")
Back to Journals » Drug Design, Development and Therapy » Volume 9
Investigation of cyclooxygenase and signaling pathways involved in human platelet aggregation mediated by synergistic interaction of various agonists
Authors Khan N, Farooq A, Sadek B
Received 10 March 2015
Accepted for publication 29 April 2015
Published 6 July 2015 Volume 2015:9 Pages 3497—3506
DOI https://doi.org/10.2147/DDDT.S84335
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Shu-Feng Zhou
Nadia Khan,1,2 Ahsana Dar Farooq,1 Bassem Sadek2
1Dr Panjwani Center for Molecular Medicine and Drug Research, International Center for Chemical and Biological Sciences, University of Karachi, Karachi, Pakistan; 2Department of Pharmacology and Therapeutics, College of Medicine and Health Sciences, United Arab Emirates University, Al Ain, United Arab Emirates
Abstract: In the present study, the mechanism(s) of synergistic interaction of various platelet mediators such as arachidonic acid (AA) when combined with 5-hydroxytryptamine (5-HT) or adenosine diphosphate (ADP) on human platelet aggregation were examined. The results demonstrated that 5-HT had no or negligible effect on aggregation but it did potentiate the aggregation response of AA. Similarly, the combination of subeffective concentrations of ADP and AA exhibited noticeable rise in platelet aggregation. Moreover, the observed synergistic effect of AA with 5-HT on platelets was inhibited by different cyclooxygenase (COX) inhibitors, namely ibuprofen and celecoxib, with half maximal inhibitory effect (IC50) values of 18.0±1.8 and 15.6±3.4 µmol/L, respectively. Interestingly, the synergistic effect observed for AA with 5-HT was, also, blocked by the 5-HT receptor blockers cyproheptadine (IC50=22.0±7 µmol/L), ketanserin (IC50=152±23 µmol/L), phospholipase C (PLC) inhibitor (U73122; IC50=6.1±0.8 µmol/L), and mitogen activated protein kinase (MAPK) inhibitor (PD98059; IC50=3.8±0.5 µmol/L). Likewise, the synergism of AA and ADP was, also, attenuated by COX inhibitors (ibuprofen; IC50=20±4 µmol/L and celecoxib; IC50=24±7 µmol/L), PLC inhibitor (U73122; IC50=3.7±0.3 µmol/L), and MAPK inhibitor (PD98059; IC50=2.8±1.1 µmol/L). Our observed data demonstrate that the combination of subthreshold concentrations of agonists amplifies platelet aggregation and that these synergistic effects largely depend on activation of COX/thromboxane A2, receptor-operated Ca2+ channels, Gq/PLC, and MAPK signaling pathways. Moreover, our data revealed that inhibition of COX pathways by using both selective and/or non-selective COX inhibitors blocks not only AA metabolism and thromboxane A2 formation, but also its binding to Gq receptors and activation of receptor-operated Ca2+ channels in platelets. Overall, our results show that PLC and MAPK inhibitors proved to inhibit the synergistic activation of platelets by several/multiple agonists.
Keywords: synergism, platelet aggregation, cyclooxygenase, signaling pathway, arachidonic acid, 5-hydroxytryptophan, adenosine-5-diphosphate
Introduction
Platelets are prime modulators of hemostasis. Circulating platelets express cyclooxygenase (COX), a membrane-bound glycoprotein that rapidly undergo oxygenation of membrane phospholipid arachidonic acid (AA) to release bioactive substances in response to damaged vessel. During hemostasis, many important platelet interactions are surface related and depend on AA.1,2 In platelets, this flexible fatty acid helps in maintaining cell membrane, their correct fluidity, and integrity, and regulates the synthesis and release of granular contents in circulation at physiological temperatures.2 Activation of platelets causes AA to metabolize into short-lived intermediates, eg, prostaglandin G2 and prostaglandin H2. The latter is converted into different bioactive prostaglandins such as prostaglandin F2, prostaglandin D2, prostaglandin I2, and thromboxane A2 (TXA2), which are involved in regulation of human physiological functions including immune system, vascular modulation, inflammation, neurostimulation, and regulation of body temperature (Figure 1).3–6 Among these molecules, TXA2 is an important metabolite that possesses two major activities; first, it acts as a potent vasoconstrictor, which induces turbulent shear stress and decreases blood flow in the vessels, resulting in cardiovascular disorders. Second, it causes activation of platelets multistep process involving adhesion, shape change, extrusion of pseudopodia, and exocytosis of stored granular contents (adenosine diphosphate [ADP], platelet activating factor, TXA2, and 5-hydroxytryptamine [5-HT]).7 Upon vascular injury, the primary adhesion of platelets with subendothelial extracellular matrix is mediated by adhesive molecules under high shear stress to form a monolayer.8 This is followed by subsequent recruitment of additional platelets from circulation by releasing stored dense granules to form a platelet plug. It has been shown that most of these diffusible agonists act via G protein-coupled receptors, particularly the phosphoinositide C-linked G-protein receptors (GqRs) (Figure 2). Activation of GqRs signaling pathway consecutively increase their own formation and release, and therefore, acting as a positive feedback mechanism that amplifies platelet activation, adhesion, aggregation followed by thrombus formation.9,10 The synergistic effect of these agonists through GqRs involves the effector protein phospholipase C (PLC) that catalyzes the metabolism of phosphatidylinositol-4,5-bisphosphate into two second messengers, namely diacylglycerol (DAG) and inositol triphosphate (IP3). IP3 increases intracellular mobilization of Ca2+ ions by non-voltage gated Ca2+ channels or receptor-operated Ca2+ channels (ROCCs), whereas DAG activates protein kinase C (PKC). Consequently, the PKC catalyzes and phosphorylates many proteins and initiate intracellular responses. Both DAG and PKC signaling molecules stimulate mitogen activated protein kinases (MAPKs) in MAPK pathway (Figure 2).11 Interestingly, an elevation of cytosolic Ca2+ by ROCCs and activation of PKC and Ca2+-regulated MAPKs initiate molecular mechanisms in which COX, ROCCs, and 5-HT cause a decrease in contraction of cardiomyocytes, impaired vascular integrity and high shear stress, exposure of subendothelial cells, and release of pro-inflammatory cytokines thus, may accelerate progression of peripheral vascular diseases, myocardial ischemia, and atherosclerosis.12
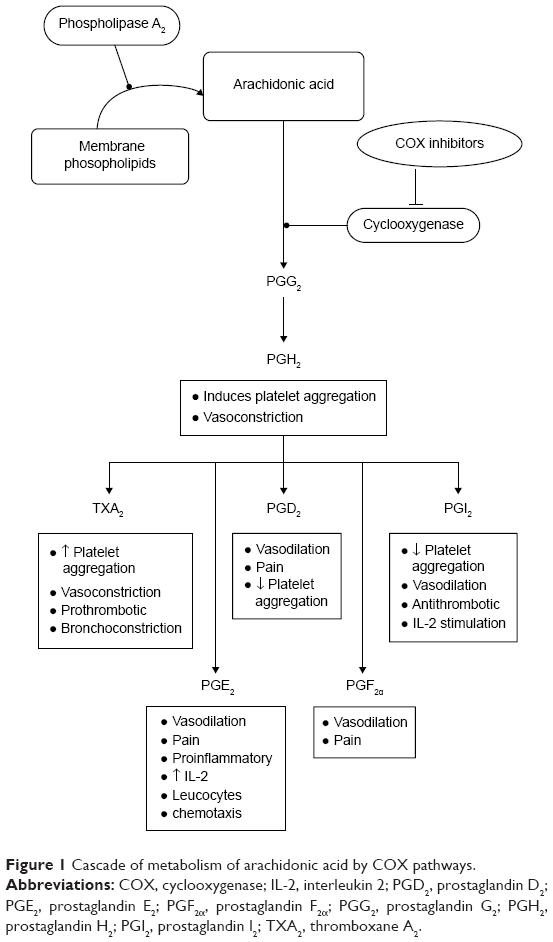 | Figure 1 Cascade of metabolism of arachidonic acid by COX pathways. |
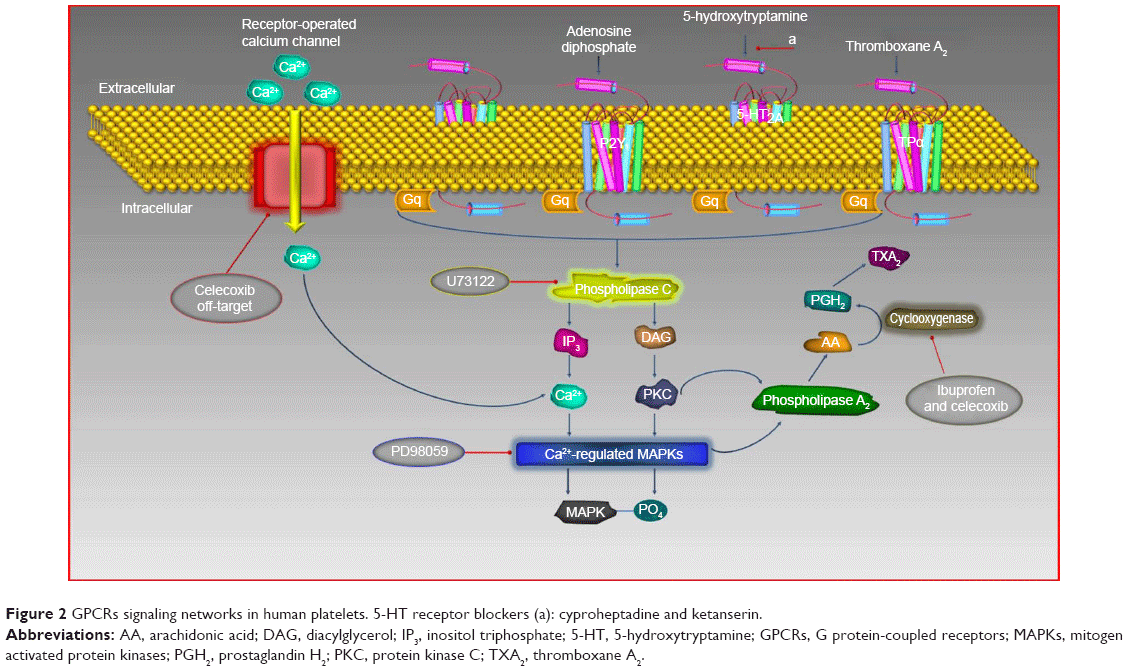 | Figure 2 GPCRs signaling networks in human platelets. 5-HT receptor blockers (a): cyproheptadine and ketanserin. |
This study was conducted to determine the interaction between AA with 5-HT and AA with ADP, and to elucidate the possible molecular mechanism(s) involved in synergism. Moreover, the involvement of multiple intracellular signaling pathways including COX, Gq/PLC, and MAPK was evaluated to identify downstream cellular and molecular events in synergism of platelet aggregation by various agonists.
Materials and methods
Chemicals
AA, ADP, ibuprofen, celecoxib, acetylsalicylic acid, 5-HT, and PD98059 were purchased form Sigma Chemicals (St Louis, MO, USA). Cyproheptadine and ketanserin were purchased from MP Biomed (Santa Ana, CA, USA). U73122 was obtained from Santa Cruz Biotechnology (Heidelberg, Germany). All other chemicals used were of the highest purity grade available.
Preparation of human platelets
This was carried out by taking blood via venipuncture from normal human volunteers aged 22–38 years, and reported to be free of medication for 7 days. Blood was drawn from the antecubital vein and was mixed with anticoagulant 3.8% (w/v) sodium citrate solution in the ratio of (9:1) in 15 mL tube and allowed to settle for 10 minutes. After 10 minutes, it was centrifuged at 1,000 rpm for 15 minutes at 20°C–25°C to obtain platelet rich plasma (PRP). The PRP was carefully taken out in a separate 15 mL tube, marked PRP, and stored at room temperature. The remaining sample was centrifuged at 3,000 rpm for 15 minutes at 20°C–25°C to obtain platelet poor plasma. Platelet count was determined by phase contrast microscopy and all aggregation studies were carried out at 37°C with PRP having platelet counts between 2.5 and 3.0×108/mL of plasma.13 All experiments were performed within 3 hours of PRP preparation.
Measurement of platelet aggregation
Aggregation assay was monitored using a Dual Channel Lumi-Aggregometer (Model 400VS, Chrono-log Corporation, Chicago, IL, USA), based on turbidimetric principle14 in which a beam of light is passed through the PRP sample continuously stirred in a test cuvette, at a fixed speed (1,100 rpm) and temperature maintained at 37°C. The change in light transmission was recorded by a dual pen chart recorder indicating a decrease in the number of platelets. Prior to running the experiment, 0% and 100% light transmission was adjusted with PRP and platelet poor plasma, respectively. In a test cuvette, 0.45 mL of PRP was preincubated for 2–3 minutes prior to challenge with an aggregating agent. The final volume was made up to 0.5 mL with normal saline or the test drug dissolved in an appropriate vehicle known to be devoid of any effect on aggregation. Aggregation was induced by AA (1.7 mmol/L) and ADP (5 μmol/L).
To examine the synergistic effect of AA plus 5-HT and AA plus ADP, subthreshold concentrations of these agonists added together in PRP and a maximal effect was achieved. The anti-aggregatory effects of various inhibitors and receptor blockers were studied by pretreating PRP with an inhibitor for 1–3 minute(s), followed by addition of subthreshold concentration of AA plus 5-HT and AA plus ADP. The test drug was dissolved either in normal saline or in appropriate vehicle known to be devoid of any effects on aggregation. The resulting aggregation was recorded for 5 minutes after challenge by the change in light transmission as a function of time. Once the anti-platelet activity of various inhibitors was established, dose–response curves were constructed to calculate 50% inhibitory concentration of the inhibitor.
Analysis of data
Half maximal inhibitory concentration (IC50) (μmol/L) producing 50% inhibition of platelet aggregation (control response taken as 100%). The 50% inhibitory concentration values were calculated as mean ± standard error of the mean (SEM) of three to five independent experiments. Statistical differences between control and test measurements were analyzed by Student’s t-test and P<0.05 was considered as significant.
Results
Concentration-dependent effect of various agonists on platelet aggregation
When PRP was treated with different concentrations of AA (0.2–1.7 mmol/L) (Figures 3B and 6A) and ADP (0.2–5.0 μmol/L) (Figure 6B), a dose-dependent aggregative effect was achieved with maximal aggregation at the highest dose. However, treatment of platelets with 5-HT (5.0–200 μmol/L) had no effect on aggregation (Figure 3A).
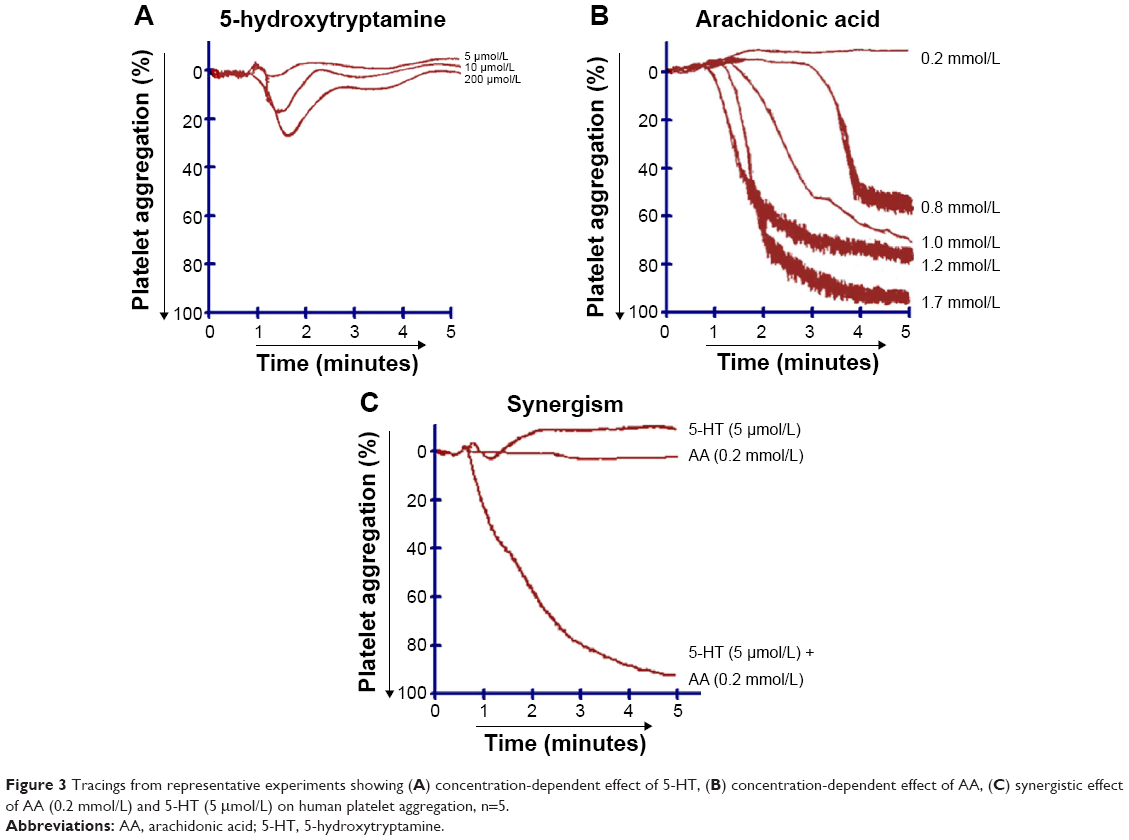 | Figure 3 Tracings from representative experiments showing (A) concentration-dependent effect of 5-HT, (B) concentration-dependent effect of AA, (C) synergistic effect of AA (0.2 mmol/L) and 5-HT (5 μmol/L) on human platelet aggregation, n=5. |
Effect of subthreshold concentration of various agonists on platelet aggregation
When subthreshold concentration of AA (0.2 mmol/L) plus 5-HT (5 μmol/L) and AA (0.2 mmol/L) plus ADP (0.2 μmol/L) was combined together in platelet suspension, they amplify the effect of each other and a marked synergism was observed with regard to aggregation. This synergism of AA with 5-HT (Figure 3C) and AA with ADP (Figure 6C) is mediated through activation of multiple signaling pathways, which are intracellularly connected to each other in platelet aggregation.
Effect of COX inhibitors on synergism of AA with 5-HT and AA with ADP
The AA derived from membrane phospholipid is actively metabolized by COX to form short-lived biomolecules including TXA2 (Figure 1). To determine the effect of COX on TXA2 formation in the synergism of AA with 5-HT and AA with ADP, we used various COX inhibitors. The pretreatment of PRP with ibuprofen (5–40 μmol/L) attenuated the synergism of AA with 5-HT in a dose-dependent fashion (Figure 4A). Likewise, celecoxib (12–30 μmol/L) addition in PRP before challenge with subthreshold concentration of agonists also elicited similar inhibitory effect (Figure 4B). The IC50 values are given in Table 1. The synergism of AA with ADP was found to be inhibited in PRP when it was pretreated with different concentrations of ibuprofen (10–50 μmol/L) and celecoxib (15–45 μmol/L). The results showed that a dose-dependent inhibitory effect was recorded (Figure 7A and B, respectively). The IC50 values of ibuprofen and celecoxib are given in Table 2. From the above results, it is obvious that TXA2 is the common aggregating molecule synthesized by COX in the synergistic pathway of both AA with 5-HT and AA with ADP. Using different COX inhibitors, the formation and release of TXA2 in the synergism is inhibited in a dose-dependent manner.
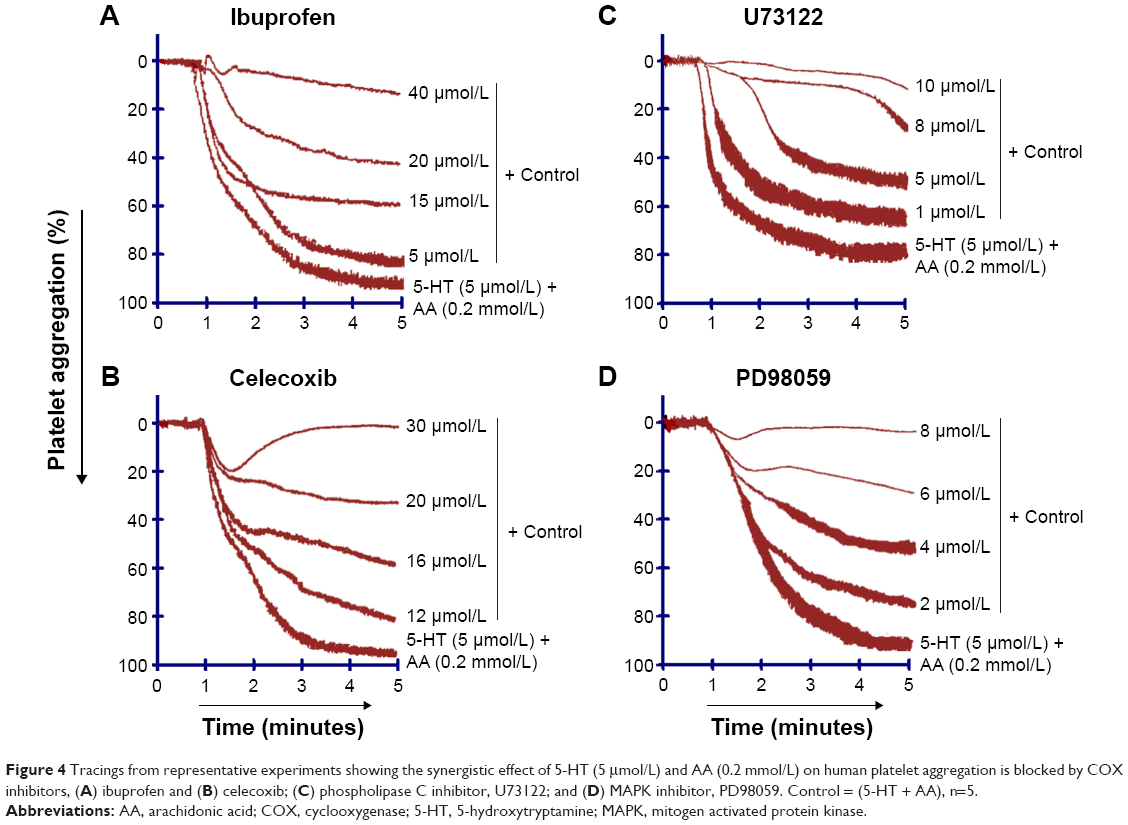 | Figure 4 Tracings from representative experiments showing the synergistic effect of 5-HT (5 μmol/L) and AA (0.2 mmol/L) on human platelet aggregation is blocked by COX inhibitors, (A) ibuprofen and (B) celecoxib; (C) phospholipase C inhibitor, U73122; and (D) MAPK inhibitor, PD98059. Control = (5-HT + AA), n=5. |
 | Table 1 Synergism between AA and 5-HT |
 | Table 2 Synergism between AA and ADP |
Effect of 5-HT2 receptor blockers in synergism of AA with 5-HT and AA with ADP
The role of 5-HT receptors in the synergism of AA and 5-HT induced by co-addition of subthreshold concentrations was investigated. Our findings showed that pretreatment of PRP with 5-HT receptor antagonist such as cyproheptadine (10–50 μmol/L) completely blocks the receptor at maximum concentration (Figure 5A), whereas ketanserin (50–350 μmol/L) inhibits the synergism by blocking the receptor in a dose-dependent manner (Figure 5B). The IC50 value of cyproheptadine is 22±7 μmol/L and ketanserin is 152±23 μmol/L (Table 1).
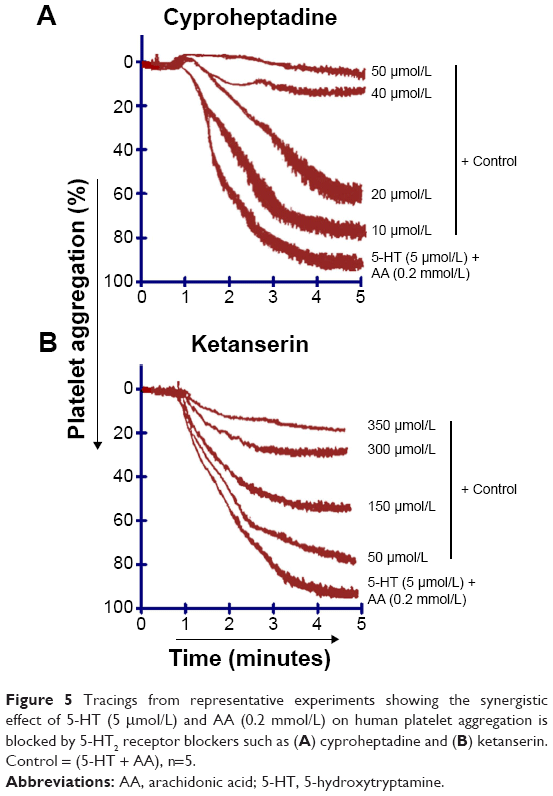 | Figure 5 Tracings from representative experiments showing the synergistic effect of 5-HT (5 μmol/L) and AA (0.2 mmol/L) on human platelet aggregation is blocked by 5-HT2 receptor blockers such as (A) cyproheptadine and (B) ketanserin. Control = (5-HT + AA), n=5. |
Effect of PLC inhibitor in synergism of AA with 5-HT and AA with ADP
A PLC inhibitor was used to figure out whether the synergistic effect of both pairs, AA with 5-HT and AA with ADP, is involved in the activation of a common PLC pathway. Our observed results showed that when U73122 was added in platelet suspension, a dose-dependent decrease in aggregation was recorded, suggesting that the synergism of AA with 5-HT, and AA with ADP was suppressed by U73122 (Figures 4C and 7C, respectively). The IC50 values for AA with 5-HT and AA with ADP are shown in Tables 1 and 2, respectively.
Effect of MAPK inhibitor in synergism of AA with 5-HT and AA with ADP
As stimulation of the G protein/PLC leads to activation of downstream MAPK (Figure 2), we further elaborated our investigation using selective MAPK inhibitor. Our results demonstrate that pretreatment of PRP with low doses (2–8 μmol/L) of PD98059 causes strong inhibition of aggregation induced by co-addition of subthreshold concentration of AA with 5-HT, suggesting its role in the synergism of AA with 5-HT (Figure 4D). Similarly, in a dose-dependent inhibition of synergism of AA with ADP was observed when PRP was treated with low doses (1–7 μmol/L) of PD98059 (Figure 7D). The IC50 values for AA with 5-HT and AA with ADP are shown in Tables 1 and 2, respectively.
Discussion
Multifunctional blood platelets express membrane-bound COX and transmembrane G protein-coupled receptors. Studies have shown that various platelet agonists at subthreshold concentrations elicit synergistic interactions.15–17 Results of our study demonstrated that platelet aggregation was entirely concentration dependent when AA (Figure 3B) and ADP (Figure 6B) were used alone with the exception of 5-HT (Figure 3A), which had no significant effect on aggregation. Interestingly, when subthreshold concentrations of AA plus ADP were added together in PRP, the effect of each component was amplified and maximum platelet aggregation was induced (Figure 6C). Similarly, the combined effect of subthreshold concentrations of AA plus 5-HT also showed a marked increase in aggregation (Figure 3C). The molecular mechanisms of these synergisms are intricate as the agonists used in the current study stimulate diverse intracellular responses.
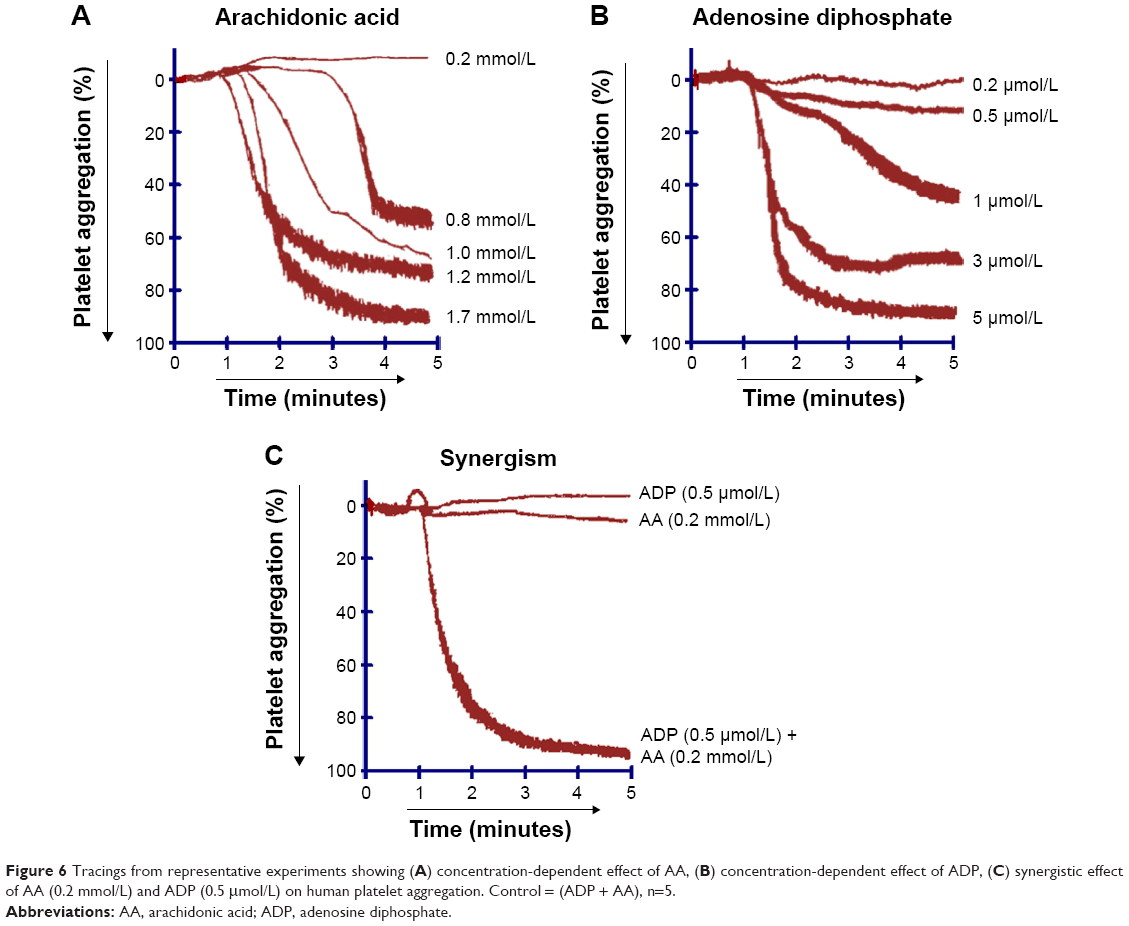 | Figure 6 Tracings from representative experiments showing (A) concentration-dependent effect of AA, (B) concentration-dependent effect of ADP, (C) synergistic effect of AA (0.2 mmol/L) and ADP (0.5 μmol/L) on human platelet aggregation. Control = (ADP + AA), n=5. |
It is well established that the molecular mechanisms of aggregation begin when AA is liberated from membrane phospholipids by phospholipase A2 (PLA2) in the cytosol through different stimuli (Figure 1)9 and is metabolized by COX pathway to generate TXA2. To investigate the involvement of COX in the synergism of both AA with 5-HT and AA with ADP, different COX inhibitors were used. Results showed that both ibuprofen (Figure 4A) and celecoxib (Figure 4B) blocked platelet aggregation induced by AA plus 5-HT in a dose-dependent manner by inhibiting the forward mechanism of AA conversion into most potent TXA2 (Table 1). It is well known that platelets lack L-type voltage-activated Ca2+ channels but they possess ROCCs on their membrane (Figure 2), which are activated by different platelet agonists acting on GqRs.18–20 Several studies21–24 have reported that celecoxib reversibly inhibit voltage-activated Ca2+ channels in different cell types. Our present findings show that the synthesis and effect of TXA2 on platelet aggregation mediated by AA and 5-HT are largely dependent on COX and modulation of ROCCs. We found that celecoxib exerts dual action on TXA2 formation, ie, inhibition of its proper target COX and off-target ROCCs in platelets, as Ca2+ ions are known secondary players in aggregation, potentiating the effect of agonists binding to GqRs. The inhibitory effect of celecoxib on ROCCs is independent of its effect on COX; thus, celecoxib has proved its dose-dependent dual independent inhibitory effects in the mechanism of synergistic effect of AA and 5-HT (Figure 2).
Similarly, ibuprofen and celecoxib were found to be effective in a dose-dependent inhibition of platelet aggregation induced by co-addition of subthreshold concentration of AA and ADP (Figure 7A and B, respectively; Table 2). Our results confirmed that celecoxib inhibits COX and Ca2+ influx by modulating non-voltage activated calcium channels or ROCCs in platelets, which leads to inhibition of TXA2 formation and its predominant effects on aggregation (Figure 2). Results clearly point the involvement of two different edges in the mechanism of synergism of platelet aggregation; one is COX cascade and the other is ROCCs which are shared by both pairs of agonists; AA plus ADP and AA plus 5-HT, through which subthreshold concentration of these agonists exerts synergism in platelet aggregation by synthesizing TXA2.
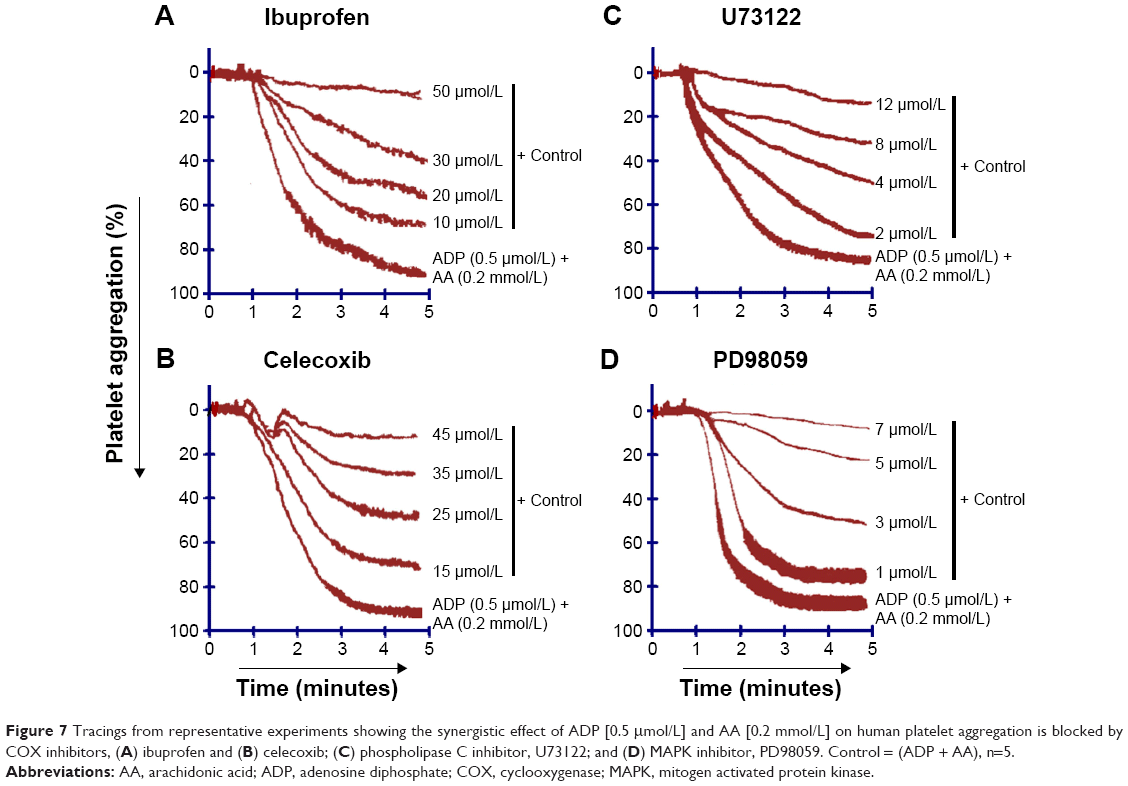 | Figure 7 Tracings from representative experiments showing the synergistic effect of ADP [0.5 μmol/L] and AA [0.2 mmol/L] on human platelet aggregation is blocked by COX inhibitors, (A) ibuprofen and (B) celecoxib; (C) phospholipase C inhibitor, U73122; and (D) MAPK inhibitor, PD98059. Control = (ADP + AA), n=5. |
As there are other pathways involved in platelet aggregation, we further elaborated our study using specific and non-specific 5-HT2 receptor blockers to find out whether the synergism of AA and 5-HT is inhibited by receptor blockers. It is well known that in the presence of low concentrations of TXA2, 5-HT released from platelets potentiates the platelet response to aggregation and this effect is mediated by 5-HT2A receptors.25,26 Previous finding by De Clerck et al27 showed that increased platelet aggregation induced by threshold concentration of 5-HT with ADP, collagen, nor-epinephrine, or epinephrine was decreased by ketanserin causing an irreversible aggregation in human platelets. Ogawa et al28 reported that in cat platelets low concentration of 5-HT with U46619 (a TXA2 analog) caused full platelet aggregation and this effect was completely blocked by ketanserin in a concentration-dependent manner indicating the involvement of 5-HT2 and TXA2 receptors in the synergism of 5-HT and U46619. In another study, Saeed et al20 used cyproheptadine and SQ29548 (5-HT2 and TXA2 receptor blocker, respectively) and found dose-dependent inhibition of Gq linked 5-HT2 and TXA2 receptors in human platelets. In our study, we used ketanserin to investigate its anti-aggregatory effect on subthreshold concentration of 5-HT and AA (a TXA2 precursor) in human platelets. The results show that platelet 5-HT2A receptors are antagonized by ketanserin in a dose-dependent manner and it also inhibited TXA2 formation by regulating PKC and PLA2. Ketanserin inhibited the second phase of aggregation induced directly by 5-HT2A receptors and indirectly by TXA2 formation from AA in human platelets. Thus, previous finding supports our current results for 5-HT2 receptor blockers for cyproheptadine (Figure 5A) and ketanserin (Figure 5B) being involved in the inhibition of the amplified response of 5-HT and AA in a dose-dependent manner by blocking the GqRs signaling pathway and its intracellular products (Table 1).
It has been established that upon activation of GqRs, intracellular PLC is activated that catalyzes the conversion of phosphatidylinositol-4,5-bisphosphate into two second messengers, namely IP3 and DAG, which cause influx of Ca2+ concentration in the cell (Figure 2). The role of PLC in the downstream signaling pathways of synergism was elucidated in the presence of AA plus ADP and AA plus 5-HT along with a PLC inhibitor. Results revealed that pretreatment of PRP with U73122 potently inhibited platelet aggregation in a dose-dependent manner against synergism induced by the combined effect of AA plus 5-HT (Figure 4C) and AA plus ADP (Figure 7C). The IC50 values are shown in Tables 1 and 2, respectively. Earlier it was reported that low concentrations of U73122 inhibited the synergism induced by 5-HT with epinephrine and 5-HT with PAF in a dose-dependent manner.29,30 Hence, our results are in agreement with these observations regarding the synergism of AA with 5-HT and AA with ADP, and that PLC is an important signaling molecule involved in inducing platelet aggregation. Since, platelets downstream signaling networks involve second messenger formation followed by Ca2+-dependent activation of PKC, MAPKs, and tyrosine kinases (Figure 2).31–33 Therefore, we further investigated the involvement of MAPKs in the activation of PLA2 and COX pathway in platelet aggregation. It is known that 5-HT2A receptors are linked to these intracellular effector molecules. Activation of these receptors by 5-HT in human platelets stimulated phosphorylation of tyrosine, threonine, and MAPK, which is prerequisite in initiation of both tyrosine and MAPK cascade. Previously, Saeed et al20 used selective inhibitor of MAPK and tyrosine light chain kinase, PD98059 and Herbimycin, respectively, in the synergism of AA and 5-HT. PD98059 and Herbimycin independently inhibit phosphorylation/activation of MAPK and tyrosine amino acid in platelet signaling pathway. In our experiment, we used only MAPK inhibitor to see the inhibition of platelet aggregation induced by synergism of AA and 5-HT. Our results confirmed that in the synergism of AA plus 5-HT (Figure 4D; Table 1), and AA plus ADP (Figure 7D; Table 2), activation of these two kinases is interconnected to each other and that inhibition of MAPK by specific inhibitor, PD98059 also reduces phosphorylation of tyrosine and initiation of tyrosine kinase cascade in platelets in a dose-dependent manner.
In conclusion, the synergistic effect among various platelet agonists and activation of GqRs seems to derive from stimulation of PLC, MAPK, COX, and ROCC-dependent signaling pathways. Activation of platelets through these pathways potentiates the effects of aggregation, thereby altering cardiovascular physiology. Inhibition of COX by using selective and/or non-selective COX inhibitors blocks not only AA metabolism and TXA2 formation, but also their binding to GqRs. Additionally, celecoxib appeared to exert dual independent inhibitory effect by inhibiting its off-target ROCCs in platelets. Celecoxib inhibit calcium-dependent platelet aggregation by ROCCs and TXA2 formation, activation and recruitment of pro-inflammatory cytokines, and expression of COX-2 in damaged vessels by platelets. These protective effects may account for the lower risk of cardiovascular events in patients treated with celecoxib. The synergism of AA and 5-HT is also inhibited by selective 5-HT2A receptors blocker ketanserin, which potently inhibit release of serotonin, interaction with PKC and PLA2. It blocks conversion of AA and activation of Gq/TXA2 receptors in the synergism. Additionally, it also inhibits the 5-HT induced atherogenic effect, plaque rupture, and proliferation of vascular smooth muscle in platelets. In a study of stable-angina pectoris, ketanserin increased coronary collateral blood flow and decreased myocardial ischemia in patients with single-vessel disease.34 PD98059, a specific MAPK inhibitor blocks phosphorylation of tyrosine, which leads to inhibition of MAPK and tyrosine cascade in the synergism of various platelet agonists. Taken together, all these inhibitors have proved to provide protective effects by blocking TXA2 formation and GqRs in the pathophysiology of cardio and arterio-venous diseases induced by synergism by various platelet agonists. Our study further demonstrates that the molecular mechanisms in which COX-2, ROCCs, and 5-HT-dependent decrease in contraction of cardiomyocytes, impaired vascular integrity and high shear stress, exposure of subendothelial cells, and release of pro-inflammatory cytokines may accelerate progression of peripheral vascular diseases, ischemia, and atherosclerosis.
Acknowledgments
The authors are very grateful to Sheikh Arshad Saeed (deceased November 2009), Karachi, for mentoring, provision of laboratory capacity and funds, and for long-standing excellent collaboration. Also, the authors like to cordially acknowledge Ms Fozia S Khan for her continuous support and guidance during this work. The financial support provided in form of Grants by College of Medicine and Health Sciences, United Arab Emirates University, and by Panjwani Center for Molecular Medicine and Drug Research (PCMD), University of Karachi, Pakistan, is gratefully acknowledged.
Disclosure
The authors report no conflicts of interest in this work.
References
Aaron JM. The role of lipids in platelet function: with particular references to the arachidonic acid pathway. J Lipid Res. 1978;19:793–826. | ||
Alan RB. Arachidonic acid as a bioactive molecule. J Clin Invest. 2001;107:1339–1344. | ||
Shad KF, Saeed SA. The metabolism of serotonin in neuronal cells in culture and platelets. Exp Brain Res. 2007;183:411–416. | ||
Aoki T, Nishimura M, Matsuoka T, et al. PGE2-EP2 signaling in endothelium is activated by haemodynamic stress and induced cerebral aneurysm through an amplifying loop via NFκB. Br J Pharmacol. 2011;163:1237–1249. | ||
Modi CM, Mody SK, Patel HB, Dudhatra GB, Kumar A, Avale M. Toxicopathological overview of analgesic and anti-inflammatory drugs in animals. J Appl Pharm Sci. 2012;2:149–157. | ||
Dorris SL, Peebles RS. PGI2 as a regulator of inflammatory diseases. Mediators Inflamm. 2012;2012:926968. | ||
Hechler B, Gachet C. P2 receptors and platelet function. Purinergic Signal. 2011;7:293–303. | ||
Rivera J, Luisa ML, Nunenz-Navarro L, Vicente V. Platelet receptors and signaling in the dynamics of thrombus formation. Haematologica. 2009;94:700–711. | ||
Offermanns S. Activation of platelet function through G protein-coupled receptors. Circ Res. 2006;99:1293–1304. | ||
Cimmino G, Golino P. Platelet biology and receptor pathways. J Cardiovasc Transl Res. 2013;6:299–309. | ||
Rasheed H, Trimizi AH, Salahuddin F, et al. Calcium signaling in human platelet aggregation mediated by platelet activating factor and calcium ionophore. J Biol Sci. 2004;4:117–121. | ||
Saeed SA, Rasheed H, Wahed FA, et al. Signaling mechanisms mediated by G-protein coupled receptors in human platelets. Acta Pharmacol Sin. 2004;25:887–892. | ||
Saeed SA, Ahmed N, Ahmed S. Dual inhibition of cyclooxygenase and lipoxygenase by human haptoglobin: its polymorphism and relation to hemoglobin binding. Biochem Biophys Res Commun. 2007;353:915–920. | ||
Born GVR. Quantitative investigations into the aggregation of blood platelets. J Physiol. 1962;162:67–68. | ||
Shah BH, Saeed SA. Phosphotidylinositol 3-kinase inhibitor, wortmannin, inhibits 5-hydroxytryptamine-mediated potentiation of platelet aggregation induced by epinephrine. Res Commun Mol Pathol Pharmacol. 1995;89:157–164. | ||
Saeed SA, Rasheed H, Kumar S, et al. Involvement of cyclooxygenase, phospholipase C and MAP kinase pathways in human platelet aggregation mediated by the synergistic interaction of platelet activating factor and arachidonic acid. Pak J Biol Sci. 2003;6:918–924. | ||
Francesconi M, Scapin M, Casonato A, Girolami A, Deana R. Adrenaline potentiates type 2B von Willebrand factor-induced activation of human platelets by enhancing both the formation and action of thromboxanes. Thromb Res. 2000;100:293–303. | ||
Doyle VM, Ruegg. Lack of evidence for voltage dependent calcium channels on platelets. Biochem Biophys Res Commun. 1985;127:161–167. | ||
Pannocchia A, Pralon N, Arduino C, Della-Dora N, Bazzan M, Schinco P. Absence of (-) [3H]desmethoxyverapamil binding sites on human platelets and lack of evidence for voltage-dependent calcium channels. Eur J Pharmacol. 1987;142:83–91. | ||
Saeed SA, Rasheed H, Gilani AH. Synergism interaction between arachidonic acid by 5-hydroxytryptamine in human platelet aggregation is mediated through multiple signaling pathways. Acta Pharmacol Sin. 2003;24:958–964. | ||
Zhang Y, Tao J, Huang H, Ding G, Cheng Y, Sun W. Effects of celecoxib on voltage-gated calcium channel currents in rat pheochromocytoma (PC12) cells. Pharmacol Res. 2007;56:267–274. | ||
Brueggemann LI, Mackie AR, Mani KB, Cribbs LL, Byron KL. Differential effects of selective cyclooxygenase-2 inhibitors on vascular smooth muscle ion channels may account for differences in cardiovascular risk profiles. Mol Pharmacol. 2009;76:1053–1061. | ||
Frolov RV, Singh S. Celecoxib and ion channels: a story of unexpected discoveries. Eur J Pharmacol. 2014;730:61–71. | ||
Frolov RV, Singh S. Evidence of more ion channels inhibited by celecoxib: Kv 1.3 and L-type Ca2+ channels. BMC Res Notes. 2015;8:62–65. | ||
Li N, Wallén NH, Ladjevardi M, Hjemdahl P. Effects of serotonin on platelet activation in whole blood. Blood Coagul Fibrinolysis. 1997;8:517–523. | ||
Martin GR. Vascular receptors for 5-hydroxytryptamine: distribution, function and classification. Pharmacol Ther. 1994;62:283–324. | ||
De Clerck F, David JL, Janssen PA. Inhibition of 5-hydroxytryptamine-induced and-amplified human platelet aggregation by ketanserin (R 41468), a selective 5-HT2-receptor antagonist. Agents Action. 1982;12:388–397. | ||
Ogawa T, Sugidachi A, Asai F, Koike H. Involvement of platelet-derived 5-hydroxytryptamine in thromboxane A2-induced aggregation in cat platelets. Blood Coagul Fibrinolysis. 1998;3:233–240. | ||
Shah BH, Siddique A, Qureshi KA, et al. Co-activation of Gi and Gq proteins exerts synergistic effect on human platelet aggregation through activation of phospholipase C and Ca2+ signaling pathways. Exp Mol Med. 1999;31:42–46. | ||
Shah BH, Rasheed H, Rahman IH, et al. Molecular mechanisms involved in human platelet aggregation by synergistic interaction of platelet-activating factor and 5-hydroxytryptamine. Exp Mol Med. 2001;33:226–233. | ||
Neil GA, Maller JL, Tonks NK, Sturgill TW. Requirement for integration of signals from two distinct phosphorylation pathways for activation of MAP kinase. Nature. 1990;343:651–653. | ||
Tournois C, Mutel V, Manivet P, Launay JM, Kellermann O. Cross-talk between 5-hydroxytryptamine receptors in a serotonergic cell line. Involvement of arachidonic acid metabolism. J Biol Chem. 1998;273:17498–17503. | ||
Saeed SA, Rasheed H. Calcium-dependent synergistic interaction of platelet activating factor and epinephrine in human platelet aggregation. Acta Pharmacol Sin. 2003;24:31–36. | ||
Kyriakides ZS, Sbarouni E, Nikolaou N, Antoniadis A, Kremastinos DT. Intracoronary ketanserin augments coronary collateral blood flow and decreases myocardial ischemia during ballon angioplasty. Cardiovasc Drugs Ther. 1999;13:415–442. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.