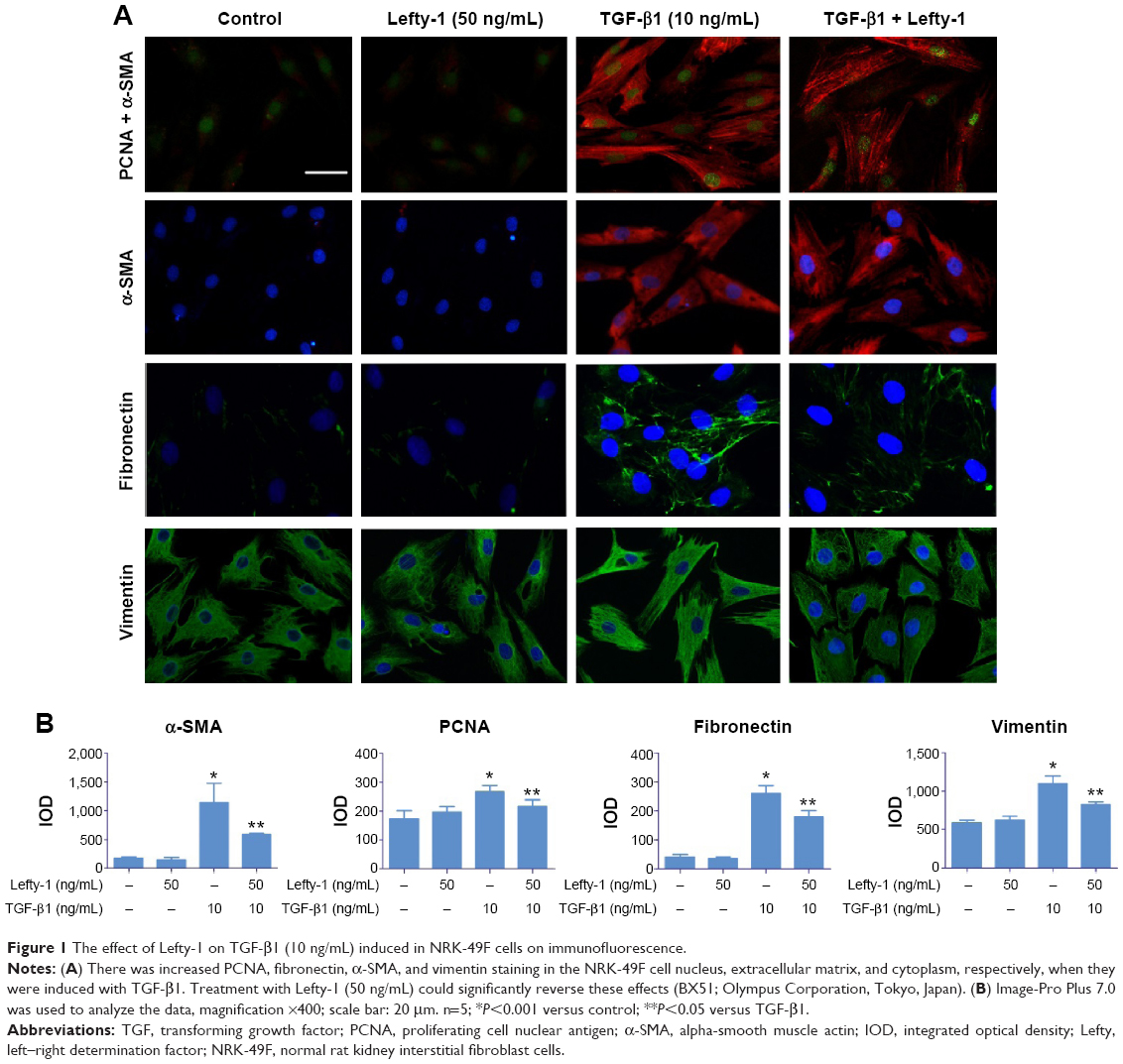 | Figure 1 The effect of Lefty-1 on TGF-β1 (10 ng/mL) induced in NRK-49F cells on immunofluorescence. |
Lefty-1 inhibits proliferation of TGF-β1-induced NRK-49F cells
The effect of Lefty-1 on the cell viability of TGF-β1-induced NRK-49F was examined by the CCK-8 assay (Figure 2A). The viability of TGF-β1 (10 ng/mL)-stimulated cells increased significantly compared with the control group (P<0.001), consistent with our previous results. Lefty-1 significantly inhibited a TGF-β1-induced increase in NRK-49F cell viability in a dose-dependent manner compared with the control. Immunofluorescence staining was performed after 48 hours of stimulation, and quantitative analysis of integrated option density values showed a significant increase in PCNA compared with the control (Figure 1). In parallel experiments, Lefty-1 significantly attenuated changes initiated by TGF-β1, thereby limiting PCNA induction. Furthermore, we found that NRK-49F cell proliferation treated with Lefty-1 (50 ng/mL) was significantly reduced; there was no difference between the treatment with Lefty-1 alone and the control sample in the BrdU incorporation assay (Figures 2B and C).
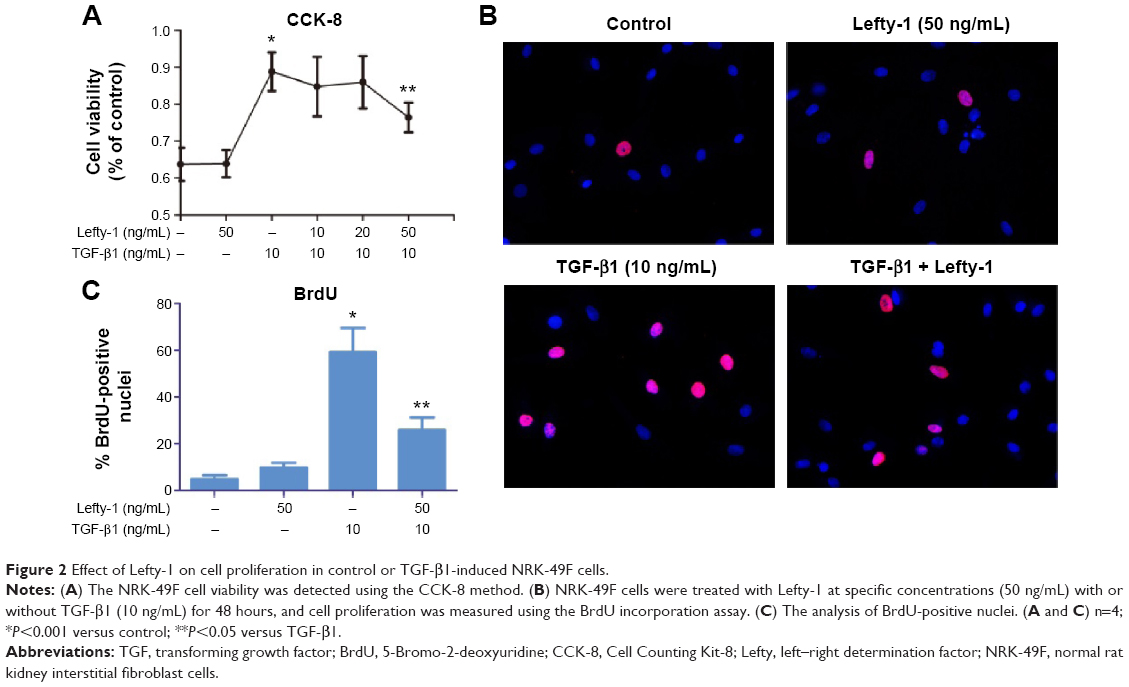 | Figure 2 Effect of Lefty-1 on cell proliferation in control or TGF-β1-induced NRK-49F cells. |
Flow cytometric cell cycle analysis was used to determine the effect of Lefty-1 on the cell cycle. Compared with the control group, the percentage of G2/M and G1 phase cells decreased significantly and the percentage of S-phase cells clearly increased with Lefty-1 treatment (P<0.05) 48 hours after TGF-β1 (10 ng/mL) stimulation. After treatment with Lefty-1 at specific concentrations (10–50 ng/mL), the percentage of S-phase cells decreased and the percentage of G1-phase cells increased. These results suggest that Lefty-1 can inhibit transition to the G1-phase of the cell cycle in TGF-β1-induced NRK-49F cells (Figure 3). Expression of related proteins and genes were further assayed by immunoblotting and quantitative real-time PCR, respectively. Lefty-1 treatment significantly decreased PCNA and cyclin D1 levels in NRK-49F cells (Figure 4). Consistent with earlier findings, no significant changes were observed upon treatment with Lefty-1 alone.
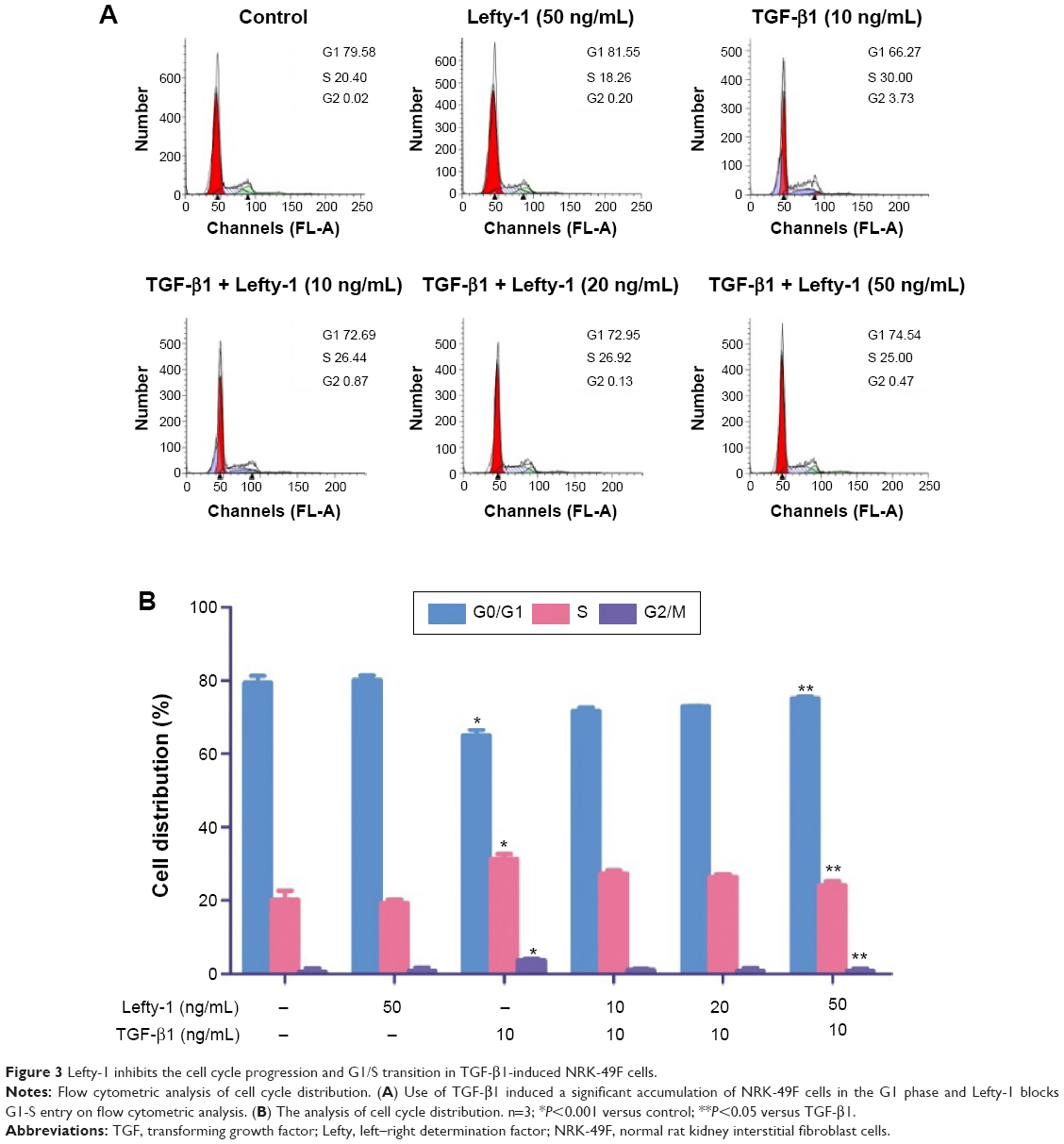 | Figure 3 Lefty-1 inhibits the cell cycle progression and G1/S transition in TGF-β1-induced NRK-49F cells. |
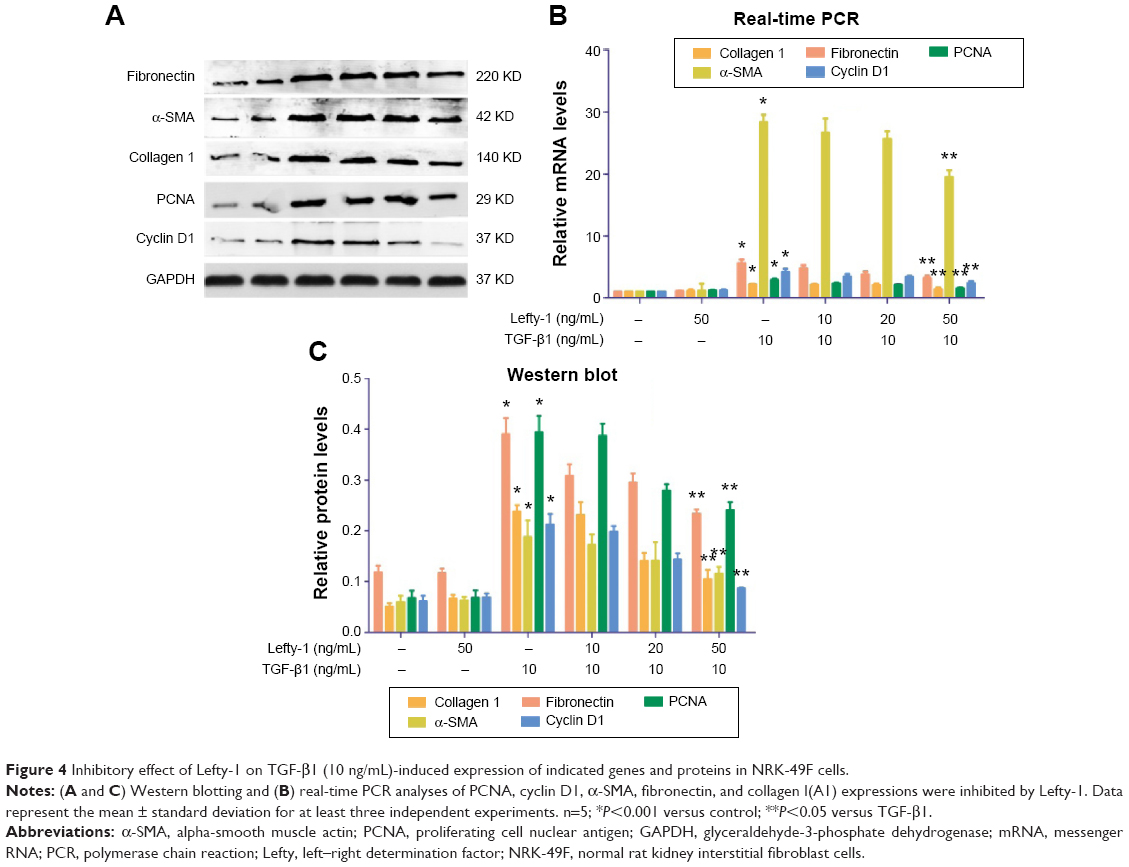 | Figure 4 Inhibitory effect of Lefty-1 on TGF-β1 (10 ng/mL)-induced expression of indicated genes and proteins in NRK-49F cells. |
Lefty-1 prevents TGF-β1-induced NRK-49F cell activation
To determine whether Lefty-1 affects fibroblast–myofibroblast transdifferentiation, we investigated the effects of Lefty-1 on TGF-β1 (10 ng/mL)-induced NRK-49F cell transdifferentiation. Lefty-1 (0 ng/mL, 10 ng/mL, 20 ng/mL, and 50 ng/mL) was added to the TGF-β1-induced NRK-49F cell system for 48 hours, and cells were analyzed by quantitative real-time PCR and Western blotting. The messenger (m)RNA and protein (Figure 4) expression levels of α-SMA, collagen I (A1), and fibronectin were upregulated by TGF-β1 compared with the control. Expressions of these genes were inhibited by Lefty-1 in a dose-dependent manner, and significant inhibition was observed at 50 ng/mL. Immunostaining and quantitative analysis confirmed that Lefty-1 (50 ng/mL) significantly attenuated such changes (Figure 1). Expressions of these genes remained constant with Lefty-1 treatment alone based on quantitative real-time PCR and Western blot results.
Lefty-1 modulates activities of Smad3, JNK-3, and BMP-5 in the TGF-β/BMP signaling pathway
TGF-β signaling plays important role in renal fibrosis through downstream Smad and non-Smad signaling pathways. In this study, Lefty-1 was observed to inhibit TGF-β1-induced NRK-49F cell transdifferentiation via these pathways. To probe this further, we screened the expression of various genes in the TGF-β/BMP signaling pathways and further verified the results by real-time PCR. A gene microarray consisting of 88 representative genes related to these signal transduction pathways was used. The results showed that 82 genes were differentially expressed in the Lefty-1 treatment group (50 ng/mL) relative to the TGF-β1-induced group. Among these genes, 57 were upregulated and 25 were downregulated (data not shown). We found that Lefty-1 significantly inhibited the expression of Smad3 and mitogen-activated protein kinase (MAPK)-10/(JNK-3) and enhanced BMP-5 expression. Real-time PCR and Western blotting analyses validated these results; mRNA and protein expression levels of Smad3 and MAPK-10/JNK-3 decreased, whereas that of BMP-5 increased in the Lefty-1-treated group (Figure 5).
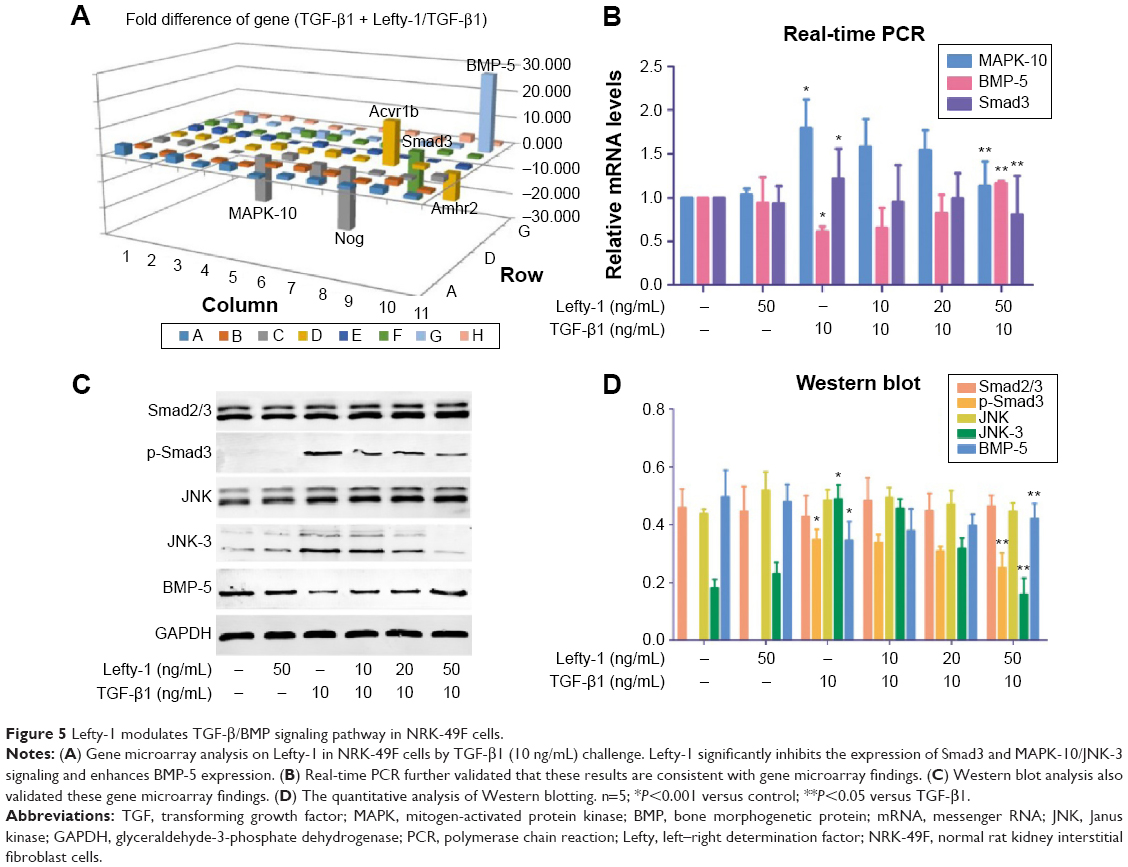 | Figure 5 Lefty-1 modulates TGF-β/BMP signaling pathway in NRK-49F cells. |
Discussion
In this study, we provide evidence that Lefty-1 alleviates TGF-β1-mediated fibroblast–myofibroblast transdifferentiation in NRK-49F cells and suggest a possible molecular mechanism mediating this effect. In our experiments, TGF-β1 stimulation upregulated the expression of activation- and proliferation-related markers in NRK-49F cells; this change was accompanied by a transition to the characteristic phenotype observed during fibroblast–myofibroblast transdifferentiation, which is directly related to the progression of renal fibrosis.16,17 While the importance of fibroblast–myofibroblast transdifferentiation in renal interstitial fibrosis has been widely disputed,18–20 increasing evidence suggests that activated fibroblasts are likely to be the major sources of myofibroblasts during the pathological process of renal fibrosis.21 Our study has revealed that fibroblasts undergo morphological and functional changes to acquire a myofibroblastic phenotype, which is ultimately related to pathological development of renal interstitial fibrosis. Thus, blocking fibroblast–myofibroblast transdifferentiation may effectively alleviate the progression of renal failure, thereby enabling intervention of kidney disease.
Lefty is an important regulator of TGF-β signaling that is involved in stem cell differentiation and embryonic development. In this study, various Lefty-1 concentrations (0–50 ng/mL) with or without TGF-β1 (10 ng/mL) induction of NRK-49F cells for 48 hours were used to identify the function of Lefty-1 in fibroblast–myofibroblast transdifferentiation. In the absence of Lefty-1, TGF-β1 significantly promoted the differentiation of fibroblast cells into myofibroblasts. Our results also showed that Lefty-1 treatment can partially prevent TGF-β1-induced activation and proliferation of NRK-49F cells. In a number of ways, activated fibroblasts and smooth muscle cells resemble myofibroblasts owing to their expression of α-SMA, contractility, and excessive production of ECM.22 Our results suggest that preventing the fibroblast–myofibroblast transdifferentiation may be a potential mechanism through which Lefty-1 inhibits the proliferation and activation of fibroblast responses to TGF-β1 induction.
The signaling pathways involved in fibroblast–myofibroblast transdifferentiation are complex and poorly understood so far. To the best of our knowledge, Smads signaling is considered the most important among the TGF-β/BMP pathways. In the present study, we identified previously unknown pathways of TGF-β/BMP signaling using gene microarrays and examined these pathways further by real-time PCR and Western blotting. Previous studies indicated that Lefty negatively modulates phosphorylation of receptor-activated Smads (R-Smads) and R-Smad/co-Smad copolymer in the TGF-β1/Smad pathway.12,23 However, Lefty-1 treatment in our experiment failed to downregulate Smad2 and Smad4, the canonical TGF-β/Smads signaling components. Instead, we found that Lefty-1 significantly reduced the expression of Smad3 and enhanced BMP-5 expression. BMPs, which make up the largest subfamily of the TGF-β family, include growth and differentiation factors and activin. These proteins have multiple functions associated with antifibrosis, growth, differentiation, and cell death during embryonic development.24,25 A possible explanation for the observed results is that the molecules mediating intracellular signaling may vary depending on specific cellular contexts. Moreover, increasing evidence suggests that besides the canonical Smads pathway, TGF-β1 signals through the non-Smads pathway, particularly MAPKs-ERKs, p38, and JNKs.26,27
Recent studies have identified the function of JNK signaling in other mesenchymal cell types, including rat cardiac fibroblasts and human synovial fibroblasts.28,29 Similar to these observations, our results indicated that Lefty-1 treatment has no effect on TGF-β1-induced p38-MAPK and ERK1/2 activation in NRK-49F cells. This finding is in sharp contrast with the changes we observed in MAPK-10/JNK-3 signaling upon Lefty-1 treatment. These results suggest the need to evaluate the biological characteristics of the TGF-β signaling pathway in different species and cells with diverse functions. It is noteworthy that, without TGF-β1, Lefty-1 had no influence on Smad3 and JNK-3 activation and fibroblast–myofibroblast transdifferentiation. We propose that Lefty-1 regulates a scaffolding molecule involved in TGF-β1-induced fibroblast–myofibroblast transdifferentiation. Lefty-1 predominantly inhibits activation of Smad3, and recent studies argue that Smad3, rather than other Smad-signaling molecules, is the major mediator of TGF-β1-induced fibrosis.30,31 Data also show that Lefty-1 may regulate the cell cycle by modulating the expressions of P57 and cyclin D1 to inhibit the decidualization process.32 As observed in the present study, Lefty-1 regulates the cell cycle, which may be an important part of its suppression of the activation and proliferation of TGF-β1-induced NRK-49F cells. Overall, the results indicate that Lefty-1 is a crucial mediator of activation of Smad3, JNK-3 and BMP-5, all of which are critical signaling molecules for fibroblast–myofibroblast transdifferentiation in response to TGF-β1.
In this study, TGF-β1-induced transdifferentiation of fibroblasts to myofibroblasts was attenuated by the exogenous Lefty-1 in cell lines. It is necessary to further investigate whether equivalent effects were observed in in vivo studies and clinical trials. In addition, our study only investigated the Smad, non-Smad, and BMP signaling pathways; the effects on other signaling pathways and cross-talk between these pathways require further study. In summary, this study primarily examined the protective roles of Lefty-1 in alleviating TGF-β1-induced activation and proliferation of NRK-49F cells via the Smad3 and JNK-3 signaling pathways and enhancing the antifibrotic factor of BMP-5 expression. Elevated Lefty-1 expression was found to partially inhibit TGF-β1-induced fibroblast–myofibroblast transdifferentiation. Thus, our findings highlight the importance of Lefty-1 in the prevention of renal interstitial fibrosis and may help identify potential targets for therapeutic intervention of renal diseases.
Acknowledgment
This work was financially supported by the National Natural Science Foundation of China (number 81170710).
Disclosure
The authors report no conflicts of interest in this work.
References
Hewitson TD. Renal tubulointerstitial fibrosis: common but never simple. Am J Physiol Renal Physiol. 2009;296(6):F1239–F1244. | ||
Liu Y. Cellular and molecular mechanisms of renal fibrosis. Nat Rev Nephrol. 2011;7(12):684–696. | ||
Baum J, Duffy HS. Fibroblasts and myofibroblasts: what are we talking about? J Cardiovasc Pharmacol. 2011;57(4):376–379. | ||
Asada N, Takase M, Nakamura J, et al. Dysfunction of fibroblasts of extrarenal origin underlies renal fibrosis and renal anemia in mice. J Clin Invest. 2011;121(10):3981–3990. | ||
Meran S, Steadman R. Fibroblasts and myofibroblasts in renal fibrosis. Int J Exp Pathol. 2011;92(3):158–167. | ||
Meng XM, Chung AC, Lan HY. Role of the TGF-β/BMP-7/Smad pathways in renal diseases. Clin Sci (Lond). 2013;124(4):243–254. | ||
Lan HY. Tubular epithelial-myofibroblast transdifferentiation mechanisms in proximal tubule cells. Curr Opin Nephrol Hypertens. 2003;12(1):25–29. | ||
Xiong M, Jiang L, Zhou Y, et al. The miR-200 family regulates TGF-β1-induced renal tubular epithelial to mesenchymal transition through Smad pathway by targeting ZEB1 and ZEB2 expression. Am J Physiol Renal Physiol. 2012;302(3):F369–F379. | ||
Kosaki K, Bassi MT, Kosaki R, et al. Characterization and mutation analysis of human LEFTY A and LEFTY B, homologues of murine genes implicated in left-right axis development. Am J Hum Genet. 1999;64(3):712–721. | ||
Makridakis M, Roubelakis MG, Vlahou A. Stem cells: insights into the secretome. Biochim Biophys Acta. 2013;1834(11):2380–2384. | ||
Barroso-delJesus A, Lucena-Aguilar G, Sanchez L, Ligero G, Gutierrez-Aranda I, Menendez P. The Nodal inhibitor Lefty is negatively modulated by the microRNA miR-302 in human embryonic stem cells. FASEB J. 2011;25(5):1497–1508. | ||
Ulloa L, Tabibzadeh S. Lefty inhibits receptor-regulated Smad phosphorylation induced by the activated transforming growth factor-beta receptor. J Biol Chem. 2001;276(24):21397–21404. | ||
Cavallari C, Fonsato V, Herrera MB, Bruno S, Tetta C, Camussi G. Role of Lefty in the anti tumor activity of human adult liver stem cells. Oncogene. 2013;32(7):819–826. | ||
Li Y, Zhang J, Fang L, Luo P, Peng J, Du X. Lefty A attenuates the TGF-beta1-induced epithelial to mesenchymal transition of human renal proximal epithelial tubular cells. Mol Cell Biochem. 2010;339(1–2):263–270. | ||
Zheng RP, Bai T, Zhou XG, et al. Lefty A protein inhibits TGF-β1-mediated apoptosis in human renal tubular epithelial cells. Mol Med Rep. 2013;8(2):621–625. | ||
Desmoulière A, Chaponnier C, Gabbiani G. Tissue repair, contraction, and the myofibroblast. Wound Repair Regen. 2005;13(1):7–12. | ||
Poosti F, Bansal R, Yazdani S, et al. Selective delivery of IFN-γ to renal interstitial myofibroblasts: a novel strategy for the treatment of renal fibrosis. FASEB J. 2015;29(3):1029–1042. | ||
Lin SL, Kisseleva T, Brenner DA, Duffield JS. Pericytes and perivascular fibroblasts are the primary source of collagen-producing cells in obstructive fibrosis of the kidney. Am J Pathol. 2008;173(6):1617–1627. | ||
Humphreys BD, Lin SL, Kobayashi A, et al. Fate tracing reveals the pericyte and not epithelial origin of myofibroblasts in kidney fibrosis. Am J Pathol. 2010;176(1):85–97. | ||
Grgic I, Duffield JS, Humphreys BD. The origin of interstitial myofibroblasts in chronic kidney disease. Pediatr Nephrol. 2012;27(2):183–193. | ||
Hinz B, Phan SH, Thannickal VJ, Galli A, Bochaton-Piallat ML, Gabbiani G. The myofibroblast: one function, multiple origins. Am J Pathol. 2007;170(6):1807–1816. | ||
Zeisberg M, Strutz F, Müller GA. Role of fibroblast activation in inducing interstitial fibrosis. J Nephrol. 2000;13 Suppl 3:S111–S120. | ||
Yao Y, Zhang J, Ye DF, et al. Left-right determination factor is down-regulated in fibrotic renal tissue of human hydronephrosis. BJU Int. 2011;107(6):1002–1008. | ||
Miyazono K, Kamiya Y, Morikawa M. Bone morphogenetic protein receptors and signal transduction. J Biochem. 2010;147(1):35–51. | ||
Guan Q, Li S, Gao S, Chen H, Nguan CY, Du C. Reduction of chronic rejection of renal allografts by anti-transforming growth factor-β antibody therapy in a rat model. Am J Physiol Renal Physiol. 2013;305(2):F199–F207. | ||
Moustakas A, Heldin CH. Non-Smad TGF-beta signals. J Cell Sci. 2005;118(Pt 16):3573–3584. | ||
Mu Y, Gudey SK, Landström M. Non-Smad signaling pathways. Cell Tissue Res. 2012;347(1):11–20. | ||
Liu S, Li W, Xu M, Huang H, Wang J, Chen X. Micro-RNA 21Targets dual specific phosphatase 8 to promote collagen synthesis in high glucose-treated primary cardiac fibroblasts. Can J Cardiol. 2014;30(12):1689–1699. | ||
Lee WS, Lim JH, Sung MS, Lee EG, Oh YJ, Yoo WH. Ethyl acetate fraction from Angelica sinensis inhibits IL-1β-induced rheumatoid synovial fibroblast proliferation and COX-2, PGE2, and MMPs production. Biol Res. 2014;47(1):41. | ||
Tsukamoto S, Mizuta T, Fujimoto M, et al. Smad9 is a new type of transcriptional regulator in bone morphogenetic protein signaling. Sci Rep. 2014;4:7596. | ||
Sun GX, Ding R, Li M, et al. Ghrelin attenuates renal fibrosis and inflammation of obstructive nephropathy. J Urol. 2015;193(6):2107–2115. | ||
Li H, Li H, Bai L, Yu H. Lefty inhibits in vitro decidualization by regulating P57 and cyclin D1 expressions. Cell Biochem Funct. 2014;32(8):657–664. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.